An Overview on Antimalarial Peptides: Natural Sources, Synthetic Methodology and Biological Properties


Abstract
:1. Introduction
2. Isolation of Antimalarial Peptide
3. Synthesis of Antimalarial Peptides
3.1. Cyclopeptides
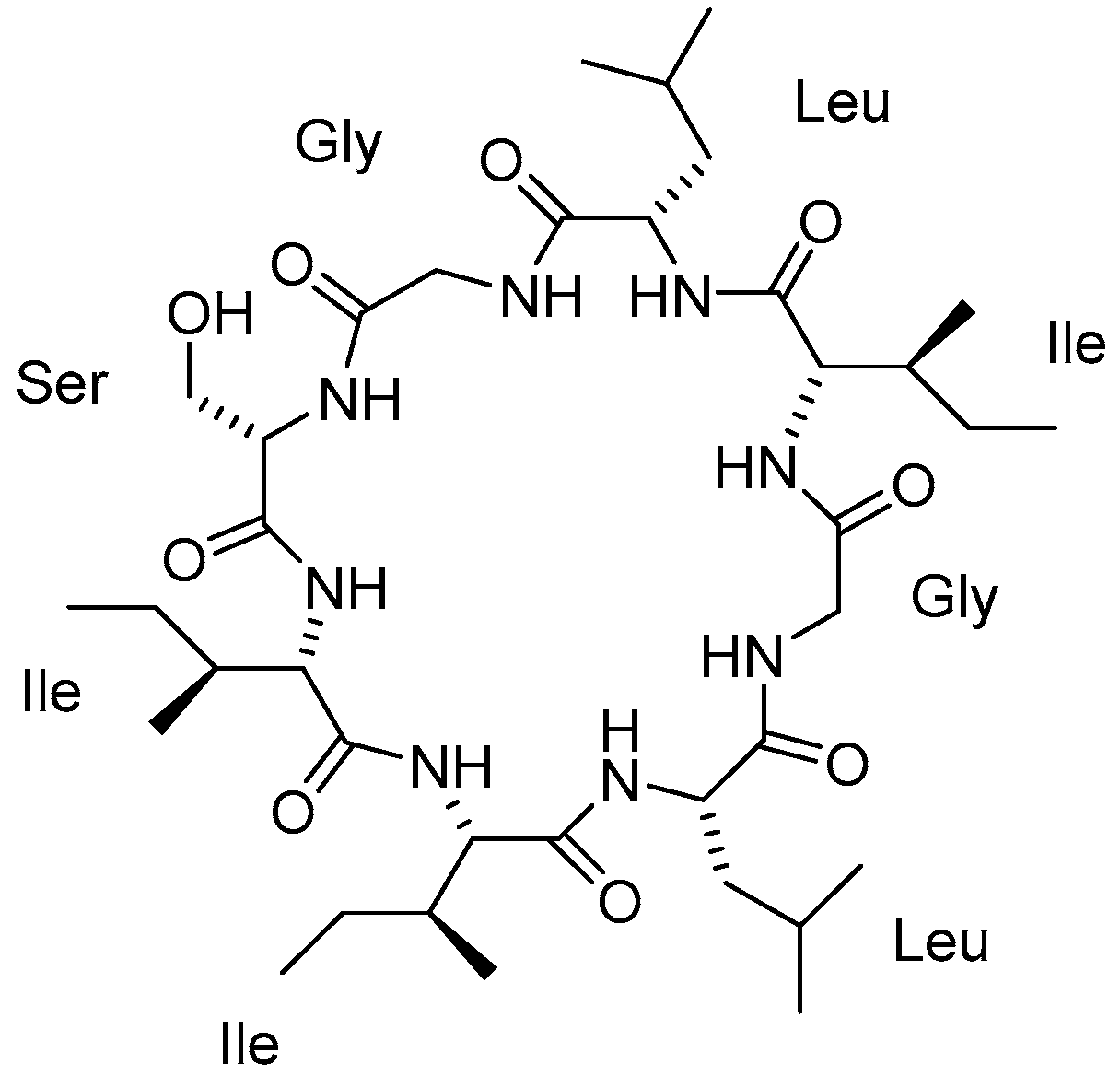
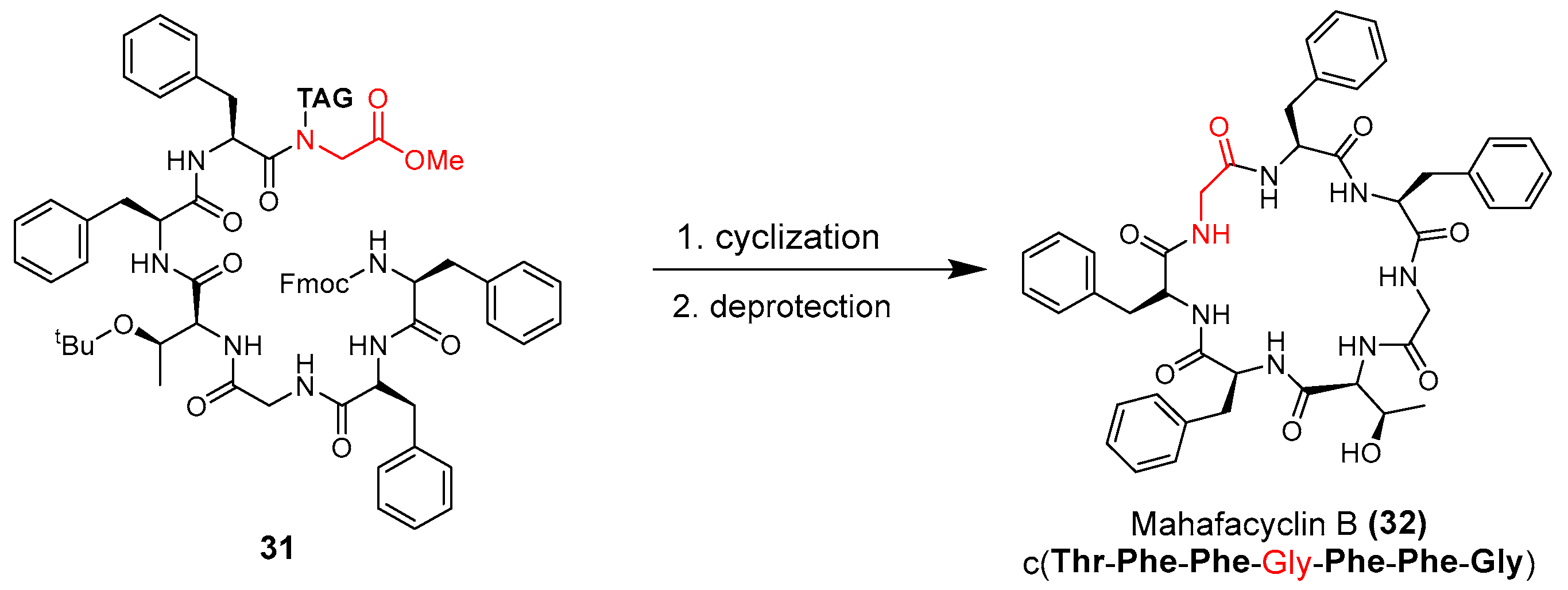
3.1.1. Mahafacyclin B
3.1.2. Aerucylamide B
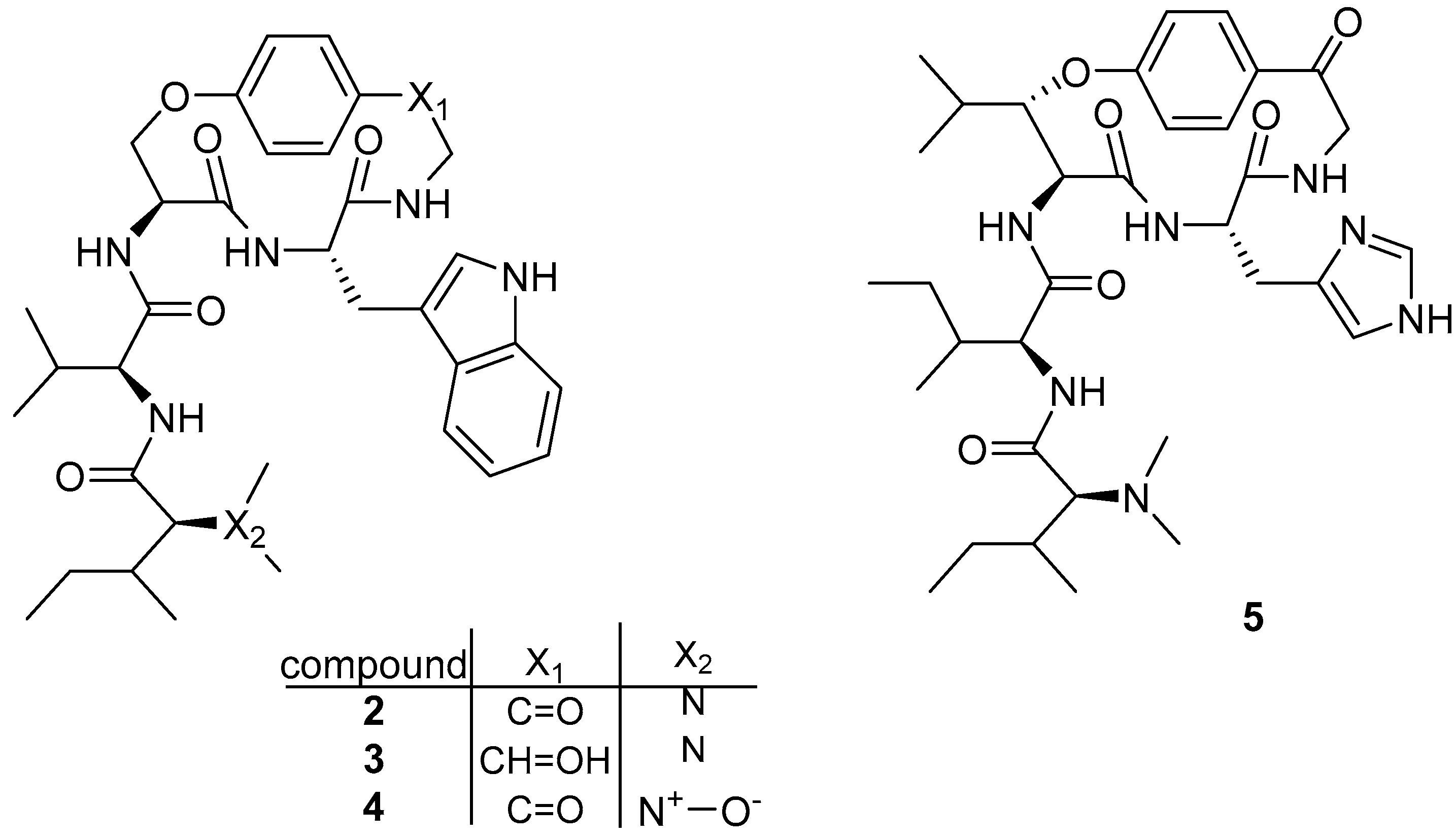
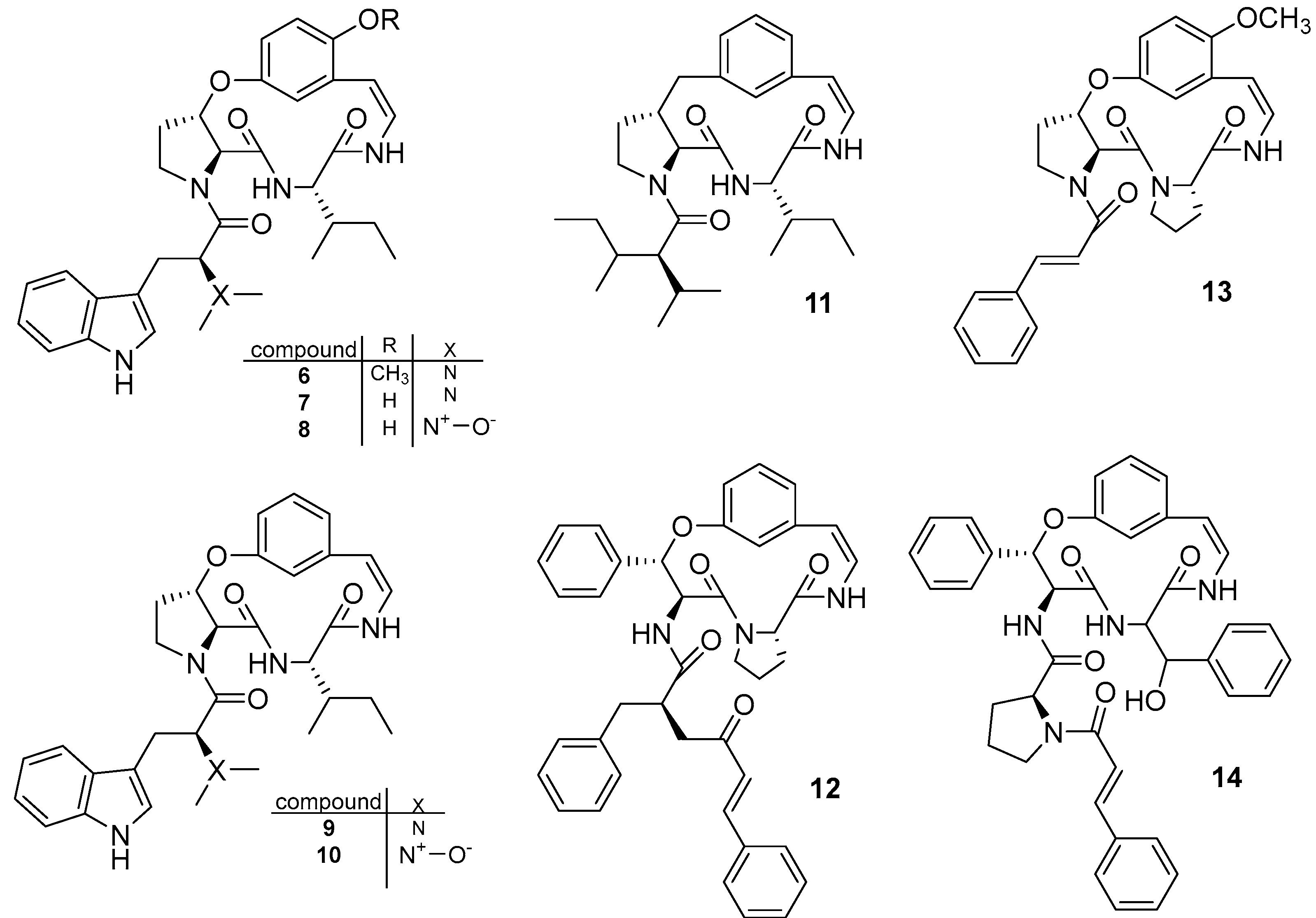
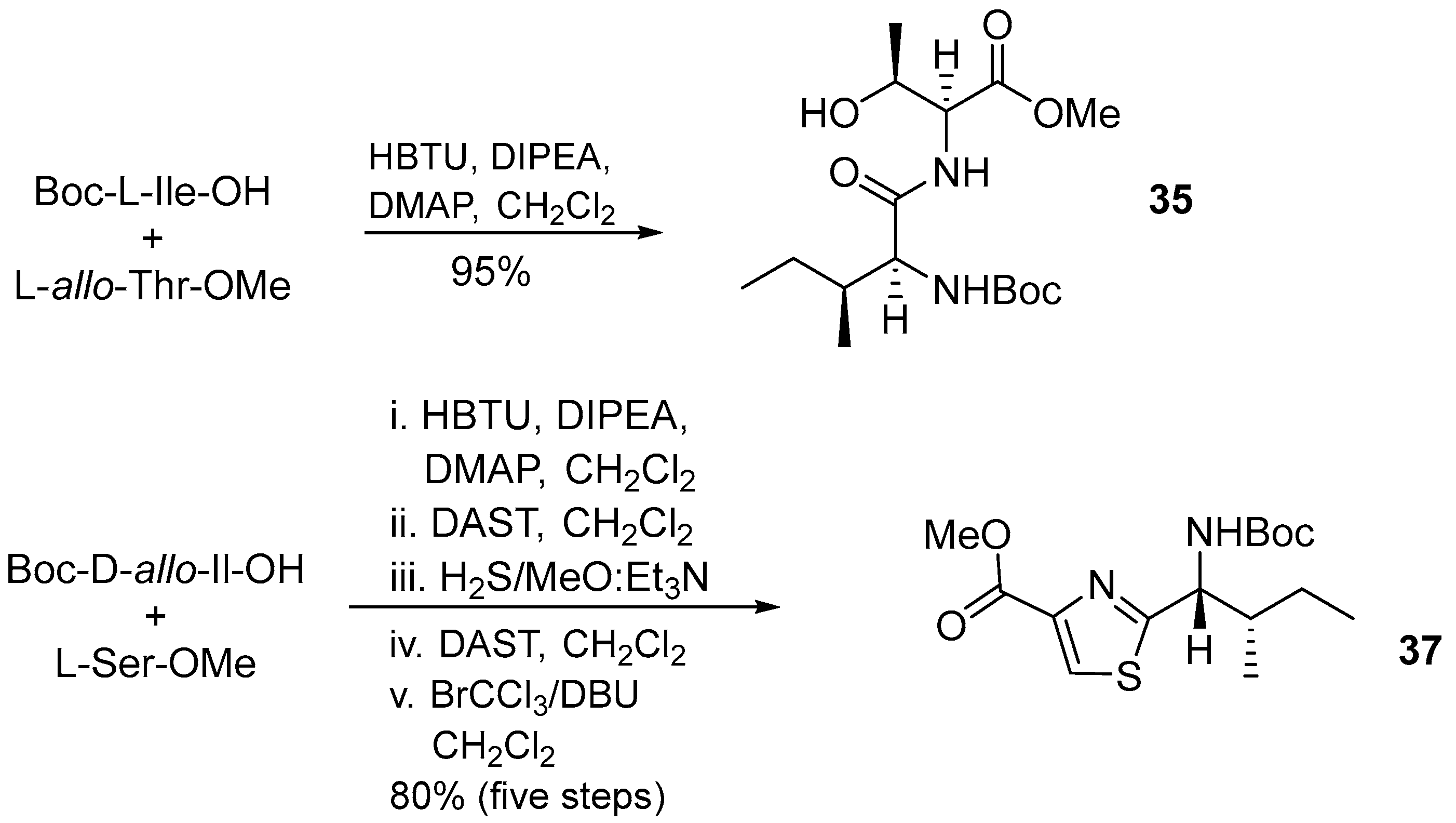
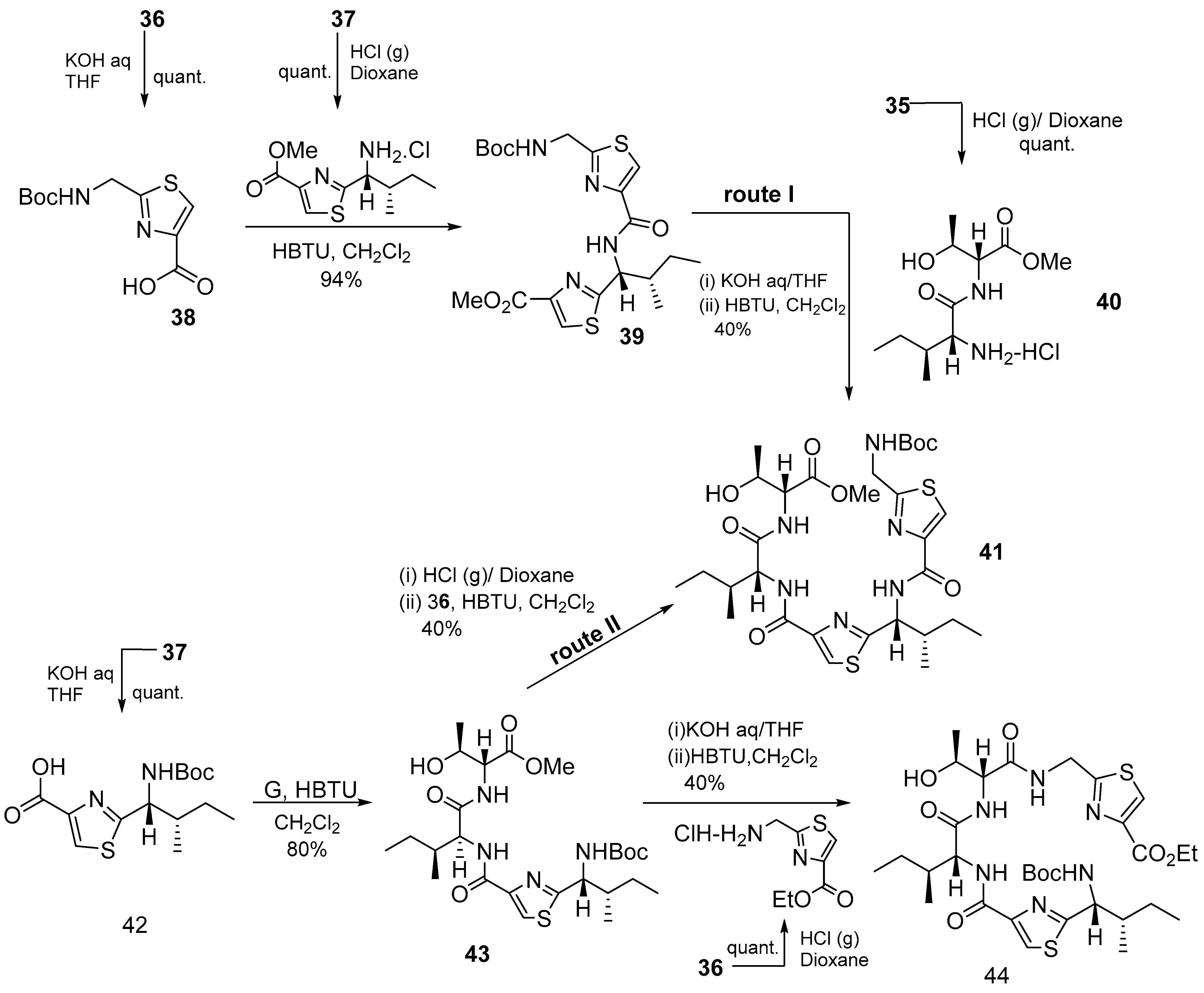
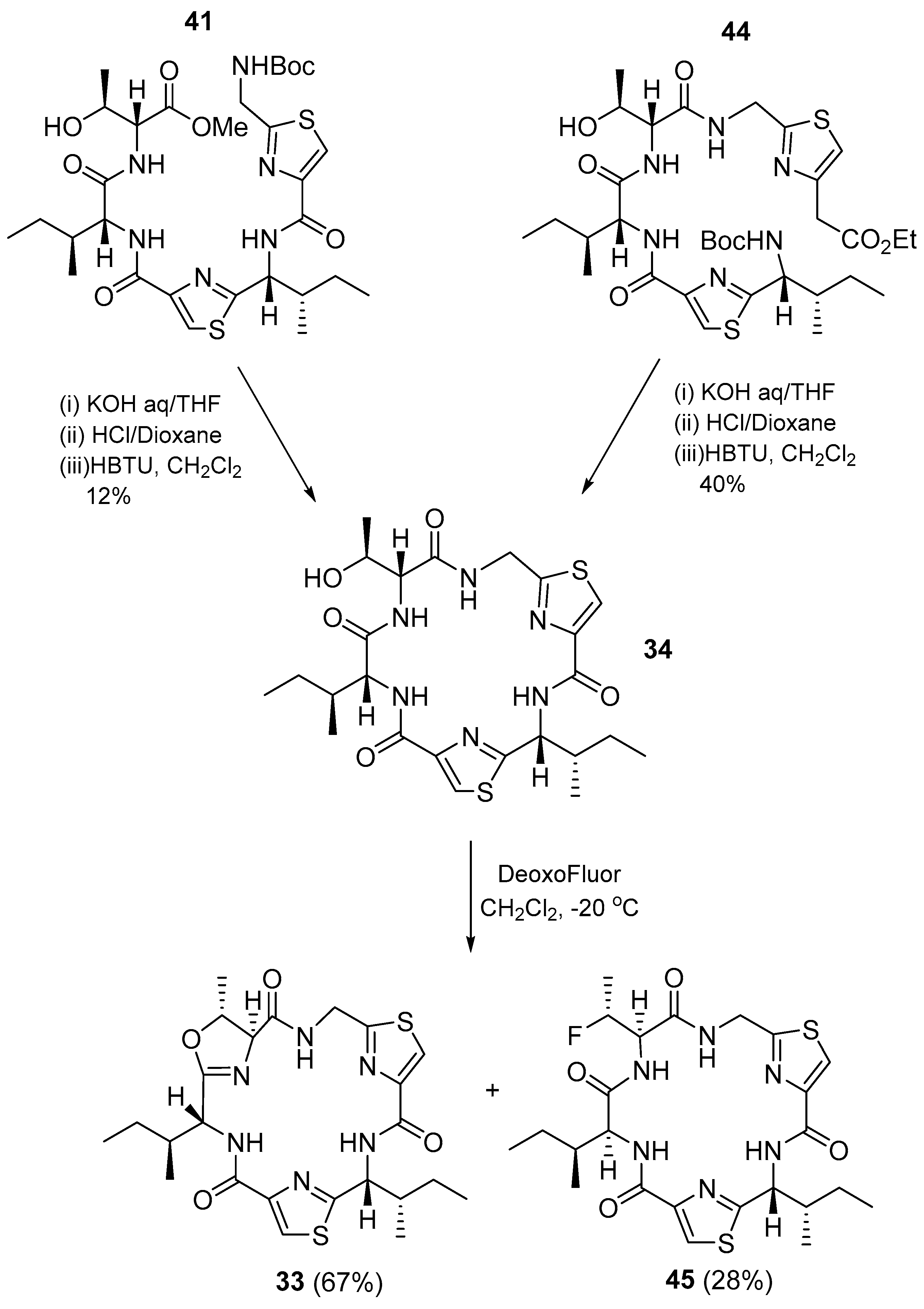
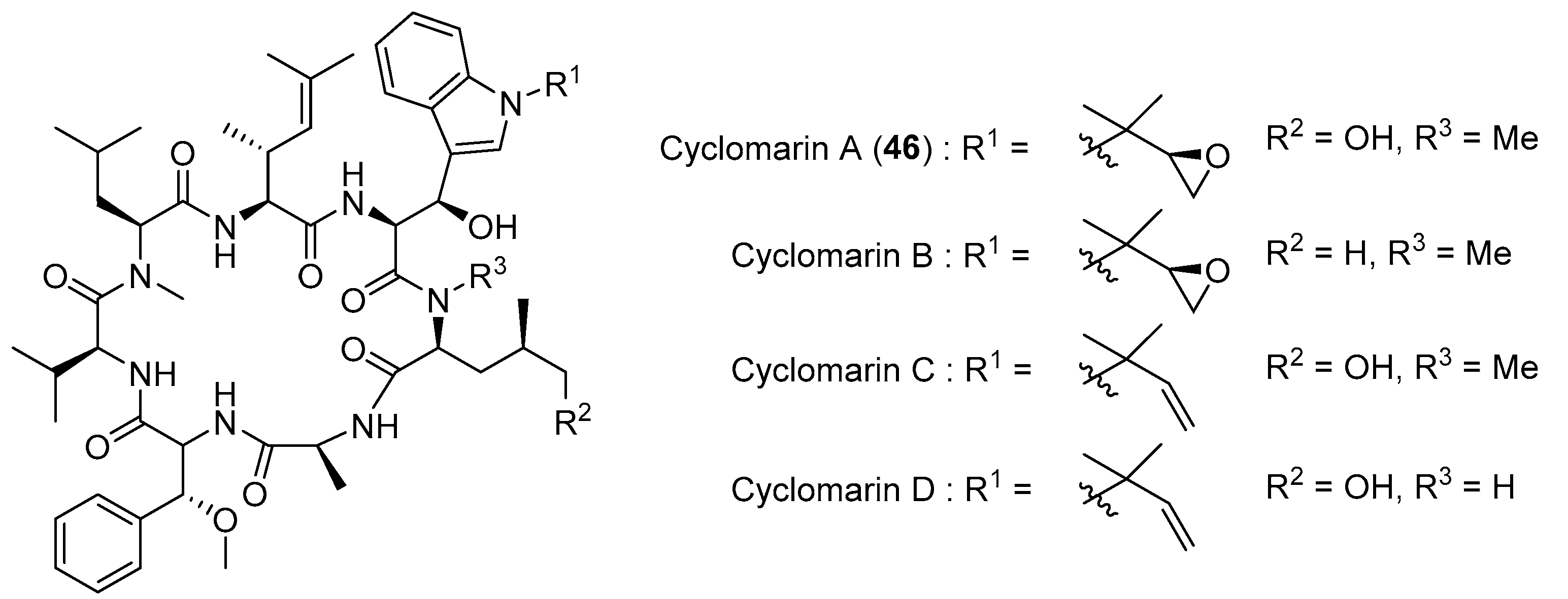
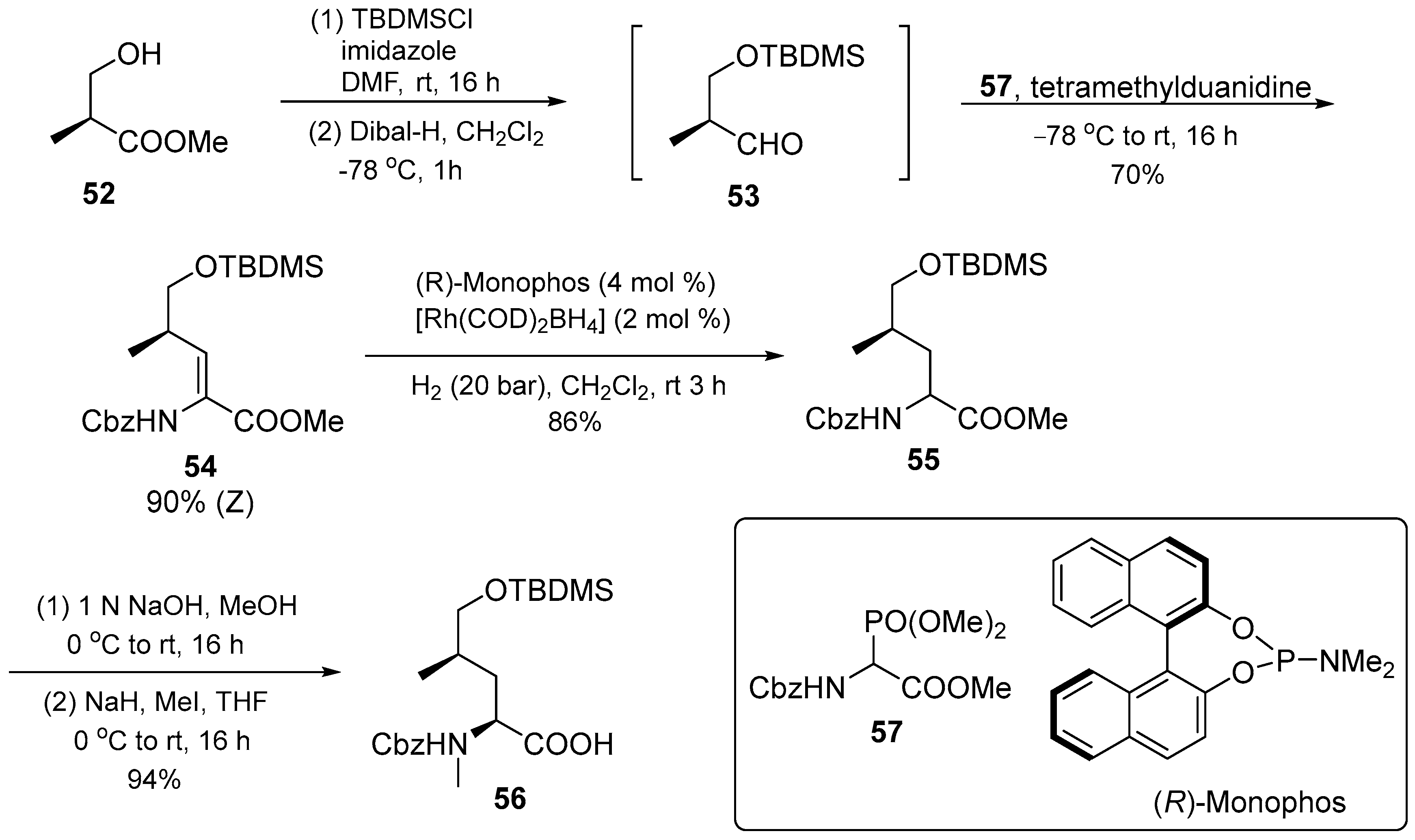
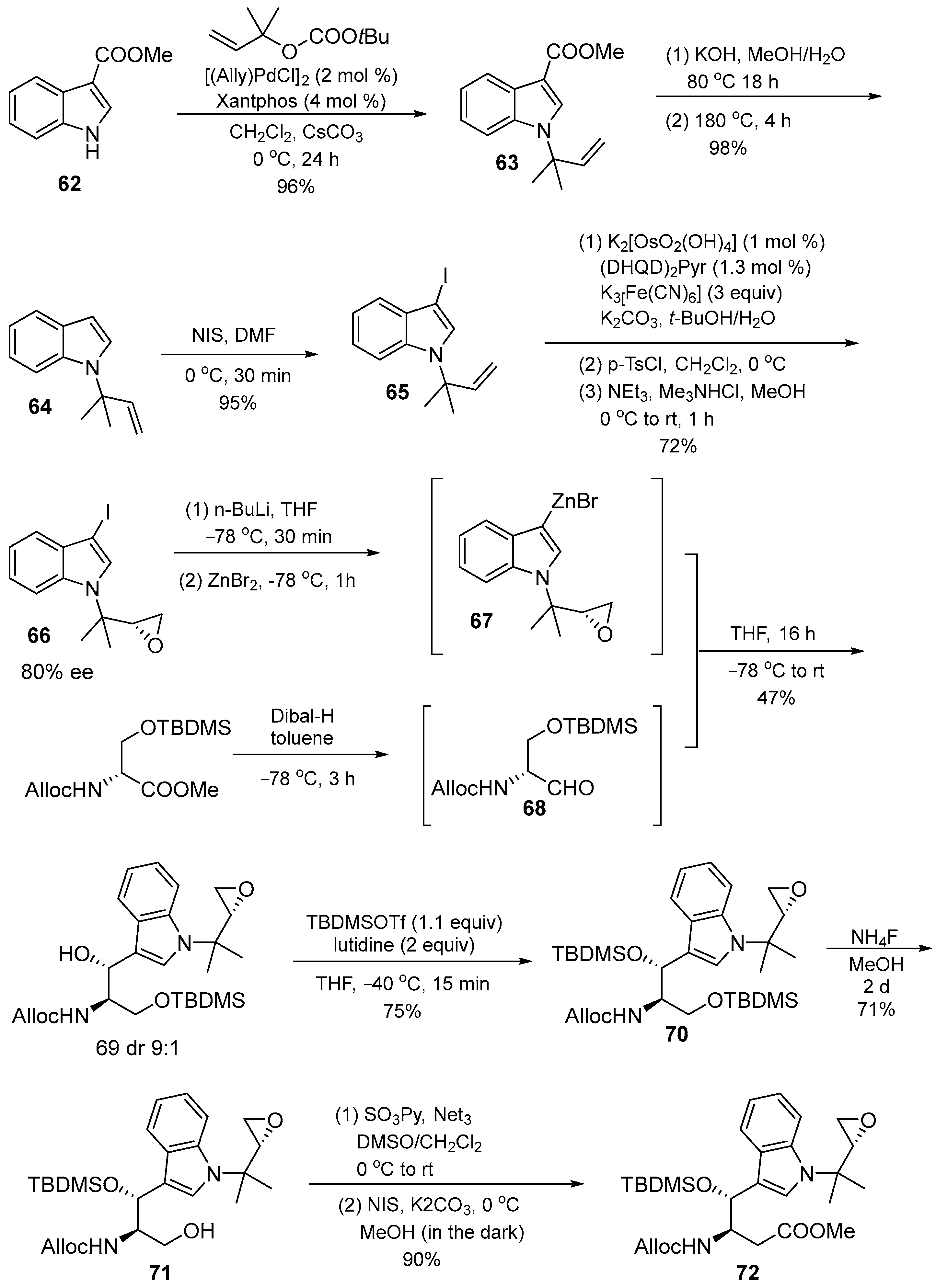
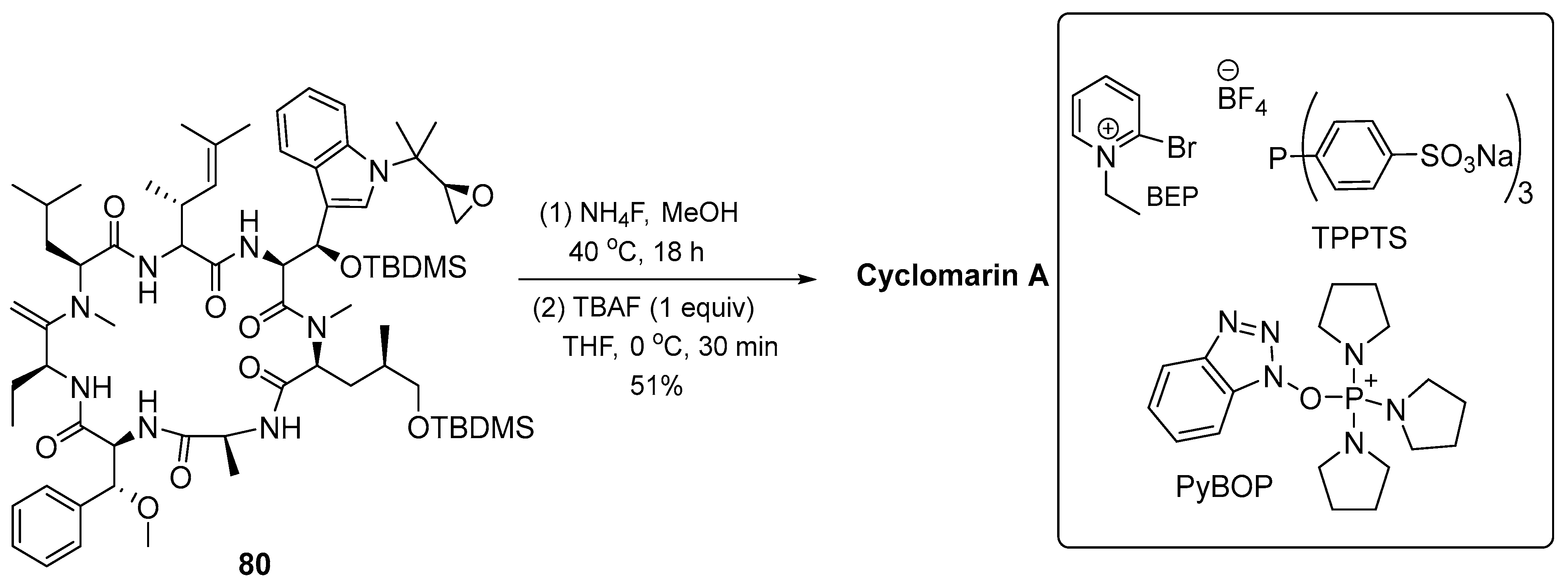
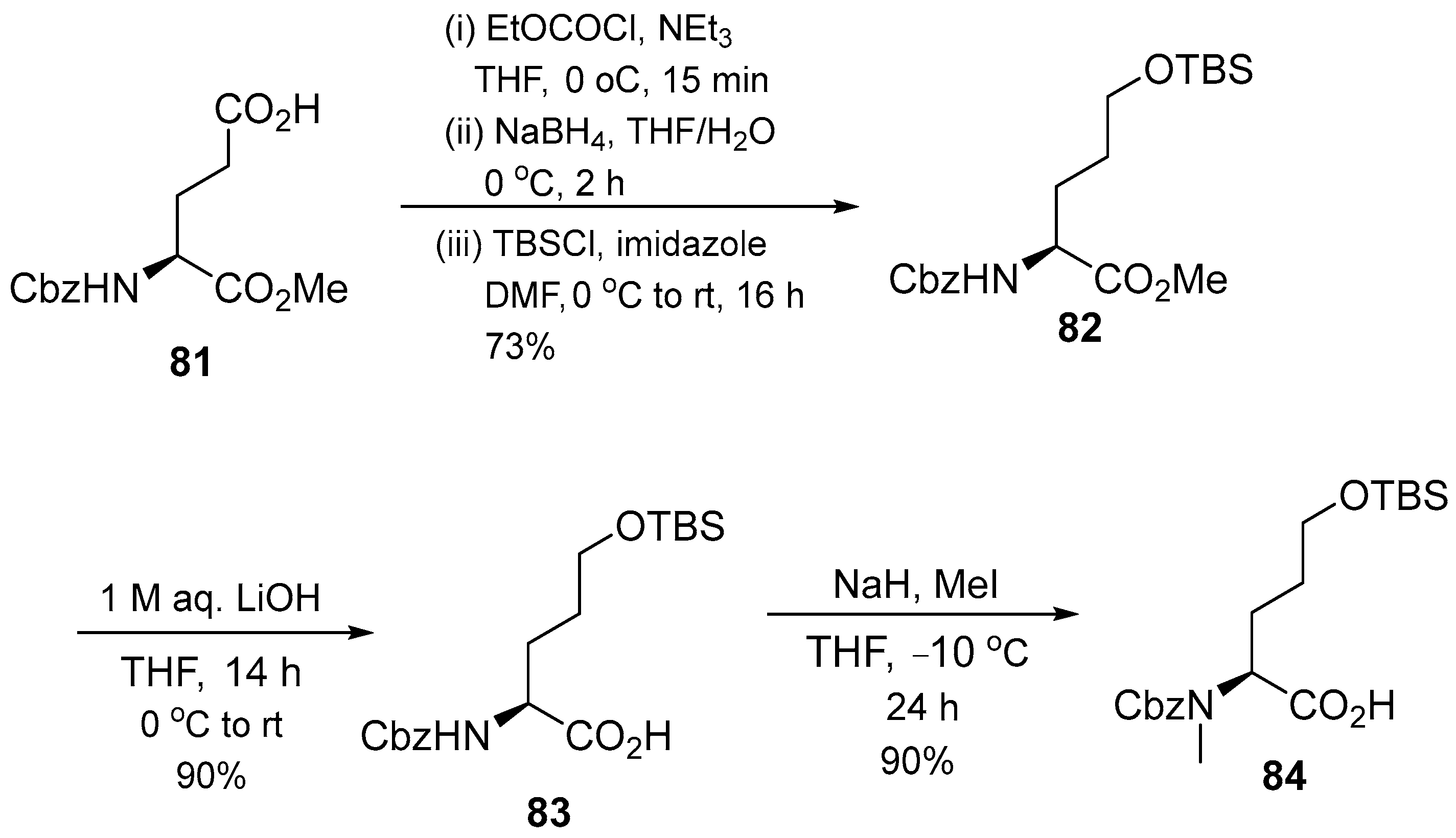
3.1.3. Cyclomarin
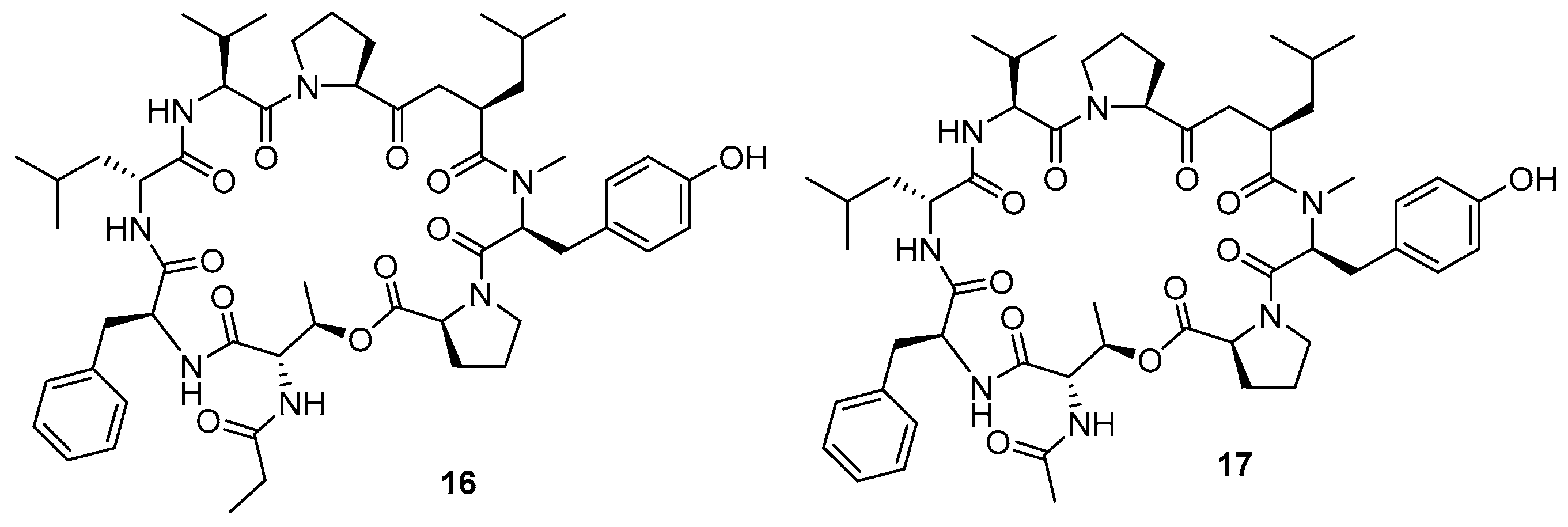
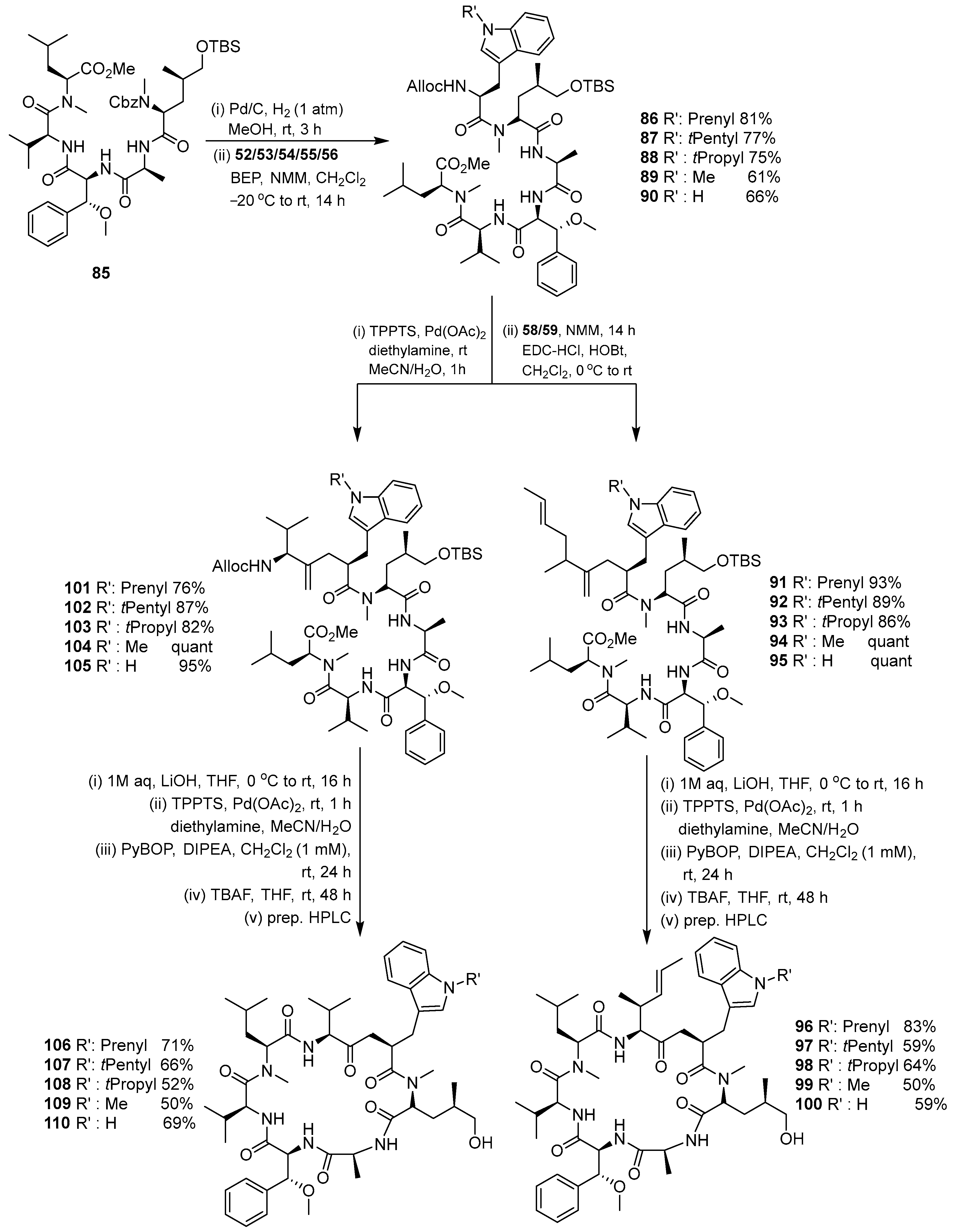
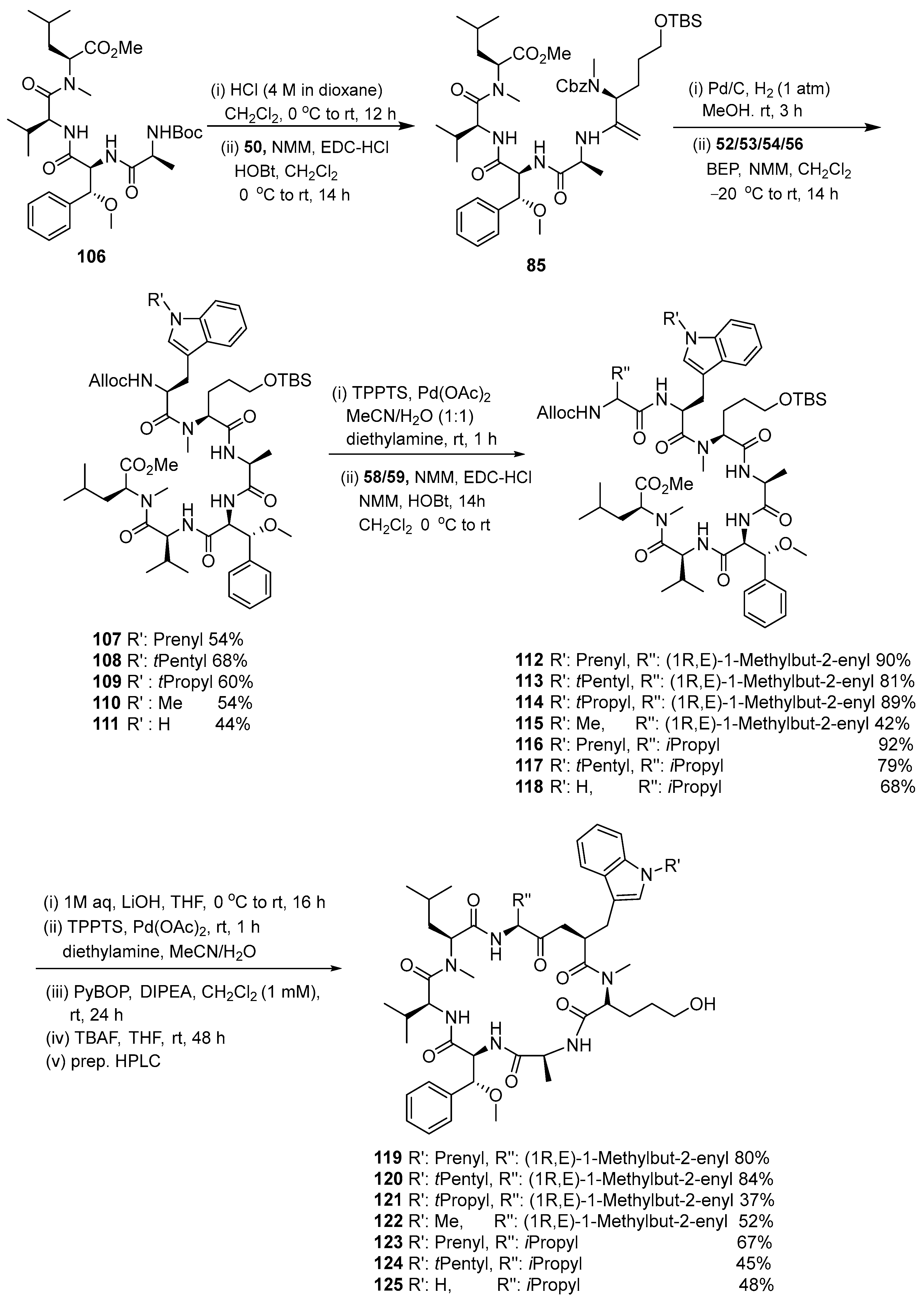
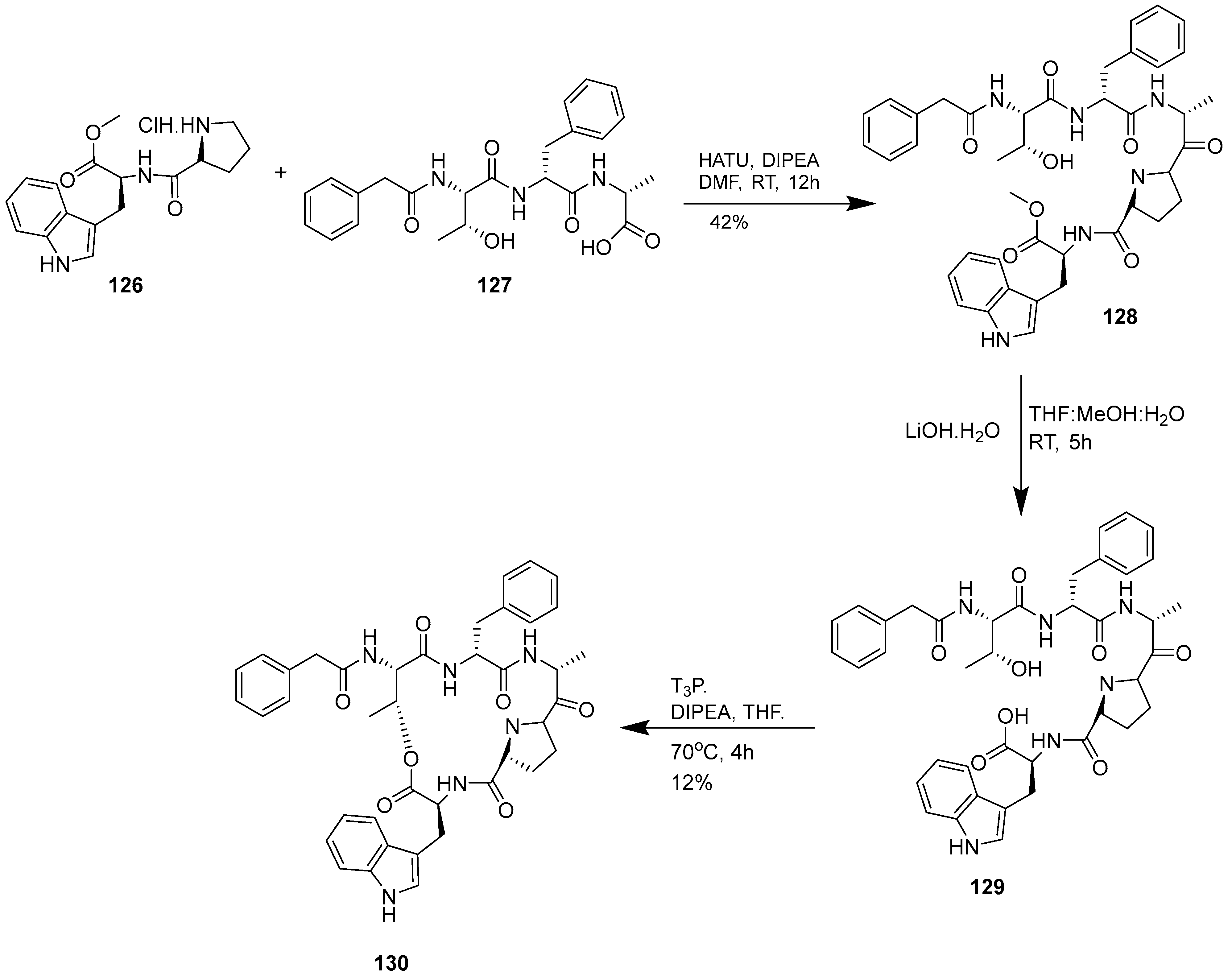
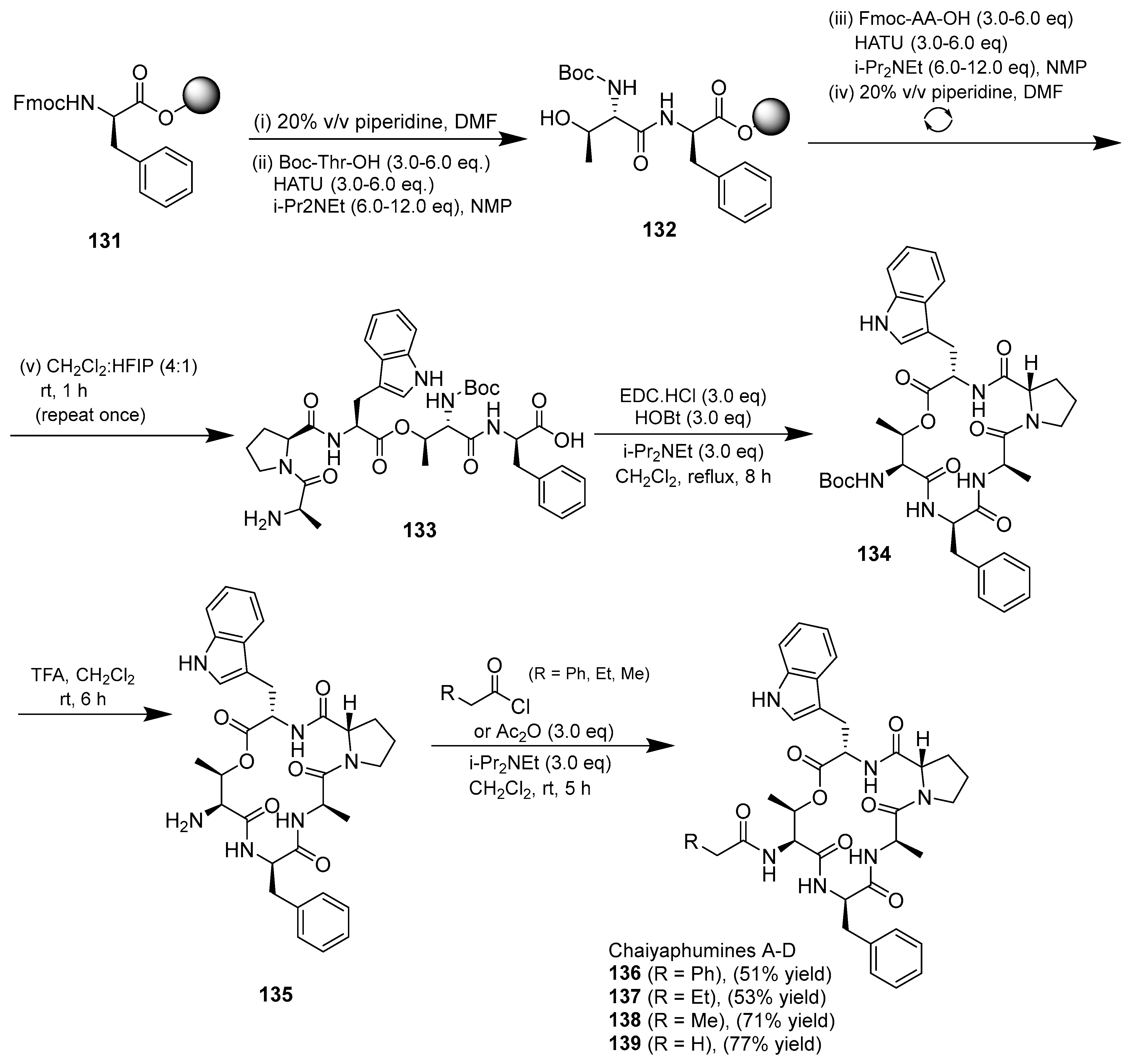
3.1.4. Chaiyaphumine
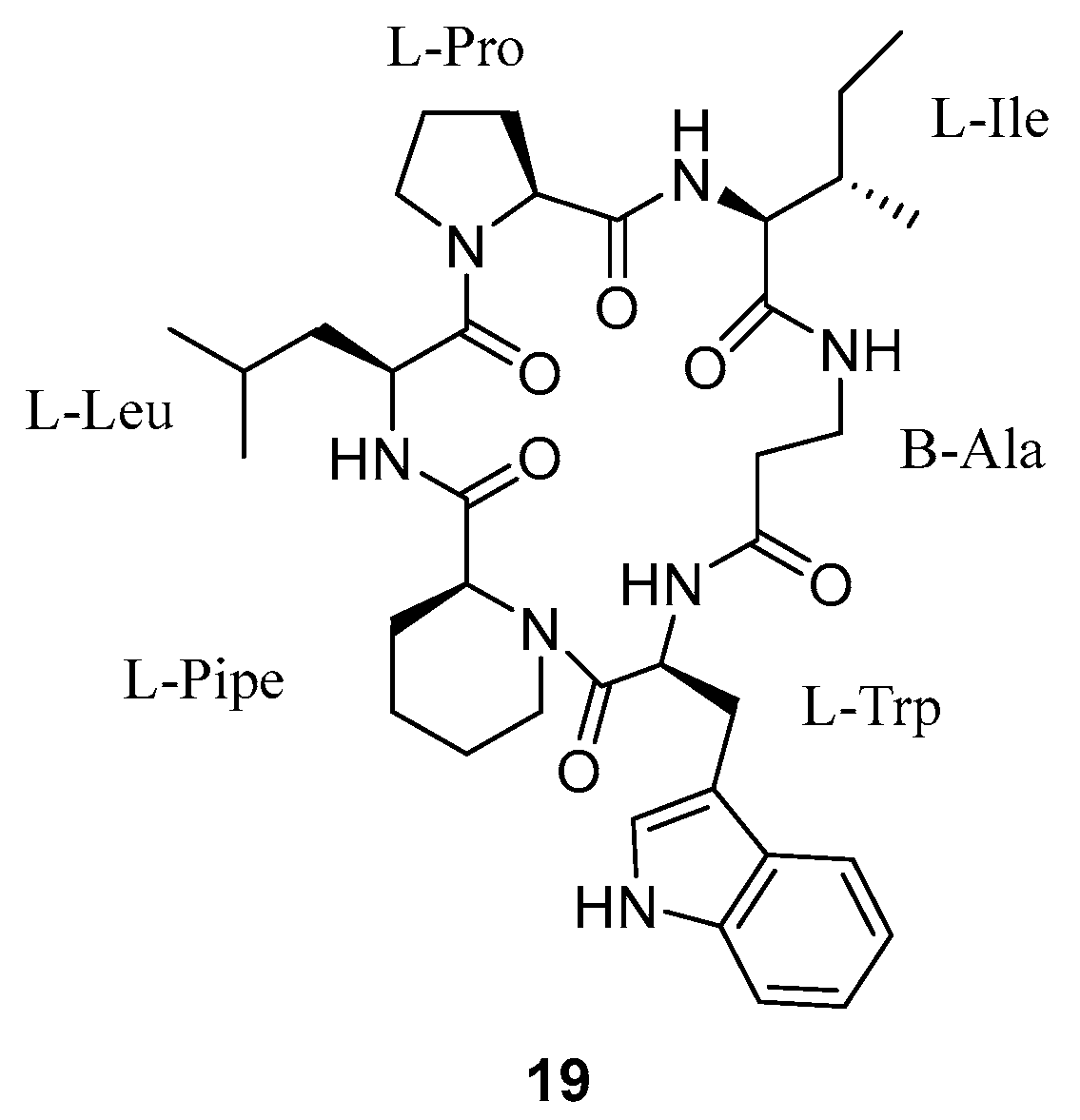
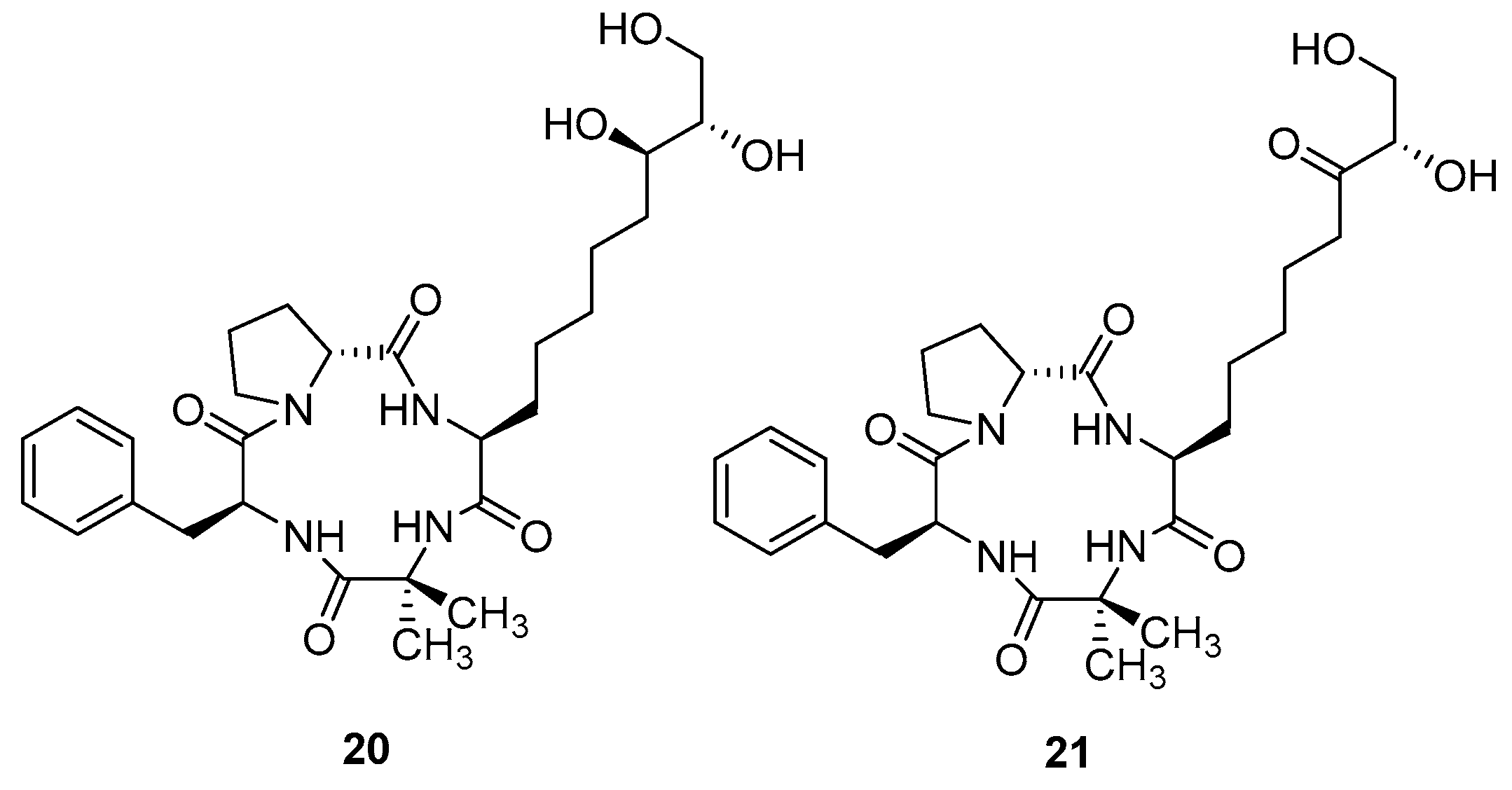
3.1.5. Cyclopeptides Anolgs Prepared by Macrocyclization
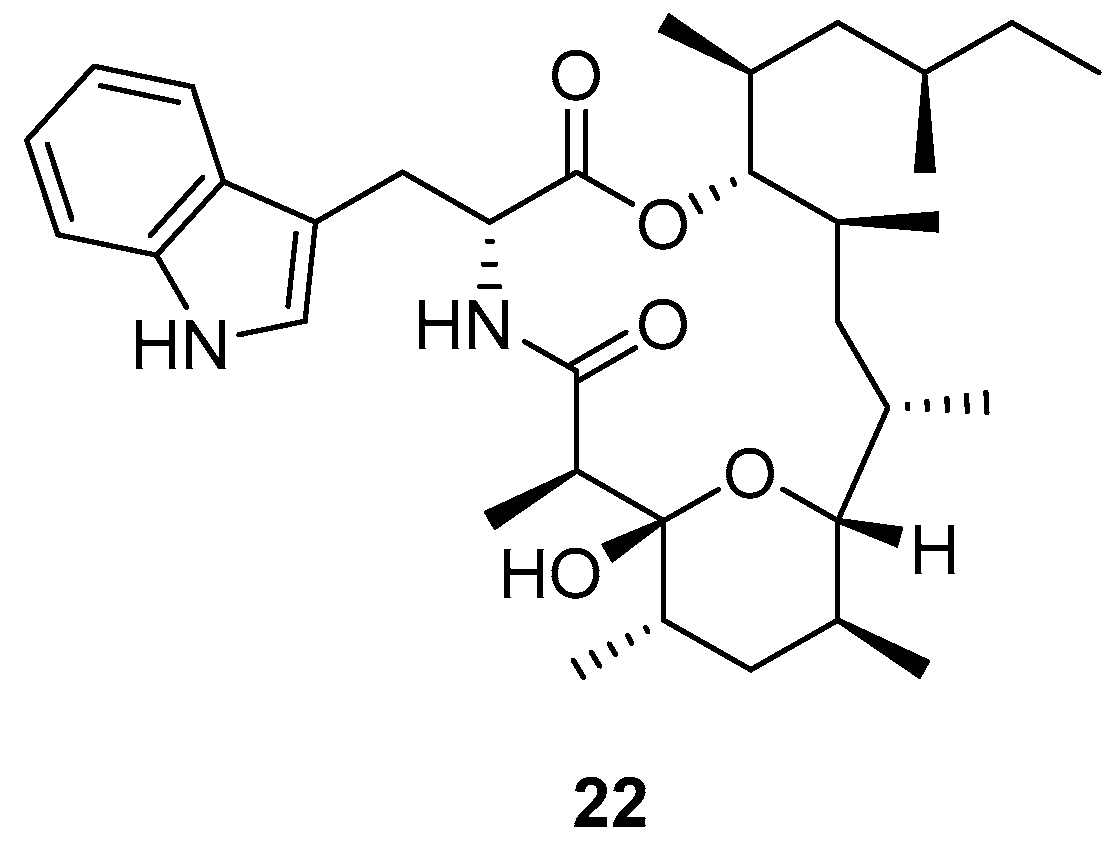
3.1.6. Hirsutellide A and Its Analogues
3.1.7. Macrocyclization Strategies
3.2. Linear Peptides
3.2.1. Falcitidin
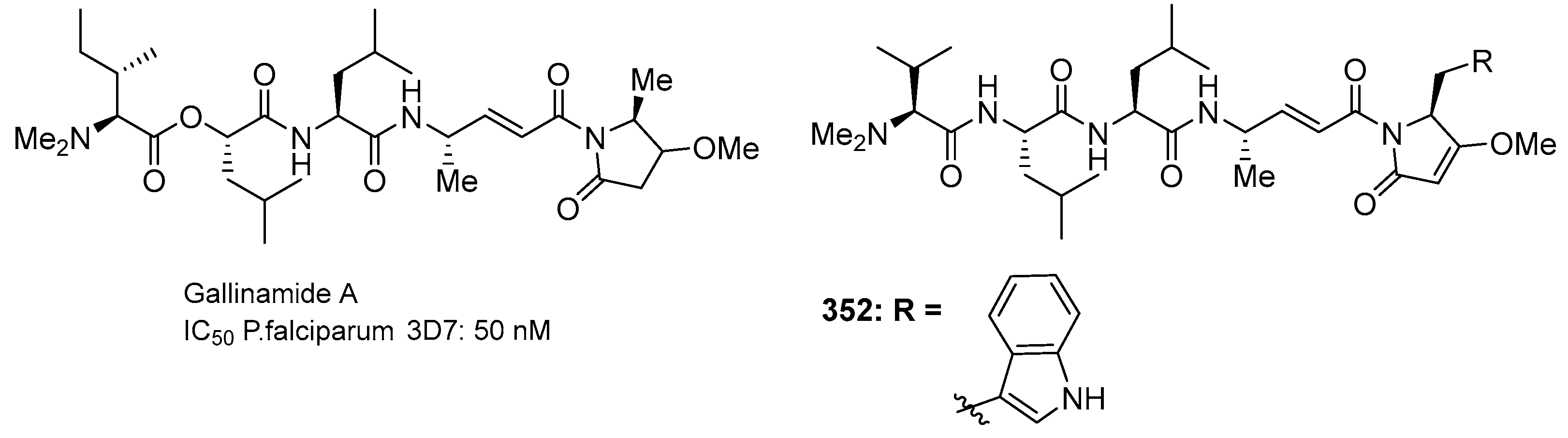
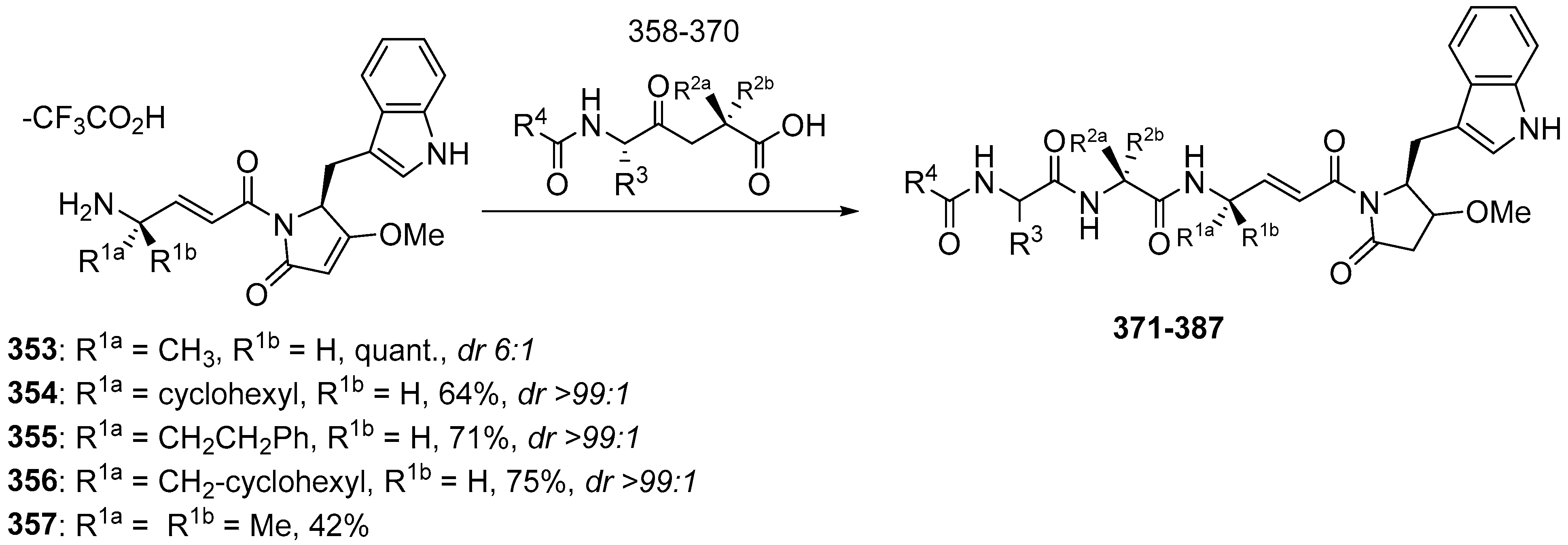
3.2.2. Gallinamide
3.2.3. PfSERA5 Analogs
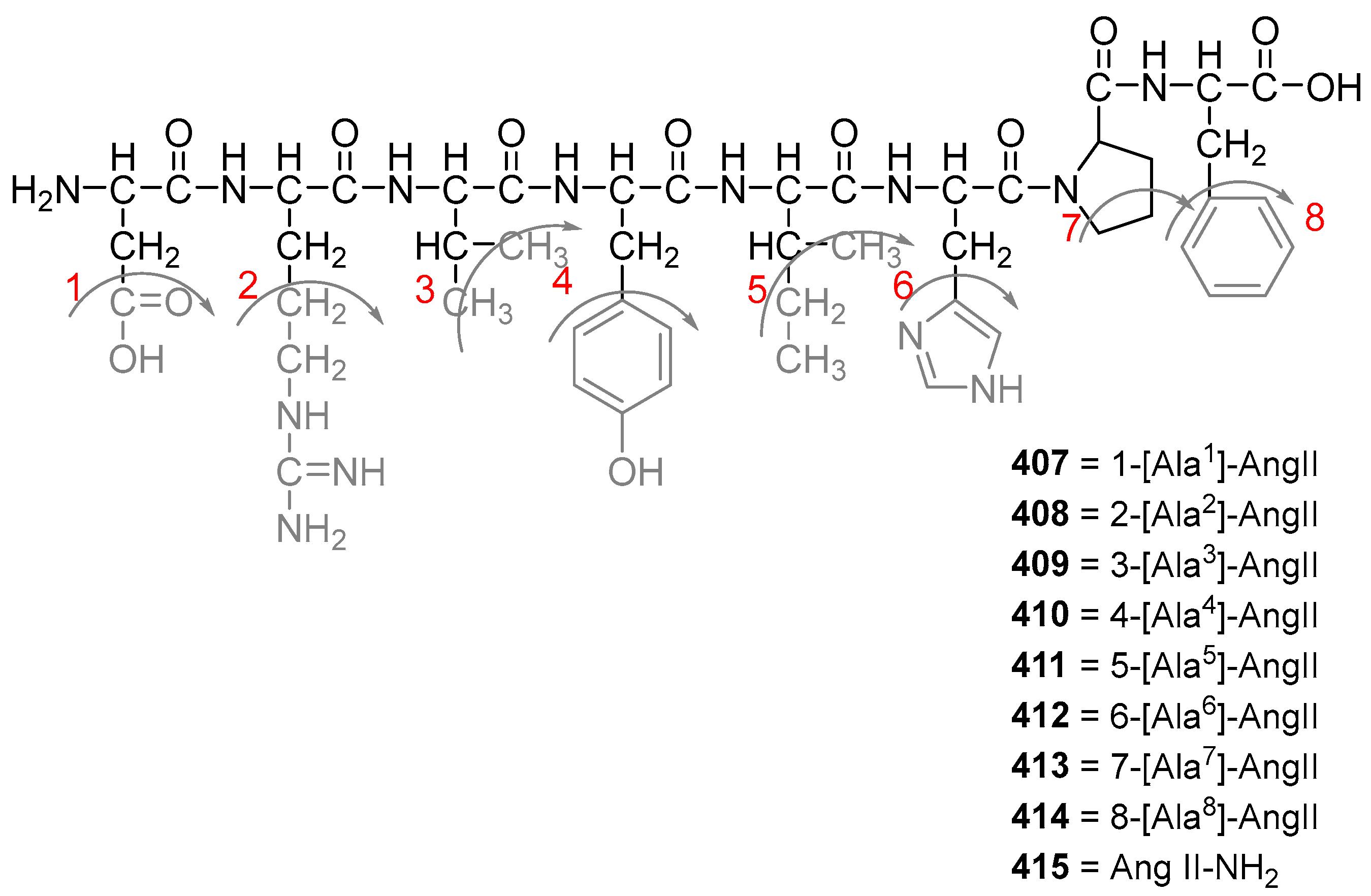
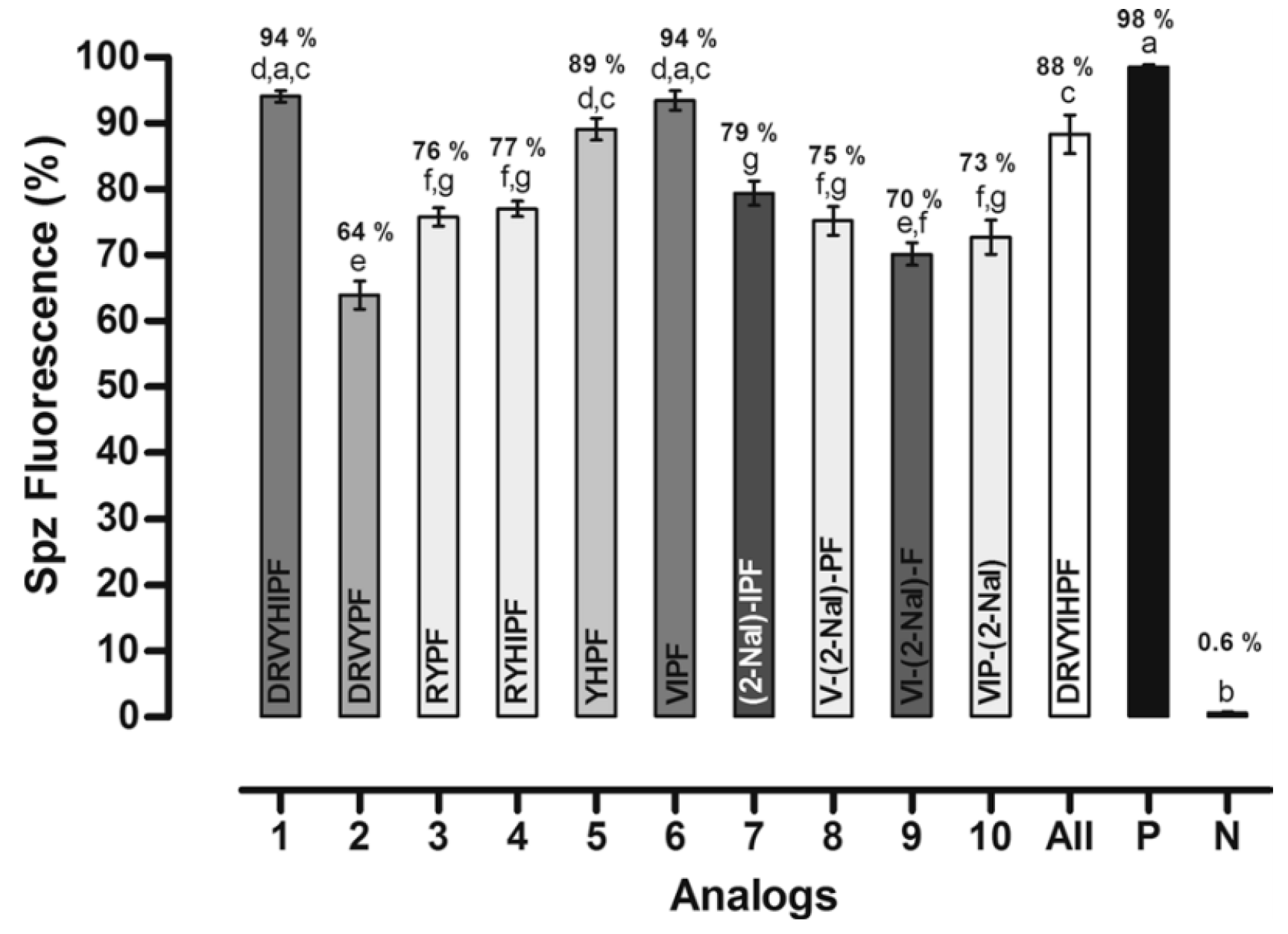
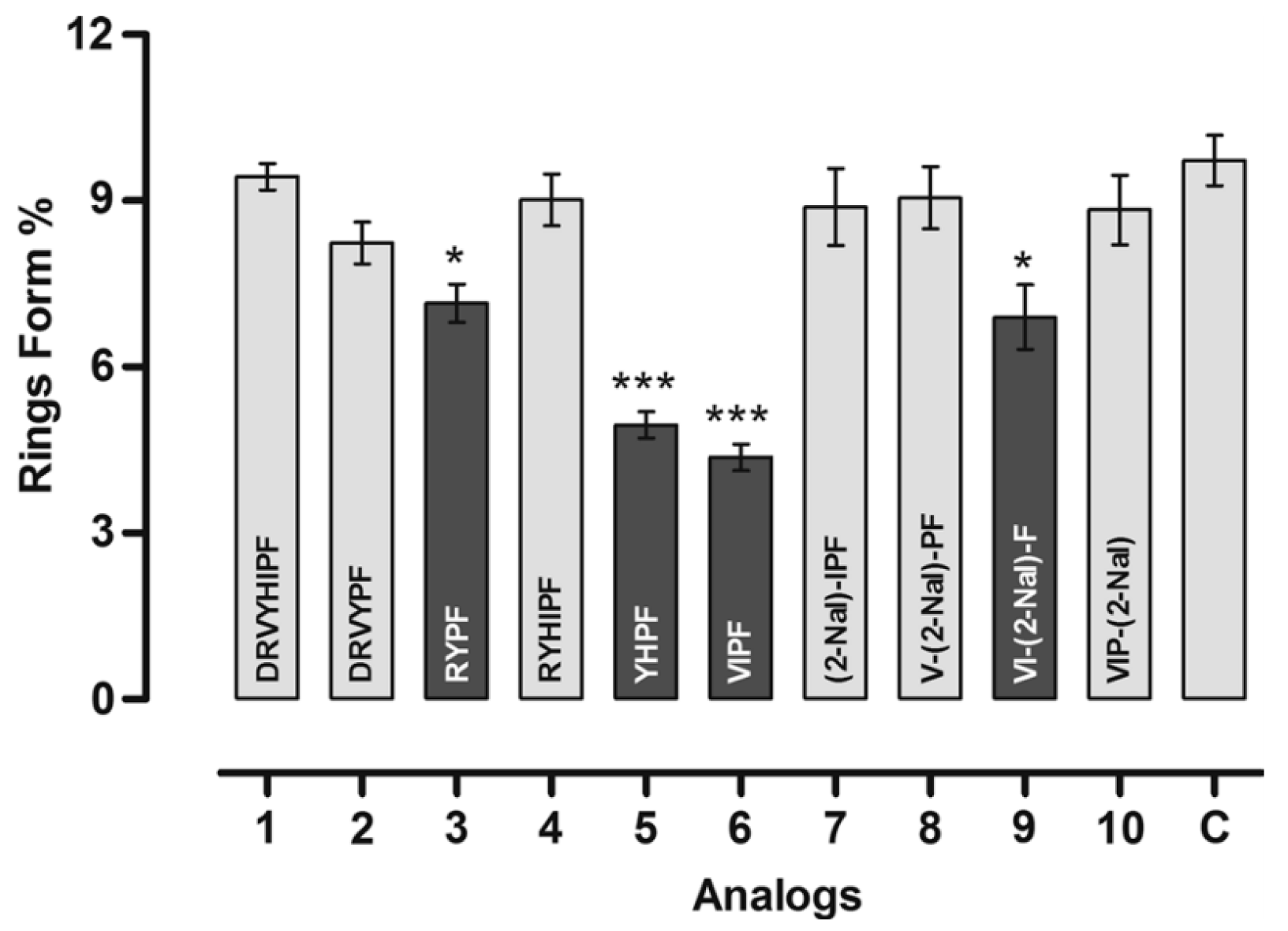
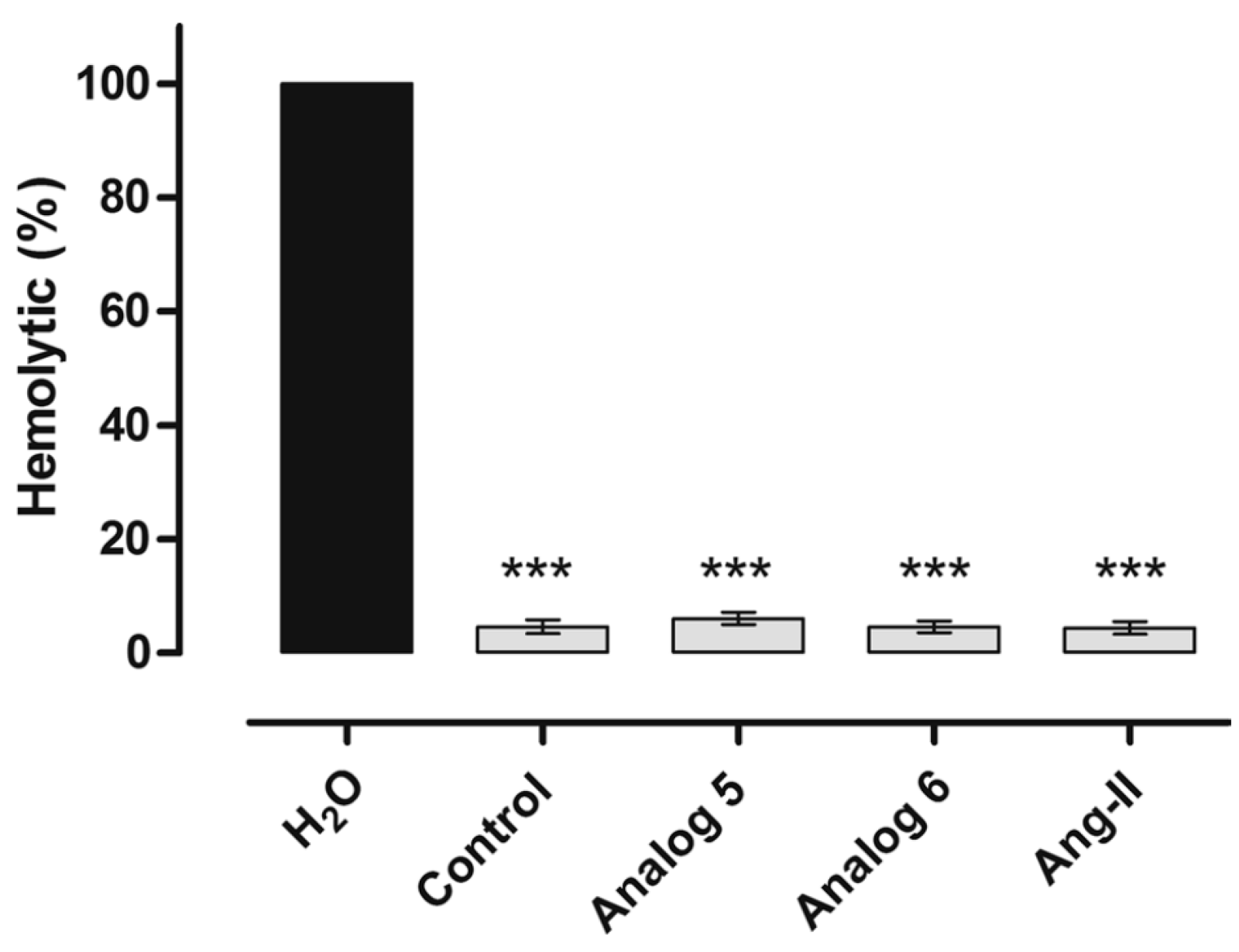
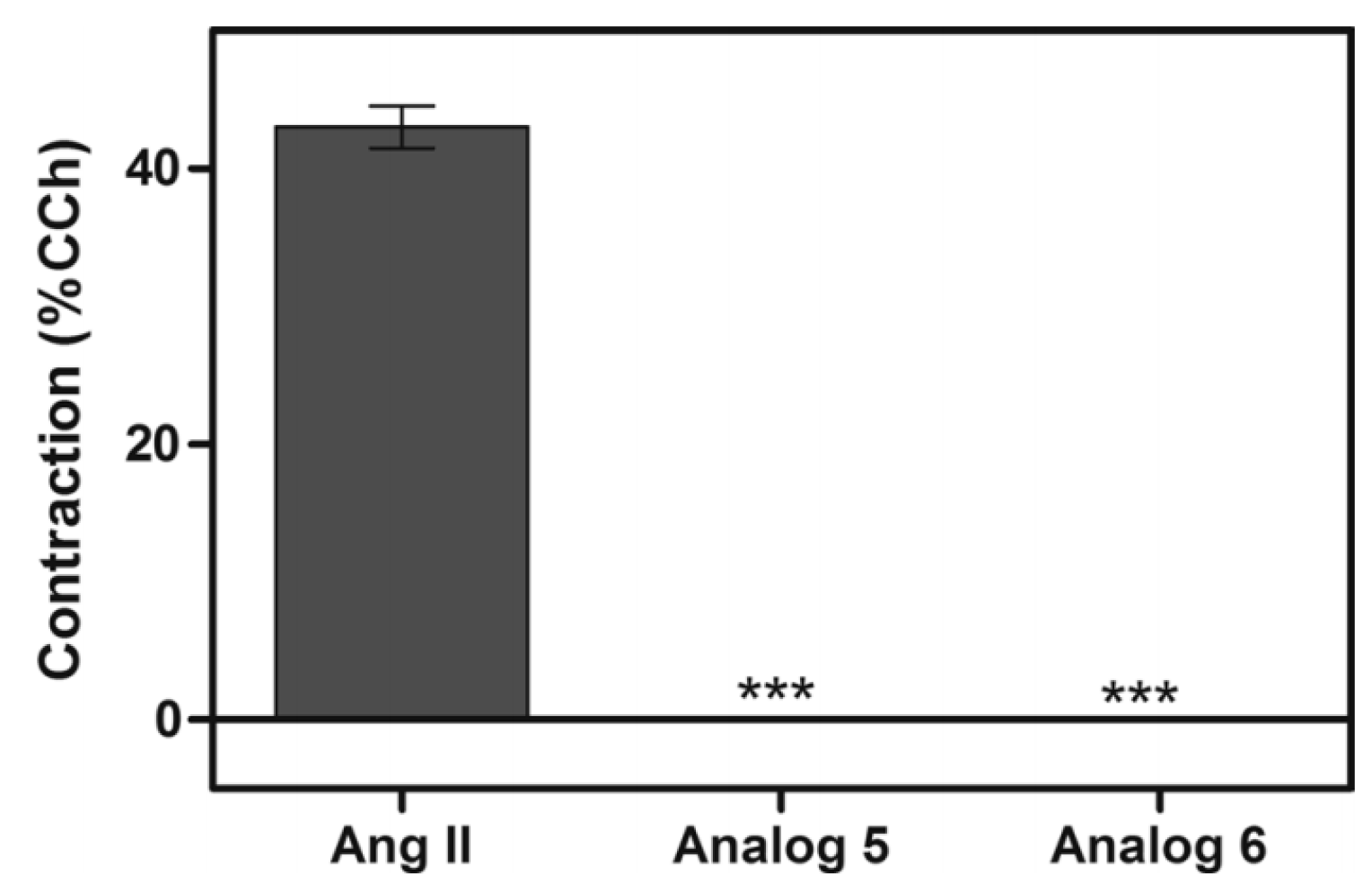
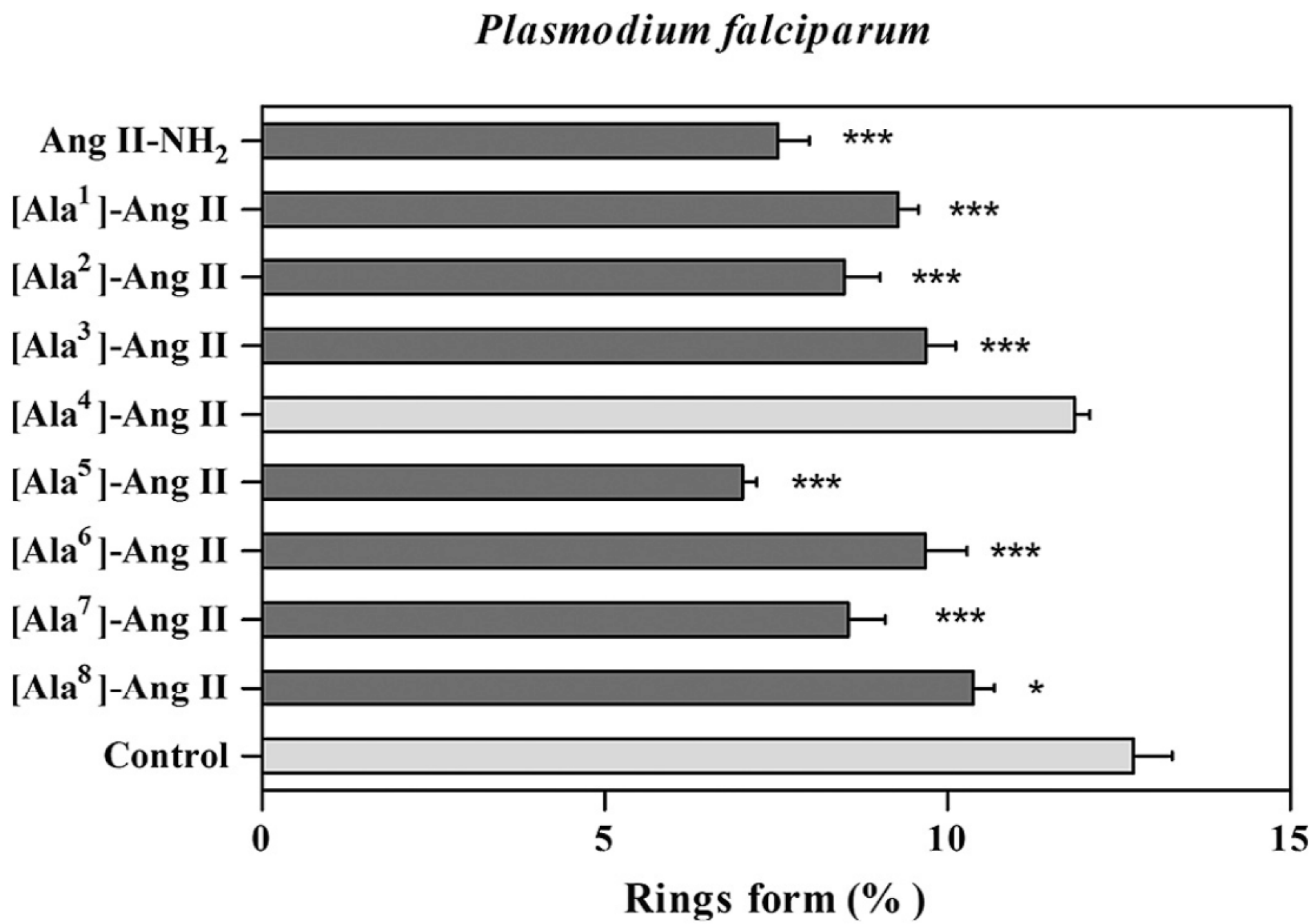
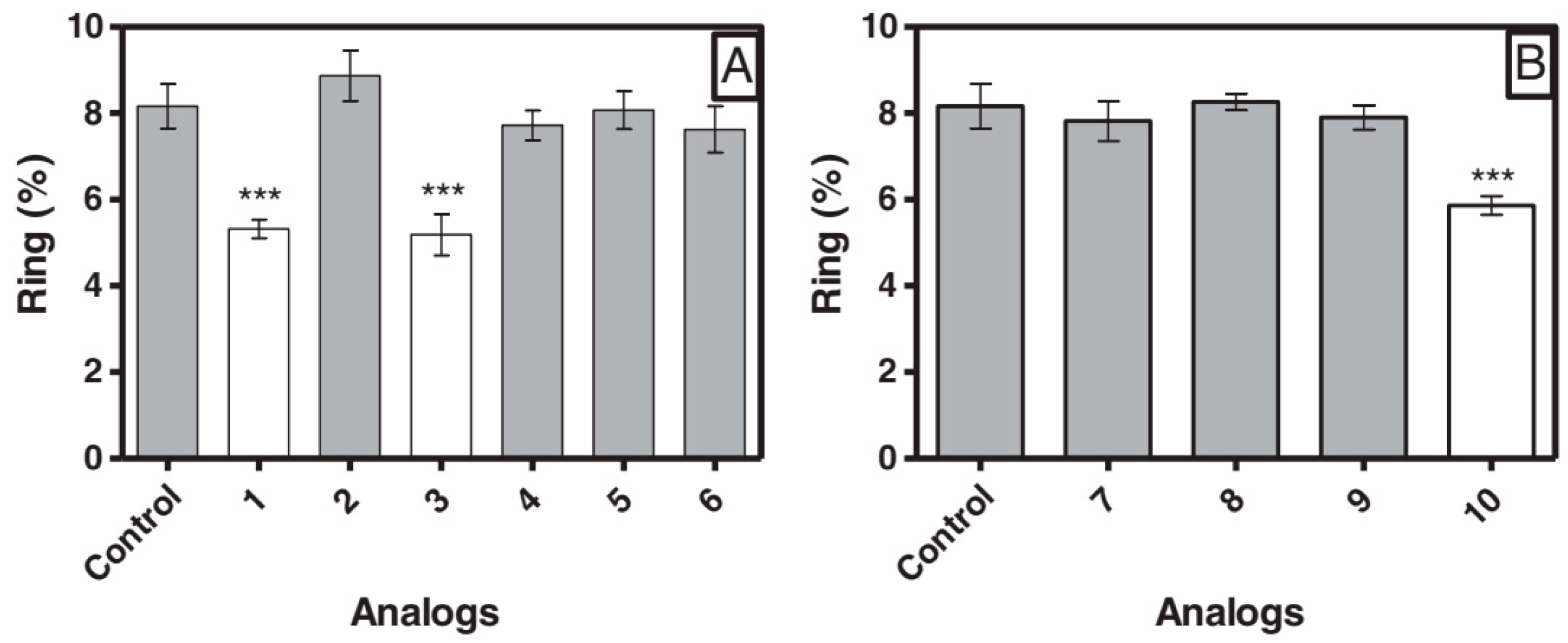
3.2.4. Angiotensin
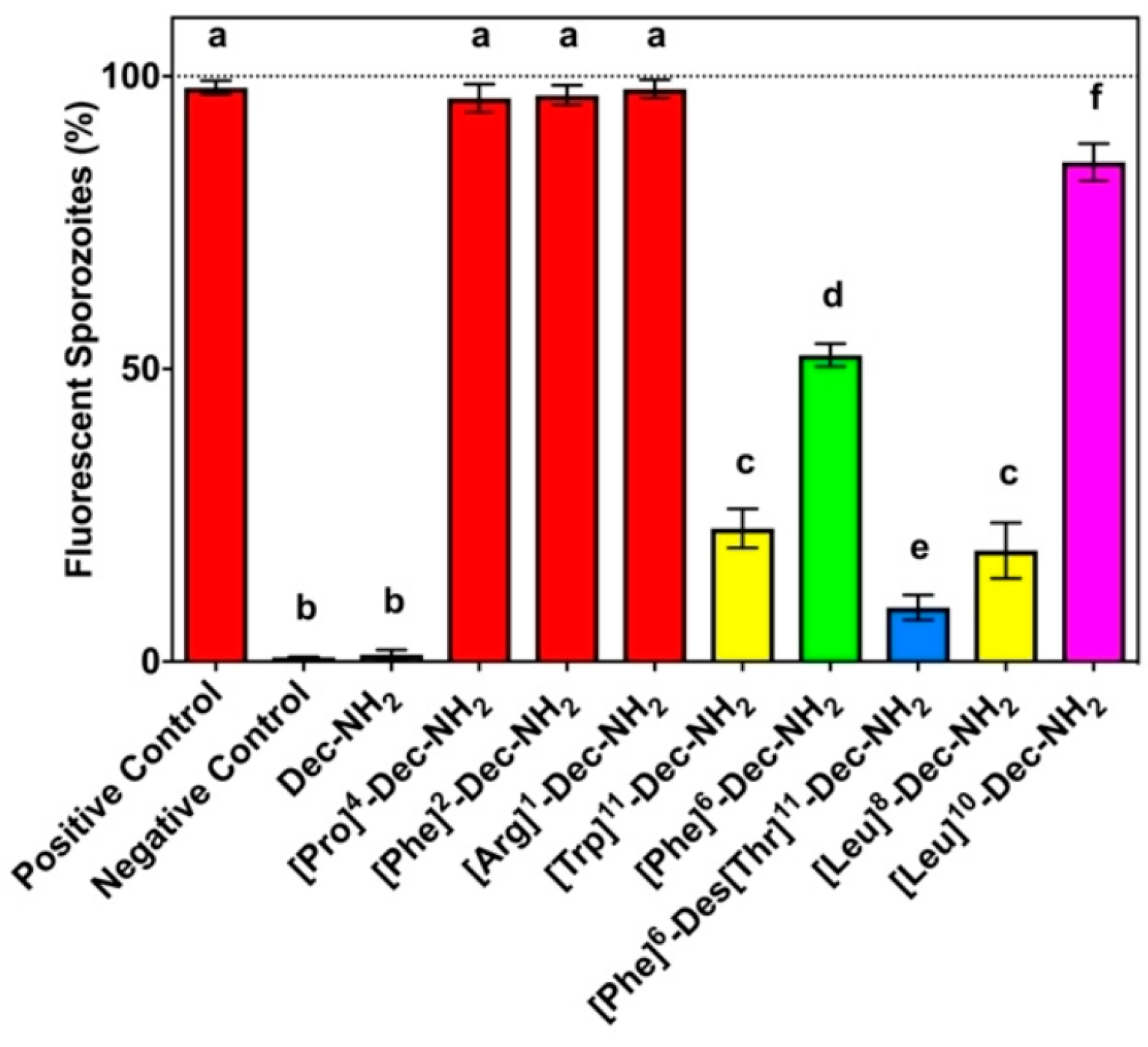
3.2.5. Decoralin
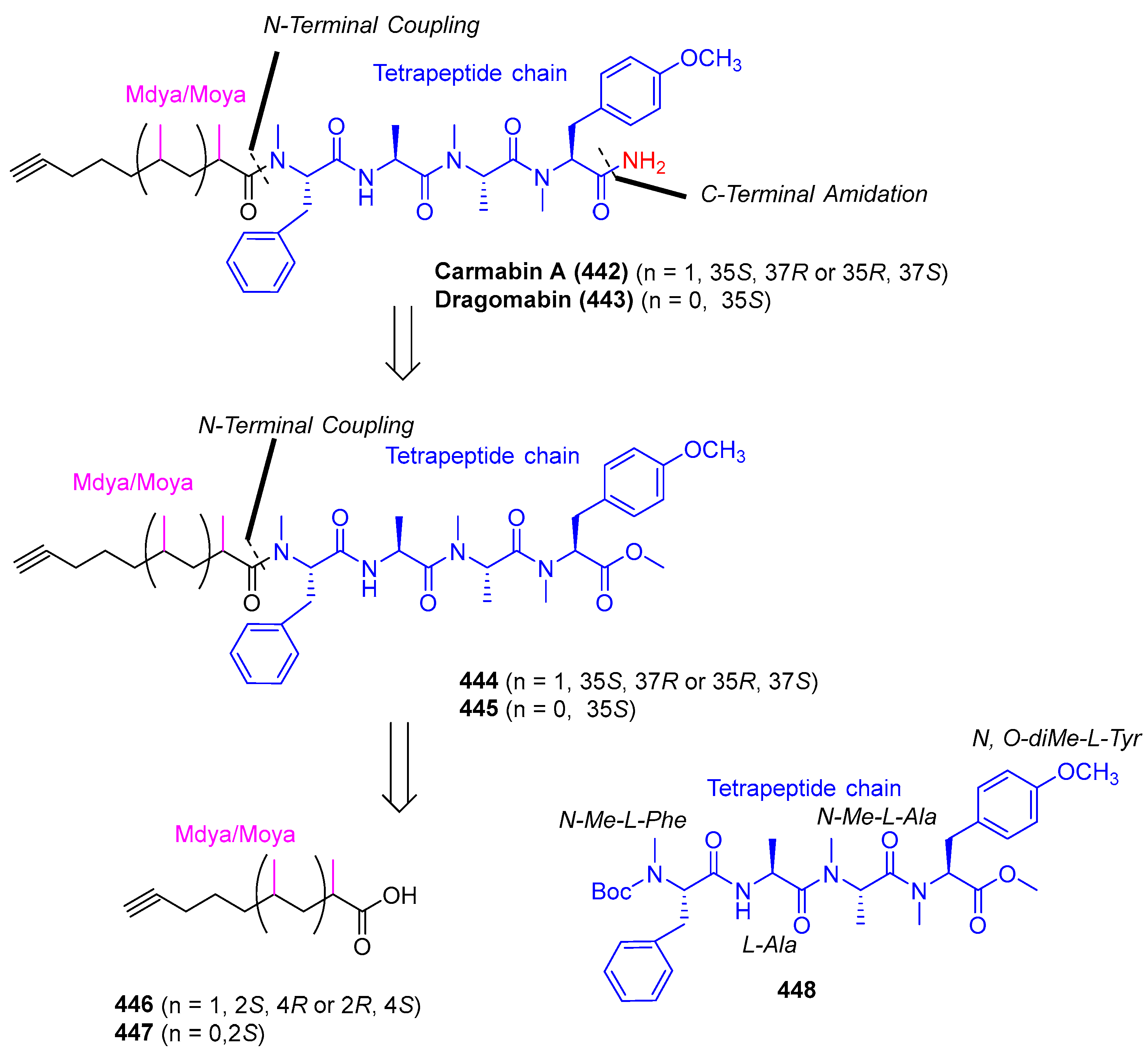
3.2.6. Carmabin and Dragomabin
3.3. Biology Activity of Isolated and Synthesized Compounds
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed on 11 September 2023).
- Talapko, J.; Škrlec, I.; Alebić, T.; Jukić, M.; Včev, A. Malaria: The past and the present. Microorganisms 2019, 7, 179. [Google Scholar] [CrossRef]
- Pinto, M.E.F.; Batista, J.M.; Koehbach, J.; Gaur, P.; Sharma, A.; Nakabashi, M.; Cilli, E.M.; Giesel, G.M.; Verli, H.; Gruber, C.W.; et al. Ribifolin, an orbitide from jatropha ribifolia, and its potential antimalarial activity. J. Nat. Prod. 2015, 78, 374–380. [Google Scholar] [CrossRef]
- Tuenter, E.; Exarchou, V.; Baldé, A.; Cos, P.; Maes, L.; Apers, S.; Pieters, L. Cyclopeptide Alkaloids from Hymenocardia acida. J. Nat. Prod. 2016, 79, 1746–1751. [Google Scholar] [CrossRef]
- Tuenter, E.; Ahmad, R.; Foubert, K.; Amin, A.; Orfanoudaki, M.; Cos, P.; Maes, L.; Apers, S.; Pieters, L.; Exarchou, V. Isolation and Structure Elucidation by LC-DAD-MS and LC-DAD-SPE-NMR of Cyclopeptide Alkaloids from the Roots of Ziziphus oxyphylla and Evaluation of Their Antiplasmodial Activity. J. Nat. Prod. 2016, 79, 2865–2872. [Google Scholar] [CrossRef]
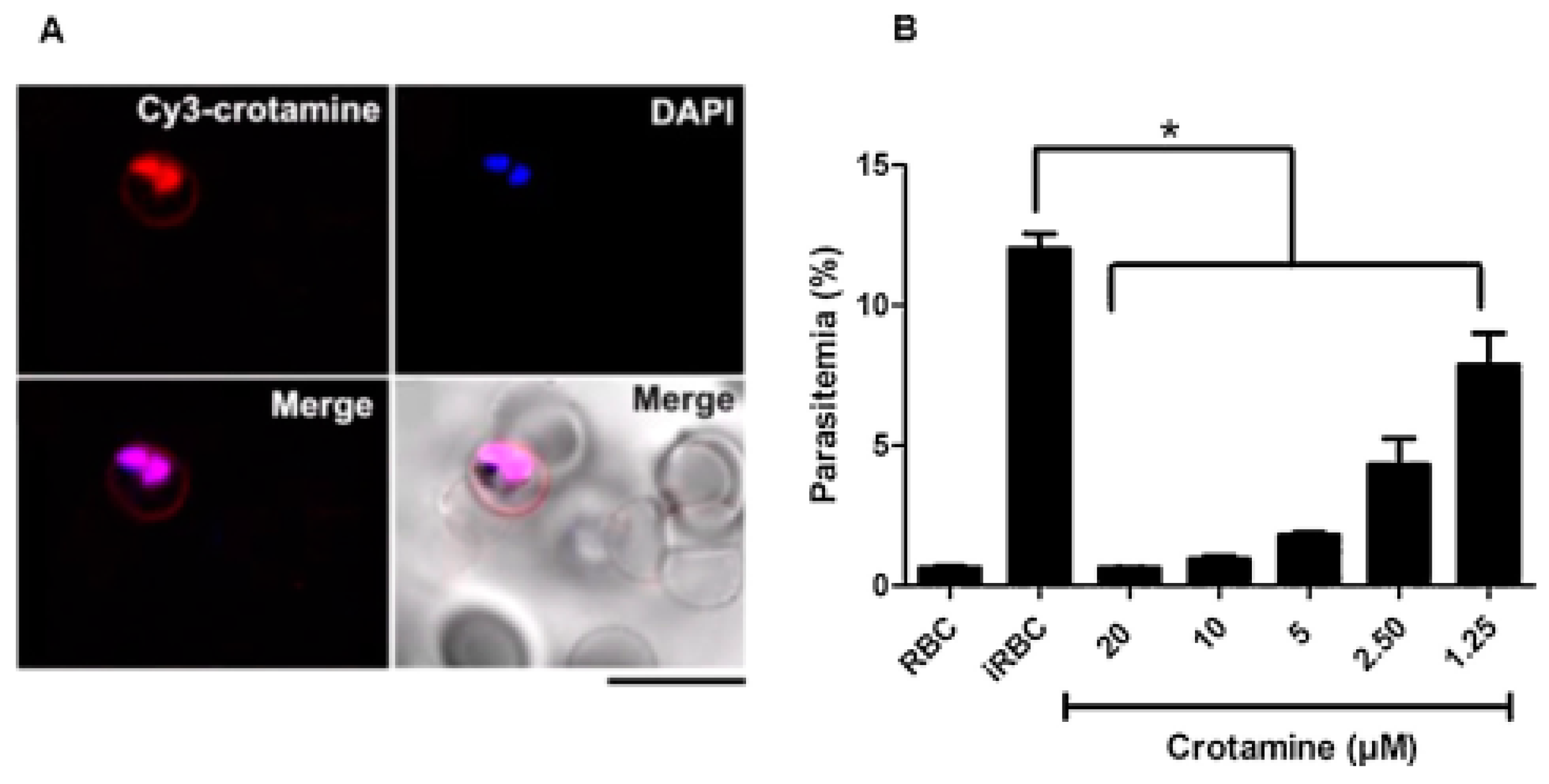
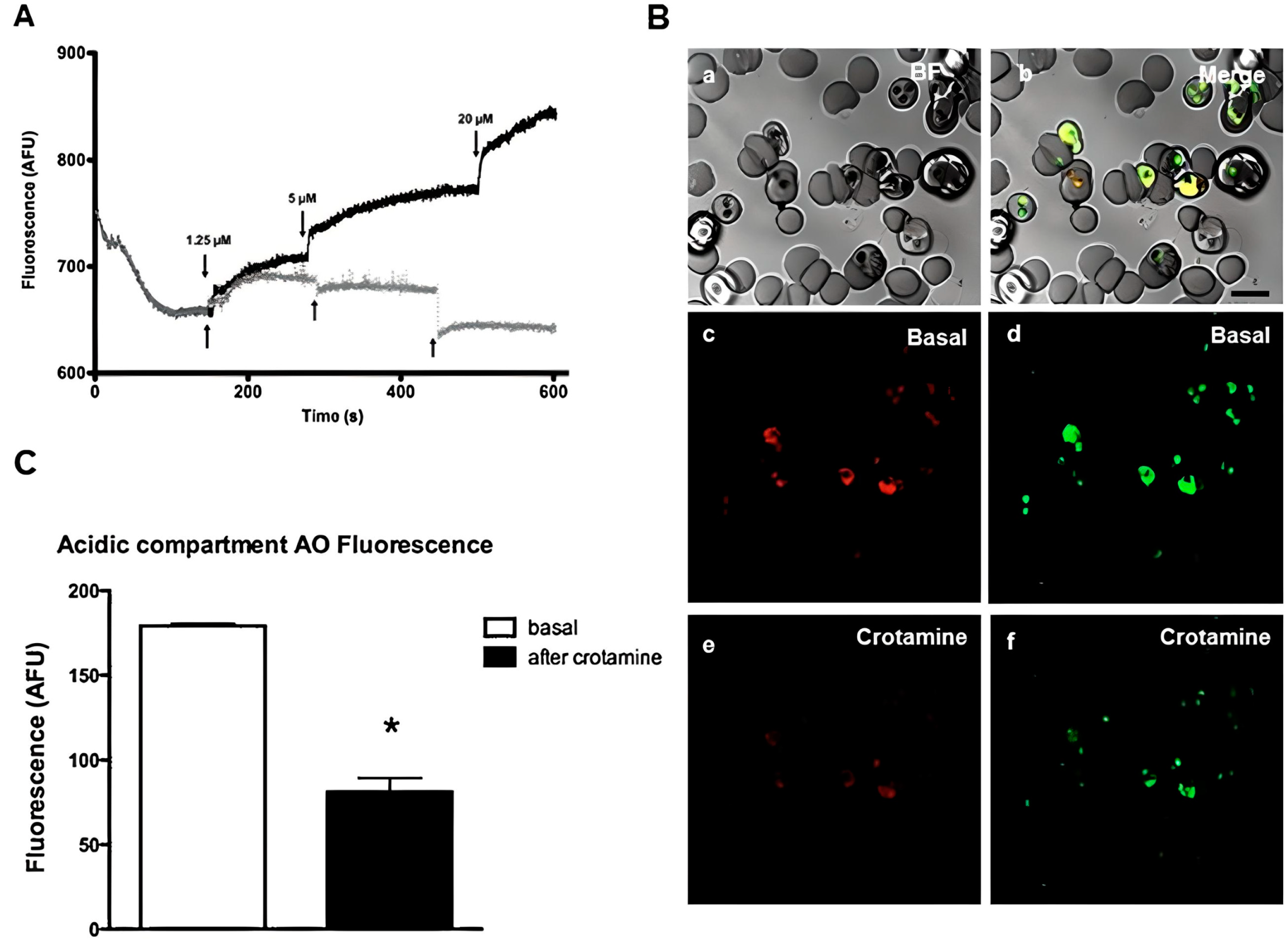
- Maluf, E.; Mas, C.; Oliveira, E.B.; Melo, P.M.; Carmona, A.K.; Gazarini, M.L.; Hayashi, M.A.F. Inhibition of malaria parasite Plasmodium falciparum development by crotamine, a cell penetrating peptide from the snake venom. Peptides 2016, 78, 11–16. [Google Scholar] [CrossRef]
- Jang, J.P.; Nogawa, T.; Futamura, Y.; Shimizu, T.; Hashizume, D.; Takahashi, S.; Jang, J.H.; Ahn, J.S.; Osada, H. Octaminomycins A and B, cyclic octadepsipeptides active against Plasmodium falciparum. J. Nat. Prod. 2017, 80, 134–140. [Google Scholar] [CrossRef]
- Sweeney-Jones, A.M.; Gagaring, K.; Antonova-Koch, J.; Zhou, H.; Mojib, N.; Soapi, K.; Skolnick, J.; McNamara, C.W.; Kubanek, J. Antimalarial peptide and polyketide natural products from the Fijian marine cyanobacterium moorea producens. Mar. Drugs 2020, 18, 167. [Google Scholar] [CrossRef]
- Fernández-Pastor, I.; González-Menéndez, V.; Annang, F.; Toro, C.; Mackenzie, T.A.; Bosch-Navarrete, C.; Genilloud, O.; Reyes, F. Pipecolisporin, a novel cyclic peptide with antimalarial and antitrypanosome activities from a wheat endophytic nigrospora oryzae. Pharmaceuticals 2021, 14, 268. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Hachiya, K.; Ikeda, A.; Nonaka, K.; Higo, M.; Muramatsu, R.; Noguchi, C.; Honsho, M.; Asami, Y.; Inahashi, Y.; et al. Koshidacins A and B, Antiplasmodial Cyclic Tetrapeptides from the Okinawan Fungus Pochonia boninensis FKR-0564. J. Nat. Prod. 2022, 85, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.M.; Richter, C.; Challinor, V.L.; Grün, P.; Girela Del Rio, A.; Kaiser, M.; Schüffler, A.; Piepenbring, M.; Schwalbe, H.; Bode, H.B. Georatusin, a Specific Antiparasitic Polyketide-Peptide Hybrid from the Fungus Geomyces auratus. Org. Lett. 2018, 20, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Bracegirdle, J.; Casandra, D.; Rocca, J.R.; Adams, J.H.; Baker, B.J. Highly N-Methylated Peptides from the Antarctic Sponge Inflatella coelosphaeroides Are Active against Plasmodium falciparum. J. Nat. Prod. 2022, 85, 2454–2460. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Muñoz, A.; Sierra-Gómez, Y.; Covaleda-Cortés, G.; Reytor, M.L.; González-González, Y.; Bautista, J.M.; Avilés, F.X.; Alonso-del-Rivero, M. Isolation and Characterization of NpCI, a New Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita peloronta with Anti-Plasmodium falciparum Activity. Mar. Drugs 2023, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, S.; Okada, Y.; Chiba, K. Soluble tag-assisted peptide head-to-tail cyclization: Total synthesis of mahafacyclin B. Org. Lett. 2013, 15, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Peña, S.; Scarone, L.; Manta, E.; Serra, G. First total synthesis of aerucyclamide B. Tetrahedron Lett. 2013, 54, 2806–2808. [Google Scholar] [CrossRef]
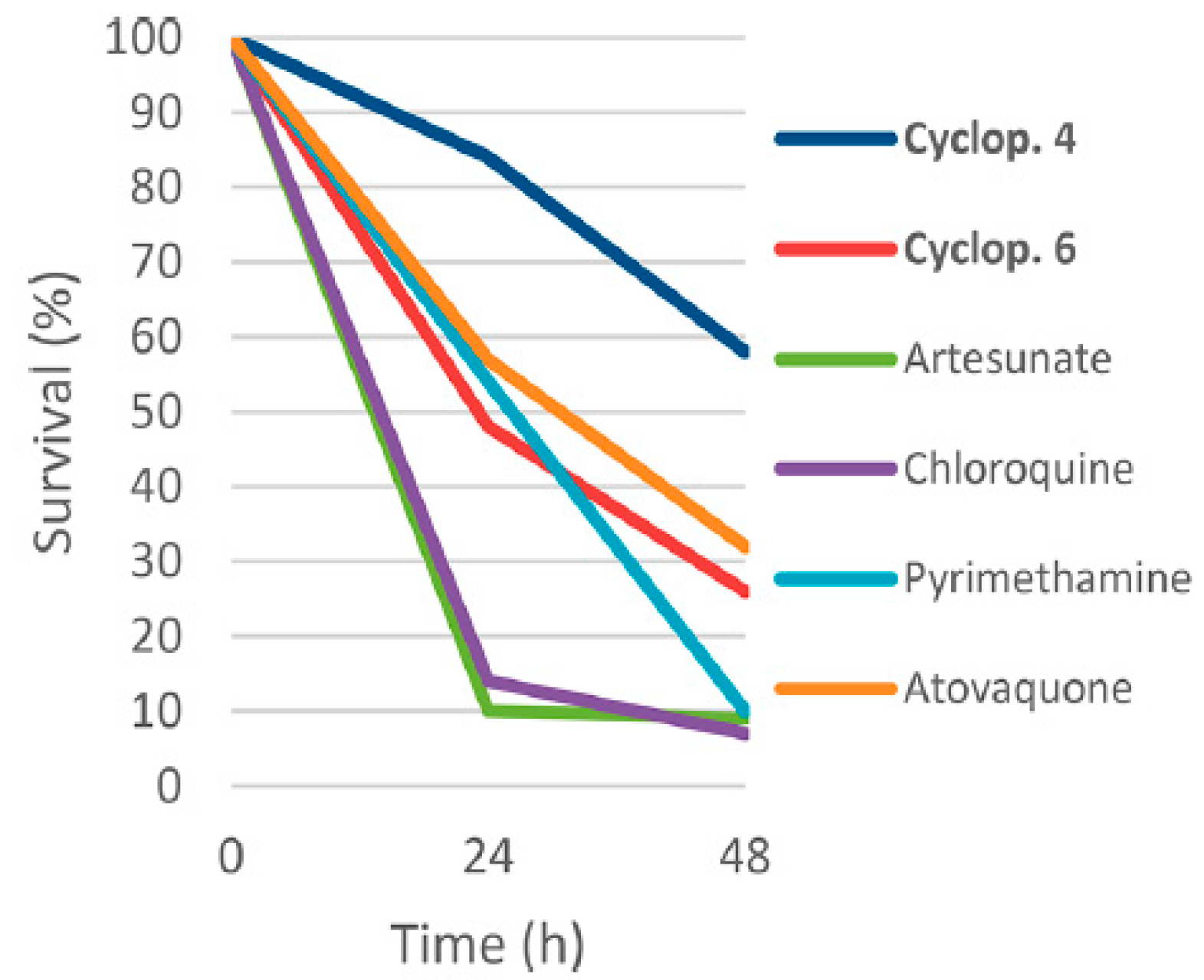
- Barbie, P.; Kazmaier, U. Total Synthesis of Cyclomarin A, a Marine Cycloheptapeptide with Anti-Tuberculosis and Anti-Malaria Activity. Org. Lett. 2016, 18, 204–207. [Google Scholar] [CrossRef]
- Kiefer, A.; Bader, C.D.; Held, J.; Esser, A.; Rybniker, J.; Empting, M.; Müller, R.; Kazmaier, U. Synthesis of New Cyclomarin Derivatives and Their Biological Evaluation towards Mycobacterium Tuberculosis and Plasmodium Falciparum. Chem.-A Eur. J. 2019, 25, 8894–8902. [Google Scholar] [CrossRef]
- Gholap, S.S.; Ugale, S.R. A Total Synthesis of the Cyclic Depsipeptide Chaiyaphumine-A. ChemistrySelect 2017, 2, 7445–7449. [Google Scholar] [CrossRef]
- Lu, H.; Batey, R.A. Total synthesis of chaiyaphumines A-D: A case study comparing macrolactonization and macrolactamization approaches. Tetrahedron Lett. 2022, 108, 154138. [Google Scholar] [CrossRef]
- Fagundez, C.; Sellanes, D.; Serra, G. Synthesis of cyclic peptides as potential anti-malarials. ACS Comb. Sci. 2018, 20, 212–219. [Google Scholar] [CrossRef]
- Fagundez, C.; Sellanes, D.; Peña, S.; Scarone, L.; Aguiar, A.C.C.; De Souza, J.O.; Guido, R.V.C.; Stewart, L.; Yardley, V.; Ottilie, S.; et al. Synthesis, Profiling, and in Vivo Evaluation of Cyclopeptides Containing N-Methyl Amino Acids as Antiplasmodial Agents. ACS Med. Chem. Lett. 2019, 10, 137–141. [Google Scholar] [CrossRef]
- Sahile, H.A.; Sahile, H.A.; Martínez-Martínez, M.S.; Dillenberger, M.; Becker, K.; Imming, P. Synthesis and Evaluation of Antimycobacterial and Antiplasmodial Activities of Hirsutellide A and Its Analogues. ACS Omega 2020, 5, 14451–14460. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, L.; Duan, X.; Meng, Y.; Jiang, L.; Li, M.; Zhao, G.; Li, Y. Total synthesis of hirsutellide A. Tetrahedron Lett. 2005, 46, 4377–4379. [Google Scholar] [CrossRef]
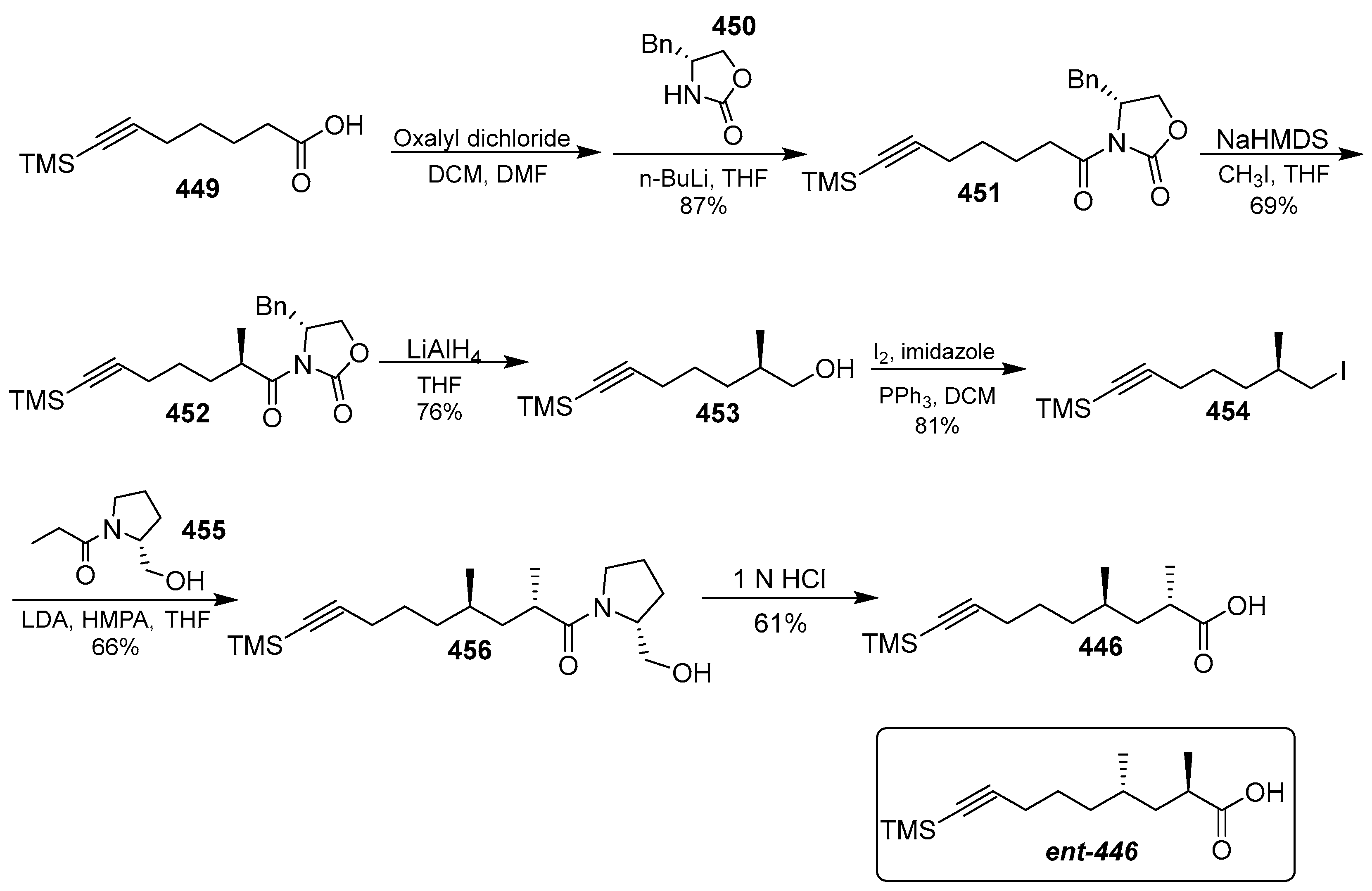
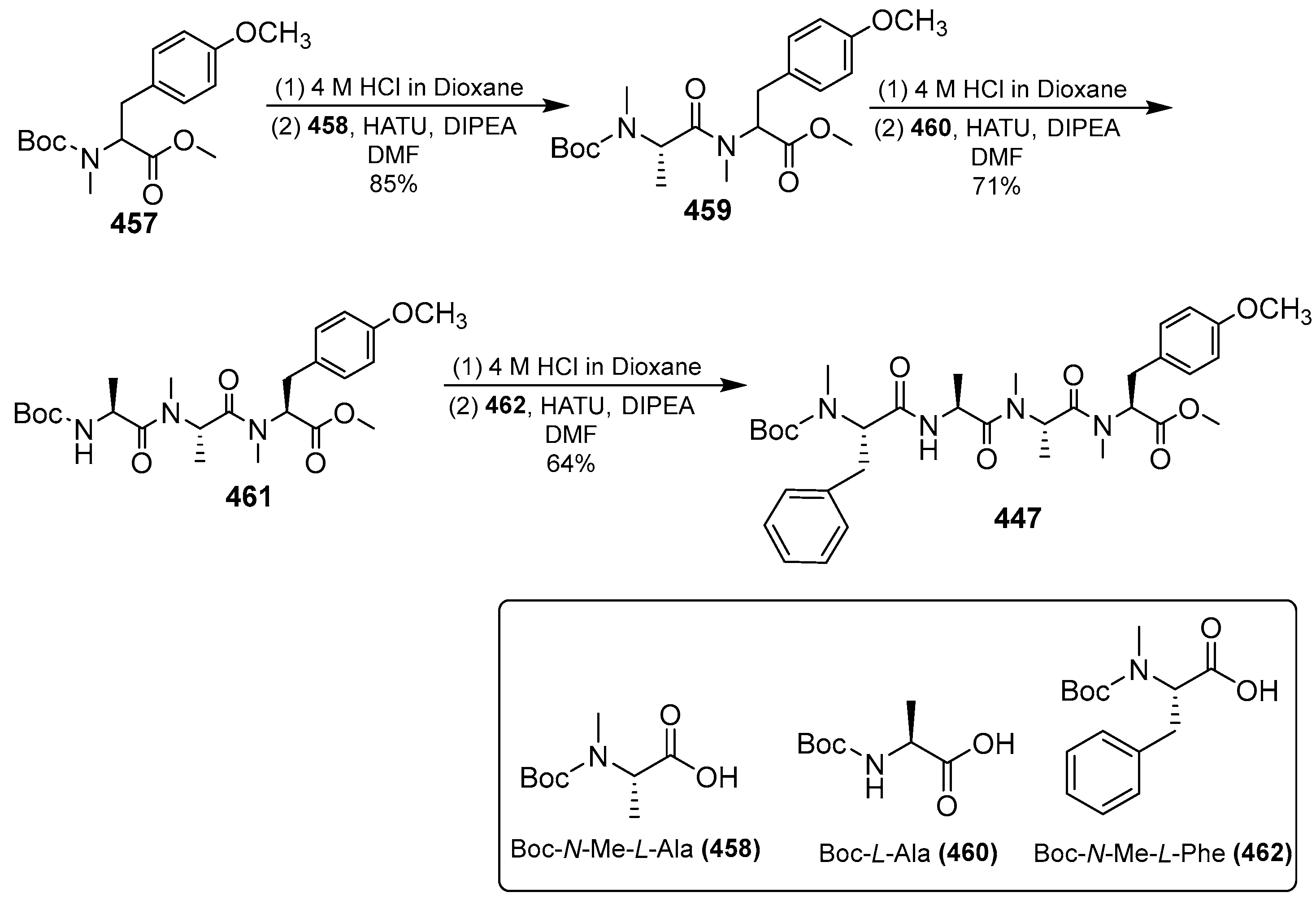
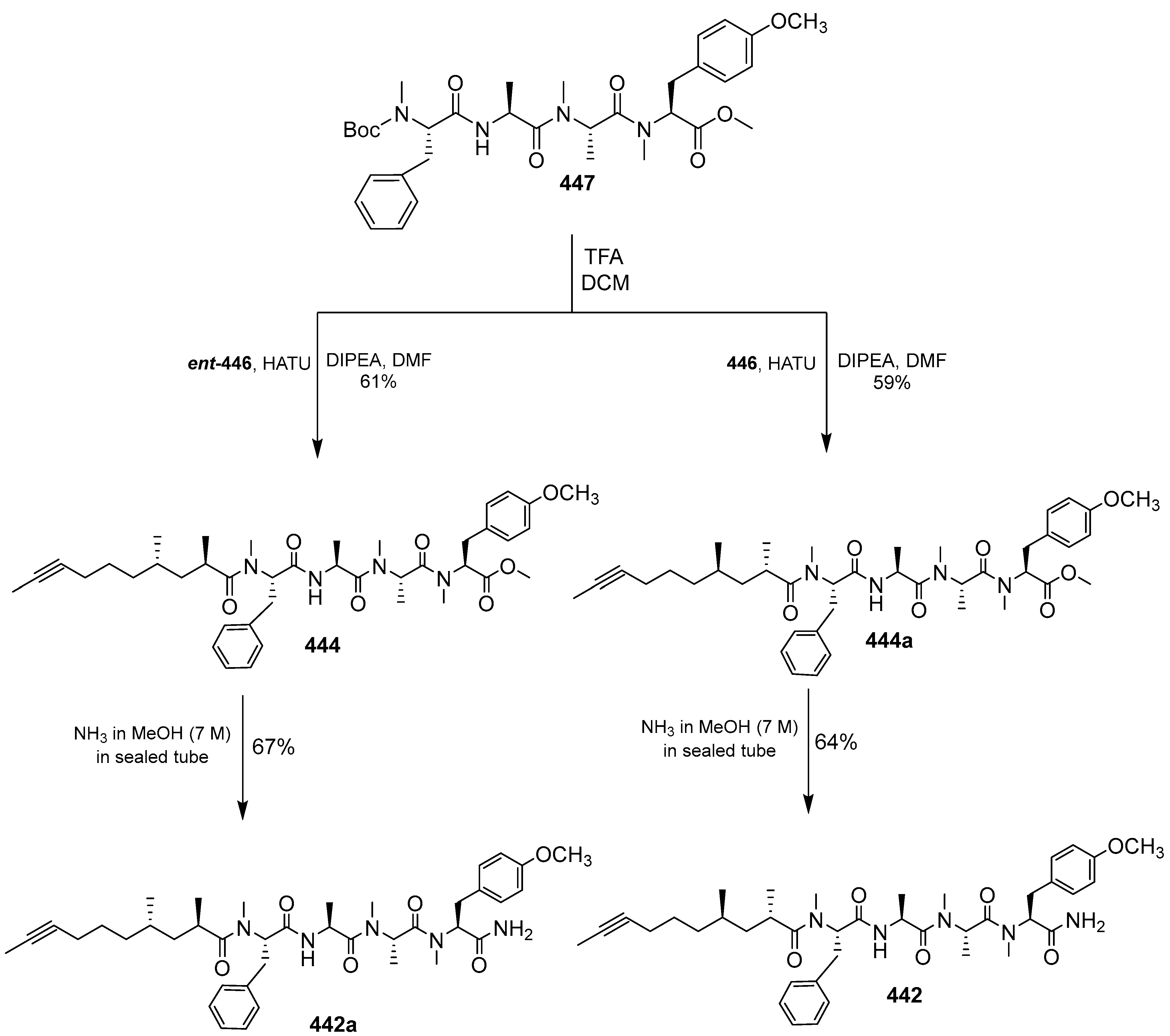
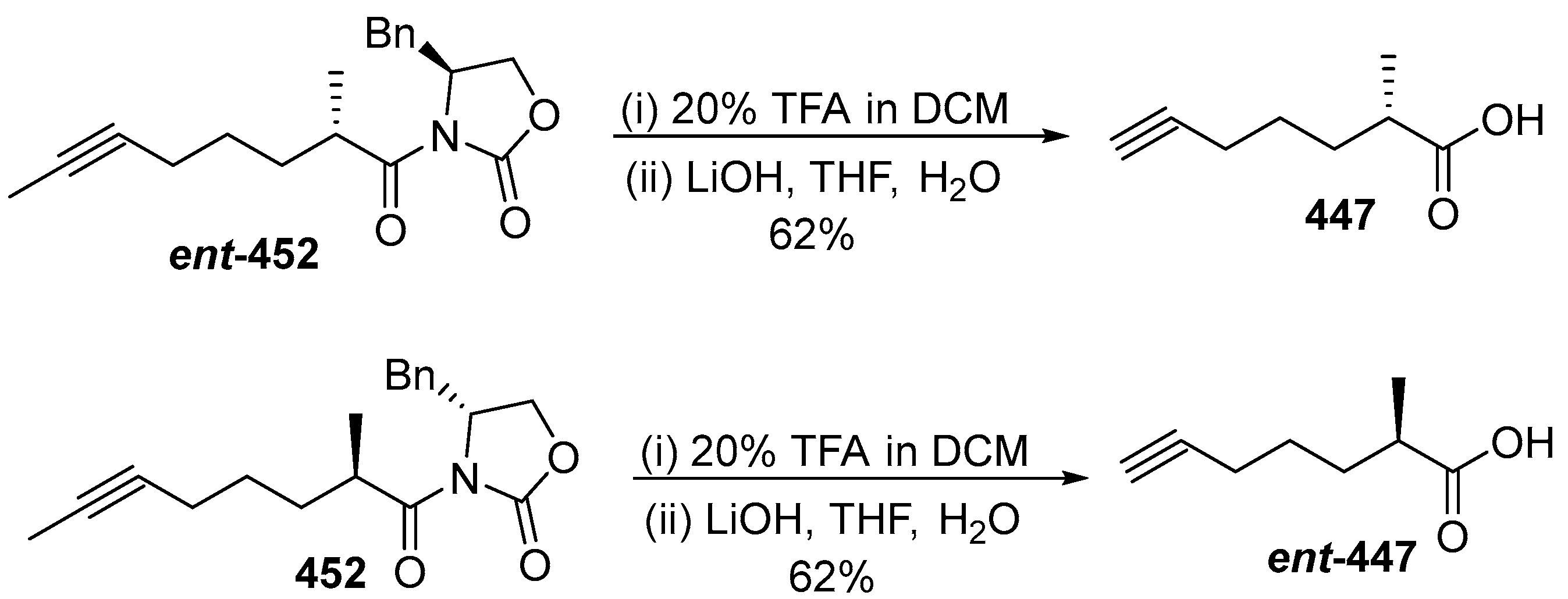
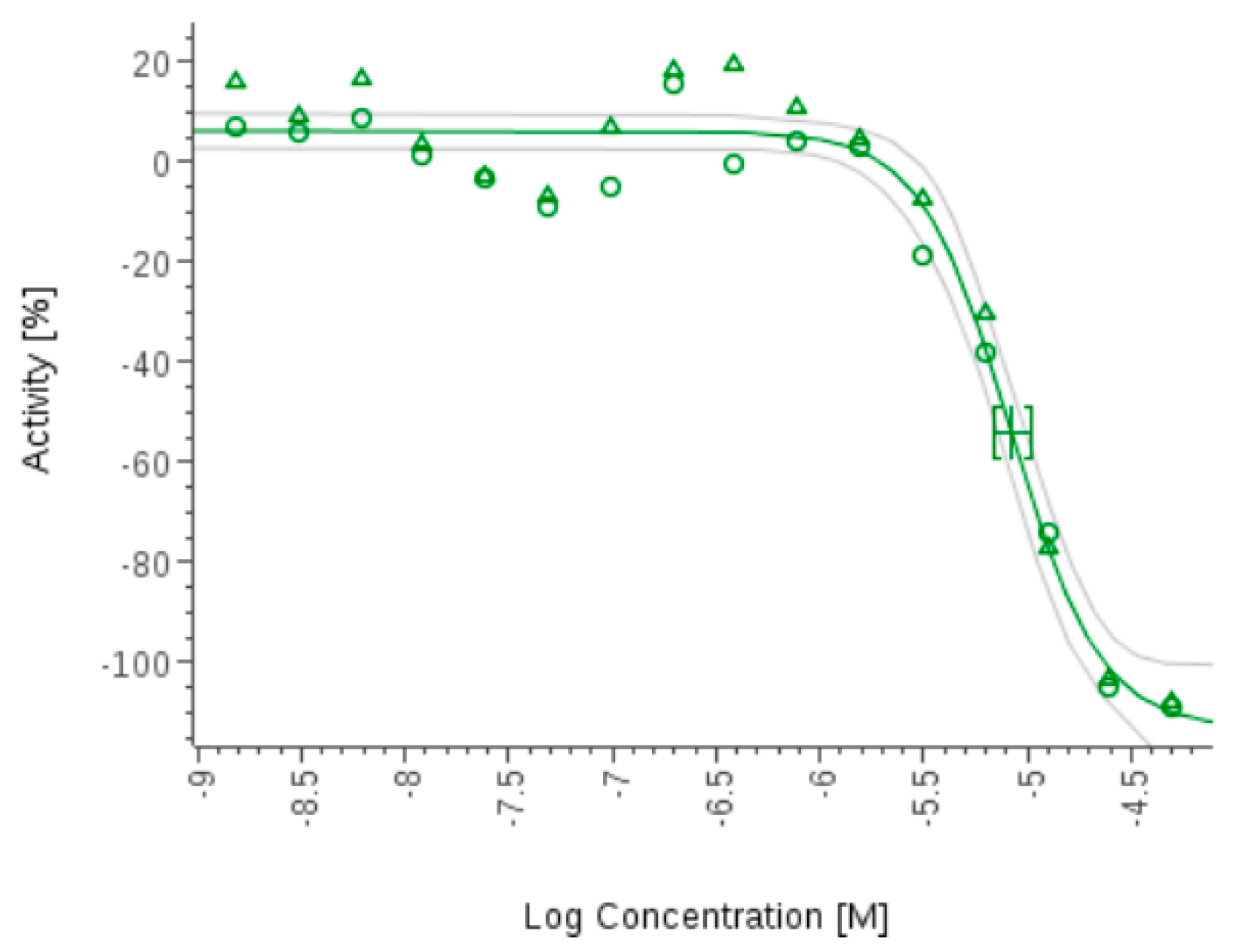
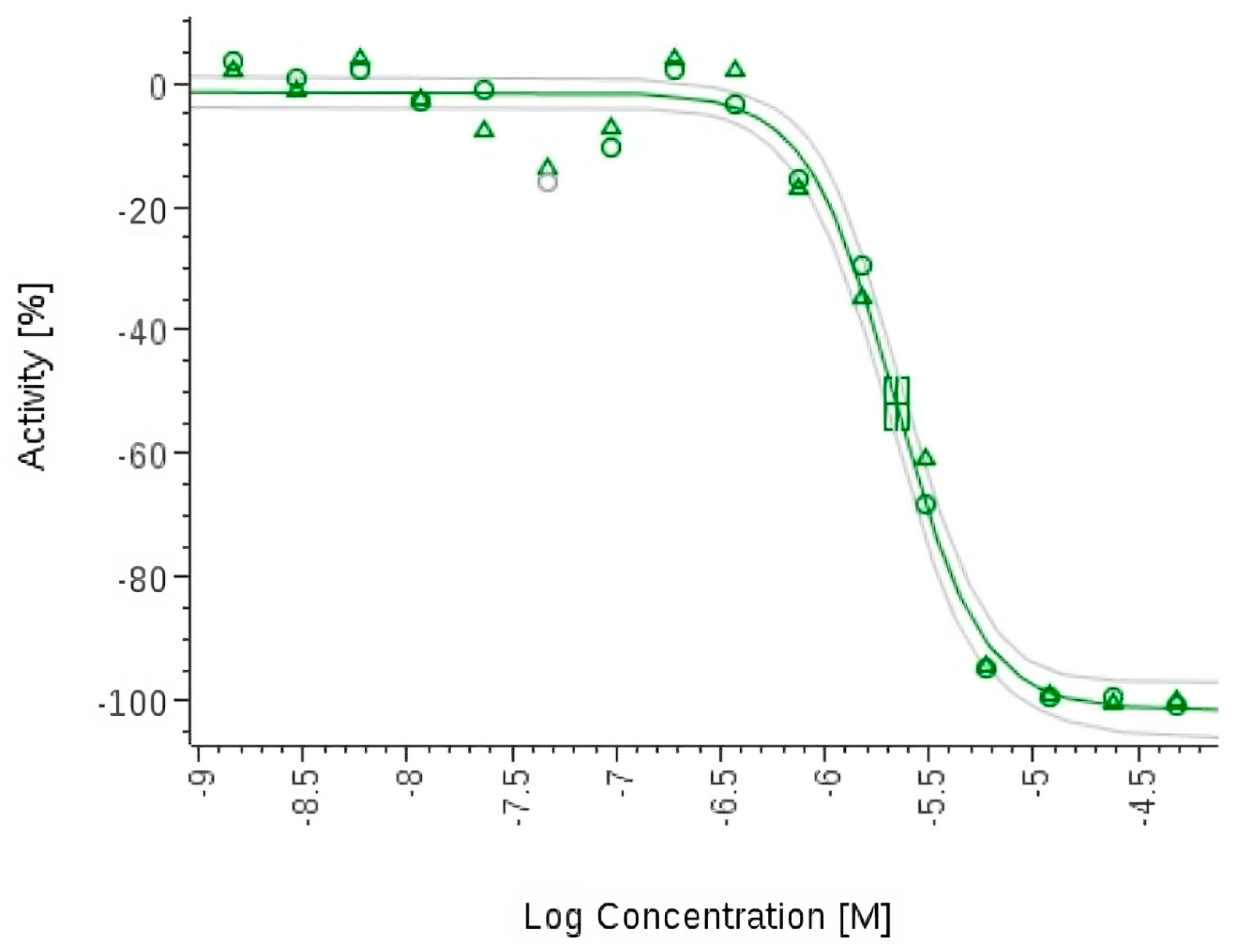
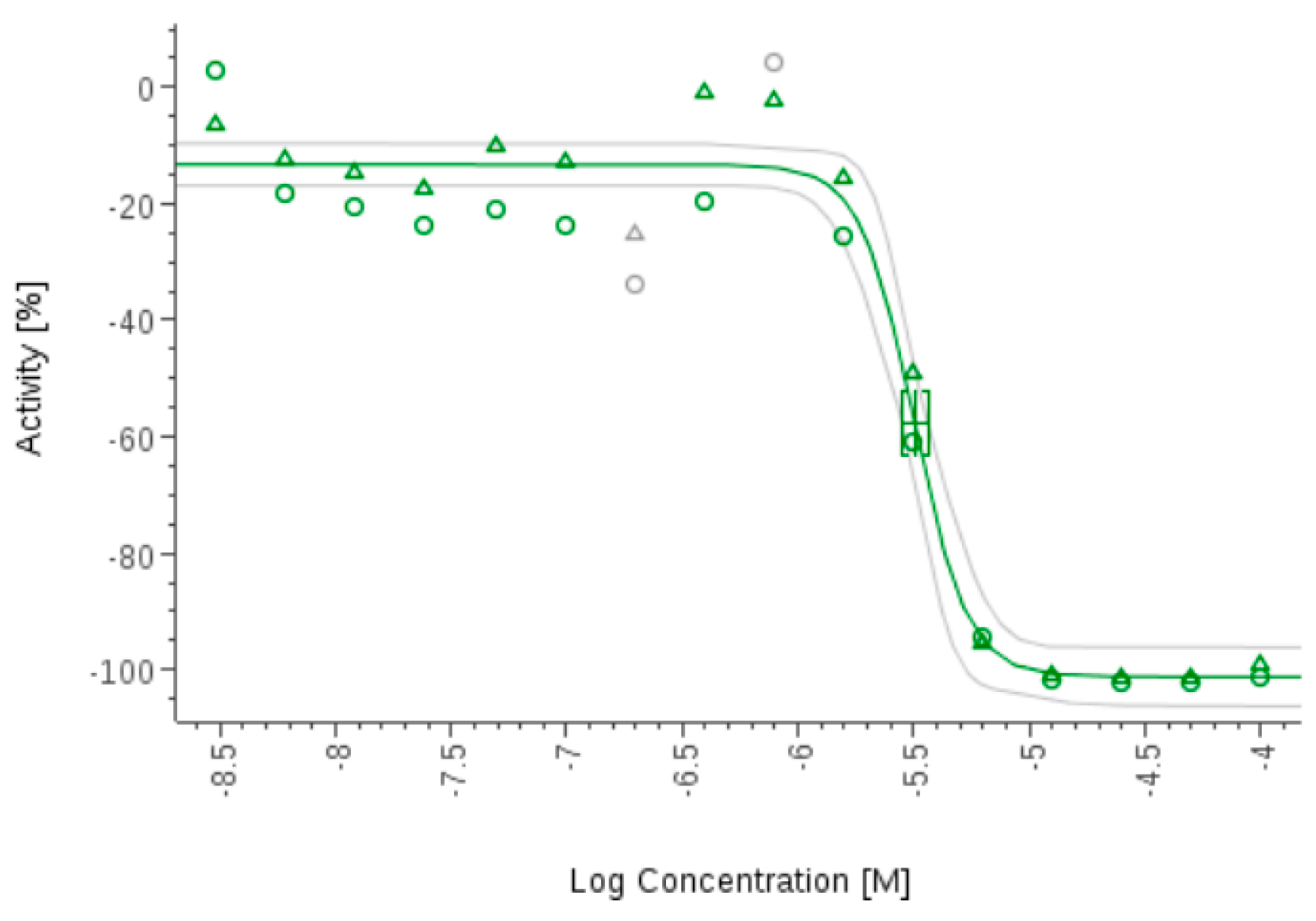
- Zhang, H.; Ginn, J.; Zhan, W.; Liu, Y.J.; Leung, A.; Toita, A.; Okamoto, R.; Wong, T.T.; Imaeda, T.; Hara, R.; et al. Design, Synthesis, and Optimization of Macrocyclic Peptides as Species-Selective Antimalaria Proteasome Inhibitors. J. Med. Chem. 2022, 65, 9350–9375. [Google Scholar] [CrossRef] [PubMed]
- Somanadhan, B.; Kotturi, S.R.; Yan Leong, C.; Glover, R.P.; Huang, Y.; Flotow, H.; Buss, A.D.; Lear, M.J.; Butler, M.S. Isolation and synthesis of falcitidin, a novel myxobacterial-derived acyltetrapeptide with activity against the malaria target falcipain-2. J. Antibiot. 2013, 66, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Kotturi, S.R.; Somanadhan, B.; Ch’Ng, J.H.; Tan, K.S.W.; Butler, M.S.; Lear, M.J. Diverted total synthesis of falcitidin acyl tetrapeptides as new antimalarial leads. Tetrahedron Lett. 2014, 55, 1949–1951. [Google Scholar] [CrossRef]
- Conroy, T.; Guo, J.T.; Elias, N.; Cergol, K.M.; Gut, J.; Legac, J.; Khatoon, L.; Liu, Y.; McGowan, S.; Rosenthal, P.J.; et al. Synthesis of gallinamide A analogues as potent falcipain inhibitors and antimalarials. J. Med. Chem. 2014, 57, 10557–10563. [Google Scholar] [CrossRef] [PubMed]
- Stoye, A.; Juillard, A.; Tang, A.H.; Legac, J.; Gut, J.; White, K.L.; Charman, S.A.; Rosenthal, P.J.; Grau, G.E.R.; Hunt, N.H.; et al. Falcipain Inhibitors Based on the Natural Product Gallinamide A Are Potent in Vitro and in Vivo Antimalarials. J. Med. Chem. 2019, 62, 5562–5578. [Google Scholar] [CrossRef]
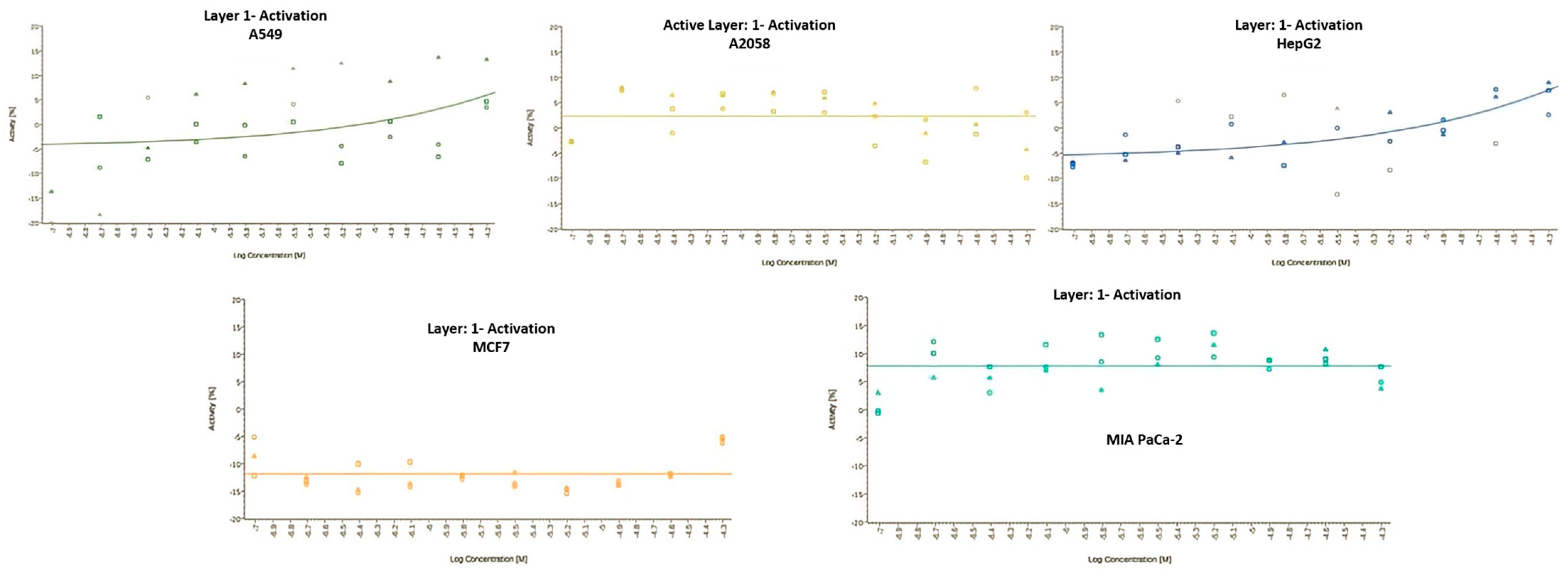
- Kanodia, S.; Kumar, G.; Rizzi, L.; Pedretti, A.; Hodder, A.N.; Romeo, S.; Malhotra, P. Synthetic peptides derived from the C-terminal 6 kDa region of Plasmodium falciparum SERA5 inhibit the enzyme activity and malaria parasite development. Biochim. Et Biophys. Acta-Gen. Subj. 2014, 1840, 2765–2775. [Google Scholar] [CrossRef]
- Silva, A.F.; Torres, M.D.T.; Silva, L.D.S.; Alves, F.L.; Pinheiro, A.A.D.S.; Miranda, A.; Capurro, M.L.; Oliveira, V.X. New linear antiplasmodial peptides related to angiotensin II. Malar. J. 2015, 14, 433. [Google Scholar] [CrossRef]
- Silva, A.F.; Silva, L.D.S.; Alves, F.L.; TorossianTorres, M.D.; de SáPinheiro, A.A.; Miranda, A.; LaraCapurro, M.; Oliveira, V.X. Effects of the angiotensin II Ala-scan analogs in erythrocytic cycle of Plasmodium falciparum (in vitro) and Plasmodium gallinaceum (ex vivo). Exp. Parasitol. 2015, 153, 1–7. [Google Scholar] [CrossRef]
- Torres, M.D.T.; Silva, A.F.; Silva, L.D.S.; de Sá Pinheiro, A.A.; Oliveira, V.X. Angiotensin II restricted analogs with biological activity in the erythrocytic cycle of Plasmodium falciparum. J. Pept. Sci. 2015, 21, 24–28. [Google Scholar] [CrossRef]
- Silva, A.F.; Torres, M.D.T.; Silva, L.S.; Alves, F.L.; de Sá Pinheiro, A.A.; Miranda, A.; Capurro, M.L.; De La Fuente-Nunez, C.; Oliveira, V.X. Angiotensin II-derived constrained peptides with antiplasmodial activity and suppressed vasoconstriction. Sci. Rep. 2017, 7, 14326. [Google Scholar] [CrossRef]
- Torres, M.D.T.; Silva, A.F.; Pedron, C.N.; Capurro, M.L.; de la Fuente-Nunez, C.; Junior, V.X.O. Peptide Design Enables Reengineering of an Inactive Wasp Venom Peptide into Synthetic Antiplasmodial Agents. ChemistrySelect 2018, 3, 5859–5863. [Google Scholar] [CrossRef]
- Ye, B.; Jiang, P.; Zhang, T.; Sun, Y.; Hao, X.; Cui, Y.; Wang, L.; Chen, Y. Total synthesis of the highly N-methylated peptides carmabin a and dragomabin. Mar. Drugs 2018, 16, 338. [Google Scholar] [CrossRef] [PubMed]
- Auvin, C.; Baraguey, C.; Blond, A.; Lezenven, F.; Pousset, J.L.; Bodo, B. Curcacycline B, a cyclic nonapeptide from Jatropha curcas enhancing rotamase activity of cyclophilin. Tetrahedron Lett. 1997, 38, 2845–2848. [Google Scholar] [CrossRef]
- Baraguey, C.; Auvin-Guette, C.; Blond, A.; Cavelier, F.; Lezenven, F.; Pousset, J.L.; Bodo, B. Isolation, structure and synthesis of chevalierins A, B and C, cyclic peptides from the latex of Jatropha chevalieri. J. Chem. Soc.-Perkin Trans. 1998, 18, 3033. [Google Scholar] [CrossRef]
- Baraguey, C.; Blond, A.; Correia, I.; Pousset, J.L.; Bodo, B.; Auvin-Guette, C. Mahafacyclin A, a cyclic heptapeptide from Jatropha mahafalensis exhibiting β-bulge conformation. Tetrahedron Lett. 2000, 41, 325–329. [Google Scholar] [CrossRef]
- Baraguey, C.; Blond, A.; Cavelier, F.; Pousset, J.L.; Bodo, B.; Auvin-Guette, C. Isolation, structure and synthesis of mahafacyclin B, a cyclic heptapeptide from the latex of Jatropha mahafalensis. J. Chem. Soc. Perkin Trans. 2001, 17, 2098–2103. [Google Scholar] [CrossRef]
- Auvin-Guette, C.; Baraguey, C.; Blond, A.; Xavier, H.S.; Pousset, J.L.; Bodo, B. Pohlianins A, B and C, cyclic peptides from the latex of Jatropha pohliana ssp. molissima. Tetrahedron 1999, 55, 11495–11510. [Google Scholar] [CrossRef]









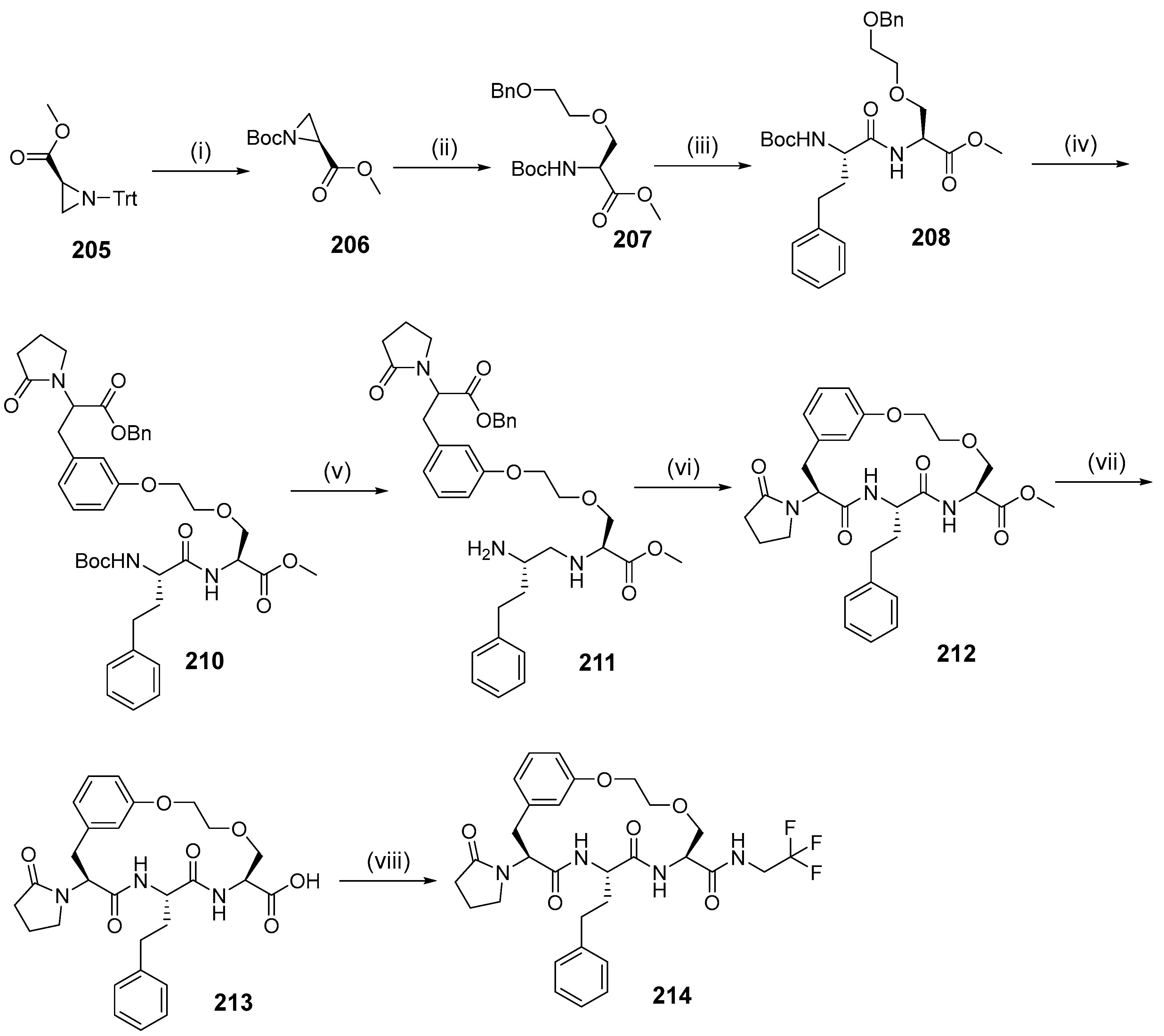
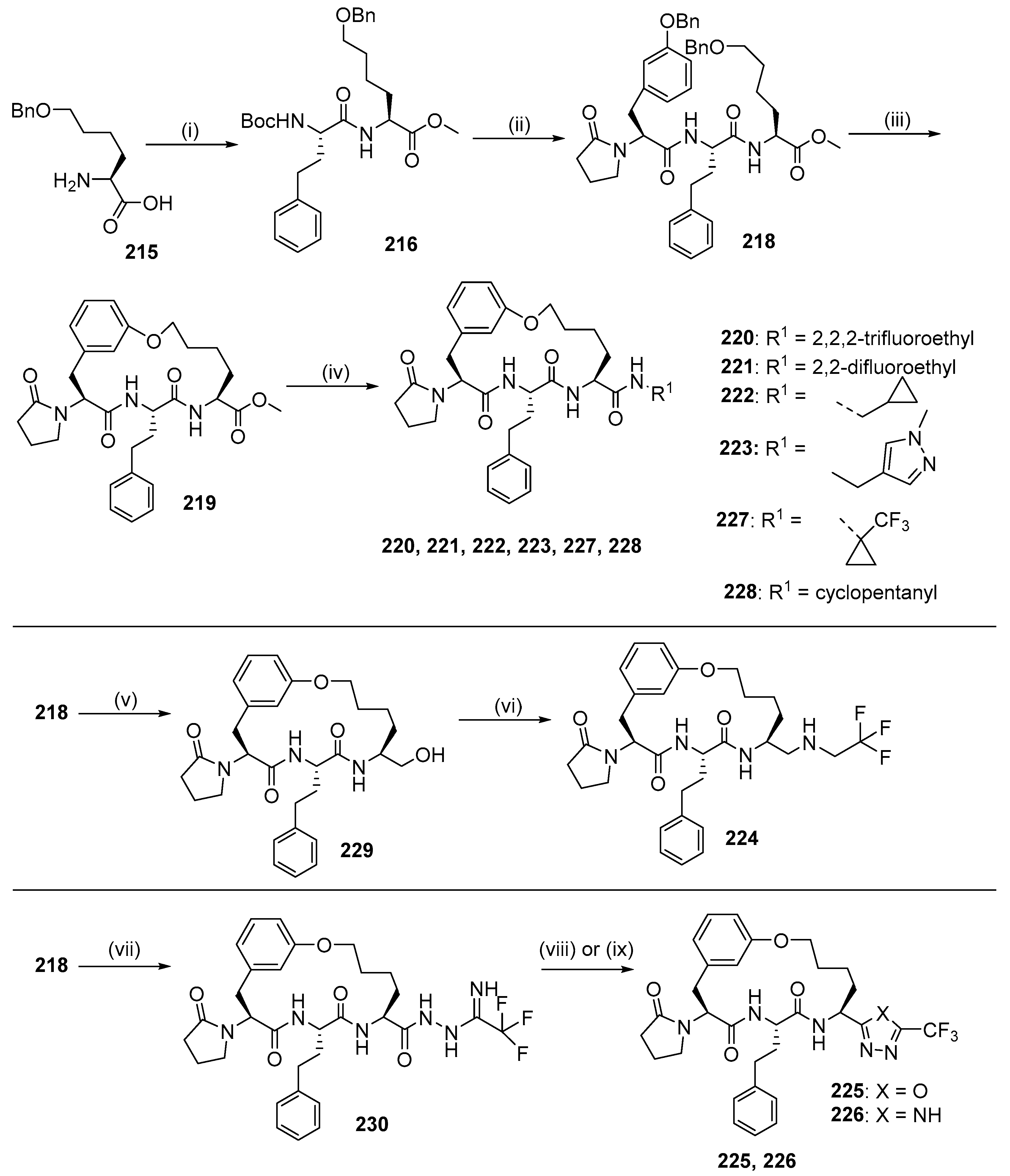


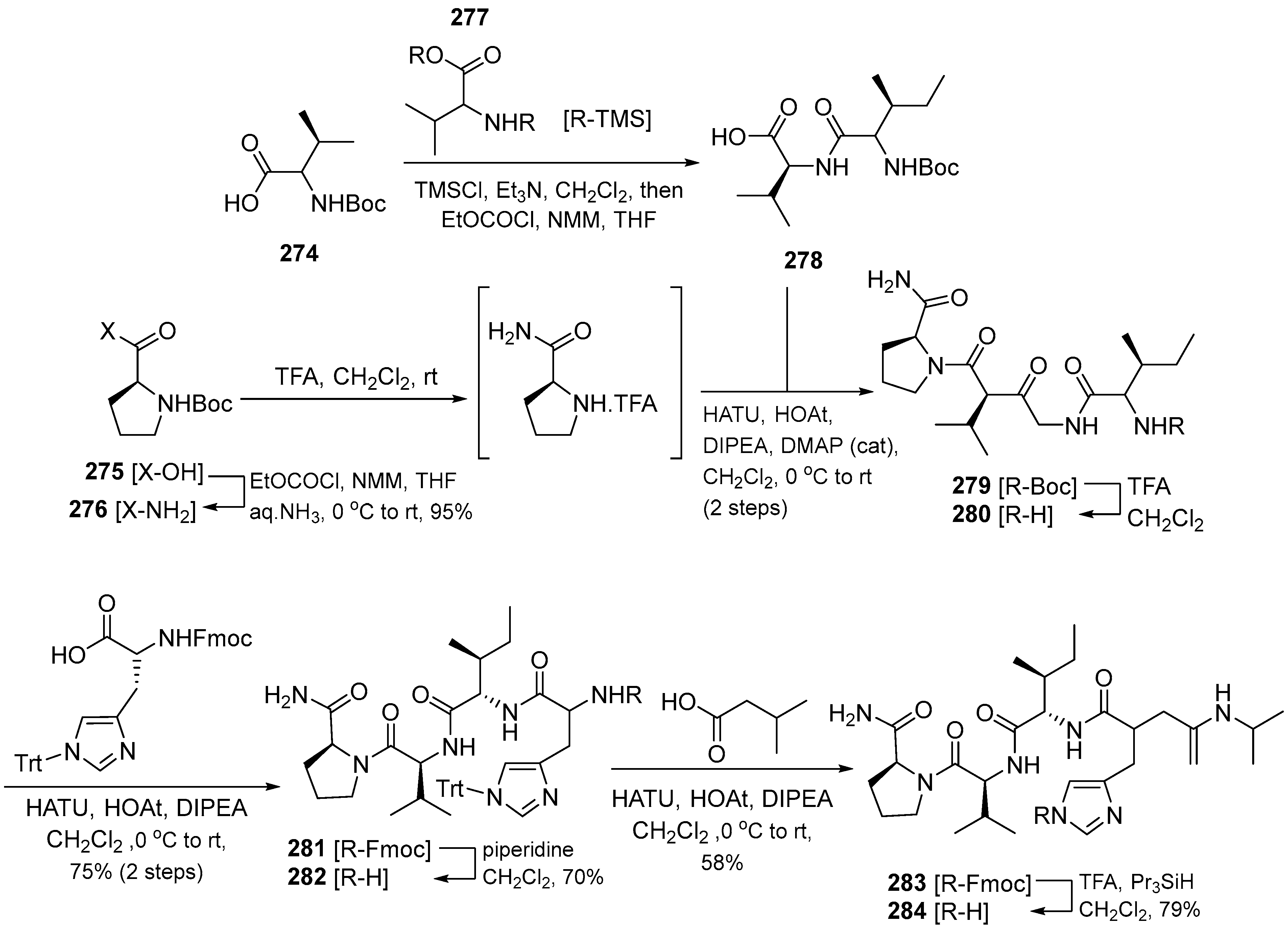
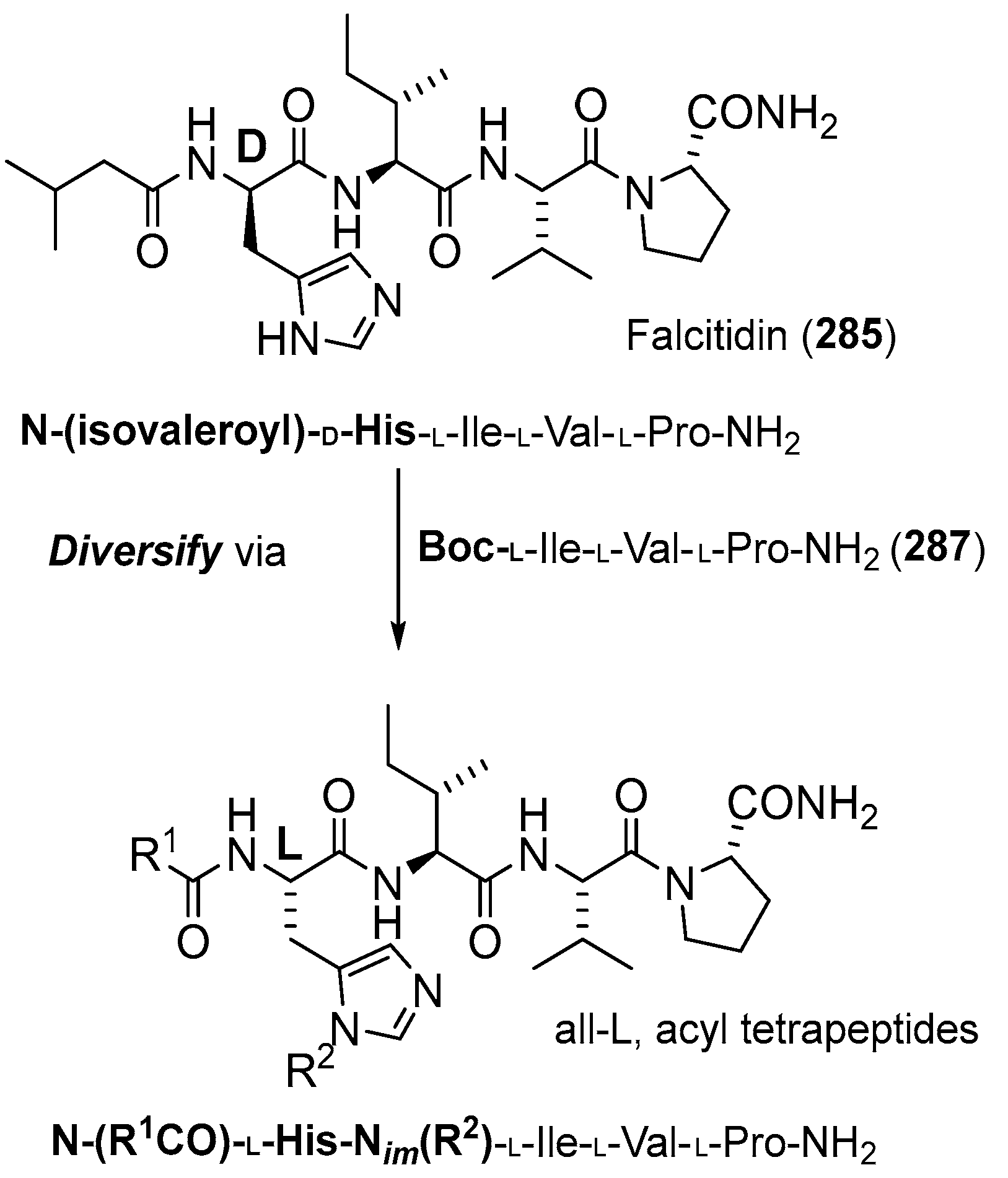
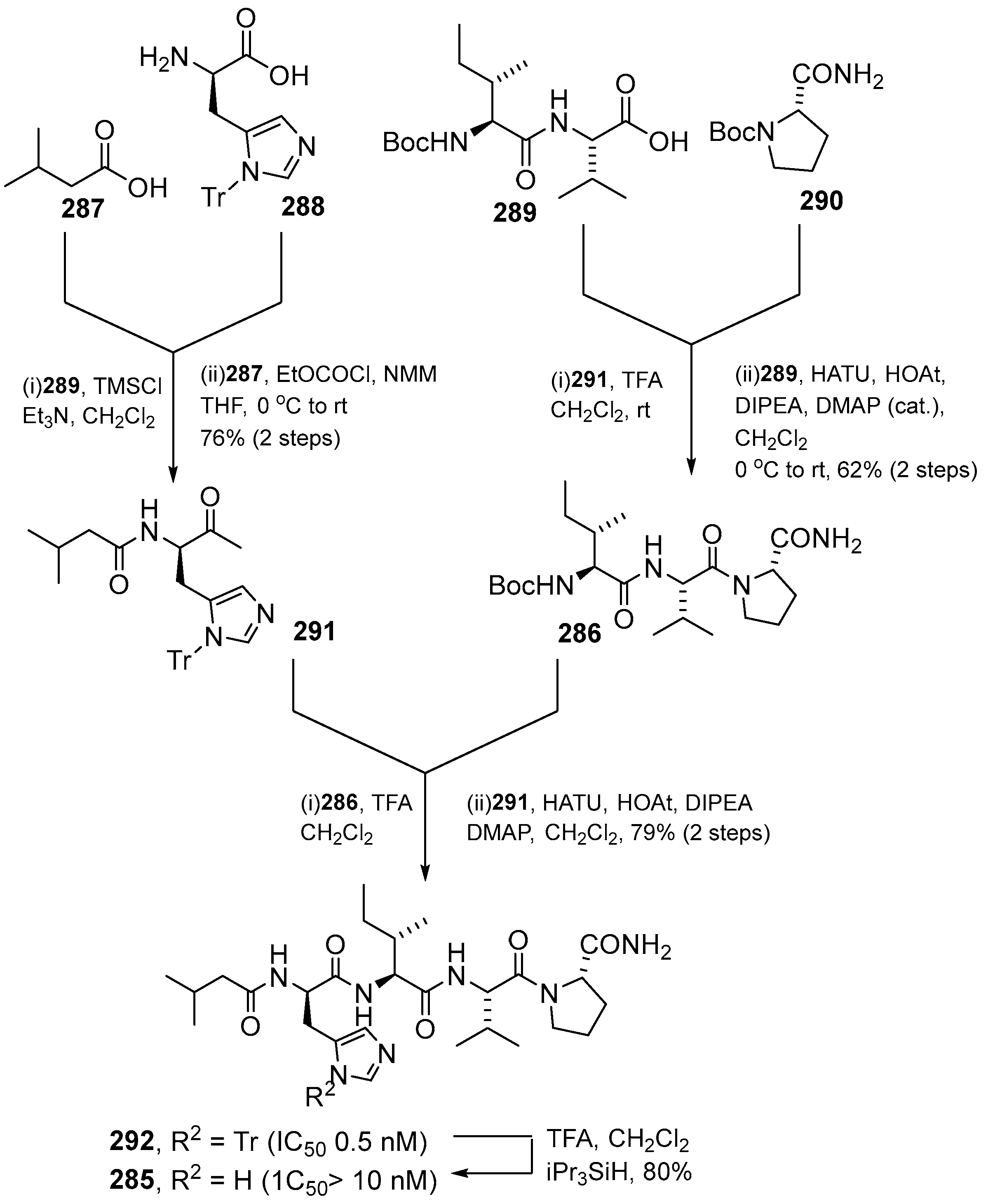
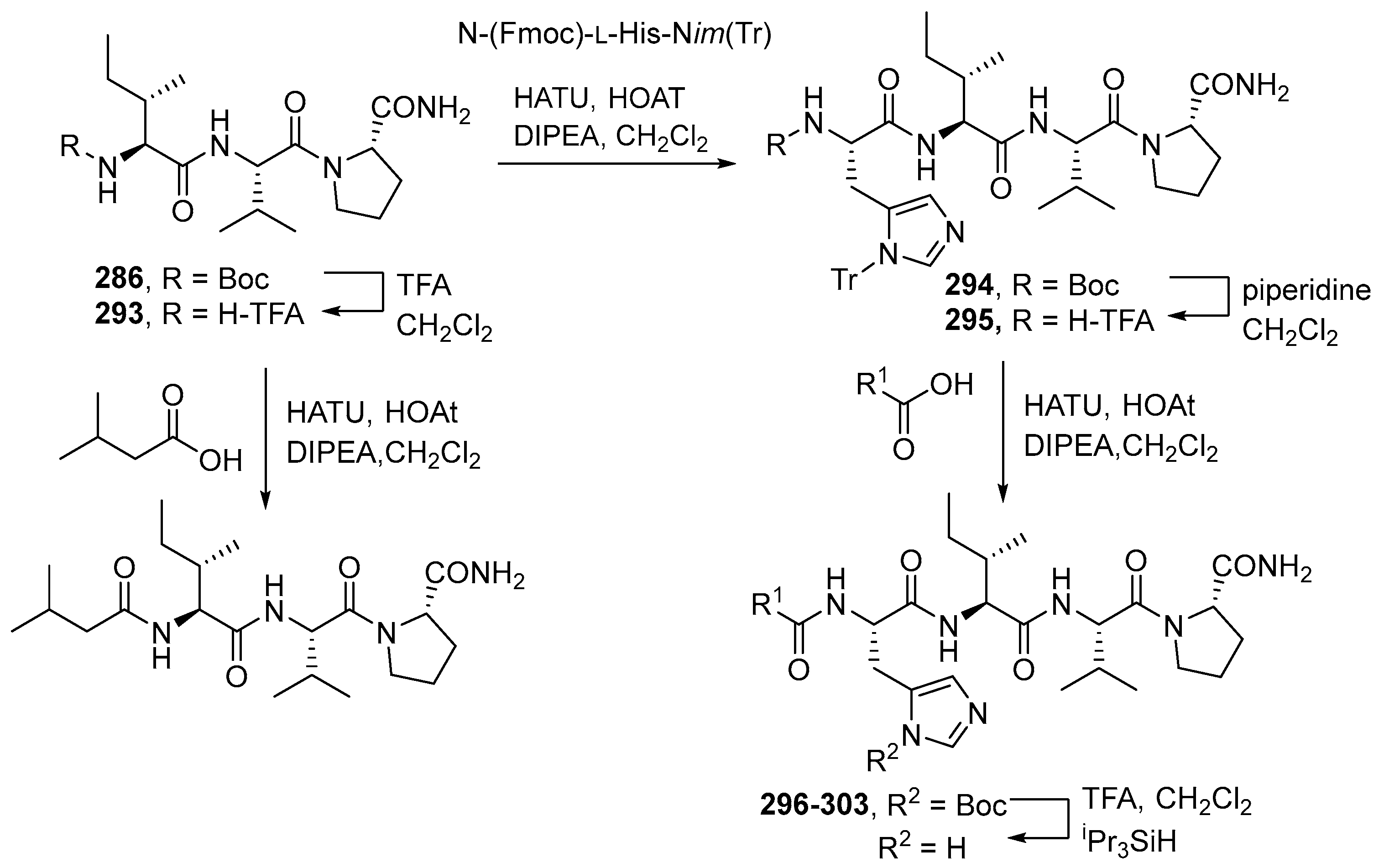
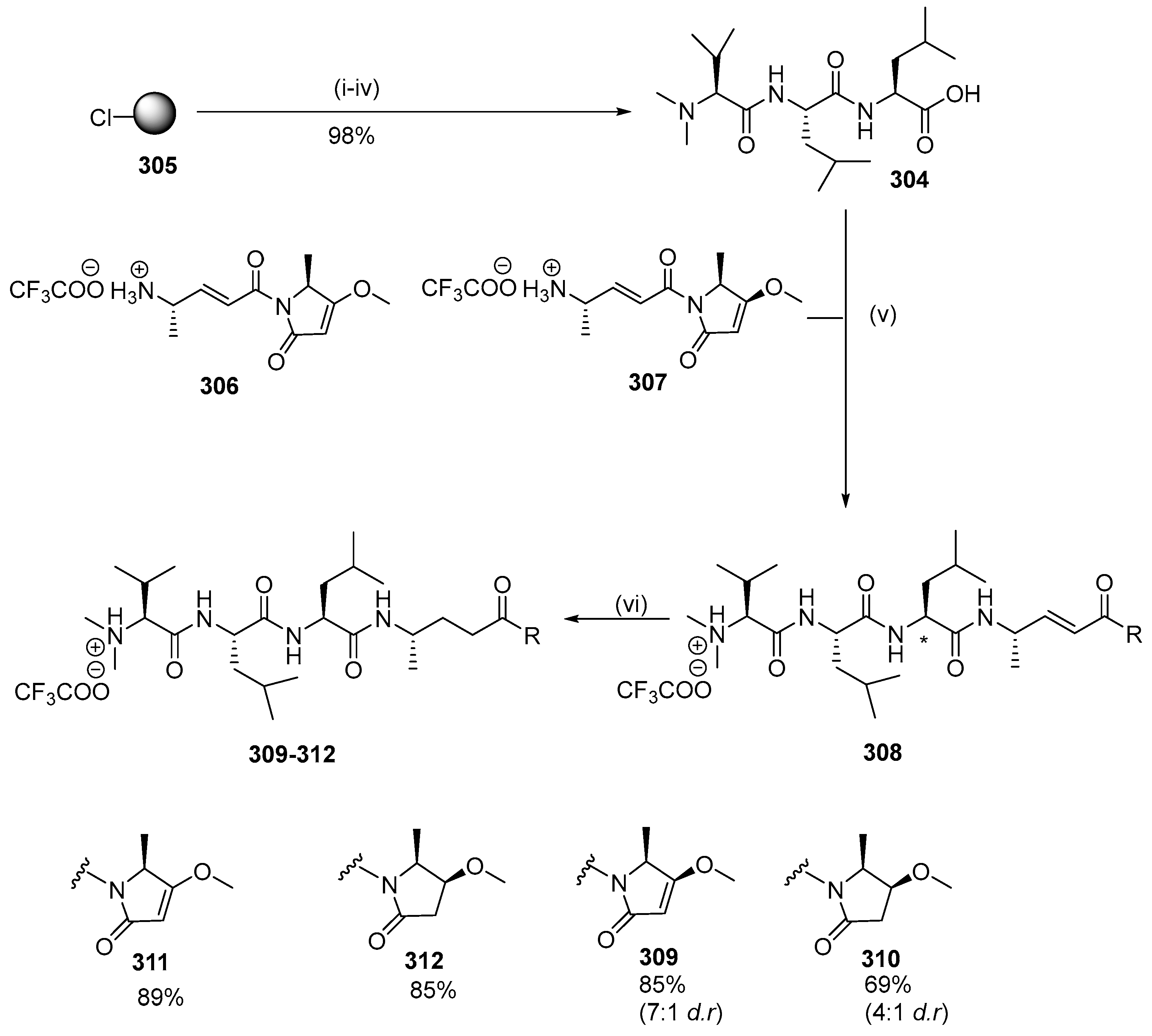
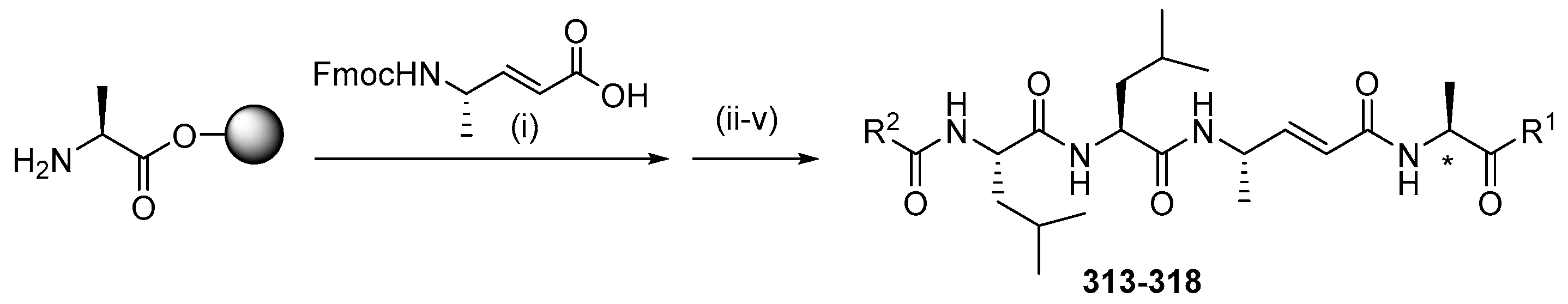






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Source | Reference | |
|---|---|---|---|
| 1 | Ribifolin | Jatropha ribifolia | [3] |
| 2 | Hymenocardine | Hymenocardia acida | [4] |
| 3 | Hymenocardinol | Hymenocardia acida | |
| 4 | Hymenocardine N-oxide | Hymenocardia acida | |
| 5 | Hymenocardine-H | Hymenocardia acida | |
| 6 | Nummularine-R | Ziziphus oxyphylla | [5] |
| 7 | O-desmethylnummularine-R | Ziziphus oxyphylla | |
| 8 | O-desmethylnummularine-R N-oxide | Ziziphus oxyphylla | |
| 9 | Hemsine-A | Ziziphus oxyphylla | |
| 10 | Hemsine-A N-oxide | Ziziphus oxyphylla | |
| 11 | Ramosine-A | Ziziphus oxyphylla | |
| 12 | Oxyphylline-C | Ziziphus oxyphylla | |
| 13 | Oxyphylline-E | Ziziphus oxyphylla | |
| 14 | Oxyphylline-F | Ziziphus oxyphylla | |
| 15 | Crotamine | rattlesnake venom | [6] |
| 16 | Octaminomycins A | a microbial metabolite fraction library of Streptomyces sp. RK85-270 | [7] |
| 17 | Octaminomycins B | a microbial metabolite fraction library of Streptomyces sp. RK85-270 | |
| 18 | Kakeromamide B | Fijian marine cyanobacterium Moorea producens | [8] |
| 19 | Pipecolisporin | Nigrospora oryzae | [9] |
| 20 | Kosidachins A | Pochonia binensis | [10] |
| 21 | Kosidachins B | Pochonia binensis | |
| 22 | Georatusin | Geomyces auratus | [11] |
| 23 | Friomaramide B | Inflatella coelosphaeroide | [12] |
| 24 | Shagamide A | Inflatella coelosphaeroide | |
| 25 | Shagamide B | Inflatella coelosphaeroide | |
| 26 | Shagamide C | Inflatella coelosphaeroide | |
| 27 | Shagamide D | Inflatella coelosphaeroide | |
| 28 | Shagamide E | Inflatella coelosphaeroide | |
| 29 | Shagamide F | Inflatella coelosphaeroide | |
| 30 | NpCI | Nerita versicolor | [13] |
| Peptide Precursor | Product | Time (Days) | Macrocycliz Yield c/Purity d (%) | Overall Yield e (%) |
|---|---|---|---|---|
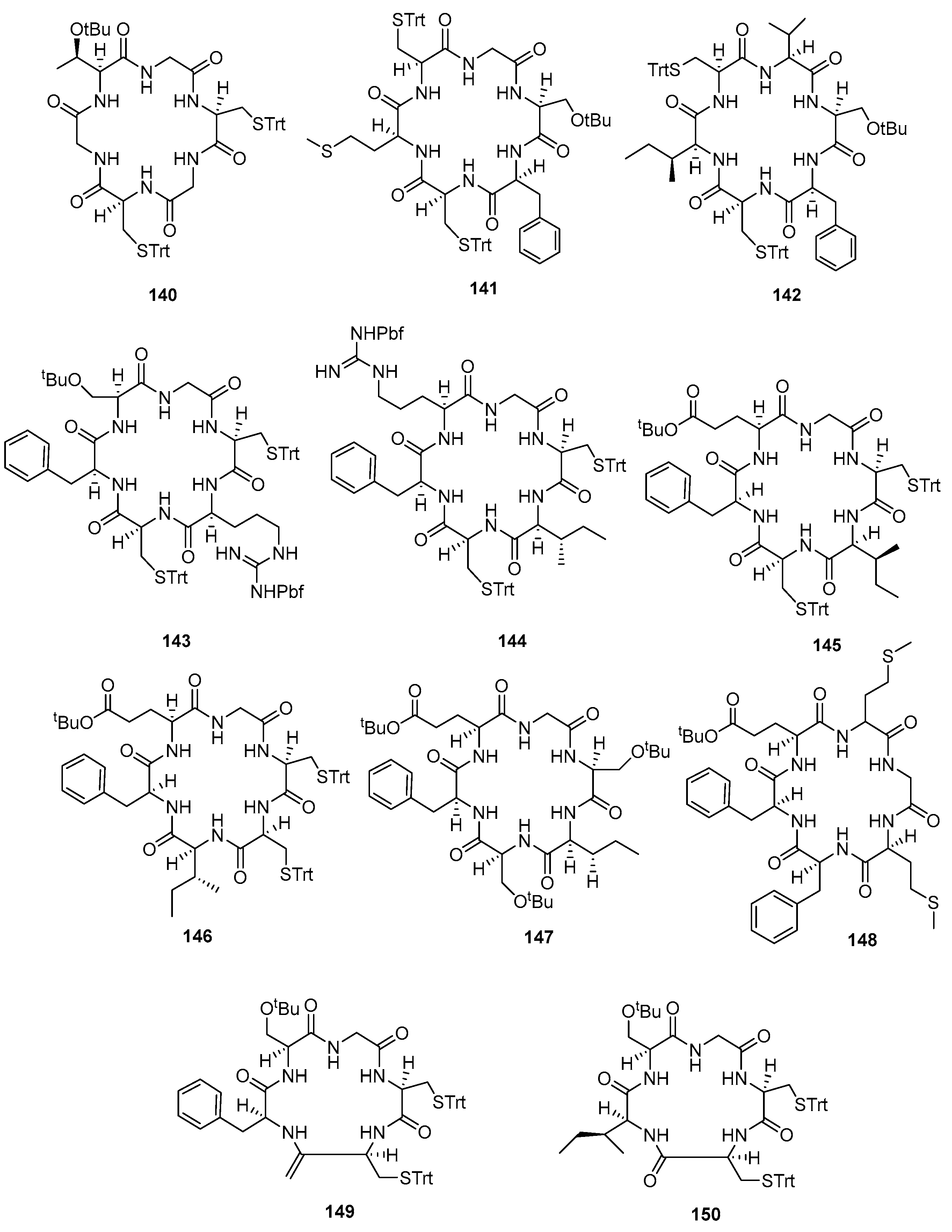
| H-L-Cys(Trt)-Gly-L-Thr(tBu)-Gly-L-Cys(Trt)-Gly-OH | 140 | 3 | 34 a/98 | 32 |
| H-L-Ser(tBu)-L-Phe-L-Cys(Trt)-L-Met-L-Cys(Trt)-Gly-OH | 141 | 3 | 38 a/85 | 31 |
| H-L-Val-Ser(tBu)-L-Phe-L-Cys(Trt)-L-Ile-L-Cys(Trt)-OH | 142 | 3 | 43 a/82 | 42 |
| H-L-Cys(Trt)-L-Arg(Pbf)-L-Cys(Trt)-L-Phe-L-Ser(tBu)-Gly-OH | 143 | 3 | 27 a/80 | 22 |
| H-L-Cys(Trt)-L-Ile-L-Cys(Trt)-L-Phe-L-Arg(Pbf)-Gly-OH | 144 | 3 | 26 a/93 | 23 |
| H-L-Cys(Trt)-L-Cys(Trt)-L-Ile-L-Phe-L-Glu(γ-tBu)-Gly-OH | 145 | 1 | 46 a/89–59 b/85 | 43 a/55 b |
| H-L-Cys(Trt)-L-Cys(Trt)-L-Ile-L-Phe-L-Glu(tBu)-Gly-OH | 146 | 2 | 59 a/83 | 51 |
| H-L-Ser(tBu)-L-Ile-L-Ser(tBu)-L-Phe-L-Glu(tBu)-Gly-OH | 147 | 3 | 34 a/93 | 32 |
| H-L-Met-L-Phe-L-Phe-L-Glu(tBu)-L-Met-Gly-OH | 148 | 2 | 80 a/93 | 63 |
| H-L-Cys(Trt)-L-Cys(Trt)-L-Phe-L-Ser(tBu)-Gly-OH | 149 | 1 | 54 a/94 | 51 |
| H-L-Cys(Trt)-L-Cys(Trt)-L-Ser(tBu)-Gly-OH | 150 | 1 | 69 a/96 | 58 |
| Peptide Precursor | Product | Crude Yield a/Purity b (%) | Isolated Macrocycle Yield a/Purity b (%) | Time (h) |
|---|---|---|---|---|
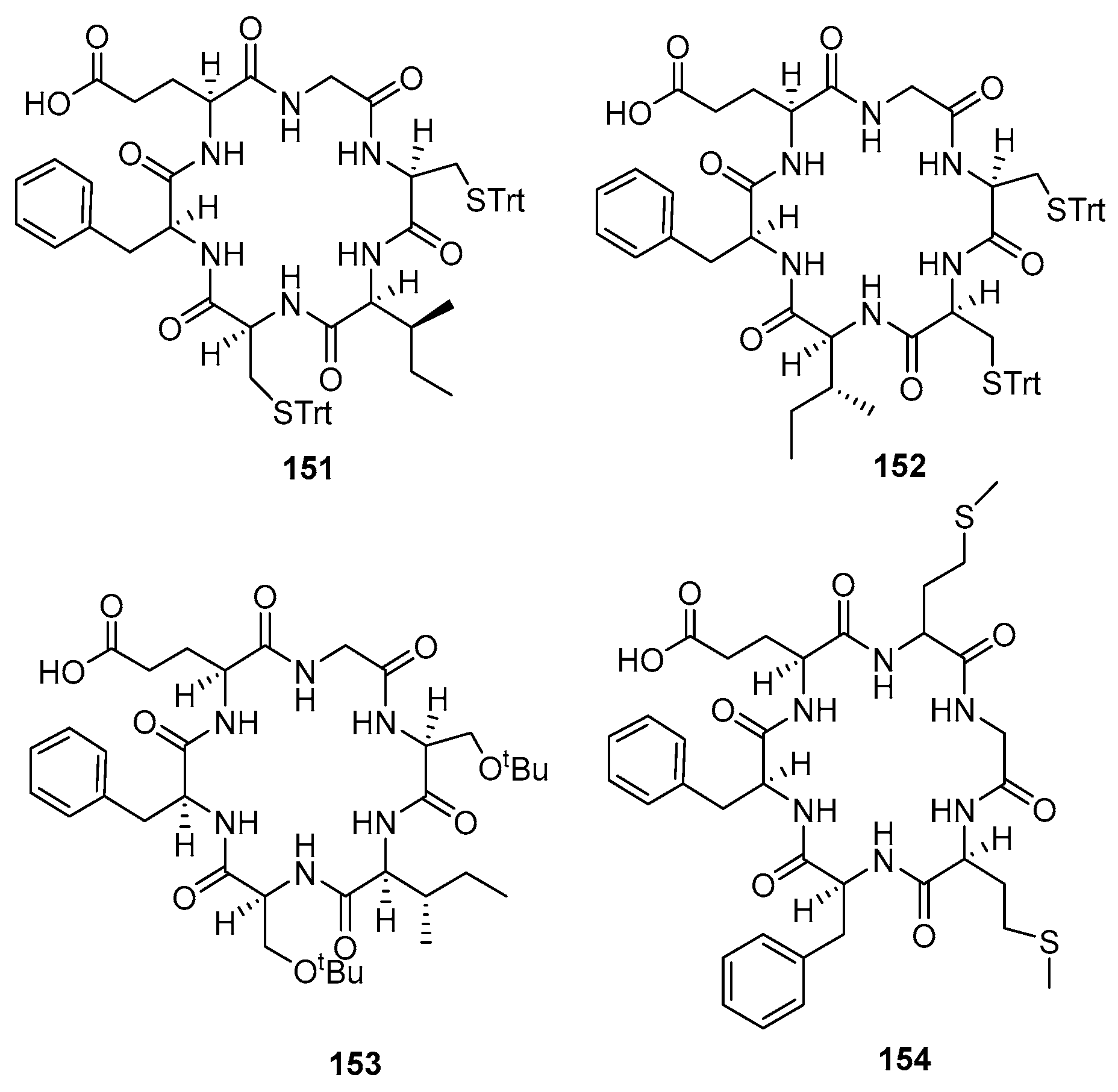
| H-Gly-L-Cys(Trt)-L-Ile-L-Cys(Trt)-L-Phe-L-Glu-OH | 151 | 68/81 | 51/95 | 16 |
| H-Gly-L-Cys(Trt)-L-Cys(Trt)-L-Ile-L-Phe-L-Glu-OH | 152 | 98/72 | 63/95 | 16 |
| H-Gly-L-Ser(tBu)-L-Ile-L-Ser(tBu)-L-Phe-L-Glu-OH | 153 | 63/78 | 46/93 | 16 |
| H-L-Met-Gly-L-Met-L-Phe-L-Phe-L-Glu-OH | 154 | 99/83 | 80/95 | 16 |
| Macrocyle Methodology A | Yield/Purity | Macrocycle Methodology B | Yield/Purity |
|---|---|---|---|
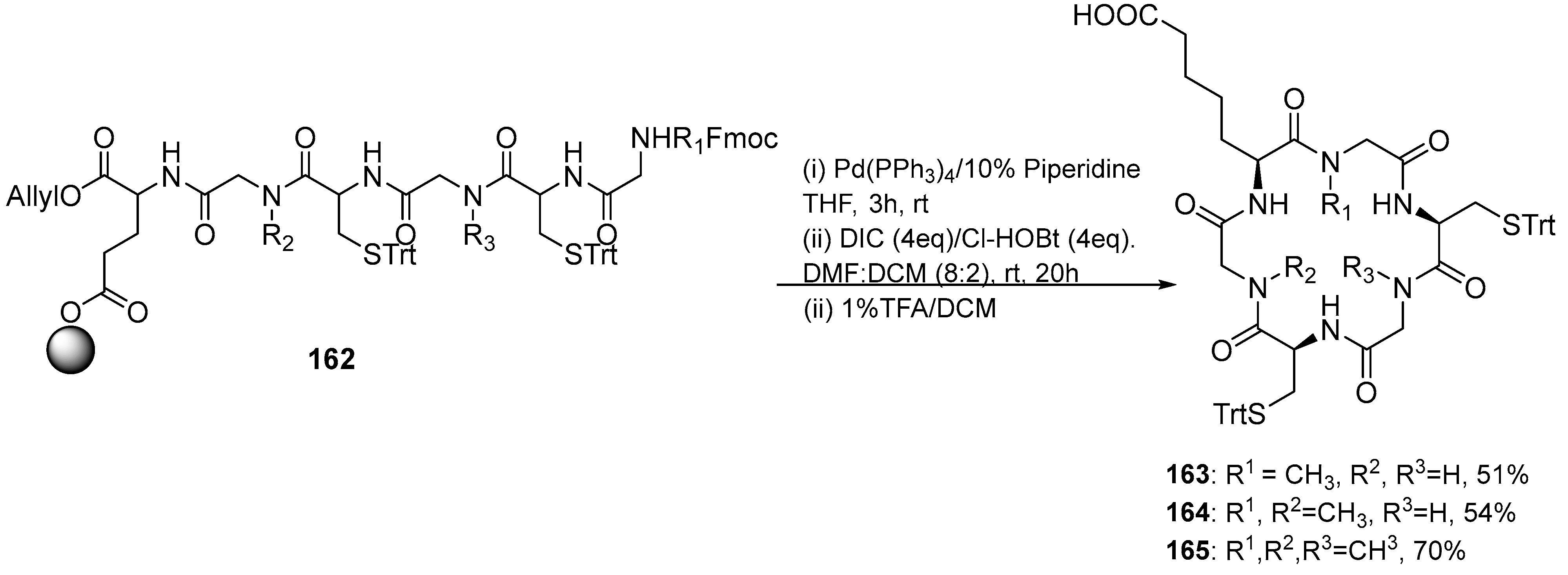
| 155 | 59 a/85 | 78 | 51/95 |
| 156 | 59/83 | 79 | 63/95 |
| 157 | 34/93 | 80 | 46/93 |
| 158 | 80/93 | 81 | 80/95 |
| cpd | I | II | III | X | R |
|---|---|---|---|---|---|
| 177 | allo-Ile | D-Phe | Gly | –O– | –CH3 |
| 178 | Leu | D-Phe | Gly | –O– | –CH3 |
| 179 | Val | D-Phe | Gly | –O– | –CH3 |
| 180 | Gly | D-Phe | Gly | –O– | –CH3 |
| 181 | Ile | D-Phe | Gly | –O– | –H |
| 182 | Ile | D-Phe | Gly | –NH– | –CH3 |
| 183 | Val | D-Phe | Gly | –NH– | –CH3 |
| 184 | allo-Ile | D-Phe | Gly | –NH– | –CH3 |
| 185 | allo-Ile | D-Phe (4-Cl) | Gly | –NH– | –CH3 |
| 186 | allo-Ile | D-Phe (4-OCH3) | Gly | –NH– | –CH3 |
| 187 | allo-Ile | D-Phe | Gly | –NH– | –CH3 |
| 188 | allo-Ile | D-Phe | Pro | –NH– | –CH3 |
| Entry | R1C(O) | R2 | Yield (%) |
|---|---|---|---|
| 296 | 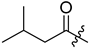 | Tr | 79 (from 295) |
| 297 | 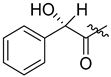 | H | 80 (from 296) |
| 298 | Tr | 67 (from 295) | |
| 299 | H | 20 (from 298) | |
| 300 | 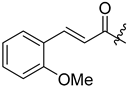 | H | 37 (from 295) |
| 301 |  | H | 33 (from 295) |
| 302 | Tr | 89 (from 295) | |
| 303 | 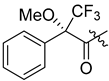 | H | 70 (from 302) |
| Analogue | R2 | R1 | Yield (Based on Original Resin Loading) |
|---|---|---|---|
| 313 |  | 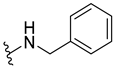 | 40% (5:1 d.r.) |
| 314 | 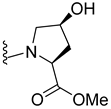 | 33% (1:1 d.r.) | |
| 315 | 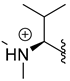 | 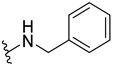 | 65% (7:1 d.r.) |
| 316 | 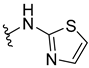 | 62% (1:1 d.r.) | |
| 317 | 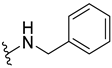 | 78% (6:1 d.r.) | |
| 318 | 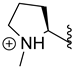 | 75% (5:1 d.r.) |
| cpd | R4 | R3 | R2a | R2b | Yield (%) |
|---|---|---|---|---|---|
| 358 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | 99 |
| 359 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2CH(CH3)2 | –H | 98 |
| 360 | 4-(N-Me)-piperidine– | –CH2-cyclohexyl | –CH2CH(CH3)2 | –H | 94 |
| 361 | 4-(N-Me)-piperidine– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | 99 |
| 362 | Me2NCH(iPr)– | –CH2Ph | –CH2CH(CH3)2 | –H | 91 |
| 363 | Me2NCH(iPr)– | –CH2CH2Ph | –CH2CH(CH3)2 | –H | 98 |
| 364 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH2Ph | –H | 94 |
| 365 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2CH2Ph | –H | 90 |
| 366 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2-cyclohexyl | –H | 90 |
| 367 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2-cyclohexyl | –H | 95 |
| 368 | 4-(N-Me)-piperidine– | –CH2-cyclohexyl | –CH2-cyclohexyl | –H | 81 |
| 369 | 4-(N-Me)-piperidine– | –CH2CH(CH3)2 | –CH2-cyclohexyl | –H | 98 |
| 370 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH3 | –CH3 | 93 |
| cpd | R4 | R3 | R2a | R2b | R1a | R1b | Yield (%) |
|---|---|---|---|---|---|---|---|
| 352 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | –CH3 | –H | 91 |
| 371 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2CH(CH3)2 | –H | –CH3 | –H | 72 |
| 372 | 4-(N-Me)-piperidine– | –CH2-cyclohexyl | –CH2CH(CH3)2 | –H | –CH3 | –H | 86 |
| 373 | 4-(N-Me)-piperidine– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | –CH3 | –H | 78 |
| 374 | Me2NCH(iPr)– | –CH2Ph | –CH2CH(CH3)2 | –H | –CH3 | –H | 92 |
| 375 | Me2NCH(iPr)– | –CH2CH2Ph | –CH2CH(CH3)2 | –H | –CH3 | –H | 83 |
| 376 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH2Ph | –H | –CH3 | –H | 90 |
| 377 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2CH2Ph | –H | –CH3 | –H | 87 |
| 378 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2-cyclohexyl | –H | –CH3 | –H | 93 |
| 379 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2-cyclohexyl | –H | –CH3 | –H | 71 |
| 380 | 4-(N-Me)piperidine– | –CH2-cyclohexyl | –CH2-cyclohexyl | –H | –CH3 | –H | 68 |
| 381 | 4-(N-Me)piperidine– | –CH2CH(CH3)2 | –CH2-cyclohexyl | –H | –CH3 | –H | 95 |
| 382 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH3 | –CH3 | –CH3 | –H | 81 |
| 383 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | –CH3 | –CH3 | 51 a |
| 384 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | –cyclohexyl | –H | 69 |
| 385 | Me2NCH(iPr)– | –CH2-cyclohexyl | –CH2CH(CH3)2 | –H | –cyclohexyl | –H | 65 |
| 386 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | –CH2-cyclohexyl | –H | 88 |
| 387 | Me2NCH(iPr)– | –CH2CH(CH3)2 | –CH2CH(CH3)2 | –H | –CH2CH2Ph | –H | 83 |
| Peptides | Amino Acid Sequence | Position | Mol. Mass (Da) |
|---|---|---|---|
| SE5 P1 (388) | Ac-TTKKESKIYDYYL-NH2 | 830–842 aa | 1821 |
| SE5 P2 (389) | Ac-KASPEFYHNLYFKNF-NH2 | 843–857 aa | 1946 |
| SE5 P3 (390) | Ac-NVGKKNLFSEKEDN-NH2 | 858–871 aa | 1663 |
| SE5 P4 (391) | Ac-KKYVKLPSNGTTGEQ-NH2 | 176–190 aa | 1691 |
| SE5 P5 (392) | Ac-GSSTGTVRGDTEPISDS-NH2 | 191–207 aa | 1707 |
| SE5 P6 (393) | Ac-SESLPANGPDSPTVK-NH2 | 233–247 aa | 1540 |
| SE5 P7 (394) | Ac-FKEIKAETEDDDEDDY-NH2 | 384–399 aa | 2003 |
| SE5 P8 (395) | Ac-YIIFGQDTAGSGQSGK-NH2 | 881–896 aa | 1670 |
| SE5 P9 (396) | Ac-TALESAGTSNEVSERV-NH2 | 900–915 aa | 1620 |
| Entry | Name | Sequence | HPLC Purity a (%) | Calcd Mass b (g mol−1) | Obsd Mass b (g mol−1) |
|---|---|---|---|---|---|
| 397 | [Ile6,His5]-All | DRVYHIPF | 98 | 1045.5 | 1046 |
| 398 | des-Ile5, His6-All | DRVYPF | 98 | 795.4 | 796 |
| 399 | des-Asp1-Val3,Ile5,His6-All | RYPF | 99 | 581.3 | 582 |
| 400 | [Ile4,His3]-des-Asp1,Val3-All | RYHIPF | 98 | 831.4 | 833 |
| 401 | des-Asp1,Arg2,Val3,Ile5-All | YHPF | 99 | 562.2 | 563 |
| 402 | des-Asp1,Arg2,Tyr4,His6-All | VIPF | 99 | 474.3 | 475 |
| 403 | [2-Na1]-des-Asp1-Arg2,Tyr4,His6-All | (2-NaI)-IPF | 97 | 572.3 | 574 |
| 404 | [2-Na2]-des-Asp1-Arg2,Tyr4,His6-All | V-(2-NaI)-PF | 99 | 558.3 | 559 |
| 405 | [2-Na3]-des-Asp1-Arg2,Tyr4,His6-All | VI-(2-NaI)-F | 97 | 574.3 | 575 |
| 406 | [2-Na4]-des-Asp1-Arg2,Tyr4,His6-All | VIP-(2-NaI) | 98 | 523.3 | 524 |
| Entry | Sequence | HPLC Purity a | Calculated Mass | Observed Mass b |
|---|---|---|---|---|
| 416 | Glu-Asp-Arg-Orn-Val-Tyr-Ile-His-Pro-Phe | 99% | 1272.4 | 1271.4 |
| 417 | Glu-Asp-Arg-Lys-Val-Tyr-Ile-His-Pro-Phe | 99% | 1284.4 | 1286.1 |
| 418 | Asp-Glu-Arg-Val-Orn-Tyr-Ile-His-Pro-Phe | 99% | 1272.4 | 1272.4 |
| 419 | Asp-Glu-Arg-Val-Lys-Tyr-Ile-His-Pro-Phe | 99% | 1284.4 | 1286.4 |
| 420 | DGlu-Asp-Arg-Val-Orn-Tyr-Ile-His-Pro-Phe | 98% | 1272.4 | 1272.4 |
| 421 | Glu-Asp-Arg-Val-DOrn-Tyr-Ile-His-Pro-Phe | 98% | 1272.4 | 1272.4 |
| 422 | Asp-Arg-Cys-Val-Cys-Tyr-Ile-His-Pro-Phe | 99% | 1249.4 | 1250.4 |
| 423 | Cys-Asp-Arg-Val-Cys-Tyr-Ile-His-Pro-Phe | 98% | 1249.4 | 1250.4 |
| 424 | Asp-Cys-Arg-Cys-Val-Tyr-Ile-His-Pro-Phe | 99% | 1249.4 | 1250.4 |
| 425 | Cys-Asp-Arg-Cys-Val-Tyr-Ile-His-Pro-Phe | 99% | 1249.4 | 1249.4 |
| Entry | Sequence | HPLC Purity a (%) | Calculed Mass b (g mol−1) | Observed Mass b (g mol−1) | Antiplasmodial Activity vs. P. gallinaceum (% Fluorescent Sporozoltes) | Antiplasmodial Activity vs. P. falciparum (New Ring Formation%) | Design Rationale |
|---|---|---|---|---|---|---|---|
| Ang II | DRVYIHPF | 88 | 47 | WT peptide | |||
| 426 | CDRVYIHPFC | 98 | 1249.6 | 1250 | 92 | 42 | Contrained version of Ang II |
| 427 | CDRVYHIPFC | 98 | 1249.6 | 1250 | 91 | 42 | Residues H and I were reversed, which is expected to increase antiplasmodial activity based on previously described linear variant. |
| 428 | CDRVCYHIPF | 98 | 1249.6 | 1250 | 82 | 11 | Designed based on most potent antiplasmodial peptide (CDRVCYIHPF) obtained previously, with an additional 1H reverse modification. |
| 429 | CDRVYPFC | 97 | 999.4 | 1000 | 95 | 9 | Hydrophobic patch composed of H and I was deleted to asses its influence on functions |
| 430 | CRYHIPFC | 98 | 1035.5 | 103.6 | 98 | 0 | D and V were deleted to optimize interactions between R and Y within the same plane. I and H were reversed to evaluate the effect of new pi-stack interactions in the activity. |
| 431 | CRYPFC | 99 | 765.3 | 786 | 4 | 29 | Most important residues for function within the Ang II sequence were kept to build a minimal peptide with antiplasmodial activity. |
| 432 | CYHPFC | 97 | 766.3 | 767 | 6 | 17 | Only aromatic residues were kept, inspired by the linear form (YHPF), which was one of our lead peptides previously described. |
| 433 | CVIPFC | 98 | 675.3 | 676 | 7 | 3.5 | Aliphatic and hydrophobic residue F were kept based on previous lead linear peptide (VIPF) |
| Peptide | Sequence | H | µH | q | Purity a | Calculated Mass (Da) | Observed Mass b (Da) |
|---|---|---|---|---|---|---|---|
| Dec-NH2 (434) | SLLSLIRKLIT-NH2 | 0.780 | 0.652 | +3 | 99% | 1254.8 | 1256 |
| [Leu]8-Dec-NH2 (435) | SLLSLIRLLIT-NH2 | 1.025 | 0.479 | +2 | 98% | 1239.8 | 1241 |
| [Leu]10-Dec-NH2 (436) | SLLSLIRKLLT-NH2 | 0.771 | 0.647 | +3 | 99% | 1254.8 | 1256 |
| [Pro]4-Dec-NH2 (437) | SLLPLIRKLIRT-NH2 | 0.849 | 0.583 | +3 | 99% | 1265.8 | 1267 |
| [Arg]1-Dec-NH2 (438) | RLLSLIRKLIT- NH2 | 0.692 | 0.704 | +4 | 99% | 1339.9 | 1340 |
| [Phe]2-Dec-NH2 (439) | SFLSLIRKLIT- NH2 | 0.788 | 0.659 | +3 | 99% | 1288.8 | 1290 |
| [Phe]6-Dec-NH2 (440) | SLLSLFRKLIT- NH2 | 0.779 | 0.651 | +3 | 99% | 1288.8 | 1290 |
| [Phe]6-Dec[Thr]11-Dec-NH2 (441) | SLLSLFRKLI- NH2 | 0.831 | 0.392 | +3 | 98% | 1188.8 | 1190 |
| Species | Compounds | Sequences | Antiplasmodial Activity (IC50 (µM)) | References |
|---|---|---|---|---|
| Jatropha ribofilia | Ribifolin (1) | cyclo ILGSIILG | 42 | [3] |
| Jatropha curcas | Curcacyline B | cyclo ILGSPILLG | 10,000 | [3,36] |
| Jatropha chevalieri | Chevalierin A | cyclo IMGIPILA | 9 | [3,37] |
| Jatropha mahafalensis | Mahafacyclin A | cyclo VFGTILG | 16 | [3,38] |
| Jatropha mahafalensis | Mahafacyclin B | cyclo FFGTFFG | 2 | [3,39] |
| Jatropha pohliana | Pohlianin A | cyclo VLLYPLG | 57 | [3,40] |
| Jatropha pohliana | Pohlianin B | cyclo LLLYPLG | 25 | [3,40] |
| Jatropha pohliana | Pohlianin C | cyclo FGGTIIFG | 16 | [3,40] |
| Compound | P. falciparum KI | MRC-5 |
|---|---|---|
| 2 | 16.4 ± 7 | 51.1 ± 17 |
| 3 | 17.5 ± 9 | >64.0 |
| 4 | 12.2 ± 7 | >64.0 |
| 5 | 27.9 ± 17 | >64.0 |
| chloroquine | 0.2 ± 0.1 |
| Compound | P. falciparum KI | MRC-5 |
|---|---|---|
| 6 | 3.2 ± 3 | 30.6 ± 4 |
| 7 | 7.1 ± 2 | >64.0 |
| 9 | 13.6 ± 9 | >64.0 |
| 11 | >32.0 | >64.0 |
| 14 | 7.4 ± 3 | 31.2 ± 1 |
| chloroquine | 0.3 ± 0.2 |
| Compound | P. falciparum 3D7 | P. falciparum Dd2 | P. falciparum KI |
|---|---|---|---|
| 16 | 1.5 | 1.6 | 1.3 |
| 17 | 1.5 | 1.1 | 0.83 |
| chloroquine | 0.041 | 0.52 | 0.46 |
| Compound | Blood-Stage P. falciparum | Liver-Stage P. berghei | HEK2931 Cytotoxicity | HepG2 Cytotoxicity |
|---|---|---|---|---|
| 18 | 8.9 | 11, >12 | >23 | >23 |
| Atovaquone (+control) | 0.0061 | <0.00028, 0.0017, 0.0037 | >2.0 | not tested |
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| Antiplasmodial Activity | Cytotoxicity | Selectivity Index | |||
| pf KI a | pf FCR3 b | MRC-5 | MRC-5/pf KI | MRC-5/pf FCR3 | |
| 20 | 12.5 ± 1 | 17.1 ± 1.0 | 6.8 ± 2 | 0.5 | 0.4 |
| 21 | 0.83 ± 0.04 | 0.89 ± 0.04 | 14.7 ± 4 | 18.4 | 16.3 |
| artemisinin c | 0.022 ± 0.001 | 0.026 ± 0.003 | >25 | >1136 | >961 |
| Compound | Inhibition (%) |
|---|---|
| 20 | 18.3 ± 6 |
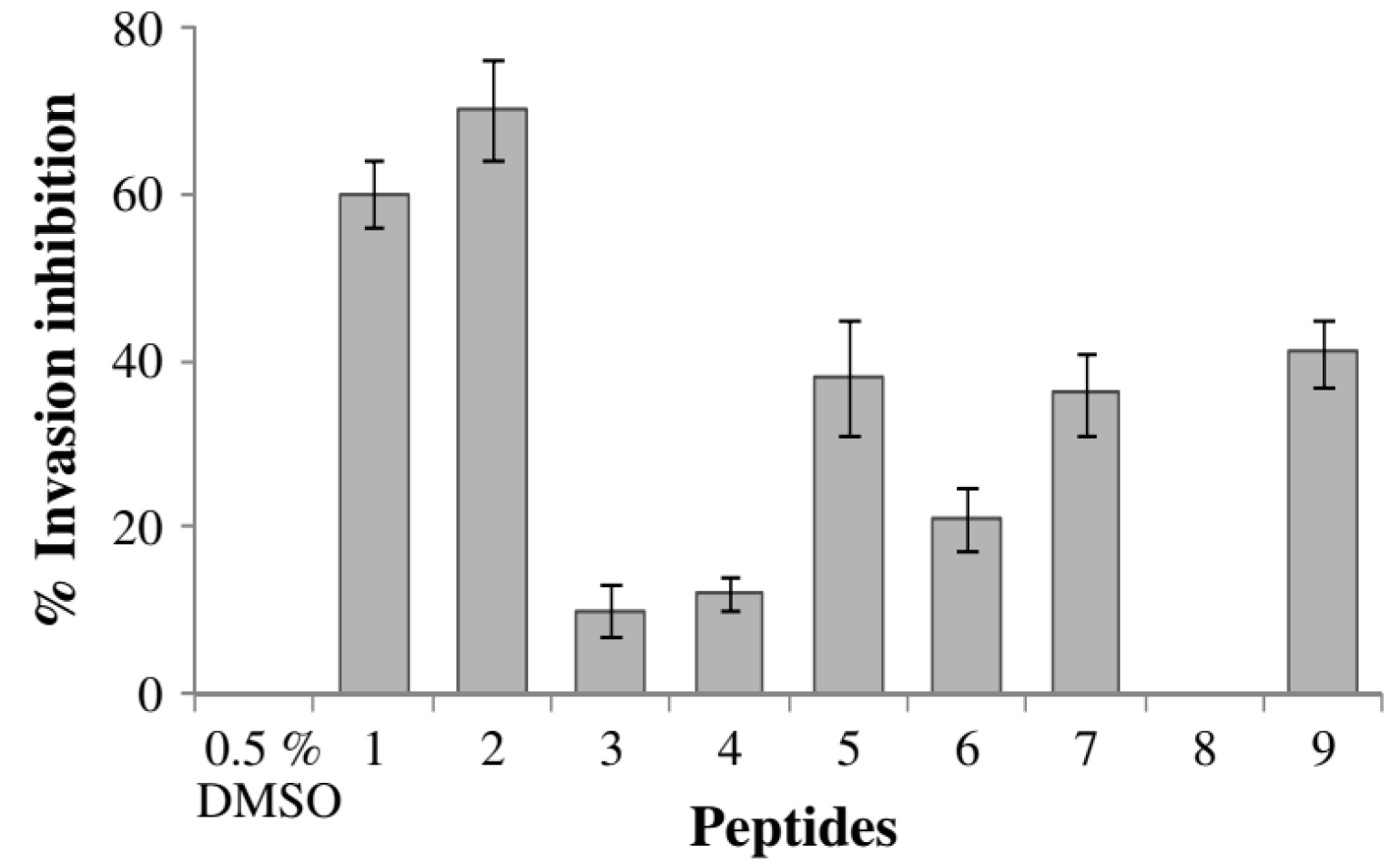
| 21 | 41.0 ± 2 |
| Artesunate a | 99.7 ± 0.10 |
| Compound | NF54 | Dd2 | 3D7 |
|---|---|---|---|
| 23 | 3.93 | 3.30 | 8.65 |
| 24 | 2.61 | 1.72 | 2.10 |
| 25 | >10 | 4.37 | >10 |
| 26 | 3.73 | 1.07 | 2.52 |
| 27 | 3.05 | 2.02 | >10 |
| 28 | >10 | >10 | >10 |
| 29 | >10 | >10 | >10 |
| CQ | 0.03 | 1.28 | >10 |
| Entry | Compound | R′ | R″ | R‴ | Pfalcp IC50 [nm] 3D7 | IC50 [nm] Dd2 | Mtb MIC [µM] wt | Overall Synthetic Steps |
|---|---|---|---|---|---|---|---|---|
| ref Mtb | Isoniazid | 0.9 | ||||||
| ref Pfalcp | CQ | 3.4 | 233.9 | |||||
| 1 | desoxycyclomarin C |  |  | Me | 39.8 | NA | 0.9 | 37 |
| 2 | 96 | 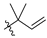 | 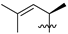 | Me | 9.0 | 12.9 | 0.5 | 34 |
| 3 | 97 |  | 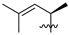 | Me | 4.4 | 6.5 | 1.7 | 31 |
| 4 | 98 | iPr | 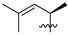 | Me | 47.8 | 76.0 | 0.4 | 36 |
| 5 | 99 | Me | 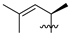 | Me | 13.4 | 17.8 | 0.9 | 31 |
| 6 | 100 | H | 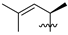 | Me | 28.1 | 27.5 | 1.5 | 31 |
| 7 | 106 | 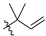 | 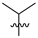 | Me | 34.4 | 71.8 | 21.2 | 32 |
| 8 | 107 | 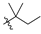 | 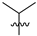 | Me | 57.6 | 200.2 | 1.7 | 33 |
| 9 | 108 | iPr |  | Me | 355.7 | 300.3 | 0.4 | 34 |
| 10 | 109 | Me |  | Me | 230.3 | 256.7 | 1.6 | 29 |
| 11 | 110 | H | 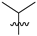 | Me | 314.2 | 421.5 | 0.5 | 29 |
| 12 | 119 | 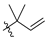 | 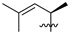 | H | 47.9 | 36.7 | 2.3 | 32 |
| 13 | 120 |  | 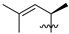 | H | 452.0 | 346.5 | >32 | 33 |
| 14 | 121 | iPr | 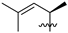 | H | 177.5 | 287.8 | 1.8 | 34 |
| 15 | 122 | Me | 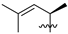 | H | 362.4 | 318.4 | 3.4 | 32 |
| 16 | 123 |  | 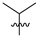 | H | 303.5 | 260.8 | 2.6 | 30 |
| 17 | 124 |  |  | H | 428.5 | 305.1 | 3.2 | 31 |
| 18 | 125 | H | 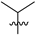 | H | 287.4 | 196.7 | 4.3 | 36 |
| Compound | EC50 P. falciparum K1 (µM) | Compound | EC50 P. falciparum K1 (µM) |
|---|---|---|---|
| 140 | 0.028 ± 0.006 | 148 | 4.6 ± 1 |
| 141 | 0.68 ± 0.04 | 149 | 8.4 ± 1 |
| 142 | 6.50 ± 0.05 | 150 | 0.73 ± 0.06 |
| 143 | 16.8 ± 1 | 151 | 0.47 ± 0.04 |
| 144 | 43.1 ± 1 | 152 | 0.42 ± 0.05 |
| 145 | 0.42 ± 0.03 | 153 | >50 |
| 146 | >10 | 154 | >50 |
| 147 | 3.8 ± 1 |
| Cyclopeptide | EC50/EC50 P. falciparum K1 (µM) b | EC50 P. falciparum 3D7 (µM) c | SI IC50 d/IC50 PfK1 | SI IC50 e/IC50 Pf3D7 |
|---|---|---|---|---|
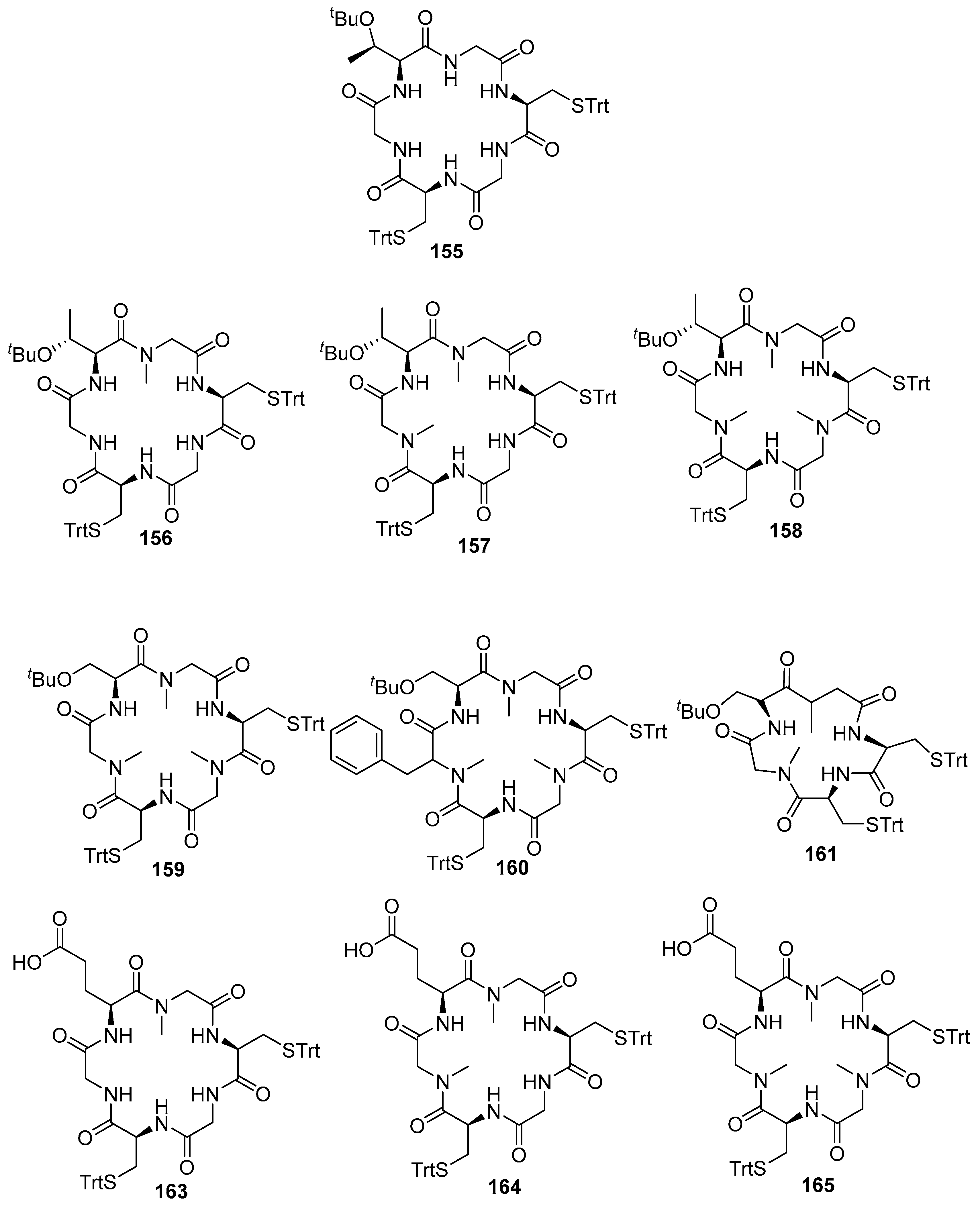
| 156 | 8.0 ± 0.5/39 ± 3 | 3.8 ± 0.1 | >1250 | >6.6 × 104 |
| 157 | 0.008 ± 0.004/1.0 ± 0.5 | 0.25 ± 0.02 | >1.3 × 107 | >1.0 × 106 |
| 158 | 0.040 ± 0.006/1.0 ± 0.3 | 1.0 ± 0.3 | >2.5 × 106 | >2.5 × 105 |
| 159 | 0.13 ± 0.04/4.0 ± 0.6 | 1.4 ± 0.6 | >7.7 × 105 | >1.8 × 105 |
| 160 | 9.0 ± 0.5/59 ± 3 | 1.8 ± 0.1 | >1.1 × 104 | > 1.4 × 105 |
| 161 | 150 ± 5/900 ± 9 | nt | nt | nt |
| 163 | nt | 5400 ± 100 | nt | >46 |
| 164 | nt | 210 ± 10 | nt | >1.2 × 103 |
| 165 | 0.20 ± 0.04/4.0 ± 0.6 | 12 ± 1 | 5 × 105 | >2 × 104 |
| Bioactivity/ADME Profile Compound 177 | Values |
|---|---|
| antimicrobial activities | |
| MIC (Mtb H73Rv) | >80 µM |
| MIC (M. vaccae) | 18.8 µM |
| IC50 (P. falciparum 3D7) | 2.3 µM |
| cytotoxicity | |
| IC50 (Hep2G cells) | >100 µM |
| membrane permeability a | 280 nm/s |
| % human serum albumin binding | 88% |
| Intrinsic microsomal clearance (Clint) | |
| Human | 46.53 mL/(min g) tissue |
| Mouse | 38.3 mL/(min g) tissue |
| Plasma stability (% remain after 2 h incubation) b | |
| Human plasma | 103% |
| Mouse plasma | 18% |
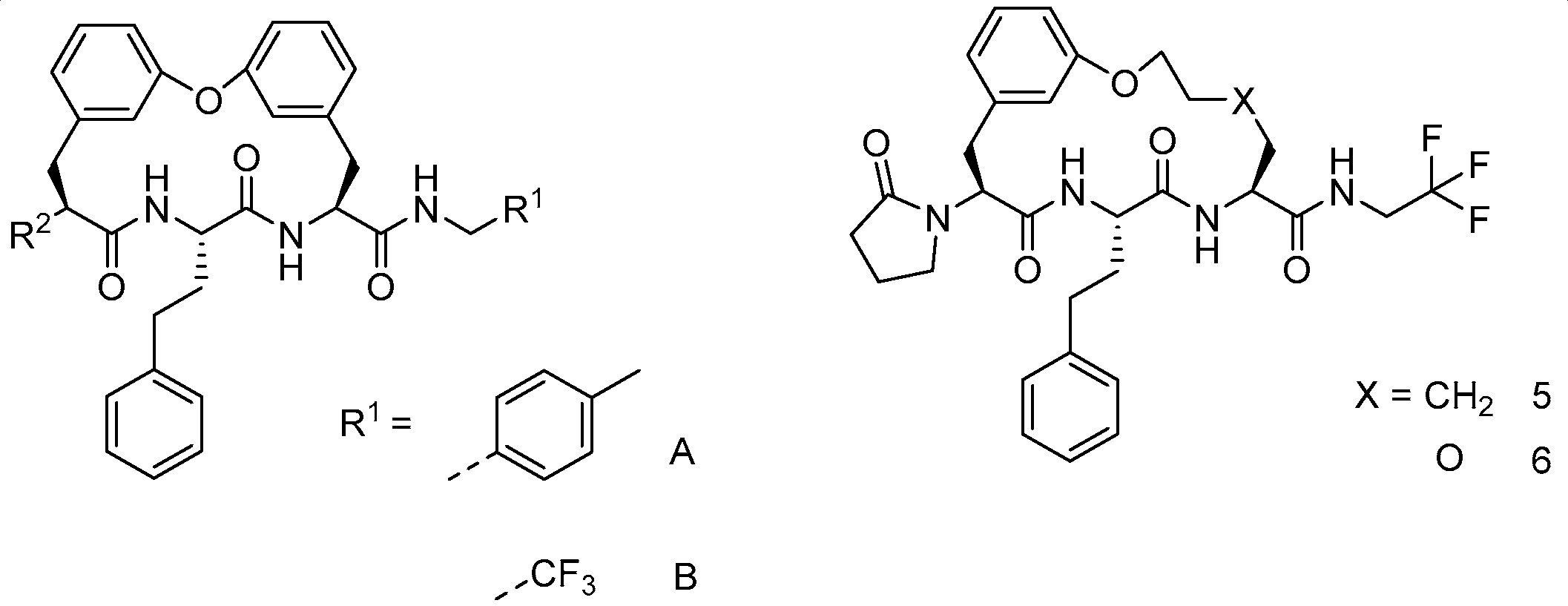 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (µM) | β5 IC50 (µM) | Permeability | Liver Microsomal Stability | Kinetic Solubility | Cytotoxicity HepG2 | Lipopholicity | ||||||
| ID | R1 | R2 | 3D7 | Pf20S | c20S | i20S | PAMPA | MDR | m/hLM | pH 6.8 | Viability % | cLogP |
| cyclic peptide 201 | A | 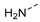 | 0.078 | 0.13 | >100 | >100 | Un-detected | >2.9/<1 | 90.6 179.4 | <1.6 | 71 | 6.3 |
| 202 | A |  | 0.002 | 0.017 | >100 | >100 | 22 | >34/1 | 171.3 N.D. | - | 93 | 6.2 |
| 203 | B |  | 0.003 | 0.014 | 4.0 | >100 | 151 | >0.71/<4 | 269.7 11.2 | <1.4 | 93 | 4.7 |
| 204 | B |  | 0.0002 | 0.003 | 26.3 | >100 | 214 | 31/5 | >768 644.3 | 3.8 | 77 | 5.3 |
| 214 | - | - | 0.002 | 0.019 | 2.4 | >100 | 187 | 53/2 | 566.6 349.8 | 76 | 97 | 4.2 |
| 220 | - | - | 0.019 | 0.080 | 6.4 | >100 | 120 | 89/2 | 397.4 275.7 | >130 | 109 | 3.0 |
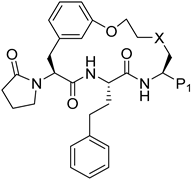 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (µM) | β5 IC50 (µM) | Permeability | Metabolic Stability | Kinetic Solubility | Cytotoxicity HepG2 | Lipopholicity | |||||
| ID | P1 | 3D7 | Pf20S | c20S | i20S | PAMPA | MDR | m/hLM | pH 6.8 | Viability % | cLogP |
| 220 | 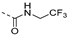 | 0.002 | 0.019 | 2.4 | >100 | 187 | 53/2 | 566.6 349.8 | 76 | 97 | 4.2 |
| 221 | 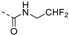 | 0.008 | 0.022 | 8.9 | >100 | 72 | >11/<5 | 288 187.7 | 35 | 101 | 4.0 |
| 222 | 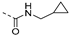 | 0.003 | 0.007 | 3.0 | >100 | 103 | 68/2 | 477.6 340.6 | 49 | 101 | 4.4 |
| 223 | 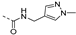 | 0.008 | 0.054 | 9.8 | >100 | 11 | >6/<1 | 261 187.7 | >120 | 107 | 3.4 |
| 224 |  | >2.77 | 23.8 | >100 | >100 | - | - | - | - | - | 4.8 |
| 225 | 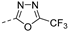 | >2.77 | 9.0 | >100 | >100 | 160 | 11/17 | >768 672.1 | 6.2 | 109 | 3.3 |
| 226 |  | >2.77 | 64.0 | >100 | >100 | 82 | >21/<1 | 59.6 28.6 | 2 | 103 | 3.7 |
| 227 |  | 0.398 | 2.5 | >100 | >100 | 145 | 44/2 | 703.4 288 | 31 | 111 | 4.5 |
| 228 | 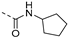 | 0.001 | 0.003 | 2.5 | >100 | 151 | 57/3 | 657.8 596.5 | 100 | 96 | 4.9 |
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (µM) | β5 IC50 (µM) | Permeability | Metabolic Stability | Kinetic Solubility | Cytotoxicity HepG2 | Lipopholicity | |||||
| ID | P3 | 3D7 | Pf20S | c20S | i20S | PAMPA | MDR | m/hLM | pH 6.8 | Viability % | cLogP |
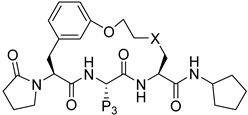
| 228 | 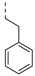 | 0.001 | 0.003 | 2.5 | >100 | 151 | 57/3 | 657.8 596.5 | 100 | 96 | 4.9 |
| 247 | 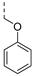 | 0.072 | 0.91 | 13.1 | >100 | 208 | 529 405 | 20 | 104 | 4.5 | |
| 254 |  | 0.011 | 0.009 | 2.7 | 51.0 | 212 | 45/3 | >768 618 | 17 | 100 | 5.0 |
| 272 | 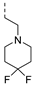 | 0.012 | 0.043 | 3.2 | 46.5 | 133 | 49/1 | 128 113 | >130 | 102 | 2.8 |
| TDI8304 | 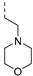 | 0.016 | 0.081 | >100 | >100 | 15 | >2/<1 | 6 19 | >130 | 103 | 2.4 |
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (µM) | β5 IC50 (µM) | Permeability | Metabo-lic Stability | Kinetic Solubility | Cytotoxicity HepG2 | Lipopholicity | TPSA | |||||
| ID | P3 | 3D7 | Pf20S | c20S | i20S | PAMPA | MDR | m/hLM | pH 6.8 | Viability % | cLogP | |
| TDI8304 | - | 0.016 | 0.081 | >100 | >100 | 15 | >2/<1 | 6 19 | >130 | 103 | 2.4 | 129.3 |
| 249 | Gemdime-thyl | 0.096 | 0.21 | 45.2 | >100 | 269 | 35/11 | 690 167 | 90 | 97 | 3.7 | 109 |
| 271 | 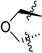 | 0.034 | 0.56 | 31.1 | >100 | 89 | >28/<1 | 5 4 | 68 | 102 | 2.4 | 118 |
| 273 |  | 0.32 | 4.9 | >100 | >100 | 0 | >2/<1 | −14 −26 | >140 | 100 | 2.2 | 121 |
| 248 | 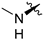 | 0.7 | 3.9 | >100 | >100 | 20 | 1.2/5 | −1 15 | >130 | 104 | 2.8 | 99.8 |
| 270 | 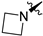 | 0.32 | 2.0 | 13.1 | 17.6 | 0 | >2/<1 | −27 6 | >140 | 100 | 2.9 | 112.2 |
| Compound | IC50 (µM) |
|---|---|
| 296 | 1.6 |
| 297 | 1.2 |
| 298 | 1.4 |
| 299 | >10 |
| 300 | 1.3 |
| 301 | 3.3 |
| 302 | 0.14 |
| 303 | 2.4 |
| Analogue | IC50 FP-2 [nM] | IC50 FP-3 [nM] | IC50 P. falciparum (3D7) [nM] |
|---|---|---|---|
| 309 | 6.78 ± 0.44 | 292 ± 1 | 8.93 ± 73 |
| 310 | 2.81 ± 0.39 | 163 ± 21 | 210 ± 80 |
| 311 | 3710 ± 420 | >50,000 | 4375 ± 690 |
| 312 | >50,000 | >50,000 | >50,000 |
| Analogue | IC50 FP-2 [nM] | IC50 FP-3 [nM] | IC50 P. falciparum (3D7) [nM] |
|---|---|---|---|
| 313 | 10,500 ± 640 | >25,000 | 540 ± 210 |
| 314 | 7700 ± 190 | >50,000 | 1900 ± 620 |
| 315 | 11,500 ± 190 | >25,000 | 320 ± 10 |
| 316 | 2500 ± 150 | 34,000 ± 460 | 1100 ± 800 |
| 317 | 3400 ± 390 | >25,000 | 5400 ± 380 |
| 318 | 6000 ± 160 | 46,000 ± 5400 | 6600 ± 3800 |
| Analogue | IC50 FP-2 [nM] | IC50 FP-3 [nM] | IC50 P. falciparum (3D7) [nM] |
|---|---|---|---|
| 309 | 6.78 ± 0.44 | 292 ± 1 | 89.3 ± 73 |
| 346 | 24.8 ± 23 | 225 ± 27 | 57.3 ± 26 |
| 347 | 9.52 ± 0.14 | 131 ± 44 | 16.6 ± 9 |
| 348 | 5.25 ± 2 | 81.4 ± 8 | 20.0 ± 8 |
| 349 | 12.0 ± 3 | 66.7 ± 25 | 9.7 ± 2 |
| 350 | 9.59 ± 0.21 | 196 ± 7 | 96.0 ± 74 |
| 351 | 6.86 ± 3 | 182 ± 16 | 62.0 ± 47 |
| CQ | 17.3 ± 3 |
| Analog | IC50 P. falciparum (Dd2) [nM] | HEK298 [nM] | AP M1, M17, and M18 [nM] |
|---|---|---|---|
| 309 | 302 ± 126 | 14 200 ± 1050 | >10,000 |
| 313 | 421 ± 2 | >50,000 | >10,000 |
| 315 | 378 ± 20 | >50,000 | >10,000 |
| 317 | 170 ± 68 | >50,000 | >10,000 |
| 346 | 419 ± 185 | 16,300 ± 1700 | >10,000 |
| 347 | 165 ± 68 | 9650 ± 1480 | >10,000 |
| 348 | 67.0 ± 30 | 18,900 ± 110 | >10,000 |
| 349 | 29.0 ± 16 | 8500 ± 124 | >10,000 |
| Compound | IC50 FP-2 [nM] | IC50 FP-3 [nM] | IC50 3D7 [nM] | IC50 W2 [nM] |
|---|---|---|---|---|
| 352 | 12 | 67 | 9.7 | ND |
| 371 | 59 | 131 | 14 | 11 |
| 372 | 31 | 117 | 26 | 28 |
| 373 | 29 | 79 | 42 | 49 |
| 374 | 60 | 851 | 24 | 10 |
| 375 | 57 | 228 | 5 | 7 |
| 376 | 3097 | 10,235 | 2593 | 691 |
| 377 | 3523 | 8788 | 1248 | 478 |
| 378 | 464 | 459 | 229 | 119 |
| 379 | 235 | 407 | 72 | 37 |
| 380 | 455 | 547 | 164 | 55 |
| 381 | 156 | 480 | 205 | 59 |
| 382 | >50,000 | >50,000 | >10,000 | 3729 |
| 383 | >50,000 | >50,000 | 6994 | 1017 |
| 384 | 84 | 484 | 9 | 10 |
| 385 | 169 | 490 | 83 | 95 |
| 386 | 270 | 2571 | 76 | 54 |
| 387 | 33 | 112 | 1 | 4 |
| CQ | ND | ND | 4 | 55 |
| ART | ND | ND | 32 | 21 |
| E64 | 68 | 136 | ND | ND |
| Degradation Half-Life (min) | ||||||
|---|---|---|---|---|---|---|
| 352 | 372 | 373 | 375 | 384 | 387 | |
| plasma | <2 | 126 | 82 | 557 | >600 | 373 |
| blood | <2 | 41 | 17 | 110 | >600 | 273 |
| Peptides | Act > 50% | Shared Volume % |
|---|---|---|
| SE5 P1 (388) | 1 | 50.1 |
| SE5 P2 (389) | 1 | 57.5 |
| SE5 P3 (390) | 0 | 27.9 |
| SE5 P4 (391) | 0 | 41.6 |
| SE5 P5 (392) | 0 | 19.9 |
| SE5 P6 (393) | 0 | 43.5 |
| SE5 P7 (394) | 0 | 45.1 |
| SE5 P8 (395) | 0 | 40.2 |
| SE5 P9 (396) | 0 | 39.2 |
| MHC [M] | MIC [M] | MHC/MIC | |
|---|---|---|---|
| Ang II | 10−5 | 10−12 | 107 |
| Peptide 401 | 10−5 | 10−14 | 109 |
| Peptide 402 | 10−6 | 10−14 | 108 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurniaty, N.; Maharani, R.; Hidayat, A.T.; Supratman, U. An Overview on Antimalarial Peptides: Natural Sources, Synthetic Methodology and Biological Properties. Molecules 2023, 28, 7778. https://doi.org/10.3390/molecules28237778
Kurniaty N, Maharani R, Hidayat AT, Supratman U. An Overview on Antimalarial Peptides: Natural Sources, Synthetic Methodology and Biological Properties. Molecules. 2023; 28(23):7778. https://doi.org/10.3390/molecules28237778
Chicago/Turabian StyleKurniaty, Nety, Rani Maharani, Ace Tatang Hidayat, and Unang Supratman. 2023. "An Overview on Antimalarial Peptides: Natural Sources, Synthetic Methodology and Biological Properties" Molecules 28, no. 23: 7778. https://doi.org/10.3390/molecules28237778
APA StyleKurniaty, N., Maharani, R., Hidayat, A. T., & Supratman, U. (2023). An Overview on Antimalarial Peptides: Natural Sources, Synthetic Methodology and Biological Properties. Molecules, 28(23), 7778. https://doi.org/10.3390/molecules28237778