
Voltammetry in Determination of Trace Amounts of Lanthanides—A Review


Abstract
:1. Introduction
2. Stripping Voltammetry Methods for the Determination of Rare Earth Elements
2.1. AdSV Procedures of REEs Determination
2.2. ASV Procedure for Lutetium(III) Determination
2.3. CSV Procedure for Cerium(III) Determination
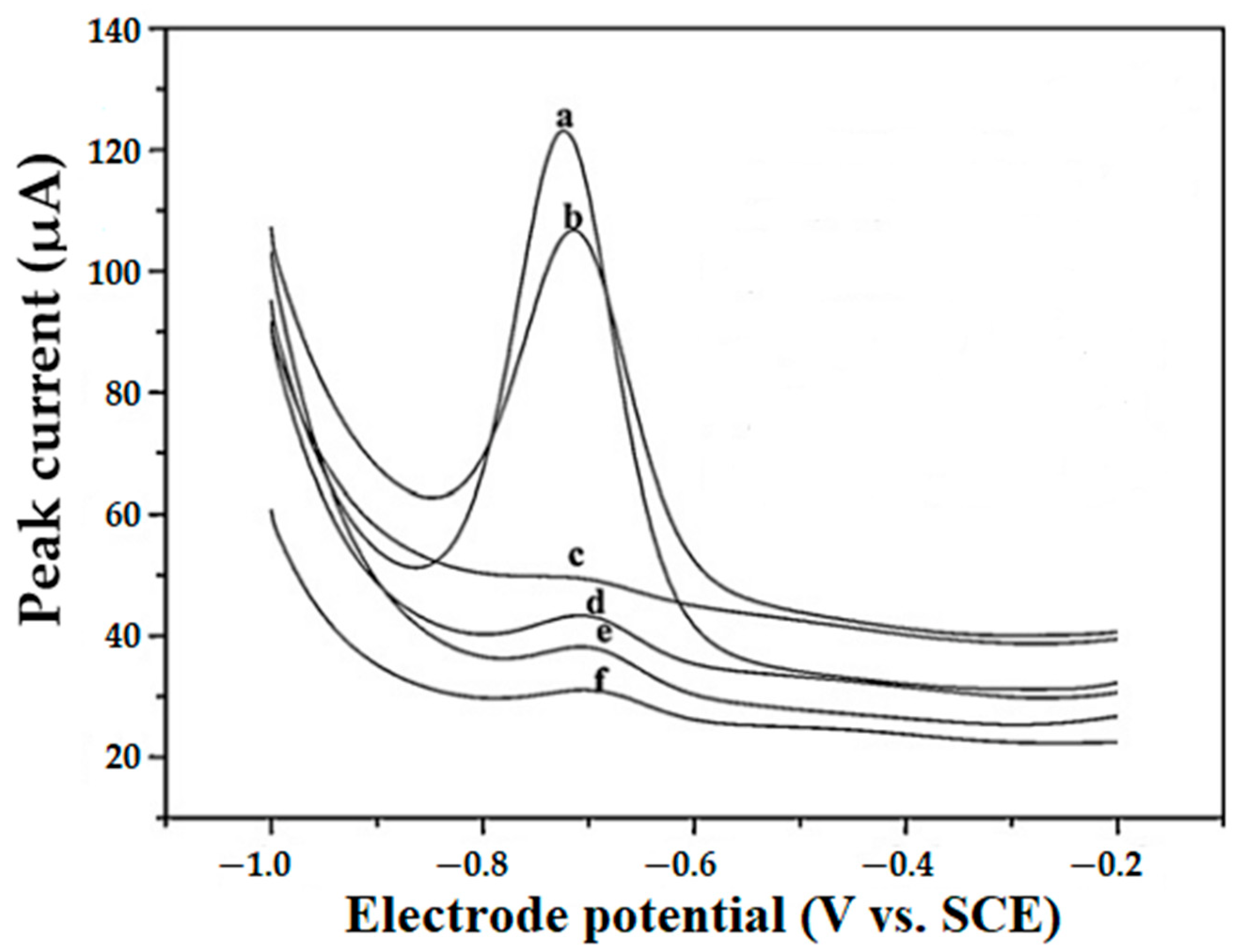
2.4. Types of Working Electrodes and Electrode Modifiers Used
2.4.1. Mercury-Based Electrodes
2.4.2. Solid Electrodes
3. Impact of Interferents on REE Determination
3.1. Impact of Co-Existing Ions
3.1.1. Impact of Co-Existing Ions on Cerium Signal in Cerium Determination Procedure
3.1.2. Impact of Co-Existing Ions on Europium Signal in Europium Determination Procedures
3.1.3. Selectivity of Other Procedures
3.2. Impact of Organic Compounds
4. Practical Application
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charewicz, W. Pierwiastki Ziem Rzadkich. Surowce, Technologia, Zastosowania; Wydawnictwo Naukowo-Techniczne: Warsaw, Poland, 1990. [Google Scholar]
- Jarosiński, A. Możliwości pozyskiwania metali ziem rzadkich w Polsce. Zesz. Nauk. IGSMiE PAN 2016, 92, 75–88. [Google Scholar]
- Connelly, N.G.; Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of inorganic chemistry: IUPAC recommendations 2005. Int. Union Pure Appl. Chem. 2005, 51, 1–366. [Google Scholar]
- Burkowicz, A.; Galos, K.; Guzik, K.; Kamyk, J.; Kot-Niewiadomska, A.; Lewicka, E.; Smakowski, T.; Szlugaj, J. Minerals Yearbook of Poland 2013, 20th ed.; Mineral and Energy Economy Research Institute of the Polish Academy of Sciences, Department of Mineral Policy: Warsaw, Poland, 2015; pp. 1–1170. [Google Scholar]
- Abre, R.D.; Morais, C. Purification of rare earth elements from monazite sulphuric acid leach liquor and the production of high-purity ceric oxide. Miner. Eng. 2010, 23, 536–540. [Google Scholar] [CrossRef]
- Yantasee, W.; Fryxell, G.E.; Addleman, R.S.; Wiacek, R.J.; Koonsiripaiboon, V.; Pattamakomsan, K.; Sukwarotwat, V.; Xu, J.; Raymond, K.N.J. Selective removal of lanthanides from natural waters, acidic streams and dialysate. Hazard. Mater. 2009, 168, 1233–1238. [Google Scholar] [CrossRef]
- Iwashita, M.; Saito, A.; Arai, M.; Furusho, Y.; Shimamura, T. Determination of rare earth elements in water collected in suburban Tokyo. Geochem. J. 2011, 45, 187–197. [Google Scholar] [CrossRef]
- Norgate, T.E.; Jahanshahi, S.; Rankin, W.J. Assessing the environmental impact of metal production processes. J. Clean. Prod. 2007, 15, 838–848. [Google Scholar] [CrossRef]
- Castor, S.B.; Hedrick, J.B. Rare Earth Elements. In Industrial Minerals and Rocks-Commodities Markets and Uses, 7th ed.; Kogel, J.E., Trivedi, N.C., Barker, J.M., Krukowski, S.T., Eds.; Society for Mining, Metallurgy and Exploration, Inc. (SME): Englewood, CO, USA, 2006; p. 76. [Google Scholar]
- Li, Y.; Li, P.; Yu, H.; Bian, Y. Recent advances (2010–2015) in studies of cerium oxide nanoparticles’ health effects. Environ. Toxicol. Chem. 2016, 44, 25–29. [Google Scholar] [CrossRef]
- Guo, Y.L.; Zhang, S.H.; Lai, L.L.; Wang, G. Rare earth elements in Oolong tea and their human health risks associated with drinking tea. J. Food Compos. Anal. 2015, 44, 122–127. [Google Scholar] [CrossRef]
- Ali, S.H. Social and environmental impact of the rare earth industries. Resources 2014, 3, 123–134. [Google Scholar] [CrossRef]
- Oliveira, S.S.; Ribeiro, V.S.; Almeida, T.S.; Araujo, R.G.O. Quantification of ytterbium in road dust applying slurry sampling and detection by high-resolution continuum source graphite furnace atomic absorption spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2020, 171, 105938. [Google Scholar] [CrossRef]
- Rajabi, N.; Masrournia, M.; Abedi, M. Measuring and Pre-concentration of Lanthanum Using Fe3O4@Chitosan Nanocomposite with Solid-phase Microextraction for ICP-OES Determination. Arab. J. Sci. Eng. 2020, 45, 121–129. [Google Scholar] [CrossRef]
- Ticová, B.; Novotný, K.; Kanický, V. Comparison of different spectral resolution ICP-OES spectrometers for the determination of rare earth elements. Chem. Pap. 2019, 73, 2913–2921. [Google Scholar] [CrossRef]
- Pradhan, S.K.; Ambade, B. Extractive separation of rare earth elements and their determination by inductively coupled plasma optical emission spectrometry in geological samples. J. Anal. At. Spectrom. 2020, 35, 1395–1404. [Google Scholar] [CrossRef]
- Sá, Í.P.; Almeida, O.N.; Lima, D.D.C.; Gonzalez, M.H.; Amorim, F.A.C. Determination of lanthanide and actinide elements by energy dispersive x-ray fluorescence spectrometry applying DLLME preconcentration and dried spot. Spectrochim. Acta Part B At. Spectrosc. 2021, 182, 106253. [Google Scholar] [CrossRef]
- Rossbach, L.M.; Brede, D.A.; Nuyts, G.; Cagno, S.; Skjervold Olsson, R.M.; Oughton, D.H. Synchrotron XRF Analysis Identifies Cerium Accumulation Colocalized with Pharyngeal Deformities in CeO2 NP-Exposed Caenorhabditis elegans. Environ. Sci. Technol. 2022, 56, 5081–5089. [Google Scholar] [CrossRef]
- Slavković-Beškoski, L.; Ignjatović, L.; Bolognesi, G.; Maksin, D.; Savić, A.; Vladisavljević, G.; Onjia, A. Dispersive Solid–Liquid Microextraction Based on the Poly(HDDA)/Graphene Sorbent Followed by ICP-MS for the Determination of Rare Earth Elements in Coal Fly Ash Leachate. Metals 2022, 12, 791. [Google Scholar] [CrossRef]
- Chen, S.; Yan, J.; Li, J.; Lu, D. Magnetic ZnFe2O4 Nanotubes for Dispersive Micro Solid-Phase Extraction of Trace Rare Earth Elements Prior to Their Determination by ICP-MS. Microchim. Acta 2019, 186, 228. [Google Scholar] [CrossRef]
- Wysocka, I. Determination of rare earth elements concentrations in natural waters–A review of ICP-MS measurement approaches. Talanta 2021, 221, 121636. [Google Scholar] [CrossRef]
- Elgendy, K.; El-didamony, A.; Abd El-wahaab, B. Analytical applications using spectrophotometric technique for the determination of uranium(VI), samarium(III) and cerium(III) by new organic reagent. J. Iran. Chem. Soc. 2020, 17, 1317–1327. [Google Scholar] [CrossRef]
- Silachyov, I.Y. Using the Internal Standard Method with a Planar Detector in the Determination of Lanthanides in Geological Samples by Neutron Activation Analysis. J. Anal. Chem. 2020, 75, 1415–1423. [Google Scholar] [CrossRef]
- Telmore, V.M.; Kumar, P.; George, J.P.; Kannan, S. Dual column HPLC method for determination of lanthanides in zirconium matrix. J. Liq. Chromatogr. Relat. 2021, 44, 171–180. [Google Scholar] [CrossRef]
- Zawisza, B.; Pytlakowska, K.; Feist, B.; Polowniak, M.; Kita, A.; Sitko, R. Determination of rare earth elements by spectroscopic techniques: A review. J. Anal. At. Spectrom. 2011, 26, 2373–2390. [Google Scholar] [CrossRef]
- Ashina, J.; Babain, V.; Kirsanov, D.; Legin, A. A Novel Multi-Ionophore Approach for Potentiometric Analysis of Lanthanide Mixtures. Chemosensors 2021, 9, 23. [Google Scholar] [CrossRef]
- Yaroshenko, I.S.; Alyapyshev, M.Y.; Babain, V.A.; Legin, A.V.; Kirsanov, D.O. Potentiometric Sensors and Multisensor Systems for the Determination of Lanthanides. J. Anal. Chem. 2019, 74, 1003–1018. [Google Scholar] [CrossRef]
- Saefurohman, A.; Buchari, B.; Noviandri, I. La(III) Ion Selective Electrode with PTFE Membrane Containing Tributyl Phosphate Ionophore. Key Eng. Matter. 2021, 874, 50–57. [Google Scholar] [CrossRef]
- Li, J.A.; Liu, S.M.; Yan, Z.H.; Mao, X.; Gao, P. Adsorptive voltammetric studies on the cerium (III)-alizarin complexon complex at a carbon paste electrode. Microchim. Acta 2006, 154, 241–246. [Google Scholar] [CrossRef]
- Liu, S.M.; Li, J.A.; Zhang, S.J.; Zhao, J.Q. Study on the adsorptive stripping voltammetric determination of trace cerium at a carbon paste electrode modified in situ with cetyltrimethylammonium bromide. Appl. Surf. Sci. 2005, 252, 2078–2084. [Google Scholar] [CrossRef]
- Adamczyk, M.; Grabarczyk, M.; Wlazłowska, E. Fast and simple differential pulse adsorptive stripping voltammetric determination of Ce(III) in natural water samples. Desalin. Water Treat. 2022, 264, 188–195. [Google Scholar] [CrossRef]
- Javanbakht, M.; Khoshsafar, H.; Ganjali, M.R.; Norouzi, P.; Badei, A.; Hasheminasab, A. Stripping voltammetry of cerium(III) with a chemically modified carbon paste electrode containing functionalized nanoporous silica gel. Electroanalysis 2008, 20, 203–206. [Google Scholar] [CrossRef]
- Javanbakht, M.; Khoshsafar, H.; Ganjali, M.R.; Norouzi, P.; Adib, M. Adsorptive stripping voltammetric determination of nanomolar concentration of cerium(III) at a carbon paste electrode modified by N′-[(2-Hydroxyphenyl)Methylidene]-2-Furohydrazide. Electroanalysis 2009, 21, 1605–1610. [Google Scholar] [CrossRef]
- Prasad, B.B.; Jauhari, D. Double-ion imprinted polymer@ magnetic nanoparticles modified screen printed carbon electrode for simultaneous analysis of cerium and gadolinium ions. Anal. Bioanal. Chem. 2015, 875, 83–91. [Google Scholar] [CrossRef]
- Alizadeh, T.; Ganjali, M.R.; Akhoundian, M.; Norouzi, P. Voltammetric determination of ultratrace levels of cerium (III) using a carbon paste electrode modified with nano-sized cerium-imprinted polymer and multiwalled carbon nanotubes. Microchim. Acta 2016, 183, 1123–1130. [Google Scholar] [CrossRef]
- Chen, J.; Bai, H.; Xia, J.; Liu, X.; Liu, Y.; Cao, Q. Trace detection of Ce3+ by adsorption strip voltammetry at a carbon paste electrode modified with ion imprinted polymers. J. Rare Earths 2018, 36, 1121–1126. [Google Scholar] [CrossRef]
- Khoo, S.B.; Zhu, J. Poly (catechol) film modified glassy carbon electrode for ultratrace determination of cerium (III) by differential pulse anodic stripping voltammetry. Electroanalysis 1999, 11, 546–552. [Google Scholar] [CrossRef]
- Chen, J.; Bai, H.; Li, Z.; Xia, J.; Cao, Q. Stripping voltammetric determination of cerium in food using an electropolymerized poly-catechol and ion-imprinted membrane modified electrode. J. Electroanal. Chem. 2018, 808, 41–49. [Google Scholar] [CrossRef]
- Ojo, K.; Zhao, D.; Rusinek, C.A.; Pixley, S.K.; Heineman, W.R. Cathodic stripping voltammetric determination of cerium using indium tin oxide (ITO). Electroanalysis 2017, 29, 1124–1130. [Google Scholar] [CrossRef]
- Abollino, O.; Aceto, M.; Mentasti, E.; Sarzanini, C.; Bergi, C.M.G. Determination of trace europium by adsorptive cathodic stripping voltammetry after complexation with cupferron. Electroanalysis 1997, 9, 444–448. [Google Scholar] [CrossRef]
- Alizadeh, T.; Amjadi, S. Synthesis of nano-sized Eu3+ imprinted polymer and its application for indirect voltammetric determination of europium. Talanta 2013, 106, 431–439. [Google Scholar] [CrossRef]
- Chen, J.; Bai, H.; Xia, J.; Li, Z.; Liu, P.; Cao, Q. Electrochemical sensor for detection of europium based on poly-catechol and ion-imprinted sol-gel film modified screen-printed electrode. J. Electroanal. Chem. 2018, 824, 32–38. [Google Scholar] [CrossRef]
- Cruickshank, L.; Officer, S.; Pollard, P.; Prabhu, R.; Stutter, M.; Fernandez, C. Rare elements electrochemistry: The development of a novel electrochemical sensor for the rapid detection of europium in environmental samples using gold electrode modified with 2-pyridinol-1-oxide. Anal. Sci. 2015, 31, 623–627. [Google Scholar] [CrossRef]
- Yantasee, W.; Fryxell, G.E.; Lin, Y.H. Voltammetric analysis of europium at screen-printed electrodes modified with salicylamide self-assembled on mesoporous silica. Analyst 2006, 131, 1342–1346. [Google Scholar] [CrossRef]
- Yuan, S.; He, Q.; Yao, S.J.; Hu, S.S. Mercury-free detection of europium (III) at a glassy carbon electrode modified with carbon nanotubes by adsorptive stripping voltammetry. Anal. Lett. 2006, 39, 373–385. [Google Scholar] [CrossRef]
- Ugo, P.; Ballarin, B.; Daniele, S.; Mazzocchin, G.A. Determination of trace amounts of Eu3+ and Yb3+ ions at Nafion-coated thin mercury film electrodes. Anal. Chim. Acta 1991, 244, 29–38. [Google Scholar] [CrossRef]
- Ma, X.; Xu, Z.; Yuan, H.; He, Y.; Xiao, D.; Choi, M.M.F. High-sensitive and selective Eu3+ electrochemical sensor based on LaB6 electrode and sodium dodecylbenzene sulfonate. Sensor. Actuat. B Chem. 2010, 147, 152–158. [Google Scholar] [CrossRef]
- Makombe, M.; van der Horst, C.; Silwana, B.; Iwuoha, E.; Somerset, V. Antimony film sensor for sensitive rare earth metal analysis in environmental samples. J. Environ. Sci. Health A 2016, 51, 597–606. [Google Scholar] [CrossRef]
- Wang, J.; Farias, P.A.; Mahmoud, J.S. Trace determination of lanthanum, cerium, and praseodymium based on adsorptive stripping voltammetry. Anal. Chim. Acta 1985, 171, 215–223. [Google Scholar] [CrossRef]
- Suyanta; Sunarto; Permana Sari, L.; Wardani, N.I.; Isa, I.M. Differential Adsorptive Stripping Voltammetric Determination of Ultra Trace Lanthanum(III) based on Carbon Paste Electrode Modified with 3-Methyl-2-hydrazinobenzothiazole. Int. J. Electrochem. Sci. 2014, 9, 7763–7772. [Google Scholar] [CrossRef]
- Li, J.; Liu, S.; Mao, X.; Gao, P.; Yan, Z. Trace determination of rare earths by adsorption voltammetry at a carbon paste electrode. J. Electroanal. Chem. 2004, 561, 137–142. [Google Scholar] [CrossRef]
- Li, J.; Yi, F.; Shen, D.; Fei, J. Adsorptive stripping voltammetric study of Scandium-Alizarin complexan complex at a carbon paste electrode. Anal. Lett. 2002, 35, 1361–1372. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; Deng, P. Adsorption voltammetry of the scandium-alizarin red S complex onto a carbon paste electrode. Talanta 2001, 54, 561–566. [Google Scholar] [CrossRef]
- Lee, S.K.; Chung, T.D.; Kim, H. Indirect Voltammetric Determination of Lanthanides in the Presence of Mordant Red 19. Electroanalysis 1997, 9, 527–532. [Google Scholar] [CrossRef]
- Mlakar, M. Determination of Ytterbium Traces by Cathodic Stripping Voltammetry. Electroanalysis 2003, 15, 27–32. [Google Scholar] [CrossRef]
- Wang, J.; Zadeii, J.M. Trace determination of yttrium and some heavy rare-earths by adsorptive stripping voltammetry. Talanta 1986, 33, 321–324. [Google Scholar] [CrossRef]
- Kumrić, K.R.; Trtić-Petrović, T.M.; Ignjatović, L.M.; Čomor, J.J. Indirect determination of lutetium by differential pulse anodic stripping voltammetry at a hanging mercury drop electrode. Cent. Eur. J. Chem. 2008, 6, 65–69. [Google Scholar] [CrossRef]
- Wang, J. Stripping Analysis: Principles, Instrumentation and Applications; VCH Publishers: Deerfield Beach, FL, USA, 1985. [Google Scholar]
- Mukherji, A.K. Amperometric determination of thorium and lanthanum in presence of nitrate. J. Electroanal. Chem. 1967, 13, 425–432. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P.; Cowell, D.C. Voltammetric studies of lead at a 1-(2-pyridylazo)-2-naphthol modified screen-printed carbon electrode and its trace determination in water by stripping voltammetry. Anal. Chim. Acta 2001, 431, 89–99. [Google Scholar] [CrossRef]
- Kalcher, K.; Kauffmann, J.M.; Wang, J.; Svancara, I.; Vytras, K.; Neuhold, C.; Yang, Z. Sensors based on carbon paste in electrochemical analysis: A review with particular emphasis on the period 1990–1993. Electroanalysis 1995, 7, 5–22. [Google Scholar] [CrossRef]
- Svancara, I.; Vytras, K.; Barek, J.; Zima, J. Carbon Paste Electrodes in Modern Electroanalysis. Crit. Rev. Anal. Chem. 2001, 31, 311–345. [Google Scholar] [CrossRef]
- Metters, J.P.; Kadara, R.O.; Banks, C.E. New directions in screen printed electroanalytical sensors: An overview of recent developments. Analyst 2011, 136, 1067–1076. [Google Scholar] [CrossRef]
- Yakabson, B.; Smally, R. Fullerene Nanotubes: C 1,000,000 and Beyond: Some unusual new molecules-long, hollow fibers with tantalizing electronic and mechanical properties-have joined diamonds and graphite in the carbon family. Am. Sci. 1997, 85, 324–337. [Google Scholar]
- Singh, D.K.; Mishra, S. Synthesis and characterization of Fe(III)-ion imprinted polymer for recovery of Fe(III) from water samples. J. Sci. Ind. Res. 2010, 69, 767–772. [Google Scholar]
- Zhao, J.; Han, B.; Zhang, Y.; Wang, D. Synthesis of Zn(II) ion-imprinted solid-phase extraction material and its analytical application. Anal. Chim. Acta 2007, 603, 87–92. [Google Scholar] [CrossRef]
- Alizadeh, T. An imprinted polymer for removal of Cd2+ from water samples: Optimization of adsorption and recovery steps by experimental design. Chin. J. Polym. Sci. 2011, 29, 658–669. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, M. Polarographic adsorptive complex wave of light rare earths with o-cresolphthalexon. Anal. Chem. 1984, 56, 1912–1916. [Google Scholar] [CrossRef]

{kind=link}
| Tested Ion | Method | Working Electrode | Complexing Agent | LOD (M) | Accumulation Time (s) | Peak Potential Ep (V) | Linear Range (M) | Investigated Interferents | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Foreign Ions (Other Than REEs)/Organics: Interfering | Foreign Ions (Other Than REEs)/Organics: No Interfering | REEs: Interfering/No Interfering | |||||||||
| La(III) | AdSV | MBTH/CPE | - | 1.0 × 10−12 | - | −0.22 (vs. Ag/AgCl, 3 M KCl) | 1.0 × 10−12– 7.0 × 10−11 | - | Al(III), Ba(II), Cu(II) | -/ Ce(III) | [50] |
| Ce(III) | AdSV | Ce-IIM/PC/GCE | - | 1.0 × 10−12 | 600 | 0.88 (vs. Ag/AgCl, 3 M KCl) | 3.0 × 10−12– 1.0 × 10−4 | Cu(II), Fe(III), Ni(II) | Co(II), Mg(II), Na(I), Zn(II) | -/ Dy(III), Er(III), Eu(III), Gd(III), Ho(III), Nd(III), Pr(III), Tb(III), Yb(III) | [38] |
| Ce(III) | AdSV | Ce-IP/MWCNT/CPE | - | 1.0 × 10−11 | - | 1.05 (vs. Ag/AgCl, 3 M KCl) | 2.5 × 10−11– 1.0 × 10−6 | - | Ag(I), Cr(III), Cd(II), Co(II), Hg(II) | Dy(III), Eu(III) / La(III), Nd(III), Sm(III), Tb(III), Yb(III) | [35] |
| Eu(III) | AdSV | HMDE | Cupferron | 6.0 × 10−11 | 60 | −0.88 (vs. Ag/AgCl, 3 M KCl) | 0–1.3 × 10−8 | Cr(III) | Al(III), Mo(VI), U(VI), V(V) | -/ Dy(III), Er(III), Eu(III), Gd(III), Ho(III), Nd(III), Pr(III), Sm(III), Tb(III), Yb(III) | [40] |
| Dy(III) Ho(III) Er(III) Tm(III) Yb(III) Lu(III) | AdSV | CPE | Alizarin | 1.0 × 10−10 | 60 | 0.586 0.588 0.588 0.584 0.582 0.580 (vs. SCE) | 1.0 × 10−9– 2.0 × 10−7 | Co(II), Cu(II), Ni(II), Pb(II), Zn(II) | Ca(II), Ba(II), Cr(III), Se(IV), B(III), Ge(IV), As(III), Ag(I), Mn(II),Mg(II),Cd(II), Al(III), V(V), Hg(II), Ti(IV), Sb(III), Sn(IV), Fe(II), Ga(III), Fe(III), Th(IV), Zr(IV), In(III), SO42−, PO43−, F− | -/ La(III), Ce(III), Pr(III), Nd(III), Sc(III) | [51] |
| La(III) Ce(III) Pr(III) | AdSV | SMDE | OCP | 1.2 × 10−10 1.7 × 10−10 1.4 × 10−10 | 1200 | −0.95 −1.00 −1.05 (vs. Ag/AgCl, 3 M KCl) | 2.5 × 10−9– 2.5 × 10−8 | gelatin, albumin | Ca(II), Mg(II), Al(III), Cu(II), Cd(II), Hg(II), Zn(II), cholesterol, chloride | no data | [49] |
| Ce(III) | AdSV | PC/GCE | - | 2.0 × 10−10 | 10 | 0.85 (vs. Ag/AgCl, 3 M KCl) | 2.0 × 10−9– 1.0 × 10−7 | Al(III), Bi(III) | Zn(II), Cu(II), Pb(II), Cd(II), Hg(II), Tl(I), Re(II), Sb(III), Ge(IV),Te(IV), Se(IV), Ag(I), Au(I), Sn(IV), Co(II) | no data | [37] |
| La(III) Ce(III) Pr(III) | AdSV | GC/SbFE | Alizarin | 3.0 × 10−9 4.3 × 10−10 5.0 × 10−9 | 360 | 0.74 0.76 0.79 (vs. Ag/AgCl, 3 M KCl) | 7.1 × 10−9– 1.8 × 10−7 | - | Co(II), Fe(II), Mn(II), Ni(II), Pb(II), Zn(II) | La(III)/- | [48] |
| Ce(IV) Gd(III) | AdSV | DIIP@MNPs/SPCE | - | 5.0 × 10−10 1.2 × 10−9 | 180 | 0.05 −0.37 (vs. Ag/AgCl, 3 M KCl) | 1.9 × 10−9– 3.8 × 10−8 4.6 × 10−9– 5.5 × 10−8 | - | Cr(III), As(III), Ca(II), Mg(II), Al(III), Fe(III), SO42−, PO43−, ascorbic acid | -/ Dy(III), Ho(III), Nd(III), Pr(III), Y(III) | [34] |
| La(III) Tb(III) Yb(III) | AdSV | SMDE | MR19 | 8.0 × 10−10 5.0 × 10−10 5.0 × 10−10 | 60 | −0.682 −0.754 −0.784 (vs. Ag/AgCl, 3 M KCl) | 1.0 × 10−8– 1.0 × 10−6 | no data | [54] | ||
| Y(III) Dy(III) Ho(III) Yb(III) | AdSV | HMDE | SVRS | 1.4 × 10−9 1.1 × 10−9 1.0 × 10−9 5.0 × 10−10 | 180 | −0.98 −0.98 −1.00 −1.00 (vs. Ag/AgCl, 3 M KCl) | 0–3.4 × 10−7 0–2.5.× 10−7 0–1.8 × 10−7 0–2.3 × 10−7 | no data | [56] | ||
| Ce(III) | AdSV | CTAB/CPE | Alizarin | 6.0 × 10−10 | 120 | 0.73 (vs. SCE) | 8.0 × 10−10– 8.0 × 10−9 | - | Ca(II), Ba(II), B(III), As(III), Mg(II), Se(IV), Ge(IV), Mn(II), Zn(II), Cr(III), Ni(II), Hg(II), Cd(II), Co(II), Fe(II), Pb(II), Cu(II), Al(III), Bi(III), Fe(III), Zr(IV), In(III), Ga(III), HCr2O7−, MnO4−, AuCl4−, SO42−, PO43−, F−, ascorbic acid | -/ La(III), Pr(III), Nd(III), Sm(III), Eu(III), Y(III), Gd(III), Tb(III), Sc(III), Dy(III), Ho(III), Er(III), Yb(III), Tm(III) | [30] |
| Sc(III) | AdSV | CPE | Alizarin | 6.0 × 10−10 | 60 | −0.60 (vs. SCE) | 1.0 × 10−9– 6.0 × 10−7 | F−, C2O42−, citrate | Ca(II), Mg(II), Zn(II), Cd(II), Mn(II), Ag(I), As(III), Au(III), Ba(II), Co(II), Cr(III), Hg(II), Ni(II); MoO42−, Pb(II), Al(III), Sn(II), Ga(III) Cu(II), Fe(III), Sb(III), V(V), In(III); Bi(III), Th(IV), Zr(IV), Ti(IV), SO42−, PO43− | -/ Ce(III), Dy(III), Er(III), Eu(III), Gd(III), Ho(III), La(III), Nd(III), Pr(III), Tb(III), Yb(III) | [52] |
| Sc(III) | AdSV | CPE | Alizarin S | 6.0 × 10−10 | 180 | −0.58 (vs. SCE) | 1.0 × 10−9– 4.0 × 10−7 | Fe(III), Zr(IV) | Zn(II), Pb(II), Ni(II), Li(I), Co(II), Mn(II), Cr(III), As(III), Se(IV), Ag(I), Au(III), Be(II), Bi(III), Cd(II), Ga(III), Fe(II), Mo(VI), Sn(II), Cu(II), Ba(II), V(V), CNS−, SO42−, PO43−, F−, CN− | Eu(III), Gd(III), Tb(III), Dy(III), Ho(III), Er(III), Tm(III), Yb(III), Lu(III) / La(III), Ce(III), Pr(III), Nd(III), Sm(III) | [53] |
| Ce(III) | AdSV | NHMF/CPE | - | 8.0 × 10−10 | 350 | 0.55 (vs. Ag/AgCl, 3 M KCl) | 5.0 × 10−9– 9.0 × 10−8 | - | Cd(II), Cr(III), Cu(II), Mn(II), Ni(II), Pb(II), Th(IV), Zn(II), Br−, Cl−, SO42−, CH3COO−, UO22+ | La(III), Sm(III) / Er(III), Ho(III) | [33] |
| Ce(III) | AdSV | CPE | Alizarin | 2.0 × 10−9 | 120 | 0.69 (vs. SCE) | 6.0 × 10−9– 3.0 × 10−7 | Th(IV) | Ba(II), Ca(II), Cr(III), Mg(II), As(III), Se(IV), B(III), Ge(IV), Mn(II), Cd(II), Pb(II), In(III) Co(II), Zn(II), V(V), Hg(II), Fe(III), Fe(II), Ni(II), Sn(IV), Sb(III), Ti(IV), Al(III), Zr(IV), Cu(II), Bi(III) Ga(III), SO42−, PO43−, ascorbic acid | -/ La(III), Pr(III), Nd(III), Sm(III), Eu(III), Y(III), Gd(III), Tb(III), Sc(III), Dy(III), Ho(III), Er(III), Yb(III), Tm(III) | [29] |
| Lu(III) | ASV | HMDE | - | 2.1 × 10−9 | 120 | −0.995 (vs. Ag/AgCl, 3 M KCl) | 2.1 × 10−9– 7.3 × 10−6 | no data | [57] | ||
| Ce(III) | AdSV | DPNSG/CPE | - | 2.3 × 10−9 | 600 | 0.27 (vs. Ag/AgCl, 3 M KCl) | 2.30 × 10−9– 6.45 × 10−8 | - | Cd(II), Cr(III), Cu(II), Mn(II), Ni(II), Pb(II), Th(IV), Zn(II), Br−, Cl−, CH3COO−, SO42−, UO22+, | La(III) / Er(III), Ho(III), Sm(III) | [32] |
| Ce(III) | CSV | ITO electrode | - | 5.8 × 10−9 | 300 | 0.55 (vs. Ag/AgCl, 3 M KCl) | 1.0 × 10−7– 7.0 × 10−7 | Mn(II) | Bi(III), Cu(II), Zn(II), Sn(II), Mg(II) | -/ Eu(III) | [39] |
| Eu(III) | AdSV | SDBS/LaB6 electrode | - | 6.0 × 10−9 | 120 | −0.70 (vs. SCE) | 1.0 × 10−8– 2.0 × 10−6 | Fe(II), Mg(II), Mn(II), Pb(II), SDBS, SDS, CTAB | Na(I), Ca(II), Zn(II), Triton X-100, CPB | Ce(III), Er(III), La(III) / Sm(III), Yb(III) | [47] |
| Yb(III) | AdSV | HMDE | TTA-PAG ligand | - | 180 | −1.65 (vs. Ag/AgCl, 3 M KCl) | 5.0 × 10−9– 1.0 × 10−7 | - | Cr(III), Co(II), Mn(II), Mo(VI), U(VI), V(V), Triton X-100 | -/ Eu(III), La(III), Y(III) | [55] |
| Eu(III) | AdSV | N/MWCNTs/GCE | - | 1.0 × 10−8 | 60 | −0.70 (vs. SCE) | 4.0 × 10−8– 1.0 × 10−4 | Bi(III), Cr(III) | Mn(II), Co(II), Pd(II), Mg(II), Zn(II), Fe(II), Ba(II), Ni(II) | Er(III), La(III), Sm(III), Yb(III) /- | [45] |
| Eu(III) | AdSV | Sal-SAMMS/SPCE | - | 1.0 × 10−8 | 300 | −0.75 (vs. Ag/AgCl, 3 M KCl) | 7.5 × 10−8– 5.0 × 10−7 | no data | [44] | ||
| Eu(III) Yb(III) | AdSV | NCTMFE | - | 3.0 × 10−8 2.0 × 10−8 | 300 | −0.62 −1.46 (vs. SCE) | X– 2.0 × 10−6 | no data | La(III)/no data | [46] | |
| Ce(III) | AdSV | GCE | Alizarin S | 6.0 × 10−8 | 30 | 0.60 (vs. Ag/AgCl, 3 M KCl) | 2.0 × 10−7– 8.0 × 10−6 | Cr(III), Fe(II), Sb(III), V(V) | Al(III), As(III), As(V), Cd(II), Co(II), Cr(VI), Hg(II), K(I), Mg(II), Mn(II), Na(I), Ni(II), Pt(IV), Se(IV), Se(VI), Sn(II), Ti(IV), U(VI), Zn(II), Bi(III), Ga(III), Cu(II), Mo(VI), CTAB, rhamnolipid, humic acid, Triton X-100, SDS, fulvic acid, natural organic matter | no data | [31] |
| Eu(III) | AdSV | IIM/PC/SPE | - | 1.0 × 10−7 | 300 | −1.00 (vs. Ag/AgCl, 3 M KCl) | 3.0 × 10−7– 1.0 × 10−3 | - | Ca(II), Co(II), Cu(II), Fe(III), Mg(II), Na(I), Ni(II), Zn(II) | -/ Dy(III), Er(III), Ce(III), Gd(III), Ho(III), Nd(III), Pr(III), Tb(III), Yb(III) | [42] |
| Eu(III) | AdSV | IIPs-CPE | - | 1.5 × 10−7 | 20 | −0.18 (vs. Ag/AgCl, 3 M KCl) | 5.0 × 10−7– 3.0 × 10−5 | Cd(II), Cu(II) | Ag(I), Ca(II), Hg(II), Mg(II), Pt(II), Zn(II) | Ce(III), Gd(III), Sm(III) / Er(III), Dy(III), La(III) | [41] |
| Ce(III) | AdSV | IIPs-CPE | - | 1.5 × 10−7 | 20 | 0.93 (vs. Ag/AgCl, 3 M KCl) | 1.0 × 10−6– 1.0 × 10−5 | no data | -/ Dy(III), Er(III), Eu(III), Gd(III), Ho(III), Nd(III), Pr(III), Tb(III), Yb(III) | [36] | |
| Eu(III) | AdSV | PO/GE | - | 3.0 × 10−7 | - | 1.10 (vs. Ag/AgCl, 3 M KCl) | 1.0 × 10−6– 8.0 × 10−5 | - | Al(III), Fe(III) | no data | [43] |
| Interferent | Interferent Concentration (M) | Peak Current of 1 × 10−6 M Ce(III) (µA) |
|---|---|---|
| - | - | 2.60 |
| Na+ | 5.0 × 10−4 | 2.60 |
| Ca2+ | 5.0 × 10−4 | 2.60 |
| Mg2+ | 5.0 × 10−4 | 2.55 |
| Zn2+ | 5.0 × 10−5 | 2.48 |
| Ni2+ | 5.0 × 10−5 | 2.35 |
| Fe3+ | 5.0 × 10−5 | 2.10 |
| Co2+ | 5.0 × 10−5 | 2.50 |
| Cu2+ | 5.0 × 10−5 | 2.35 |
| Ho3+ | 2.0 × 10−5 | 2.60 |
| Dy3+ | 2.0 × 10−5 | 2.60 |
| Er3+ | 2.0 × 10−5 | 2.50 |
| Eu3+ | 2.0 × 10−5 | 2.60 |
| Gd3+ | 2.0 × 10−5 | 2.55 |
| Pr3+ | 2.0 × 10−5 | 2.60 |
| Nd3+ | 2.0 × 10−5 | 2.55 |
| Tb3+ | 2.0 × 10−5 | 2.60 |
| Yb3+ | 2.0 × 10−5 | 2.60 |
| Interferent | Interferent Concentration (M) | Peak Current of 1 × 10−5 M Ce(III) (µA) |
|---|---|---|
| - | - | 0.880 |
| Ho3+ | 5.0 × 10−3 | 0.810 |
| Dy3+ | 5.0 × 10−3 | 0.790 |
| Er3+ | 5.0 × 10−3 | 0.822 |
| Eu3+ | 5.0 × 10−3 | 0.820 |
| Gd3+ | 5.0 × 10−3 | 0.821 |
| Pr3+ | 5.0 × 10−3 | 0.821 |
| Nd3+ | 5.0 × 10−3 | 0.817 |
| Tb3+ | 5.0 × 10−3 | 0.808 |
| Yb3+ | 5.0 × 10−3 | 0.820 |
| Interferent | Interferent Concentration (M) | Peak Current of 1 × 10−5 M Eu(III) (µA) |
|---|---|---|
| - | - | 1.50 |
| Na+ | 5.0 × 10−3 | 1.50 |
| Ca2+ | 5.0 × 10−3 | 1.50 |
| Mg2+ | 5.0 × 10−3 | 1.50 |
| Zn2+ | 1.0 × 10−3 | 1.50 |
| Ni2+ | 1.0 × 10−3 | 1.50 |
| Fe3+ | 1.0 × 10−3 | 1.50 |
| Co2+ | 1.0 × 10−3 | 1.49 |
| Cu2+ | 1.0 × 10−3 | 1.48 |
| Ho3+ | 5.0 × 10−4 | 1.50 |
| Dy3+ | 5.0 × 10−4 | 1.48 |
| Er3+ | 5.0 × 10−4 | 1.50 |
| Eu3+ | 5.0 × 10−4 | 1.50 |
| Gd3+ | 5.0 × 10−4 | 1.49 |
| Pr3+ | 5.0 × 10−4 | 1.50 |
| Nd3+ | 5.0 × 10−4 | 1.49 |
| Tb3+ | 5.0 × 10−4 | 1.50 |
| Yb3+ | 5.0 × 10−4 | 1.50 |
| Interferent | Ce(IV) | Gd(III) | ||||
|---|---|---|---|---|---|---|
| Peak Current (µA) | Recovery (%) | RSD (%) (n = 3) | Peak Current (µA) | Recovery (%) | RSD (%) (n = 3) | |
| Ho3+ | 1.078 | 100 | 0.28 | 6.80 | 99.6 | 2.5 |
| Nd3+ | 1.000 | 92.8 | 2.00 | 6.40 | 93.7 | 4.02 |
| Y3+ | 1.082 | 100.4 | 0.34 | 6.78 | 99.3 | 0.65 |
| Dy3+ | 0.959 | 89.0 | 1.20 | 6.52 | 95.5 | 1.36 |
| Pr3+ | 1.020 | 94.6 | 1.06 | 6.62 | 96.9 | 0.68 |
| Cr3+ | 1.050 | 97.4 | 0.39 | 6.43 | 94.1 | 0.79 |
| As3+ | 1.040 | 96.5 | 0.58 | 6.82 | 99.8 | 1.44 |
| Ca2+ | 1.060 | 98.3 | 1.23 | 6.79 | 99.4 | 2.36 |
| Mg2+ | 1.070 | 99.3 | 2.25 | 6.69 | 97.9 | 0.39 |
| Al3+ | 1.098 | 101.8 | 1.10 | 6.80 | 99.6 | 2.58 |
| Fe3+ | 0.998 | 98.3 | 0.35 | 6.43 | 94.1 | 3.58 |
| Ascorbic acid | 1.072 | 99.4 | 2.63 | 6.78 | 99.4 | 0.84 |
| PO43− | 1.068 | 99.1 | 1.56 | 6.80 | 99.7 | 1.56 |
| SO42− | 1.088 | 100.9 | 0.71 | 6.72 | 98.5 | 2.94 |
| Mixture of interferents | 1.050 | 97.4 | 0.39 | 6.40 | 93.7 | 4.02 |
| Element | Sample | Certified Reference (%) | Found (%) | Ref. |
|---|---|---|---|---|
| Ce | BH1902-1 | 0.0102 | 0.0098 ± 0.0004 | [29] |
| BH1905-1 | 0.0115 | 0.0118 ± 0.0003 | ||
| Ce | BH1902-1 BH1905-1 | 0.0102 0.0115 | 0.0107 ± 0.0005 0.0112 ± 0.0004 | [30] |
| Ce | nodular cast iron 1 | 0.0151 | 0.0146 ± 0.0015 | [32] |
| nodular cast iron 2 | 0.0109 | 0.0118 ± 0.0012 | ||
| Ce | nodular cast iron 1 | 0.0151 | 0.0148 ± 0.0008 | [33] |
| nodular cast iron 2 | 0.0109 | 0.0112 ± 0.0006 | ||
| Element | Sample | Recovery (%) | Found (%) | Ref. |
| Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu | BH1902-1 | 97.8 | 0.082 ± 0.004 | [51] |
| BH1905-1 | 95.4 | 0.053 ± 0.002 | ||
| JC 79-18 | 92.9 | 0.039 ± 0.002 | ||
| Element | Sample | Certified reference (nM) | Found (nM) | Ref. |
| Eu | NIES CRM no. 8 vehicle exhaust particulates A | 106.0 ± 5.1 | 102 ± 5.0 | [45] |
| NIES CRM no. 8 vehicle exhaust particulates B | 126.0 ± 5.8 | 123 ± 3.9 | ||
| NIES CRM no. 8 vehicle exhaust particulates C | 134.0 ± 5.2 | 131 ± 4.4 | ||
| NIES CRM no. 8 vehicle exhaust particulates D | 143.0 ± 6.8 | 146 ± 6.5 |
| Element | Unit | Sample | Added | Found | RSD (%) | Recovery (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Sc | µg | Mineral sample 1 Mineral sample 2 Mineral sample 3 | 0.0080 0.0060 0.0040 | 0.0076 0.0055 0.0042 | 1.32 3.56 4.78 | 95.0 91.7 105 | [52] | |
| Sc | µg | Mineral sample 1 Mineral sample 2 Mineral sample 3 | 0.0080 0.0060 0.0050 | 0.0072 0.0058 0.0052 | 0.89 4.07 5.00 | 90.0 96.7 104 | [53] | |
| Ce | nM | Drink water Sea water | 100.0 1.0 100.0 1.0 | 97.3 0.95 102.7 1.04 | 3.5 3.3 3.8 4.2 | 97.3 95.0 102.7 104.0 | [35] | |
| Ce | nM | Bystrzyca river water Lake Zemborzyce | 300.0 600.0 300.0 600.0 | 294.6 614.4 292.2 604.8 | 3.7 4.2 4.0 4.9 | 98.2 102.4 97.4 100.8 | [31] | |
| Ce | nM | Human urine | 0 2.0 | 2.14 4.3 | 5.12 1.91 | - 108 | [37] | |
| Ce La Pr Ce La Pr | nM | Tap water 01 Tap water 02 | 71.4 72 71 357 360 355 | 69.0 66.0 60.2 347.0 338.0 289.1 | 2.75 3.33 7.00 1.10 2.66 6.70 | 96.60 91.67 84.79 97.20 93.88 81.44 | [48] | |
| Eu | nM | Red tribasic fluorescent powder sample | 0 150 | 210 350 | 3.4 4.0 | - 93 | [47] | |
| Eu | µM | Tap water Greenlake water Panlong river water | 0 2.0 50 150 0 2 .0 50 150 0 2.0 50 150 | 0 2.0 50 150 0 2.0 50 150 0 2.0 50 150 | - 2.6 2.3 1.8 - 3.0 1.2 2.5 - 3.6 2.8 1.5 | - 98.5 97.6 101.5 - 99.5 98.6 100.6 - 102.5 101.6 99.7 | [42] | |
| Element | Unit | Sample | Linear range | Recovery (%) | LOD | RSD (%) | ||
| Ce Gd Ce Gd Ce Gd | ng mL−1 | Aqueous Waste water Human blood serum | 0.25–6.23 0.74–9.47 0.25–6.23 0.78–8.96 0.25–5.72 0.76–9.23 | 98.2–103.3 97.4–103.2 98.2–103.3 97.5–102.9 98.8–102.5 98.4–101.7 | 0.073 0.196 0.075 0.173 0.068 0.178 | 0.51 0.76 1.23 0.98 0.82 1.02 | [34] | |
| Element | Unit | Sample | Added | Found | RSD (%) | Recovery (%) | Ref. | |
| AdSV | ICP | |||||||
| Eu | nM | Synthetic water (Na+, Ca2+, Mg2+, SO42−, Cl−) Tap water River water | 2000 5000 1000 700 | 2070 4700 900 820 | 1880 5210 1100 780 | 3.7 3.5 4.2 4.6 | 103.5 94.0 90.0 108.7 | [41] |
| Element | Unit | Sample | Found by AdSV Method | Found by ICP-OES Method | Δ% between the Two Methods | Ref. |
|---|---|---|---|---|---|---|
| Ce | µg g−1 | Spinach Mushroom Rice Pu’er tea | 0.428 2.885 0.545 0.338 | 0.436 2.905 0.552 0.341 | 1.9 0.7 1.3 0.9 | [38] |
| La | µg g−1 | Sand sample 1 Sand sample 2 Sand sample 3 | 1.06 ± 0.011 1.27 ± 0.024 1.47 ± 0.026 | 1.43 ± 0.015 1.52 ± 0.027 1.88 ± 0.031 | 34.9 19.7 27.9 | [50] |
| Ce | µg g−1 | Phosphate Sample 1 Phosphate Sample 2 | 1785 ± 65 1470 ± 54 | 1864 ± 57 1461 ± 43 | 4.4 0.6 | [33] |
| Ce | M | Catalyst sample 1 Catalyst sample 2 | 1.59 × 10−5 4.99 × 10−5 | 1.50 × 10−5 5.0 × 10−5 | 5.7 0.2 | [36] |
| Ce | M | Waste water Ce(III)-polluted water | 5.88 × 10−8 3.62 × 10−8 | 5.64 × 10−8 3.55 × 10−8 | 4.1 1.9 | [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarczyk, M.; Fialek, M.; Wlazlowska, E. Voltammetry in Determination of Trace Amounts of Lanthanides—A Review. Molecules 2023, 28, 7755. https://doi.org/10.3390/molecules28237755
Grabarczyk M, Fialek M, Wlazlowska E. Voltammetry in Determination of Trace Amounts of Lanthanides—A Review. Molecules. 2023; 28(23):7755. https://doi.org/10.3390/molecules28237755
Chicago/Turabian StyleGrabarczyk, Malgorzata, Marzena Fialek, and Edyta Wlazlowska. 2023. "Voltammetry in Determination of Trace Amounts of Lanthanides—A Review" Molecules 28, no. 23: 7755. https://doi.org/10.3390/molecules28237755
APA StyleGrabarczyk, M., Fialek, M., & Wlazlowska, E. (2023). Voltammetry in Determination of Trace Amounts of Lanthanides—A Review. Molecules, 28(23), 7755. https://doi.org/10.3390/molecules28237755