
New Application of cycloSaligenyl Prodrugs Approach for the Delivery of Fosfoxacin Derivatives in Mycobacteria

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
3. Experimental Section
3.1. Chemistry
3.1.1. General Methods
General Procedure A—Reduction of Carboxylic Acid to Alcohol
General Procedure B—Synthesis of cycloSalphosphochloridate
General Procedure C—Deprotection of Alcohol
General Procedure D—Synthesis of cycloSalphosphostriester
General Procedure E—Synthesis of cycloSalphosphostriester
General Procedure F—Deprotection of Dimethoxybenzyle
- 2-(Hydroxymethyl)-4-methylphenol (11b). The general procedure A was applied to synthesize the compound 11b from the corresponding carboxylic acid (1.0 g, 6.57 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 35:65 → EtOAc/petroleum ether, 75:25) to give 11b as a colorless solid (695 mg, 77%). Rf = 0.57 (EtOAc/petroleum ether, 3:7); 1H-NMR (500 MHz, CDCl3): 2.45 (3H, s, CH3), 5.01 (2H, d, 4J = 4.7 Hz, OCH2Ph), 6.98 (1H, d, 3J = 8.0 Hz, CHAr), 7.04 (1H, s, CHAr), 7.20 (1H, bdd, 3J = 8.2 Hz, 4J = 1.6 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 20.6 (CH3), 64.9 (OCH2Ph), 116.5 (CHAr), 124.6 (CAr), 128.6 (CHAr), 129.5 (CAr), 130.1 (CHAr), 153.9 (CArOH).
- 4-Chloro-2-(hydroxymethyl)phenol (11c). The general procedure A was applied to synthesize the compound 11c from the corresponding carboxylic acid (1.0 g, 5.79 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 20:80 → EtOAc/petroleum ether, 30:70) to give 11c as a colorless solid (1.32 g, 88%). Rf = 0.43 (EtOAc/petroleum ether, 2:8; 1H-NMR (500 MHz, CDCl3): 4.83 (2H, s, OCH2Ph), 6.82 (1H, d, 3J = 8.6 Hz, CHAr), 7.02 (1H, d, 4J = 2.4 Hz, CHAr), 7.20 (1H, dd, 3J = 8.7 Hz, 4J = 2.6 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 64.4 (OCH2Ph), 118.2 (CHAr), 124.9 (CAr), 126.1 (CAr), 127.6 (CHAr), 129.9 (CHAr), 154.9 (CArOH).
- 2,4-Dichloro-6-(hydroxymethyl)phenol (11d). The general procedure A was applied to synthesize the compound 11d from the corresponding carboxylic acid (2.20 g, 10.6 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 20:80) to give 11d as a colorless solid (1.45 g, 71%). Rf = 0.53 (EtOAc/petroleum ether, 2:8); 1H-NMR (400 MHz, CDCl3): 4.77 (2H, s, OCH2Ph), 7.12 (1H, d, 4J = 2.4 Hz, CHAr), 7.28 (1H, d, 4J = 2.4 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 62.4 (OCH2Ph), 121.2 (CAr), 125.4 (CAr), 126.9 (CHAr), 128.4 (CHAr), 128.6 (CAr), 149.1 (CArOH).
- 4-Bromo-2-(hydroxymethyl)phenol (11e). The general procedure A was applied to synthesize the compound 11e from the corresponding carboxylic acid (1.76 g, 8.10 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 20:80 → EtOAc/petroleum ether, 40:60) to give 11e as a colorless solid (1.21 g, 74%). Rf = 0.47 (EtOAc/petroleum ether, 2:8); 1H-NMR (500 MHz, CD3OD): 4.60 (2H, s, OCH2Ph), 6.68 (1H, d, 3J = 8.6 Hz, CHAr), 7.18 (1H, dd, 3J = 8.5 Hz, 4J = 2.5 Hz, CHAr), 7.40 (1H, d, 4J = 2.5 Hz, CHAr); 13C-NMR (125.8 MHz, CD3OD): 60.3 (OCH2Ph), 112.2 (CAr), 117.6 (CHAr), 131.5 (CHAr), 131.6 (CAr), 131.8 (CHAr), 155.4 (CArOH).
- 2-(Hydroxymethyl)-4-(trifluoromethyl)phenol (11f) Step1: Iodocyclohexane (5 mL, 45.4 mmol) was added to a solution of 2-(methoxy)-5-(trifluoromethyl)benzoic acid (1 g, 4.54 mmol) in DMF (5 mL). The mixture was refluxed for 4 h, and then the DMF was evaporated under reduced pressure. The resulting oil was dissolved in DCM (40 mL) and washed with a saturated aqueous solution of NaHCO3 (2 × 50 mL). The aqueous layer was acidified with a 10% HCl solution until a pH = 2 was obtained. The aqueous layer was then extracted with DCM (3 × 70 mL). The organic layer was dried over anhydrous Na2SO4, filtered and evaporated to dryness under reduced pressure. The crude product (1.95 g) was not purified and directly reduced with LiAlH4. Rf = 0.68 (EtOAc/petroleum ether, 5:5); 1H-NMR (400 MHz, CD3OD): 7.05 (1H, d, 3J = 8.6 Hz, CHAr), 7.74 (1H, dd, 3J = 8.8 Hz, 4J = 2.2 Hz, CHAr), 8.13 (1H, d, 4J = 2.0 Hz, CHAr); 13C-NMR (125.8 MHz, CD3OD): 114.3 (CAr), 119.5 (CHAr), 122.5 (q, 2JC-F = 32.9 Hz, CArCF3), 125.6 (q, 1JC-F = 269.4 Hz, CArCF3), 129.1 (q, 3JC-F = 4.0 Hz, CHAr), 133.1 (q, 3JC-F = 3.0 Hz, CHAr), 165.9 (CAr), 172.6 (CO); 19F-NMR (282.4 MHz, CD3OD): −64.4.
- 2-(Hydroxymethyl)-4-methoxyphenol (11g). To a solution of 2-hydroxy-5-methoxybenzoic acid (3 g, 17.8 mmol), NaBH4 (1.55, 41 mmol) in THF (47 mL) was added a solution of BF3-Et2O (3.3 mL, 26.7 mmol) in THF (12 mL) and refluxed overnight. The reaction mixture was cooled, poured into H2O (50 mL) and extracted with EtOAc (3 × 100 mL). The aqueous layer was saturated with NaCl and extracted with EtOAc (3 × 100 mL). The organic layers were collected, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by automated chromatography (petroleum ether → EtOAc/petroleum ether, 5:5) to give a colorless solid (2.18 g, 86%). Rf = 0.38 (EtOAc/petroleum ether, 3:7); 1H-NMR (500 MHz, CDCl3): 3.75 (3H, s, OCH3), 4.81 (2H, s, OCH2Ph), 6.61 (1H, d, 4J = 2.9 Hz, CHAr), 6.76 (1H, dd, 3J = 9.0 Hz, 4J = 2.7 Hz, CHAr), 6.81 (1H, d, 3J = 8.7 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 56.0 (OCH3), 64.9 (OCH2Ph), 113.6 (CHAr), 114.6 (CHAr), 117.4 (CHAr), 125.6 (CAr), 150.0 (CAr), 153.3 (CAr).
- 2-Chloro-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12a). The product was obtained as a colorless oil according to general procedure B. Rf = 0.91 (EtOAc/petroleum ether, 5:5); 1H-NMR (400 MHz, CDCl3): 5.44–5.53 (2H, m, OCH2Ph), 7.11 (2H, dd, 3J = 7.9 Hz, 3J = 7.9 Hz, CHAr), 7.23 (1H, dd, 3J = 7.9 Hz, 3J = 7.7 Hz, CHAr), 7.37 (1H, dd, 3J = 7.9 Hz, 3J = 7.9 Hz, CHAr); 31P-NMR (162.0 MHz, CDCl3): −6.03.
- 2-Chloro-6-methyl-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12b). The product was obtained as a slightly yellow oil according to general procedure B. Rf = 0.93 (EtOAc/petroleum ether, 5:5); 1H-NMR (400 MHz, CDCl3): 2.34 (3H, s, CH3), 5.39–5.48 (2H, m, OCH2Ph), 6.91 (1H, s, CHAr), 6.98 (1H, d, 3J = 8.4 Hz, CHAr), 7.15 (1H, d, 3J = 8.4 Hz, CHAr); 31P-NMR (162.0 MHz, CDCl3): −5.84.
- 2,6-Dichloro-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12c). The product was obtained as a colorless oil according to general procedure B. Rf = 0.88 (EtOAc/petroleum ether, 3:7); 1H-NMR (400 MHz, CDCl3): 5.40–5.50 (2H, m, OCH2Ph), 6.91 (1H, d, 3J = 8.7 Hz, CHAr), 7.13 (1H, d, 4J = 2.4 Hz, CHAr), 7.33–7.36 (1H, m, CHAr); 31P-NMR (162.0 MHz, CDCl3): −6.66.
- 2,6,8-Trichloro-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12d). The product was obtained as a yellow oil according to general procedure B. Rf = 0.33 (EtOAc/petroleum ether, 15:85); 1H-NMR (400 MHz, CDCl3): 5.41–5.49 (2H, m, OCH2Ph), 7.05 (1H, m, CHAr), 7.46 (1H, s, CHAr); 31P-NMR (162.0 MHz, CDCl3): −6.81.
- 6-Bromo-2-chloro-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12e). The product was obtained as a yellow oil according to general procedure B. 1H-NMR (300 MHz, CDCl3): 5.38–5.54 (2H, m, OCH2Ph), 7.00 (1H, d, 3J = 8.7 Hz, CHAr), 7.27–7.28 (1H, s, CHAr), 7.46–7.50 (1H, m, CHAr); 31P-NMR (121.5 MHz, CDCl3): −6.48.
- 2-Chloro-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12f). The product was obtained as a yellow oil according to general procedure B. 1H-NMR (400 MHz, CDCl3): 5.49–5.58 (2H, m, OCH2Ph), 7.24 (1H, d, 3J = 8.6 Hz, CHAr), 7.43 (1H, s, CHAr), 7.66 (1H, d, 3J = 8.2 Hz, CHAr); 31P-NMR (162.0 MHz, CDCl3): −6.99; 19F-NMR (282.4 MHz, CDCl3): −63.4.
- 2-Chloro-6-methoxy-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (12g). The product was obtained as a yellow oil according to general procedure B. 1H-NMR (400 MHz, CDCl3): 5.40–5.50 (2H, m, OCH2Ph), 6.60 (1H, d, 4J = 3.3 Hz, CHAr), 6.87–6.89 (1H, m, CHAr), 7.03 (1H, d, 3J = 8.9 Hz, CHAr); 31P-NMR (121.5 MHz, CDCl3): −5.87.
- O-(2,4-Dimethoxybenzyl)hydroxylamine Step 1: N-hydroxyphtalimide (5.00 g, 29.6 mmol) and 2,4-dimethoxybenzyl alcohol (4.96 g, 30.4 mmol) were stirred in dichloromethane (220 mL) at 0 °C. Triphenyl phosphine (12.2 g, 46.5 mmol) was added, followed by diisopropylazodicarboxylate (9.0 mL, 45.7 mmol). The resulting solution was stirred at room temperature for 24 h. The dichloromethane was removed under reduced pressure, and the resulting oil was recrystallized in boiling ethanol (200 mL) to give colorless crystals (6.32 g, 68%). Rf = 0.55 (EtOAc/petroleum ether, 3:7); 1H-NMR (500 MHz, CDCl3): 3.72 (3H, s, OCH3), 3.79 (3H, s, OCH3), 5.21 (2H, s, CH2), 6.39–6.44 (2H, m, Ar-H), 7.31 (1H, d, 3J = 8.4 Hz, Ar-H), 7.70–7.79 (4H, m, Ar-H); 13C-NMR (75.5MHz, CDCl3): 55.4 (OCH3), 55.7 (OCH3), 74.5 (CH2), 98.6–134.3 (CHAr and CAr), 160.1 (H3CO-CAr), 162.4 (H3CO-CAr), 163.7 (C=O); MS (EI)+: m/z calculated for C17H15NO5Na [M + Na]+ 336.08, found 336.08.
- 2-((t-Butyldimethylsilyl)oxy)ethan-1-ol (13). Sodium hydride (475 mg, 18.2 mmol) was suspended in THF (36 mL). The resulting mixture was cooled to 0 °C, the ethylene glycol (1 mL, 17.7 mmol) was added dropwise, and the mixture was stirred for 1 h. A solution of t-butyldimethylsilyl chloride (2.74 g, 18.0 mmol) in THF (8 mL) was added over a period of 10 min. The resulting mixture was stirred for 4 h at room temperature. A saturated aqueous solution of NaHCO3 (40 mL) was added, and the mixture was extracted with EtOAc (2 × 40 mL). The aqueous layer was saturated with NaCl and extracted with EtOAc (2 × 40 mL). The collected organic layers were dried over anhydrous Na2SO4, filtered, and solvents were removed under reduced pressure. The crude was purified by flash chromatography (EtOAc/petroleum ether, 2:8) to give the compound 13 as a colorless oil (2.48 g, 80%). Rf = 0.25 (EtOAc/petroleum ether, 1:9); 1H-NMR (300 MHz, CDCl3): 0.08 (6H, s, Si-CH3), 0.91 (9H, s, Si-t-Bu), 1.99 (1H, bs, OH), 3.62–3.65 (2H, m, CH2OTBDMS), 3.70–3.73 (2H, m, CH2OH); 13C-NMR (75.5 MHZ, CDCl3): −5.1 (Si-CH3), 18.5 (C4° of t-Bu), 26.1 (CH3 of tBu), 63.9 (CH2OTBDMS), 64.3 (CH2OH).
- N-(2-((t-butyldimethylsilyl)oxy)ethyl)-O-(2,4-dimethoxybenzyl) (14). To a solution of 13 (500 mg, 2.8 mmol) in DCM (35 mL), 2,6-lutidine (0.40 mL, 3.4 mmol) was added. The solution was cooled down to −78 °C, and the trifluoromethanesulfonic anhydride (470 μL, 2.8 mmol) was added dropwise. The resulting mixture was stirred at −78 °C for 1 h, and O-(2,4-dimethoxybenzyl)hydroxylamine (770 mg, 4.2 mmol) in DCM (20 mL) was added dropwise. The solution was stirred at −78 °C for 1 h, warmed up to room temperature, and stirred for another 2 h. The reaction mixture was diluted with DCM (30 mL) and washed with a saturated aqueous solution of NH4Cl (60 mL), a saturated solution of NaHCO3 (60 mL), water (60 mL) and brine (60 mL). The organic layer was dried over anhydrous Na2SO4 and filtered, and the solvents were removed under reduced pressure to give a pale yellow oil. The crude was purified by flash chromatography (petroleum ether → EtOAc/petroleum ether, 15:85) to give the product 14 as a colorless oil (424 mg, 44%). Rf = 0.46 (EtOAc/petroleum ether, 1:9); 1H-NMR (300 MHz, CDCl3): 0.04 (6H, s, Si-CH3), 0.87 (9H, s, tBu), 3.03 (2H, t, 3J = 5.3 Hz, CH2N), 3.73 (2H, t, 3J = 5.3 Hz, CH2OTBDMS), 3.80 (3H, s, OCH3), 3.81 (3H, s, OCH3), 4.70 (2H, s, OCH2DMP), 6.45–6.48 (2H, m, CHAr), 7.23 (1H, m, CHAr); 13C-NMR (75.5 MHZ, CDCl3): −5.2 (Si-CH3), 18.5 (C4° of tBu), 26.1 (CH3 of tBu), 53.9 (CH2N), 55.6 (OCH3), 55.7 (OCH3), 59.6 (CH2OTBDMS), 70.1 (OCH2DMP), 98.7 (CHAr), 104.0 (CHAr), 118.6 (CAr), 131.5 (CHAr), 159.1 (CArOCH3), 161.1 (CArOCH3); MS (EI)+: m/z calculated for C17H31NO4SiNa [M + Na]+ 364.19 found 364.19.
- N-(2-((t-Butyldimethylsilyl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)formamide (15a). A solution of formic acid (1.11 mL, 29.5 mmol) and acetic anhydride (0.56 mL, 5.9 mmol) was stirred at room temperature for 30 min. The solution was then cooled to 0 °C, and a solution of protected hydroxylamine 14 (200 mg, 0.59 mmol) in a minimum of THF was added dropwise. The reaction mixture was stirred at 0 °C for 10 min, allowed to warm up at room temperature, and stirred overnight. The reaction mixture was diluted with EtOAc, and the organic layer was washed twice with water and twice with a 0.1 M aqueous solution of KOH. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated to dryness. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 2:8) to give 15a as a colorless oil (180 mg, 83%) and as a mixture of the two Z and E conformers in a 30:70 ratio. Rf = 0.43 (EtOAc/petroleum ether, 2:8); 1H-NMR (300 MHz, CDCl3): 0.06 (6H, s, Si-CH3), 0.88 (9H, s, tBu), 3.44–3.83 (10H, m, OCH2CH2N and OCH3), 4.86 (7/10H of 2H, bs, OCH2DMP), 4.96 (3/10H of 2H, bs, OCH2DMP), 6.45–6.47 (2H, m, CHAr), 7.17 (1H, d, 2J = 7.0 Hz, CHAr), 7.92 (3/10H of 1H, bs, CHO), 8.19 (7/10H of 1H, bs, CHO); 13C-NMR (75.5 MHZ, CDCl3): −5.2 (Si-CH3), 18.5 (C4° of tBu), 26.1 (CH3 of tBu), 47.4 (CH2N), 51.8 (CH2N), 55.6 (OCH3), 55.7 (OCH3), 58.8 (CH2OTBDMS), 59.3 (CH2OTBDMS), 71.4 (OCH2DMP), 72.9 (OCH2DMP), 98.8 (CHAr), 104.3 (CHAr), 115.4 (CAr), 133.0 (CHAr), 159.0 (CHO), 159.7 (CArOCH3), 162.2 (CArOCH3), 163.6 (CHO); MS (EI)+: m/z calculated for C18H31NO5SiNa [M + Na]+ 392.19, found 392.18.
- N-(2-((t-Butyldimethylsilyl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)acetamide (15b). To a solution of N-H hydroxylamine 14 (116 mg, 0.34 mmol) in acetic anhydride (4 mL/mmol) was added dropwise pyridine (0.08 mL, 1.02 mmol equiv). The reaction mixture was stirred at room temperature overnight. The solvent was evaporated to dryness under reduced pressure. The product 15b was obtained without purification as a colorless oil (128 mg, 98%) and as the sole E conformer. Rf = 0.29 (EtOAc/petroleum ether, 1:9); 1H-NMR (300 MHz, CDCl3): 0.05 (6H, s, Si-CH3), 0.88 (9H, s, tBu), 2.83 (3H, s, COCH3) 3.78–3.83 (10H, m, OCH2CH2N and OCH3), 4.83 (2H, s, OCH2DMP), 6.45–6.48 (2H, m, CHAr), 7.17 (1H, d, 2J = 9.0 Hz, CHAr); 13C-NMR (75.5 MHZ, CDCl3): −5.2 (Si-CH3), 18.5 (C4° of tBu), 22.4 (COCH3), 26.1 (CH3 of tBu), 49.1 (CH2N), 55.5 (OCH3), 55.6 (OCH3), 59.6 (CH2OTBDMS), 71.4 (OCH2DMP), 98.8 (CHAr), 104.3 (CHAr), 115.7 (CAr), 132.7 (CHAr), 159.6 (CArOCH3), 162.1 (CArOCH3), 166.6 (COCH3); MS (EI)+: m/z calculated for C19H33NO5SiNa [M + Na]+ 406.20, found 406.20.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-hydroxyethyl)formamide (9a). The general procedure C was applied to synthesize the compound 9a from protected alcohol 15a (200 mg, 0.59 mmol). The crude product was purified by flash chromatography (EtOAc) to give 9a as a colorless oil (97 mg, 76%) and as a mixture of the two Z and E conformers in a 30:70 ratio. Rf = 0.37 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.70 (1H, bs, OH), 3.42–3.84 (10H, m, OCH2CH2N and OCH3), 4.87 (7/10H of 2H, bs, OCH2DMP), 5.02 (3/10H of 2H, bs, OCH2DMP), 6.45–6.47 (2H, m, CHAr), 7.17 (1H, d, 2J = 8.1 Hz, CHAr), 7.96 (3/10H of 1H, bs, CHO), 8.22 (7/10H of 1H, bs, CHO); 13C-NMR (75.5 MHZ, CDCl3): 48.7 (CH2N), 53.5 (CH2N), 55.6 (OCH3), 55.7 (OCH3), 60.8 (CH2OH), 71.5 (OCH2DMP), 73.2 (OCH2DMP), 98.8 (CHAr), 104.6 (CHAr), 114.9 (CAr), 132.2 (CHAr), 159.7 (CArOCH3), 162.4 (CArOCH3), 164.2 (CHO); MS (EI)+: m/z calculated for C12H17NO5Na [M + Na]+ 278.0999, found 278.0980.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-hydroxyethyl)acetamide (9b). The general procedure C was applied to synthesize the compound 9b from protected alcohol 15b (200 mg, 0.59 mmol). The crude product was purified by flash chromatography (EtOAc) to give 9b as a colorless oil (133 mg, 80%) and as the sole E conformer. Rf = 0.48 (EtOAc); 1H-NMR (400 MHz, CDCl3): 2.13 (3H, s, COCH3) 3.80–3.84 (10H, m, OCH2CH2N and OCH3), 4.83 (2H, s, OCH2DMP), 6.46–6.49 (2H, m, CHAr), 7.19 (1H, d, 2J = 9.0 Hz, CHAr); 13C-NMR (75.5 MHZ, CDCl3): 20.4 (COCH3), 50.4 (CH2N), 55.6 (OCH3), 55.7 (OCH3), 61.8 (CH2OH), 71.9 (OCH2DMP), 98.9 (CHAr), 104.5 (CHAr), 115.1 (CAr), 133.0 (CHAr), 159.7 (CArOCH3), 162.4 (CArOCH3), 174.2 (COCH3); MS (EI)+: m/z calculated for C13H19NO5Na [M + Na]+ 292.12, found 292.12.
- N-((2,4-Dimethoxybenzyl)oxy)-3-hydroxypropanamide (10a). A stirred suspension of O-protected hydroxylamine (3.06 g, 16.7 mmol) in dry THF (4.5 mL/mmol) at −78 °C was treated with 1 M solution of LiHMDS (55.5 mL, 55.5 mmol). After 1 h, a solution of β-propiolactone (0.70 mL, 11.1 mmol) in a minimum of THF was added. The resulting solution was stirred overnight at room temperature. The reaction was cooled at −78 °C, quenched with a saturated aqueous solution of NH4Cl, warmed to room temperature and extracted several times with EtOAc. The collected organic layers were dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The crude product was dissolved in THF and treated with TBAF.(H2O)3 (1.5 equiv). After total consumption of the starting material, the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (EtOAc → EtOAc/MeOH, 90:10) to give the product 10a (1.45 g, 51%) as a colorless solid and as a mixture of the two Z and E conformers in a 60:40 ratio. Rf = 0.24 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.30 (6/10H of 2H, bs, CH2CO), 2.63 (4/10H of 2H, bs, CH2CO), 2.91 (1H, bs, OH), 3.79–3.84 (8H, m, OCH3 and CH2OH), 4.80 (4/10H of 2H, bs, OCH2DMP), 4.91 (6/10H of 2H, bs, OCH2DMP), 6.44–6.48 (2H, m, CHAr), 7.22 (1H, m, CHAr), 8.16 (4/10 of 1H, bs, NH), 8.58 (6/10 of 1H, bs, NH); 13C-NMR (75.5 MHz, CDCl3): 33.5 (CH2CO), 35.6 (CH2CO), 55.5 (OCH3), 55.6 (OCH3), 58.1 (CH2OH), 58.6 (CH2OH), 73.2 (OCH2DMP), 74.6 (OCH2DMP), 98.6 (CHAr), 104.1 (CHAr), 114.9 (CAr), 155.9 (CAr), 132.8 (CHAr), 159.5 (CArOCH3), 161.8 (CArOCH3), 162.3 (CArOCH3), 169.8 (CO), 176.6 (CO); MS (EI)+: m/z calculated for C12H17NO5Na [M + Na]+ 278.10, found 278.10.
- N-((2,4-Dimethoxybenzyl)oxy)-3-hydroxy-N-methylpropanamide (10b). To a solution of 10a (267 mg, 1.05 mmol), anhydrous K2CO3 (290 mg, 2.1 mmol) in anhydrous acetone (10.5 mL/mmol) was added iodomethane (0.33 mL, 5.25 mmol). The resulting mixture was refluxed overnight. The mixture was filtered, and acetone was removed under reduced pressure. The resulting oil was dissolved in ether. The organic layer was washed with water, and then the aqueous layer was saturated with NaCl and extracted with EtOAc. The organic layers were collected, dried over anhydrous Na2SO4, filtered and evaporated. The crude product was purified by automated flash chromatography (EtOAc/petroleum ether, 7:3 → EtOAc) to give 10b as a yellow oil (243 mg, 86%) as a sole E conformer. Rf = 0.44 (EtOAc/petroleum ether, 7:3); 1H-NMR (500 MHz, CDCl3): 2.63 (2H, t, 3J = 5.6 Hz, CH2CO), 3.23 (4H, ds, NCH3 and OH), 3.79–3.84 (8H, m, OCH3 and CH2OH), 4.80 (2H, s, OCH2DMP), 6.46–6.48 (2H, m, CHAr), 7.18 (1H, d, 3J = 8.9 Hz, CHAr); 13C-NMR (75.5 MHz, CDCl3): 33.1 (NCH3), 34.0 (CH2CO), 55.6 (OCH3), 58.8 (CH2OH), 71.1 (OCH2DMP), 98.8 (CHAr), 104.4 (CHAr), 115.1 (CAr), 132.9 (CHAr), 159.67 (CArOCH3), 162.3 (CArOCH3), 174.7 (CO); MS (EI)+: m/z calculated for C13H19NO5Na [M + Na]+ 292.12, found 292.12.
- N-((2,4-Dimethoxylbenzyl)oxy)-N-(2-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)formamide (17a). The general procedure D was applied to synthesize the compound 17aa from alcohol 9a (172 mg, 0.67 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 8:2) to give 17aa as a colorless oil (187 mg, 66%) and as a mixture of the two Z and E conformers in a 20:80 ratio. Rf = 0.49 (EtOAc/cyclohexane, 8:2); 1H-NMR (400 MHz, CDCl3): 3.57–3.95 (8H, m, NCH2 and OCH3), 4.38 (2H, bs, POCH2), 4.80 (8/10H of 2H, bs, OCH2DMP), 4.96 (2/10 of 2H, bs, OCH2DMP), 5.25–5.39 (2H, m, ArCH2OP), 6.43–6.45 (2H, m, CHAr(DMB)), 7.03 (2H, t, 3J = 8.2 Hz, CHAr(cycloSal)), 7.11 (2H, m, CHAr(cycloSal) and CHAr(DMB)), 7.28 (1H, m, CHAr(cycloSal)), 7.87 (2/10H of 1H, bs, CHO), 8.06 (8/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CDCl3): 44.6 (d, 3JCP = 7.0 Hz, NCH2), 55.6 (OCH3), 55.7 (OCH3), 63.9 (d, 2JCP = 4.4 Hz, POCH2), 69.0 (d, 2JCP = 6.9 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.4 (CHAr(DMB)), 114.9 (CAr(DMB)), 118.9 (d, 3JCP = 8.7 Hz, CHArCArOP), 120.7 (d, 3JCP = 8.7 Hz, CArCH2OP), 124.5 (CHAr(cycloSal)), 125.4 (CHAr(cycloSal)), 129.9 (CHAr(cycloSal)), 133.1 (CHAr(DMB)), 150.1 (d, 2JCP = 6.6 Hz, CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 163.7 (CHO); 31P-NMR (162.0 MHz, CDCl3): −9.89; MS (EI)+: m/z calculated for C19H22NO8PNa [M + Na]+ 446.10, found 446.09.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-((6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)formamide (17ab). The general procedure D was applied to synthesize the compound 17ab from alcohol 9a (61 mg, 0.24 mmol). The crude product was purified by automated flash chromatography (EtOAc/petroleum ether, 6:4 → EtOAc) to give 17ab as a colorless oil (30 mg, 46%) and as a mixture of two Z and E conformers in a 20:80 ratio. Rf = 0.35 (EtOAc/petroleum ether, 7:3); 1H-NMR (400 MHz, CDCl3): 2.30 (3H, s, CH3Ar), 3.78–3.88 (8H, m, NCH2 and OCH3), 4.35–4.38 (2H, bs, POCH2), 4.80 (8/10H of 2H, bs, OCH2DMP), 4.97 (2/10 of 2H, bs, OCH2DMP), 5.20–5.31 (2H, m, ArCH2OP), 6.42–6.46 (2H, m, CHAr(DMB)), 6.83 (1H, bs, CHAr(cycloSal)), 6.90 (1H, d, 3J = 8.4 Hz, CHAr(cycloSal)),7.07 (1H, d, 3J = 8.4 Hz, CHAr(cycloSal)), 7.12–7.15 (1H, m, CHAr(DMB)), 7.86 (2/10H of 1H, bs, CHO), 8.07 (8/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CDCl3): 20.9 (CH3Ar), 44.7 (d, 3JCP = 5.8 Hz, NCH2), 55.6 (OCH3), 55.7 (OCH3), 63.9 (POCH2), 69.1 (d, 2JC-P = 7.0 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.5 (CHAr(DMB)), 114.9 (CAr(DMB)), 118.7 (d, 3JC-P = 7.9 Hz, CHArCArOP), 120.3 (d, 3JC-P = 8.9 Hz, CArCH2OP), 125.7 (CHAr(cycloSal)), 130.4 (CHAr(cycloSal)), 133.1 (CHAr(DMB)), 134.2 (CArCH3), 148.1 (CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 163.8 (CHO); 31P-NMR (162.0 MHz, CDCl3): −9.7; HRMS (EI)+: m/z calculated for C20H24NO8PNa [M + Na]+ 460.1132, found 460.1116.
- N-(2-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)formamide (17ac). The general procedure D was applied to synthesize compound 17ac from alcohol 9a (169 mg, 0.66 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 8:2) to give 17ac as a colorless oil (155 mg, 51%) and as a mixture of the two Z and E conformers in a 20:80 ratio. Rf = 0.51 (EtOAc/cyclohexane, 8:2); 1H-NMR (400 MHz, CDCl3): 3.82–3.88 (8H, m, NCH2 and OCH3), 4.39 (2H, bs, POCH2), 4.80 (8/10H of 2H, bs, OCH2DMP), 4.96 (2/10 of 2H, bs, OCH2DMP), 5.18–5.34 (2H, m, ArCH2OP), 6.43–6.46 (2H, m, CHAr(DMB)), 6.94 (1H, d, 3J = 8.7 Hz, CHAr(cycloSal)), 7.03 (1H, s, CHAr(cycloSal)), 7.12 (1H, d, 3J = 6.9 Hz, CHAr(DMB)), 7.25 (1H, d, 3J = 8.8 Hz, CHAr(cycloSal)) 7.89 (2/10H of 1H, bs, CHO), 8.07 (8/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CDCl3): 44.6 (d, 3JCP = 6.0 Hz, NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.2 (POCH2), 68.5 (d, 2JCP = 6.9 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.5 (CHAr(DMB)), 114.9 (CAr(DMB)), 120.4 (d, 3JCP = 9.5 Hz, CHArCArOP), 122.1 (d, 3JCP = 10.2 Hz, CArCH2OP), 125.4 (CHAr(cycloSal)), 129.7 (CArCl), 129.9 (CHAr(cycloSal)), 133.1 (CHAr(DMB)), 147.7 (d, 2JCP = 6.4 Hz, CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 163.7 (CHO); 31P-NMR (162.0 MHz, CDCl3): −10.4; HRMS (EI)+: m/z calculated for C19H21ClNO8PNa [M + Na]+ 480.0556, found 480.0556.
- N-(2-((6,8-Dichloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)formamide (17ad). The general procedure D was applied to synthesize compound 17ad from alcohol 9a (185 mg, 0.73 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 8:2) to give 17ad as a colorless oil (177 mg, 49%) and as a mixture of two Z and E conformers in a 20:80 ratio. Rf = 0.51 (EtOAc/cyclohexane, 8:2); 1H-NMR (400 MHz, CDCl3): 3.82–3.90 (8H, m, NCH2 and OCH3), 4.41 (2H, bs, POCH2), 4.80 (8/10H of 2H, bs, OCH2DMP), 4.94 (2/10 of 2H, bs, OCH2DMP), 5.19–5.34 (2H, m, ArCH2OP),6.46 (2H, bs, CHAr(DMB)), 6.94 (1H, bs, CHAr(cycloSal)), 7.14 (1H, m, CHAr(DMB)), 7.35 (1H, bs, CHAr(cycloSal)) 7.91 (2/10H of 1H, bs, CHO), 8.06 (8/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CDCl3): 44.4 (d, 3JCP = 6.0 Hz, NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.5 (POCH2), 68.4 (d, 2JCP = 6.9 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.5 (CHAr(DMB)), 114.8 (CAr(DMB)), 123.4 (d, 3JCP = 9.5 Hz, ClCArCArOP), 123.7 (CHAr(cycloSal)) 124.9 (CArCH2OP), 129.6 (CArCl), 130.2 (CHAr(cycloSal)), 133.1 (CHAr(DMB)), 145.0 (CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 163.7 (CHO); 31P-NMR (162.0 MHz, CDCl3): −10.9; MS (EI)+: m/z calculated for C19H20Cl2NO8PNa [M + Na]+ 514.02, found 514.02.
- N-(2-((6-Bromo-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)formamide (17ae). The general procedure D was applied to synthesize compound 17ae from alcohol 9a (60 mg, 0.24 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc/petroleum ether, 7:3) to give 17ae as a colorless oil (44 mg, 37%) and as a mixture of the two Z and E conformers in a 20:80 ratio. Rf = 0.57 (EtOAc/petroleum ether, 7:3); 1H-NMR (400 MHz, CDCl3): 3.76–3.95 (8H, m, NCH2 and OCH3), 4.38 (2H, bs, POCH2), 4.80 (8/10H of 2H, bs, OCH2DMP), 4.95 (2/10 of 2H, bs, OCH2DMP), 5.22 (1H, dd, 2J = 14.7 Hz, 3JP-H = 18.3 Hz, ArCH2OP), 5.31 (1H, dd, 2J = 14.3 Hz, 3JP-H = 8.0 Hz, ArCH2OP), 6.44–6.46 (2H, m, CHAr(DMB)), 6.89 (1H, d, 3J = 8.7 Hz, CHAr(cycloSal)), 7.11–7.17 (2H, m, CHAr(DMB) and CHAr(cycloSal)), 7.38 (1H, d, 3J = 8.7 Hz, CHAr(cycloSal)), 7.89 (2/10H of 1H, bs, CHO), 8.06 (8/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CDCl3): 44.5 (d, 3JCP = 6.9 Hz, NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.1 (POCH2), 64.2 (d, 2JC-P = 4.5 Hz, POCH2) 68.3 (d, 2JC-P = 6.7 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.5 (CHAr(DMB)), 114.8 (CAr(DMB)), 117.0 (CArBr), 120.7 (d, 3JC-P = 8.6 Hz, CHArCArOP), 122.6 (d, 3JC-P = 9.4 Hz, CArCH2OP), 128.3 (CHAr(cycloSal)), 132.8 (CHAr(cycloSal)), 133.1 (CHAr(DMB)), 149.3 (d, 2JC-P = 6.5 Hz, CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 163.7 (CHO); 31P-NMR (162.0 MHz, CDCl3): −10.5; HRMS (EI)+: m/z calculated for C19H21BrNO8PNa [M + Na]+ 524.0080, found 524.0138.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-((2-oxido-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)formamide (17af). The general procedure D was applied to synthesize the compound 17af from alcohol 9a (96 mg, 0.38 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 7:3 → EtOAc/cyclohexane, 8:2) to give 17af as a colorless oil (98 mg, 52%) and as a mixture of two Z and E conformers in a 20:80 ratio. Rf = 0.50 (EtOAc/cyclohexane, 8:2); 1H-NMR (400 MHz, CDCl3): 3.76–3.94 (8H, m, NCH2 and OCH3), 4.42 (2H, bs, POCH2), 4.80 (8/10 of 2H, bs, OCH2DMP), 4.96 2/10 of 2H, bs, OCH2DMP), 5.31 (1H, dd, 2J = 14.4 Hz, 3JP-H = 18.6 Hz, ArCH2OP), 5.40 (1H, dd, 2J = 14.4 Hz, 3JP-H = 8.7 Hz, ArCH2OP), 6.44–6.46 (2H, m, CHAr(DMB)), 7.10–7.13 (2H, m, CHAr(DMB) and CHAr(cycloSal)), 7.33 (1H, bs, CHAr(cycloSal)), 7.55 (1H, d, 3J = 8.6 Hz, CHAr(cycloSal)), 7.90 (2/10 of 1H, bs, CHO), 8.06 (8/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CDCl3): 44.5 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.4 (POCH2), 68.6 (d, 2JC-P = 6.8 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.5 (CHAr(DMB)), 114.8 (CAr(DMB)), 119.7 (d, 3JC-P = 9.4 Hz, CHArCArOP), 121.3 (d, 3JC-P = 9.1 Hz, CArCH2OP), 123.1 (CHAr(cycloSal)), 127.2 (CHAr(cycloSal)), 133.1 (CHAr(DMB)), 152.6 (d, 2JC-P = 6.1 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 163.7 (CHO); 31P-NMR (162.0 MHz, CDCl3): −10.7; 19F-NMR (282.4 MHz, CDCl3): −63.2; HRMS (EI)+: m/z calculated for C20H21F3NO8PNa [M + Na]+ 514.0849, found 514.0876.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-((6-methoxy-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)formamide (17ag). The general procedure D was applied to synthesize compound 17ag from alcohol 9a (96 mg, 0.38 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 7:3 → EtOAc) to give 17ag as a colorless oil (110 mg, 64%) and as a mixture of the two Z and E conformers in a 20:80 ratio. Rf = 0.28 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 3.54–3.89 (11H, m, NCH2 and OCH3), 4.34 (2H, bs, POCH2), 4.78 (8/10H of 2H, bs, OCH2DMP), 4.95 (2/10 of 2H, bs, OCH2DMP), 5.20–5.32 (2H, m, ArCH2OP), 6.41–6.43 (2H, m, CHAr(DMB)), 6.51 (1H, s, CHAr(cycloSal)), 6.78 (1H, d, 3J = 8.6 Hz, CHAr(cycloSal)), 6.92 (1H, d, 3J = 9.2 Hz, CHAr(cycloSal)), 7.10 (1H, m, CHAr(DMB)), 7.84 (2/10H of 1H, bs, CHO), 8.04 (8/10 of 1H, bs, CHO);13C-NMR (125.8 MHz, CDCl3): 44.7 (d, 3JCP = 6.5 Hz, NCH2), 55.6 (OCH3), 55.7 (OCH3), 55.9 (OCH3), 63.9 (POCH2), 69.1 (d, 2JC-P = 7.1 Hz, ArCH2OP), 73.2 (OCH2DMP), 98.8 (CHAr(DMB)), 104.5 (CHAr(DMB)), 110.1 (CHAr(cycloSal)), 114.9 (CAr(DMB)), 115.2 (CHAr(cycloSal)), 119.8 (d, 3JC-P = 8.1 Hz, CHArCArOP), 121.3 (CArCH2OP), 133.1 (CHAr(DMB)), 143.9 (d, 2JC-P = 7.3 Hz, CArOP), 156.2 (CArOCH3), 159.7 (CArOCH3), 162.4 (CArOCH3), 163.8 (CHO); 31P-NMR (162.0 MHz, CDCl3): −9.4.; HRMS (EI)+: m/z calculated for C20H24NO9PNa [M + Na]+ 476.1081, found 476.1109.
- N-((2,4-Dimethoxylbenzyl)oxy)-N-(2-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)acetamide (17ba). The general procedure D was applied to synthesize compound 17ba from alcohol 9b (190 mg, 0.71 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 8:2) to give 17ba as a colorless oil (227 mg, 73%) and as the sole E conformer. Rf = 0.47 (EtOAc/cyclohexane, 8:2); 1H-NMR (400 MHz, CDCl3): 1.99 (3H, s, CH3CO), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.89–3.98 (2H, m, NCH2), 4.34–4.41 (2H, m, POCH2), 4.76 (2H, s, OCH2DMP), 5.23–5.38 (2H, m, ArCH2OP), 6.43–6.47 (2H, m, CHAr(DMB)), 7.01 (2H, t, 3J = 8.6 Hz, CHAr(cycloSal)), 7.10 (1H, t, 3J = 7.1 Hz, CHAr(cycloSal)) 7.16 (1H, d, 3J = 8.1 Hz, CHAr(DMB)), 7.27 (1H, m, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.2 (CH3CO), 46.1 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.4 (d, 2JCP = 5.5 Hz, POCH2), 68.9 (d, 2JCP = 6.9 Hz, ArCH2OP), 71.6 (OCH2DMP), 98.7 (CHAr(DMB)), 104.4 (CHAr(DMB)), 115.1 (CAr(DMB)), 119.0 (d, 3JCP = 9.1 Hz, CHArCArOP), 120.7 (d, 3JCP = 10.0 Hz, CArCH2OP), 124.4 (CHAr(cycloSal)), 125.4 (CHAr(cycloSal)), 129.9 (CHAr(cycloSal)), 132.9 (CHAr(DMB)), 150.3 (d, 2JCP = 6.9 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 173.4 (CO); 31P-NMR (162.0 MHz, CDCl3): −9.90; MS (EI)+: m/z calculated for C20H24NO8PNa [M + Na]+ 460.11, found 460.11.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-((6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)acetamide (17bb). The general procedure D was applied to synthesize compound 17bb from alcohol 9b (200 mg, 0.78 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 7:3 → EtOAc) to give 17bb as a colorless oil (17 mg, 36%) and as the sole E conformer. Rf = 0.33 (EtOAc/petroleum ether, 7:3); 1H-NMR (400 MHz, CDCl3): 2.00 (3H, s, CH3CO), 2.29 (3H, s, CH3Ar), 3.82 (OCH3), 3.83 (OCH3), 3.87–3.97 (2H, m, NCH2), 4.30–4.42 (2H, m, POCH2), 4.76 (2H, s, OCH2DMP), 5.18–5.33 (2H, m, ArCH2OP), 6.43–6.46 (2H, m, CHAr(DMB)), 6.81 (1H, bs, CHAr(cycloSal)), 6.89 (1H, d, 3J = 8.3 Hz, CHAr(cycloSal)), 7.05 (1H, d, 3J = 7.9 Hz, CHAr(cycloSal)), 7.15 (1H, d, 3J = 7.9 Hz, CHAr(DMB)); 13C-NMR (125.8 MHz, CDCl3): 20.1 (CH3CO), 20.8 (CH3Ar), 46.2 (NCH2), 55.5 (OCH3), 55.6 (OCH3), 64.4 (d, 2JC-P = 5.6 Hz, POCH2), 69.0 (d, 2JC-P = 7.0 Hz, ArCH2OP), 71.6 (OCH2DMP), 98.7 (CHAr(DMB)), 104.3 (CHAr(DMB)), 115.1 (CAr(DMB)), 118.6 (d, 3JC-P = 9.3 Hz, CHArCArOP), 120.3 (d, 3JC-P = 9.5 Hz, CArCH2OP), 125.6 (CHAr(cycloSal)), 130.3 (CHAr(cycloSal)), 132.9 (CHAr(DMB)), 134.1 (CArCH3), 148.1 (d, 2JC-P = 7.1 Hz, CArOP), 159.6 (CArOCH3), 162.2 (CArOCH3), 173.5 (CO); 31P-NMR (162.0 MHz, CDCl3): −9.7; HRMS (EI)+: m/z calculated for C21H26NO8PNa [M + Na]+ 474.1288, found 474.1247.
- N-(2-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)acetamide (17bc). The general procedure D was applied to synthesize compound 17bc from alcohol 9b (180 mg, 0.67 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 8:2) to give 17bc as a colorless oil (178 mg, 56%) and as the sole E conformer. Rf = 0.40 (EtOAc/cyclohexane, 8:2); 1H-NMR (400 MHz, CDCl3): 2.00 (3H, s, CH3CO), 3.83 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.91–3.96 (2H, m, NCH2), 4.35–4.42 (2H, m, POCH2), 4.76 (2H, s, OCH2DMP), 5.16–5.33 (2H, m, ArCH2OP), 6.44–6.47 (2H, m, CHAr(DMB)), 6.93 (1H, d, 3J = 8.1 Hz CHAr(cycloSal)), 7.00 (1H, d, 4J = 2.4 Hz, CHAr(cycloSal)), 7.15 (1H, d, 3J = 8.0 Hz, CHAr(DMB)) 7.22 (1H, d, 3J = 8.9 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.2 (CH3CO), 45.9 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.7 (d, 2JCP = 5.6 Hz, POCH2), 68.4 (d, 2JCP = 7.2 Hz, ArCH2OP), 71.7 (OCH2DMP), 98.8 (CHAr(DMB)), 104.4 (CHAr(DMB)), 115.1 (CAr(DMB)), 120.4 (d, 3JCP = 9.1 Hz, CHArCArOP), 122.3 (d, 3JCP = 9.9 Hz, CArCH2OP), 125.4 (CHAr(cycloSal)),129.6 (CArCl), 129.8 (CHAr(cycloSal)), 132.9 (CHAr(DMB)), 148.8 (d, 2JCP = 7.0 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 173.4 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.4; HRMS (EI)+: m/z calculated for C20H23ClNO8Pna [M + Na]+ 494.0683, found 494.0683.
- N-(2-((6,8-Dichloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)acetamide (17bd). The general procedure D was applied to synthesize compound 17bd from alcohol 9b (214 mg, 0.79 mmol). The crude product was purified by automated flash chromatography (DCM/MeOH, 98:2) to give 17bd as a colorless oil (15 mg, 12%) and as the sole E conformer. Rf = 0.32 (DCM/MeOH, 98:2); 1H-NMR (400 MHz, CDCl3): 2.02 (3H, s, CH3CO), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.27–4.45 (2H, m, NCH2), 4.35–4.42 (2H, m, POCH2) 4.76 (2H, s, OCH2DMP), 5.17–5.32 (2H, m, ArCH2OP), 6.44–6.46 (2H, m, CHAr(DMB)), 6.92 (1H, CHAr(cycloSal)), 7.15 (1H, d, 3J = 8.3 Hz, CHAr(DMB)) 7.35 (1H, d, 3J = 8.9 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.1 (CH3CO), 45.8 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 65.1 (POCH2), 68.3 (d, 2JCP = 7.0 Hz, ArCH2OP), 71.7 (OCH2DMP), 98.7 (CHAr(DMB)), 104.4 (CHAr(DMB)), 115.1 (CAr(DMB)), 123.4 (d, 3JCP = 9.5 Hz, ClCArCArOP), 123.8 (CHAr(cycloSal)) 124.9 (d, 3JCP = 8.7 Hz,CArCH2OP), 129.5 (CArCl), 130.2 (CHAr(cycloSal)), 132.9 (CHAr(DMB)), 145.1 (d, 2JCP = 5.9 Hz, CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 173.4 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.7; MS (EI)+: m/z calculated for C20H22Cl2NO8PNa [M + Na]+ 528.04, found 528.03.
- N-(2-((6-Bromo-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-((2,4-dimethoxybenzyl)oxy)acetamide (17be). The general procedure D was applied to synthesize compound 17be from alcohol 9b (60 mg, 0.22 mmol). The crude product was purified by automated flash chromatography (EtOAc/petroleum ether, 5:5) to give 17be as a colorless oil (58 mg, 51%) and as the sole E conformer. Rf = 0.42 (EtOAc/petroleum ether, 7:3); 1H-NMR (500 MHz, CDCl3): 2.00 (3H, s, CH3CO), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3) 3.87–3.99 (2H, m, NCH2), 4.33–4.43 (2H, m, POCH2), 4.75 (2H, s, OCH2DMP), 5.20 (1H, dd, 2J = 14.4 Hz, 3JP-H = 19.1 Hz, ArCH2OP), 5.30 (1H, dd, 2J = 14.0 Hz, 3JP-H = 8.0 Hz, ArCH2OP), 6.44–6.47 (2H, m, CHAr(DMB)), 6.87 (1H, d, 3J = 8.6 Hz, CHAr(cycloSal)), 7.14–7.17 (2H, m, CHAr(DMB) and CHAr(cycloSal)), 7.37 (1H, d, 3J = 9.3 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.1 (CH3CO), 45.9 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.7 (d, 2JC-P = 5.7 Hz, POCH2), 68.3 (d, 2JC-P = 7.0 Hz, ArCH2OP), 71.7 (OCH2DMP), 98.8 (CHAr(DMB)), 104.4 (CHAr(DMB)), 115.0 (CAr(DMB)), 116.9 (CArBr), 120.7 (d, 3JC-P = 9.0 Hz, CHArCArOP), 122.7 (d, 3JC-P = 10.1 Hz, CArCH2OP), 128.3 (CHAr(cycloSal)), 132.8 (CHAr(cycloSal)), 132.9 (CHAr(DMB)), 149.4 (d, 2JC-P = 7.1 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 173.4 (CO); 31P-NMR (121.5 MHz, CDCl3): −10.3; HRMS (EI)+: m/z calculated for C20H24BrNO8P [M + H]+ 516.0417, found 516.0411.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-((2-oxido-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)acetamide (17bf). The general procedure D was applied to synthesize compound 17bf from alcohol 9b (98 mg, 0.36 mmol). The crude product was purified by automated flash chromatography EtOAc/cyclohexane, 7:3 → EtOAc) to give 17bf as a colorless oil (73 mg, 52%) and as the sole E conformer. Rf = 0.42 (EtOAc/cyclohexane, 8:2); 1H-NMR (500 MHz, CDCl3): 1.97 (3H, s, CH3CO), 3.81 (6H, s, OCH3), 3.88–3.99 (2H, m, NCH2), 4.38–4.45 (2H, m, POCH2), 4.75 (2H, bs, OCH2DMP), 5.28 (1H, dd, 2J = 14.2 Hz, 3JP-H = 18.7 Hz, ArCH2OP), 5.38 (1H, dd, 2J = 14.3 Hz, 3JP-H = 7.5 Hz, ArCH2OP), 6.44–6.47 (2H, m, CHAr(DMB)), 7.08 (1H, d, 3J = 8.4 Hz, CHAr(cycloSal)), 7.15 (1H, d, 3J = 7.9 Hz, CHAr(DMB)), 7.30 (1H, bs, CHAr(cycloSal)), 7.54 (1H, d, 3J = 8.5 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.1 (CH3CO), 44.8 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 64.8 (d, 2JC-P = 5.6 Hz, POCH2), 68.5 (d, 2JC-P = 6.9 Hz, ArCH2OP), 71.7 (OCH2DMP), 98.8 (CHAr(DMB)), 104.4 (CHAr(DMB)), 115.0 (CAr(DMB)), 119.6 (d, 3JC-P = 9.4 Hz, CHArCArOP), 121.4 (d, 3JC-P = 9.9 Hz, CArCH2OP), 123.9 (q, 3JC-F = 2.9 Hz, CHAr(cycloSal)), 125.8 (q, 1JC-F = 273.4 Hz, CArCF3), 126.7 (q, 2JC-F = 32.9 Hz, CArCF3), 127.2 (CHAr(cycloSal)), 132.9 (CHAr(DMB)), 152.7 (d, 2JC-P = 6.9 Hz, CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 173.3 (CO); 31P-NMR (121.5 MHz, CDCl3): −10.5; 19F-NMR (282.4 MHz, CDCl3): −63.2; HRMS (EI)+: m/z calculated for C21H23F3NO8PNa [M + Na]+ 528.1006, found 528.1001.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-((6-methoxy-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)acetamide (17bg). The general procedure D was applied to synthesize compound 17bg from alcohol 9b (92 mg, 0.36 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 7:3 → EtOAc) to give 17bg as a colorless oil (94 mg, 56%) and as the sole E conformer. Rf = 0.27 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.00 (3H, s, CH3CO), 3.76 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.88–3.97 (2H, m, NCH2), 4.31–4.41 (2H, bs, POCH2), 4.75 (2H, bs, OCH2DMP), 5.19–5.33 (2H, m, ArCH2OP), 6.43–6.46 (2H, m, CHAr(DMB)), 6.52 (1H, d, 4J = 2.9 Hz, CHAr(cycloSal)), 6.79 (1H, dd, 3J = 8.9 Hz, 4J = 2.5 Hz, CHAr(cycloSal)), 6.92 (1H, d, 3J = 9.1 Hz, CHAr(cycloSal)), 7.16 (1H, d, 3J = 8.1 Hz, CHAr(DMB)); 13C-NMR (125.8 MHz, CDCl3): 20.2 (CH3CO), 46.1 (NCH2), 55.6 (OCH3), 55.7 (OCH3), 55.9 (OCH3), 64.4 (d, 2JC-P = 5.6 Hz, POCH2), 69.0 (d, 2JC-P = 6.8 Hz, ArCH2OP), 71.7 (OCH2DMP), 98.7 (CHAr(DMB)), 104.4 (CHAr(DMB)), 110.1 (CHAr(cycloSal)), 115.1 (CHAr(cycloSal)), 119.8 (d, 3JC-P = 8.8 Hz, CHArCArOP), 121.1 (d, 3JC-P = 9.4 Hz, CArCH2OP), 132.9 (CHAr(DMB)), 144.0 (d, 2JC-P = 6.8 Hz, CArOP), 156.1 (CArOCH3), 159.7 (CArOCH3), 162.3 (CArOCH3), 173.5 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.7; HRMS (EI)+: m/z calculated for C21H26NO9PNa [M + Na]+ 490.1237, found 490.1250.
- N-((2,4-Dimethoxybenzyl)oxy)-3-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (16aa). The general procedure E was applied to synthesize compound 16aa from alcohol 10a (153 mg, 0.60 mmol). The crude product was purified by automated flash chromatography (EtOAc/petroleum ether, 8:2) to give 16aa as a colorless oil (205 mg, 65%) and as a mixture of the two Z and E conformers in a 60:40 ratio. Rf = 0.38 (EtOAc/cyclohexane, 9:1); 1H-NMR (400 MHz, CDCl3): 2.48 (6/10H of 2H, bs, CH2CO), 2.83 (4/10H of 2H, bs, CH2CO), 3.81 (6H, s, OCH3), 4.40–4.49 (2H, m, POCH2), 4.77 (4/10 of 2H, bs, OCH2DMP), 4.85 (6/10 of 2H, bs, OCH2DMP), 5.34 (2H, m, ArCH2OP), 6.45 (2H, m, CHAr(DMP)), 7.05 (2H, m, CHAr(cycloSal)), 7.11 (1H, pseudo-t, 3J = 7.4 Hz, CHAr(cycloSal)), 7.17 (1H, m, CHAr(DMP)), 7.29 (1H, pseudo-t, 3J = 7.8 Hz, CHAr(cycloSal)), 7.88 (4/10H of 1H, bs, NH), 8.35 (6/10H of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 32.5 (d, 3JC-P = 5.9 Hz, CH2CO), 34.6 (d, 3JC-P = 5.1 Hz, CH2CO), 55.6 (OCH3), 55.7 (OCH3), 63.6 (POCH2), 64.4 (POCH2), 68.8 (ArCH2OP), 68.9 (ArCH2OP), 73.5 (OCH2DMP), 74.9 (OCH2DMP), 98.8 (CHAr(DMP)), 104.2 (CHAr(DMP)), 104.4 (CHAr(DMP)), 114.9 (CAr(DMP)), 115.9 (CAr(DMP)), 118.9 (d, 3JC-P = 9.0 Hz, CHArCArOP), 120.7 (CArCH2OP), 124.4 (CHAr(cycloSal)) 124.6 (CHAr(cycloSal)), 125.5 (CHAr(cycloSal)), 129.9 (CHAr(DMP)), 130.0 (CHAr(DMP)), 132.9 (CHAr(cycloSal)), 133.1 (CHAr(cycloSal)), 150.2 (CArOP), 159.6 (CArOCH3), 159.7 (CArOCH3), 161.9 (CArOCH3), 162.3 (CArOCH3), 166.7 (CO), 173. (CO); 31P-NMR (121.5 MHz, CDCl3): −9.8, −9.9; MS (EI)+: m/z calculated for C19H22NO8Pna [M + Na]+ 446.10, found 446.10.
- N-((2,4-Dimethoxybenzyl)oxy)-3-((6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (16ab). The general procedure E was applied to synthesize compound 16ab from alcohol 10a (200 mg, 0.78 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 7:3 → EtOAc) to give 16ab as a colorless oil (146 mg, 43%) and as a mixture of the two Z and E conformers in a 60:40 ratio. Rf = 0.36 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.26 (3H, s, CH3Ar), 2.42–2.51 (6/10 of 2H, m, CH2CO), 2.80–2.86 (4/10 of 2H, CH2CO), 3.79 (3H, s, OCH3), 3.80 (3H, s, OCH3), 4.39–4.45 (2H, m, POCH2), 4.75 (4/10 of 2H, s, OCH2DMP), 4.80–4.85 (6/10 of 2H, s, OCH2DMP), 5.21–5.31 (2H, m, ArCH2OP), 6.42–6.44 (2H, m, CHAr(DMP)), 6.82 (1H, s, CHAr(cycloSal)), 6.90 (1H, d, 3J = 8.3 Hz, CHAr(cycloSal)), 7.05 (1H, d, 7.18 (3J = 8.5 Hz, CHAr(cycloSal)) 7.13–7.17 (1H, m, CHAr(DMP)), 7.85 (4/10H of 1H, bs, NH), 8.32 (6/10H of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 20.9 (CH3Ar), 32.5 (CH2CO), 34.7 (CH2CO), 55.6 (OCH3), 55.8 (OCH3), 63.6 (POCH2), 64.3 (POCH2), 68.9 (d, 2JC-P = 5.3 Hz ArCH2OP), 69.1 (d, 2JC-P = 6.5 Hz ArCH2OP), 73.5 (OCH2DMP), 75.0 (OCH2DMP), 98.8 (CHAr(DMP)), 104.3 (CHAr(DMP)), 104.4 (CHAr(DMP)), 116.0 (CAr(DMP)), 118.6 (d, 3JC-P = 8.8 Hz, CHArCArOP), 120.3 (CArCH2OP), 125.7 (CHAr(cycloSal)), 130.4 (CHAr(cycloSal)), 130.5 (CHAr(cycloSal)), 132.9 (CHAr(DMP)), 133.1 (CHAr(DMP)), 134.3 (CArCH3), 148.0 (CArOP), 148.2 (CArOP), 159.6 (CArOCH3), 162.0 (CArOCH3), 162.3 (CArOCH3), 166.8 (CArOCH3), 171.4 (CO), 173.4 (CO); 31P-NMR (162.0 MHz, CDCl3): −9.3, −9.5; MS (EI)+: m/z calculated for C20H24NO8PNa [M + Na]+ 460.1132, found 460.1166.
- 3-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)propanamide (16ac). The general procedure E was applied to synthesize compound 16ac from alcohol 10a (200 mg, 0.78 mmol). The crude product was purified by automated flash chromatography (EtOAc) to give 16ac as a colorless oil (103 mg, 29%) and as a mixture of the two Z and E conformers in a 60:40 ratio. Rf = 0.34 (EtOAc); 1H-NMR (400 MHz, CDCl3): 2.49 (6/10 of 2H, m, CH2CO), 2.83 (4/10 of 2H, CH2CO), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.42–4.51 (2H, m, POCH2), 4.78 (4/10 of 2H, s, OCH2DMP), 4.86 (6/10 of 2H, s, OCH2DMP), 5.22–5.38 (2H, m, ArCH2OP), 6.47 (2H, m, CHAr(DMP)), 6.98 (1H, pseudo-d, 3J = 8.7 Hz, CHAr(cycloSal)), 7.05 (1H, t, 3J = 2.1 Hz, CHAr(cycloSal)), 7.18 (1H, m, CHAr(DMP)), 7.24 (1H, bs, CHAr(cycloSal)), 7.84 (4/10H of 1H, bs, NH), 8.29 (4/10H of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 32.5 (CH2CO), 34.5 (CH2CO), 55.6 (OCH3), 55.8 (OCH3), 63.8 (POCH2), 64.6 (POCH2), 68.3 (ArCH2OP), 73.6 (OCH2DMP), 75.0 (OCH2DMP), 98.8 (CHAr(DMP)), 104.3 (CHAr(DMP)), 104.4 (CHAr(DMP)), 114.9 (CAr(DMP)), 115.9 (CAr(DMP)), 120.3 (d, 3JC-P = 9.4 Hz, CHArCArOP), 122.2 (CArCH2OP), 125.5 (CHAr(cycloSal)), 130.3 (CHAr(cycloSal)), 132.9 (CHAr(DMP)), 148.7 (CArOP), 159.7 (CArOCH3), 166.6 (CArOCH3), 173.3 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.3, −10.4; MS (EI)+: m/z calculated for C19H21ClNO8PNa [M + Na]+ 480.0586, found 480.0600.
- 3-((6,8-Dichloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)propanamide (16ad). The general procedure E was applied to synthesize compound 16ad from alcohol 10a (250 mg, 0.98 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 9:1) to give 16ad as a colorless oil (49 mg, 10%) and as a mixture of the two Z and E conformers in a 60:40 ratio. Rf = 0.47 (EtOAc/cyclohexane, 9:1); 1H-NMR (500 MHz, CDCl3): 2.50 (6/10 of 2H, m, CH2CO), 2.83 (4/10 of 2H, CH2CO), 3.81–3.83 (6H, m, OCH3), 4.42–4.55 (2H, m, POCH2), 4.77 (4/10 of 2H, s, OCH2DMP), 4.84 (6/10 of 2H, s, OCH2DMP), 5.23–5.37 (2H, m, ArCH2OP), 6.44 (2H, pseudo-s, CHAr(DMP)), 6.97 (1H, s, CHAr(cycloSal)), 7.17 (1H, m, CHAr(cycloSal)), 7.37 (1H, s, CHAr(DMP)), 8.14 (4/10H of 1H, bs, NH), 8.64 (6/10H of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 32.4 (CH2CO), 34.3 (CH2CO), 55.6 (OCH3), 55.7 (OCH3), 64.2 (POCH2), 65.0 (POCH2), 68.1 (ArCH2OP), 68.2 (ArCH2OP), 73.5 (OCH2DMP), 74.9 (OCH2DMP), 98.7 (CHAr(DMP)), 104.3 (CHAr(DMP)), 104.4 (CHAr(DMP)), 114.9 (CAr(DMP)), 115.9 (CAr(DMP)), 123.5 (ClCArCArOP), 123.9 (CHAr(cycloSal)), 124.0 (CHAr(cycloSal)), 124.8 (CArCH2OP), 129.5 (CArCl), 129.8 (CArCl), 130.3 (CHAr(cycloSal)), 132.9 (CHAr(DMP)), 133.1 (CHAr(DMP)), 144.9 (CArOP), 145.1 (CArOP), 159.6 (CArOCH3), 159.7 (CArOCH3), 161.9 (CArOCH3), 162.3 (CArOCH3), 166.5 (CO), 173.2 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.3, −10.4; MS (EI)+: m/z calculated for C19H21Cl2NO8PNa [M + Na]+ 492.04, found 492.04.
- 3-((6-Bromo-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)propanamide (16ae). The general procedure E was applied to synthesize compound 16ae from alcohol 10a (139 mg, 0.54 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 6:4 → EtOAc) to give 16ae as a colorless oil (29 mg, 11%) and as a mixture of two Z and E conformers in a 60:40 ratio. Rf = 0.37 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.48 (6/10 of 2H, bs, CH2CO), 2.81 (4/10 of 2H, bs, CH2CO), 3.81 (6H, s, OCH3), 4.37–4.54 (2H, m, POCH2), 4.76 (4/10 of 2H, bs, OCH2DMP), 4.85 (6/10 of 2H, bs, OCH2DMP), 5.24 (1H, dd, 2J = 14.5 Hz, 3JP-H = 18.0 Hz, ArCH2OP), 5.34 (1H, dd, 2J = 14.3 Hz, 3JP-H = 9.5 Hz, ArCH2OP), 6.43–6.45 (2H, m, CHAr(DMP)), 6.92 (1H, d, 3J = 8.7 Hz, CHAr(cycloSal)), 7.11–7.19 (2H, m, CHAr(DMP) and CHAr(cycloSal)), 7.39 (1H, d, 3J = 8.2 Hz, CHAr(cycloSal)). 8.11 (4/10 of AH, bs, NH), 8.63 (6/10 of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 32.4 (CH2CO), 34.4 (CH2CO), 55.6 (OCH3), 55.7 (OCH3), 63.8 (POCH2), 64.7 (POCH2), 68.1 (d, 2JC-P = 6.7 Hz, ArCH2OP), 68.2 (d, 2JC-P = 6.3 Hz, ArCH2OP), 73.5 (OCH2DMP), 74.6 (OCH2DMP), 98.8 (CHAr(DMP)), 104.3 (CHAr(DMP)), 104.4 (CHAr(DMP)), 114.9 (CAr(DMP)), 116.0 (CAr(DMP)), 116.9 (CArBr), 117.1 (CArBr), 120.6 (d, 3JC-P = 9.1 Hz, CHArCArOP), 122.5 (d, 3JC-P = 9.5 Hz, CArCH2OP), 122.7 (d, 3JC-P = 10.3 Hz, CArCH2OP), 128.3 (CHAr(cycloSal)), 128.4 (CHAr(cycloSal)), 132.9–133.1 (CHAr(cycloSal) and CHAr(DMP)), 149.2 (d, 2JC-P = 6.4 Hz, CArOP), 149.4 (d, 2JC-P = 6.4 Hz, CArOP), 159.6 (CArOCH3), 159.7 (CArOCH3), 161.9 (CArOCH3), 162.3 (CArOCH3), 166.6 (CO), 173.2 (CO); 31P-NMR (121.5 MHz, CDCl3): −10.1, −10.2; HRMS (EI)+: m/z calculated for C19H22BrNO8PNa [M + Na]+ 502.0261, found 502.0331.
- N-((2,4-Dimethoxybenzyl)oxy)-3-((2-oxido-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (16af). The general procedure E was applied to synthesize compound 16af from alcohol 10a (152 mg, 0.60 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 3:7 → EtOAc) to give 16af as a colorless oil (38 mg, 13%) and as a mixture of the two Z and E conformers in a 60:40 ratio. Rf = 0.33 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.47–2.49 (6/10 of 2H, m, CH2CO), 2.76–2.88 (4/10 of 2H, m, CH2CO), 3.80 (6H, s, OCH3), 4.42–4.54 (2H, m, POCH2), 4.76 (4/10 of 2H, bs, OCH2DMP), 4.84 (6/10 of 2H, s, OCH2DMP), 5.27–5.43 (2H, m, ArCH2OP), 6.24–6.44 (2H, m, CHAr(DMP)), 7.13–7.16 (2H, m, CHAr(cycloSal) and CHAr(DMP)), 7.33 (1H, s, CHAr(cycloSal)), 7.55 (1H, d, 3J = 6.3 Hz, CHAr(cycloSal)), 7.83 (4/10 of 1H, bs, NH), 8.28 (6/10 of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 32.5 (CH2CO), 34.5 (CH2CO), 55.6 (OCH3), 55.8 (OCH3), 64.0 (POCH2), 64.7 (POCH2), 68.3–68.5 (ArCH2OP), 73.6 (OCH2DMP), 75.0 (OCH2DMP), 98.8 (CHAr(DMP)), 104.3 (CHAr(DMP)), 104.5 (CHAr(DMP)), 114.9 (CAr(DMP)), 115.2 (CAr(DMP)), 119.6 (d, 3JC-P = 9.5 Hz, CHArCArOP), 121.4 (CArCH2OP), 123.2 (CHAr(cycloSal)), 127.4 (CHAr(cycloSal)), 132.9 (CHAr(DMP)) 133.1 (CHAr(DMP)), 152.7 (CArOP), 159.7 (CArOCH3), 162.0 (CArOCH3), 162.4 (CArOCH3), 166.5 (CArOCH3), 171.4 (CO), 173.2 (CO); 31P-NMR (121.5 MHz, CDCl3): −10.4, −10.5; 19F-NMR (282.4 MHz, CDCl3): −63.2, −63.3; HRMS (EI)+: m/z calculated for C20H22NO8P [M + H]+ 492.1030, found 492.1011.
- N-((2,4-Dimethoxybenzyl)oxy)-3-((6-methoxy-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (16ag). The general procedure E was applied to synthesize compound 16ag from alcohol 10a (175 mg, 0.69 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 3:7 → EtOAc) to give 16ag as a colorless oil (49 mg, 18%) and as a mixture of two Z and E conformers in a 60:40 ratio. Rf = 0.46 (EtOAc); 1H-NMR (300 MHz, CDCl3): 2.48–2.49 (6/10 of 2H, m, CH2CO), 2.82–2.86 (4/10 of 2H, m, CH2CO), 3.75 (3H, s, OCH3), 3.81 (3H, s, OCH3), 3.82 (3H, s, OCH3), 4.40–4.47 (2H, m, POCH2), 4.77 (4/10 of 2H, bs, OCH2DMP), 4.85 (6/10 of 2H, s, OCH2DMP), 5.21–5.33 (2H, m, ArCH2OP), 6.43–6.46 (2H, m, CHAr(DMP)), 6.55 (1H, d, 4J = 2.8 Hz CHAr(cycloSal)), 6.80 (1H, dd, 3J = 9.1 Hz, 4J = 1.8 Hz, CHAr(DMP)), 6.96 (1H, d, 3J = 8.9 Hz, CHAr(DMP)), 7.15–7.20 (1H, m, CHAr(cycloSal)), 7.91 (4/10 of 1H, bs, NH), 8.40 (6/10 of 1H, bs, NH); 13C-NMR (125.8 MHz, CDCl3): 32.8 (CH2CO), 34.7 (CH2CO), 55.6 (OCH3), 55.7 (OCH3), 55.9 (OCH3), 63.6 (POCH2), 64.4 (POCH2), 68.8–69.0 (ArCH2OP), 73.5 (OCH2DMP), 75.0 (OCH2DMP), 98.8 (CHAr(DMP)), 104.3 (CHAr(DMP)), 110.2 (CHAr(cycloSal)), 115.3 (CHAr(cycloSal)), 116.0 (CAr(DMP)), 119.7 (d, 3JC-P = 8.8 Hz, CHArCArOP), 121.3–121.5 (CArCH2OP), 132.9–133.1 (CHAr(DMP)), 143.8–144.0 (CArOP), 156.1 (CArOCH3), 156.3 (CArOCH3), 159.6 (CArOCH3), 162.0 (CArOCH3), 162.3 (CArOCH3), 166.8 (CO), 171.1 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.3, −9.5; HRMS (EI): m/z calculated for C20H24NO9PNa [M + Na]+ 476.1081, found 476.1102.
- N-((2,4-Dimethoxybenzyl)oxy)-N-methyl-3-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (16ba). The general procedure D was applied to synthesize compound 16ba from alcohol 10b (153 mg, 0.57 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 8:2) to give 16ba as a colorless oil (148 mg, 76%) and as the sole E conformer. Rf = 0.25 (EtOAc/cyclohexane, 8:2); 1H-NMR (300 MHz, CDCl3): 2.79–2.92 (2H, m, CH2CO), 3.18 (3H, s, NCH3), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.39–4.51 (2H, m, POCH2), 4.77 (2H, s, OCH2DMP), 5.27–5.42 (2H, m, ArCH2OP), 6.47 (2H, m, CHAr(DMP)), 7.02–7.06 (2H, m, CHAr(cycloSal)), 7.11 (1H, t, 3J = 7.5 Hz, CHAr(cycloSal)), 7.17 (1H, d, 3J = 8.9 Hz, CHAr(DMP)), 7.28 (1H, t, 3J = 7.6 Hz, CHAr(cycloSal)); 13C-NMR (75.5 MHz, CDCl3): 32.9 (d, 3JC-P = 7.0 Hz, CH2CO), 33.3 (NCH3), 55.6 (OCH3), 55.7 (OCH3), 64.4 (d, 2JC-P = 5.3 Hz, POCH2), 66.7 (d, 2JC-P = 6.8 Hz, ArCH2OP), 71.3 (OCH2DMP), 98.8 (CHAr(DMP)), 104.4 (CHAr(DMP)), 115.0 (CAr(DMP)), 118.9 (d, 3JC-P = 9.0 Hz, CHArCArOP), 120.9 (d, 3JC-P = 9.8 Hz, CArCH2OP), 124.3 (CHAr(cycloSal)), 125.3 ((CHAr(cycloSal)), 129.7 (CHAr(cycloSal)), 132.8 (CHAr(DMP)), 150.4 (d, 2JC-P = 6.9 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 171.2 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.5; MS (EI)+: m/z calculated for C20H24NO8PNa [M + Na]+ 460.11, found 460.11.
- N-((2,4-Dimethoxybenzyl)oxy)-N-methyl-3-((6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (16bb). The general procedure D was applied to synthesize compound 16bb from alcohol 10b (150 mg, 0.56 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 5:5 → EtOAc) to give 16bb as a colorless oil (190 mg, 75%) and as the sole E conformer. Rf = 0.47 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.30 (3H, s, CH3Ar), 2.79–2.92 (2H, m, CH2CO), 3.18 (3H, s, NCH3), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.37–4.50 (2H, m, POCH2), 4.77 (2H, s, OCH2DMP), 5.23–5.379 (2H, m, ArCH2OP), 6.45–6.47 (2H, m, CHAr(DMP)), 6.87 (1H, bs, CHAr(cycloSal)), 6.91 (1H, d, 3J = 8.5 Hz, CHAr(DMP)), 7.07 (1H, d, 3J = 8.0 Hz, CHAr(DMP)), 7.18 (1H, d, 3J = 8.9 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.9 (CH3Ar), 33.0 (d, 3JC-P = 7.1 Hz, CH2CO), 33.3 (NCH3), 55.6 (OCH3), 55.7 (OCH3), 64.4 (d, 2JC-P = 5.4 Hz, POCH2), 68.8 (d, 2JC-P = 6.5 Hz, ArCH2OP), 71.3 (OCH2DMP), 98.8 (CHAr(DMP)), 104.4 (CHAr(DMP)), 115.0 (CAr(DMP)), 118.6 (d, 3JC-P = 9.1 Hz, CHArCArOP), 120.5 (d, 3JC-P = 9.6 Hz, CArCH2OP), 125.7 (CHAr(cycloSal)), 130.3 (CHAr(cycloSal)), 133.0 (CHAr(DMP)), 134.0 (CArCH3), 148.3 (d, 2JC-P = 6.4 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 171.2 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.3; HRMS (EI)+: m/z calculated for C21H27NO8P [M + H]+ 452.1469, found 452.1508.
- 3-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)-N-methylpropanamide (16bc). The general procedure D was applied to synthesize compound 16bc from alcohol 10b (195 mg, 0.72 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 9:1) to give 16bc as a colorless oil (292 mg, 86%) and as the sole E conformer. Rf = 0.51 (EtOAc/cyclohexane, 9:1); 1H-NMR (300 MHz, CDCl3): 2.78–2.90 (2H, m, CH2CO), 3.18 (3H, s, NCH3), 3.82 (3H, s, OCH3), 3.84 (3H, s, OCH3), 4.39–4.51 (2H, m, POCH2), 4.77 (2H, s, OCH2DMP), 5.21–5.39 (2H, m, ArCH2OP), 6.47 (2H, m, CHAr(DMP)), 6.97 (1H, d, 3J = 8.9 Hz, CHAr(cycloSal)), 7.05 (1H, t, 3J = 2.4 Hz, CHAr(cycloSal)), 7.17 (1H, d, 3J = 8.6 Hz, CHAr(DMP)), 7.24 (1H, bs, CHAr(cycloSal)); 13C-NMR (75.5 MHz, CDCl3): 32.9 (d, 3JC-P = 7.3 Hz, CH2CO), 33.3 (NCH3), 55.6 (OCH3), 55.7 (OCH3), 64.4 (d, 2JC-P = 5.2 Hz, POCH2), 68.1 (d, 2JC-P = 6.8 Hz, ArCH2OP), 71.3 (OCH2DMP), 98.8 (CHAr(DMP)), 104.5 (CHAr(DMP)), 115.0 (CAr(DMP)), 118.9 (d, 3JC-P = 9.0 Hz, CHArCArOP), 122.3 (d, 3JC-P = 9.1 Hz, CArCH2OP), 125.4 (CHAr(cycloSal)), 129.5 (CArCl), 129.9 (CHAr(cycloSal)), 133.0 (CHAr(DMP)), 148.5 (d, 2JC-P = 6.5 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 171.1 (CO); 31P-NMR (121.5 MHz, CDCl3): −10.0; MS (EI)+: m/z calculated for C20H23ClNO8PNa [M + Na]+ 494.0742, found 494.0626.
- 3-((6,8-Dichloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)-N-methylpropanamide (16bd). The general procedure D was applied to synthesize compound 16bd from alcohol 10b (241 mg, 0.89 mmol). The crude product was purified by flash chromatography (EtOAc/cyclohexane, 8:2) to give 16bd as a colorless oil (175 mg, 43%) and as the sole E conformer. Rf = 0.40 (EtOAc/petroleum ether, 8:2); 1H-NMR (400 MHz, CDCl3): 2.84–2.89 (2H, m, CH2CO), 3.19 (3H, s, NCH3), 3.82 (3H, s, OCH3), 3.85 (3H, s, OCH3), 4.41–4.62 (2H, m, POCH2), 4.78 (2H, s, OCH2DMP), 5.22–5.39 (2H, m, ArCH2OP), 6.47 (2H, m, CHAr(DMP)), 6.97 (1H, d, 3J = 2.3 Hz, CHAr(cycloSal)), 7.20 (1H, t, 3J = 8.9 Hz, CHAr(DMP)), 7.38 (1H, bs, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 32.7 (d, 3JC-P = 6.9 Hz, CH2CO), 33.1 (NCH3), 55.4 (OCH3), 55.5 (OCH3), 64.7 (d, 2JC-P = 5.5 Hz, POCH2), 67.9 (d, 2JC-P = 6.9 Hz, ArCH2OP), 71.2 (OCH2DMP), 98.6 (CHAr(DMP)), 104.3 (CHAr(DMP)), 114.7 (CAr(DMP)), 123.4 (d, 3JC-P = 9.5 Hz, ClCArCArOP), 123.7 (CHAr(cycloSal)), 124.7 (d, 3JC-P = 8.8 Hz, CArCH2OP) 129.3 (CArCl), 130.0 (CHAr(cycloSal)), 132.8 (CHAr(DMP)), 145.1 (d, 2JC-P = 5.9 Hz, CArOP), 159.5 (CArOCH3), 162.2 (CArOCH3), 171.2 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.5; MS (EI)+: m/z calculated for C20H22Cl2NO8PNa [M + Na]+ 528.04, found 528.04.
- 3-((6-Bromo-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)-N-methylpropanamide (16be). The general procedure D was applied to synthesize compound 16be from alcohol 10b (150 mg, 0.56 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 5:5 → EtOAc) to give 16be as a colorless oil (215 mg, 74%) and as the sole E conformer. Rf = 0.47 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.76–2.91 (2H, m, CH2CO), 3.18 (3H, s, NCH3), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.38–4.52 (2H, m, POCH2), 4.77 (2H, s, OCH2DMP), 5.24 (1H, dd, 2J = 14.2 Hz, 3JP-H = 17.9 Hz, ArCH2OP), 5.36 (1H, dd, 2J = 14.1 Hz, 3JP-H = 8.7 Hz, ArCH2OP), 6.45–6.47 (2H, m, CHAr(DMP)), 6.91 (1H, d, 3J = 8.8 Hz, CHAr(cycloSal)), 7.16–7.20 (2H, m, CHAr(DMP) and CHAr(cycloSal)), 7. 38 (1H, d, 3J = 8.7 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 32.9 (d, 3JC-P = 7.0 Hz, CH2CO), 33.3 (NCH3), 55.6 (OCH3), 55.7 (OCH3), 64.5 (d, 2JC-P = 5.2 Hz, POCH2), 68.0 (d, 2JC-P = 6.9 Hz, ArCH2OP), 71.3 (OCH2DMP), 98.8 (CHAr(DMP)), 104.5 (CHAr(DMP)), 114.9 (CAr(DMP)), 116.8 (CArBr), 120.7 (d, 3JC-P = 9.2 Hz, CHArCArOP), 122.8 (d, 3JC-P = 9.8 Hz, CArCH2OP), 128.3 (CHAr(cycloSal)), 132.8 (CHAr(cycloSal)), 133.0 (CHAr(DMP)), 149.5 (d, 2JC-P = 6.6 Hz, CArOP), 159.7 (CArOCH3), 162.3 (CArOCH3), 171.1 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.3; HRMS (EI)+: m/z calculated for C20H23BrNO8PNa [M + Na]+ 538.0237, found 538.0255.
- 3-((6-((Difluoro-λ3-methyl)-λ2-fluoranyl)-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)-N-methylpropanamide (16bf). The general procedure D was applied to synthesize the compound 16bf from alcohol 10b (150 mg, 0.56 mmol). The crude product was purified by automated flash chromatography (EtOAc/cyclohexane, 5 5 → EtOAc) to give 16bf as a colorless oil 149 mg, 53%) and as the sole E conformer. Rf = 0.44 (EtOAc/cyclohexane, 7:3); 1H-NMR (500 MHz, CDCl3): 2.78–2.91 (2H, m, CH2CO), 3.18 (3H, s, NCH3), 3.82 (3H, s, OCH3), 3.84 (3H, s, OCH3), 4.41–4.54 (2H, m, POCH2), 4.77 (2H, s, OCH2DMP), 5.33 (1H, dd, 2J = 14.4 Hz, 3JP-H = 18.5 Hz, ArCH2OP), 5.45 (1H, dd, 2J = 14.3 Hz, 3JP-H = 8.4 Hz, ArCH2OP), 6.45–6.47 (2H, m, CHAr(DMP)), 7.14 (1H, d, 3J = 8.7 Hz CHAr(cycloSal)), 7.17 (1H, d, 3J = 8.6 Hz, CHAr(DMP)), 7.35 (1H, s, CHAr(cycloSal)), 7.57 (1H, d, 3J = 8.4 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 32.9 (d, 3JC-P = 7.0 Hz, CH2CO), 33.3 (NCH3), 55.6 (OCH3), 55.7 (OCH3), 64.7 (d, 2JC-P = 5.3 Hz, POCH2), 68.3 (d, 2JC-P = 6.7 Hz, ArCH2OP), 71.3 (OCH2DMP), 98.8 (CHAr(DMP)), 104.7 (CHAr(DMP)), 114.9 (CAr(DMP)), 119.6 (d, 3JC-P = 9.3 Hz, CHArCArOP), 121.5 (d, 3JC-P = 9.7 Hz, CArCH2OP), 123.1 (q, 3JC-F = 3.2 Hz, CHAr(cycloSal)), 123.7 (q, 1JC-F = 271.1 Hz, CArCF3), 126.8 (q, 2JC-F = 33 Hz, CArCF3), 127.2 (q, 3JC-F = 3.6 Hz, CHAr(cycloSal)), 133.0 (CHAr(DMP)), 152.9 (d, 2JC-P = 6.4 Hz, CArOP), 159.7 (CArOCH3), 162.4 (CArOCH3), 171.1 (CO); 31P-NMR (121.5 MHz, CDCl3): −10.3; 19F-NMR (282.4 MHz, CDCl3): −63.2; HRMS (EI)+: m/z calculated for C21H23F3NO8PNa [M + Na]+ 528.1006, found 528.1005.
- N-((2,4-Dimethoxybenzyl)oxy)-3-((6-methoxy-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-methylpropanamide (16bg). The general procedure D was applied to synthesize compound 16bg from alcohol 10b (150 mg, 0.56 mmol). The crude product was purified by flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc) to give 16bg as a colorless oil (160 mg, 61%) and as the sole E conformer. Rf = 0.5 (EtOAc/cyclohexane, 8:2); 1H-NMR (500 MHz, CDCl3): 2.79–2.92 (2H, m, CH2CO), 3.18 (8/10 of 3H, s, NCH3), 3.23 (2/10 of 3H, s, NCH3), 3.76 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.37–4.50 (2H, m, POCH2), 4.77 (8/10 of 2H, s, OCH2DMP), 4.80 (2/10 of 2H, s, OCH2DMP), 5.23–5.37 (2H, m, ArCH2OP), 6.45–6.47 (2H, m, CHAr(DMP)), 6.55 (1H, d, 4J = 2.8 Hz CHAr(cycloSal)), 6.80 (1H, dd, 3J = 8.8 Hz, 4J = 1.9 Hz, CHAr(cycloSal)), 6.95 (1H, d, 3J = 8.9 Hz, CHAr(DMP)), 7.18 (1H, d, 3J = 8.5 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 32.8 (d, 3JC-P = 7.1 Hz, CH2CO), 33.1 (NCH3), 55.4 (OCH3), 55.5 (OCH3), 55.8 (OCH3), 64.4 (d, 2JC-P = 5.4 Hz, POCH2), 68.6 (d, 2JC-P = 6.7 Hz, ArCH2OP), 71.1 (OCH2DMP), 98.6 (CHAr(DMP)), 104.3 (CHAr(DMP)), 110.1 (CHAr(cycloSal)), 114.8 (CAr(DMP)), 114.9 (CHAr(cycloSal)), 119.6 (d, 3JC-P = 9.2 Hz, CHArCArOP), 121.4 (d, 3JC-P = 9.6 Hz, CArCH2OP), 132.8 (CHAr(DMP)), 143.9 (d, 2JC-P = 6.7 Hz, CArOP), 155.9 (CArOCH3), 159.5 (CArOCH3), 162.1 (CArOCH3), 171.1 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.3; HRMS (EI)+: m/z calculated for C21H26NO9PNa [M + Na]+ 490.1237, found 490.1277.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)-3-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (18a). When general procedure D was applied to synthesize compound 16aa from alcohol 10a (200 mg, 0.78 mmol), a byproduct 18a was obtained. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc) to give 18a as a colorless oil (90 mg, 20%) and as a mixture of two diastereoisomers. Rf = 0.38 (EtOAc/cyclohexane, 5:5); 1H-NMR (500 MHz, CDCl3): 2.88 (2H, t, 3J = 6.3 Hz, CH2CO), 3.78 (3H, s, OCH3), 3.82 (3H, s, OCH3), 4.37–4.51 (2H, m, POCH2), 4.90–4.97 (2H, m, OCH2DMP), 5.11–5.27 (2H, m, ArCH2OP), 5.33–5.38 (1H, m, ArCH2OP), 5.44–5.50 (1H, m, ArCH2OP) 6.42–6.44 (2H, m, CHAr(DMP)), 6.88 (5/10 of 1H, d 3J = 8.4 Hz, CHAr(cycloSal)), 6.90 (5/10 of 1H, d, 3J = 8.5 Hz, CHAr(cycloSal)), 6.96 (1H, d, 3J = 7.1 Hz, CHAr(cycloSal)), 7.03 (2H, t, 3J = 7.5 Hz, CHAr(cycloSal)), 7.10 (2H, t, 3J = 7.5 Hz, CHAr(cycloSal)), 7.17 (1H, dd, 3J = 8.9 Hz, 4JP-H = 2.0 Hz, CHAr(DMP)), 7.28 (2H, m, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 32.9 (d, 3JC-P = 7.1 Hz, CH2CO), 55.6 (OCH3), 55.7 (OCH3), 63.7 (POCH2), 68.9 (d, 2JC-P = 6.9 Hz, ArCH2OP), 69.3 (d, 2JC-P = 7.1 Hz, ArCH2OP), 69.4 (d, 2JC-P = 7.3 Hz, ArCH2OP), 71.8 (OCH2DMP), 98.5 (CHAr(DMP)), 104.1 (CHAr(DMP)), 117.6 (CAr(DMP)), 118.9 (d, 3JC-P = 8.5 Hz, CHArCArOP), 119.0 (d, 3JC-P = 8.8 Hz, CHArCArOP), 120.5 (d, 3JC-P = 10.2 Hz, CArCH2OP), 120.8 (d, 3JC-P = 9.8 Hz, CArCH2OP), 124.4 (CHAr(cycloSal)), 124.7 (CHAr(cycloSal)), 125.3 (CHAr(cycloSal)), 125.4 (CHAr(cycloSal)), 129.9 (CHAr(cycloSal)), 131.6 (CHAr(DMP)), 143.0 (d, 2JC-P = 9.5 Hz, CArOP), 143.1 (d, 2JC-P = 9.7 Hz, CArOP), 150.1 (d, 2JC-P = 7.2 Hz, CArOP), 150.3 (d, 2JC-P = 7.6 Hz, CArOP), 159.0 (CArOCH3), 161.3 (CArOCH3), 171.4 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.6, −9.7, −18.7; HRMS (EI)+: m/z calculated for C26H27NO11P2Na [M + Na]+ 614.0952, found 614.0951.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)-3-((6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (18b). When general procedure D was applied to synthesize compound 16ab from alcohol 10a (200 mg, 0.78 mmol), a byproduct 18b was obtained. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc) to give 18b as a colorless oil 92 mg, 38%) and as a mixture of two diastereoisomers. Rf = 0.72 (EtOAc/petroleum ether, 7:3); 1H-NMR (500 MHz, CDCl3): 2.27 (6H, s, CH3Ar), 2.83 (2H, t, 3J = 6.2 Hz, CH2CO), 3.76 (3H, s, OCH3), 3.80 (3H, s, OCH3), 4.33–4.46 (2H, m, POCH2), 4.88–4.94 (2H, m, OCH2DMP), 5.05–5.21 (2H, m, ArCH2OP), 5.29 (1H, d, 2J = 13.9 Hz, 3JC-P = 8.1 Hz, ArCH2OP), 5.38–5.44 (1H, m, ArCH2OP) 6.40–6.42 (2H, m, CHAr(DMP)), 6.72–6.79 (3H, m, CHAr), 6.88 (5/10 of 1H, d, 3J = 6.0 Hz, CHAr(cycloSal)), 6.90 (5/10 of 1H, d, 3J = 6.1 Hz, CHAr(cycloSal)), 7.00–7.05 (2H, m, CHAr), 7.14 (5/10 of 1H, d, 3J = 7.9 Hz, CHAr), 7.15 (5/10 of 1H, d, 3J = 7.9 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 20.8 (CH3Ar), 20.9 (CH3Ar), 32.9 (d, 3JC-P = 6.7 Hz, CH2CO), 55.6 (OCH3), 55.7 (OCH3), 63.6 (POCH2), 68.9 (d, 2JC-P = 6.6 Hz, ArCH2OP), 69.4 (d, 2JC-P = 7.3 Hz, ArCH2OP), 69.5 (d, 2JC-P = 7.1 Hz, ArCH2OP), 71.8 (OCH2DMP), 98.5 (CHAr(DMP)), 104.1 (CHAr(DMP)), 117.6 (CAr(DMP)), 118.6 (CHArCArOP), 120.0 (d, 3JC-P = 10.0 Hz, CArCH2OP), 120.4 (d, 3JC-P = 10.1 Hz, CArCH2OP), 125.5 (CHAr(cycloSal)), 125.7 (CHAr(cycloSal)), 130.4 (CHAr(cycloSal)), 131.5 (CHAr(DMP)), 134.0 (CArCH3), 134.4 (CArCH3), 143.1 (d, 2JC-P = 9.2 Hz, CArOP), 143.2 (d, 2JC-P = 9.7 Hz, CArOP), 147.9 (d, 2JC-P = 7.2 Hz, CArOP), 148.2 (d, 2JC-P = 5.7 Hz, CArOP), 158.9 (CArOCH3), 161.2 (CArOCH3), 171.4 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.4, −9.5, −18.5; HRMS (EI)+: m/z calculated for C28H31NO11P2Na [M + Na]+ 642.1265, found 642.1335.
- N-(6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)-3-((6-chloro-2-oxido-4H-benzo[d][1,3,2]ioxaphosphinine-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)propanamide (18c). When general procedure D was applied to synthesize compound 16ac from alcohol 10a (212 mg, 0.78 mmol), a byproduct 18c was obtained. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc) to give 18c as a colorless oil 3 (24 mg, 84%) and as a mixture of two diastereoisomers. Rf = 0.83 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.88 (2H, t, 3J = 6.3 Hz, CH2CO), 3.77 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.35–4.54 (2H, m, POCH2), 4.87–4.97 (2H, m, OCH2DMP), 5.03–5.23 (2H, m, ArCH2OP), 5.27–5.34 (1H, m, ArCH2OP), 5.38–5.46 (1H, m, ArCH2OP), 6.43–6.46 (2H, m, CHAr(DMP)), 6.79 (5/10 of 1H, d, 3J = 6.1 Hz, CHAr(cycloSal)), 6.82 (5/10 of 1H, d, 3J = 6.3 Hz, CHAr(cycloSal)), 6.92–7.02 (3H, m, CHAr(cycloSal)), 7.14 (1H, dd, 3J = 8.8 Hz, 4J = 2.4 Hz, CHAr(cycloSal)), 7.19–7.22 (2H, m, CHAr); 13C-NMR (125.8 MHz, CDCl3): 32.7 (d, 3JC-P = 7.0 Hz, CH2CO), 55.4 (OCH3), 55.5 (OCH3), 63.7 (d, 2JC-P = 4.1 Hz, POCH2), 63.7 (d, 2JC-P = 4.6 Hz, POCH2), 68.0 (d, 2JC-P = 6.6 Hz, ArCH2OP), 68.1 (d, 2JC-P = 6.6 Hz, ArCH2OP), 68.6 (d, 2JC-P = 7.1 Hz, ArCH2OP), 68.7 (d, 2JC-P = 7.1 Hz, ArCH2OP), 71.8 (OCH2DMP), 98.4 (CHAr(DMP)), 103.9 (CHAr(DMP)), 117.0 (CAr(DMP)), 120.1 (CHArCArOP), 120.2 (CHArCArOP), 121.7 (d, 3JC-P = 9.1 Hz, CArCH2OP), 122.1 (d, 3JC-P = 9.0 Hz, CArCH2OP), 125.0 (CHAr(cycloSal)), 125.2 (CHAr(cycloSal)), 129.4 (CArCl), 129.5 (CArCl), 129.7 (CHAr(cycloSal)), 129.8 (CHAr(cycloSal)), 131.5 (CHAr(DMP)), 131.6 (CHAr(DMP)), 142.6 (d, 2JC-P = 9.5 Hz, CArOP), 142.7 (d, 2JC-P = 10.1 Hz, CArOP), 148.3 (d, 2JC-P = 7.8 Hz, CArOP), 148.2 (d, 2JC-P = 6.0 Hz, CArOP), 158.9 (CArOCH3), 161.3 (CArOCH3); 31P-NMR (121.5 MHz, CDCl3): −10.1, −10.2, −19.4, −19.5; HRMS (EI)+: m/z calculated for C26H25Cl2NO11P2Na [M + Na]+ 682.0172, found 682.0168.
- N-(6-Bromo-2-oxido-4H-benzo[d][1,3,2]ioxaphosphinin-2-yl)-3-((6-bromo-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-((2,4-dimethoxybenzyl)oxy)propanamide (18e). When general procedure D was applied to synthesize compound 16ae from alcohol 10a (200 mg, 0.78 mmol), a byproduct 18e was obtained. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc/petroleum ether, 7:3) to give 18e as a colorless oil (432 mg, 74%) and as a mixture of two diastereoisomers. Rf = 0.34 (EtOAc/petroleum ether, 5:5); 1H-NMR (400 MHz, CDCl3): 2.87 (2H, t, 3J = 6.1 Hz, CH2CO), 3.79 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.37–4.51 (2H, m, POCH2), 4.88–4.96 (2H, m, OCH2DMP), 5.04–5.22 (2H, m, ArCH2OP), 5.28–5.33 (1H, m, ArCH2OP), 5.38–5.45 (1H, m, ArCH2OP), 6.43–6.46 (2H, m, CHAr(DMP)), 6.73 (5/10 of 1H, d, 3J = 8.3 Hz, CHAr(cycloSal)), 6.75 (5/10 of 1H, d, 3J = 8.1 Hz, CHAr(cycloSal)), 6.90 (5/10 of 1H, d, 3J = 8.1 Hz, CHAr(cycloSal)), 6.92 (5/10 of 1H, d, 3J = 8.3 Hz, CHAr(cycloSal)) 7.08–7.18 (3H, m, CHAr(cycloSal) and CHAr(DMP)), 7.34–7.41 (2H, m, CHAr); 13C-NMR (125.8 MHz, CDCl3): 32.8 (d, 3JC-P = 6.4 Hz, CH2CO), 55.6 (OCH3), 55.7 (OCH3), 63.8 (d, 2JC-P = 4.7 Hz, POCH2), 63.9 (d, 2JC-P = 4.2 Hz, POCH2), 68.1 (d, 2JC-P = 6.7 Hz, ArCH2OP), 68.2 (d, 2JC-P = 6.7 Hz, ArCH2OP), 68.7 (d, 2JC-P = 7.3 Hz, ArCH2OP), 68.8 (d, 2JC-P = 7.6 Hz, ArCH2OP), 71.9 (OCH2DMP), 98.6 (CHAr(DMP)), 104.1 (CHAr(DMP)), 116.9 (CAr(DMP)), 117.0 (CAr(DMP)), 117.2 (CArBr), 117.3 (CArBr), 120.6 (CHArCArOP), 120.7 (CHArCArOP), 122.3 (d, 3JC-P = 9.2 Hz, CArCH2OP), 122.7 (d, 3JC-P = 9.9 Hz, CArCH2OP), 128.1 (CHAr), 128.2 (CHAr), 128.3 (CHAr), 131.7 (CHAr), 131.8 (CHAr), 132.8 (CHAr), 142.8 (d, 2JC-P = 9.7 Hz, CArOP), 142.9 (d, 2JC-P = 9.7 Hz, CArOP), 149.1 (d, 2JC-P = 7.2 Hz, CArOP), 149.4 (d, 2JC-P = 6.7 Hz, CArOP), 149.5 (d, 2JC-P = 6.7 Hz, CArOP), 159.1 (CArOCH3), 161.4 (CArOCH3); 31P-NMR (162.0 MHz, CDCl3): −10.4, −10.5, −19.7, −19.8; HRMS (EI)+: m/z calculated for C26H26Br2NO11P2 [M + H]+ 749.9324, found 749.9361.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(2-oxido-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)-3-((2-oxido-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (18f). When general procedure D was applied to synthesize compound 16af from alcohol 10a (152 mg, 0.60 mmol), a byproduct 18f was obtained. The crude product was purified by automated flash chromatography (EtOAc/petroleum ether, 5:5 → EtOAc/petroleum ether, 7:3) to give 18f as a colorless oil (59 mg, 15%) and as a mixture of two diastereoisomers. Rf = 0.90 (EtOAc/petroleum ether, 7:3); 1H-NMR (300 MHz, CDCl3): 2.90 (2H, t, 3J = 6.2 Hz, CH2CO), 3.77 (3H, s, OCH3), 3.82 (3H, s, OCH3), 4.20–4.55 (2H, m, POCH2), 4.88–4.95 (2H, m, OCH2DMP), 5.13–5.31 (2H, m, ArCH2OP), 5.39 (1H, d, 2J = 14.5 Hz, 3J = 7.7 Hz, ArCH2OP), 5.47–5.54 (1H, m, ArCH2OP), 6.41–6.50 (2H, m, CHAr(DMP)), 6.93–6.97 (1H, m, CHAr), 7.13–7.16 (1H, m, CHAr), 7.23 (1H, s, CHAr), 7.28–7.35 (2H, m, CHAr), 7.45–7.58 (2H, m, CHAr); 13C-NMR (125.8 MHz, CDCl3): 32.9 (d, 3JC-P = 8.0 Hz, CH2CO), 55.5 (OCH3), 55.7 (OCH3), 64.1 (d, 2JC-P = 5.6 Hz, POCH2), 68.4 (d, 2JC-P = 6.6 Hz, ArCH2OP), 69.0 (d, 2JC-P = 7.1 Hz, ArCH2OP), 72.0 (OCH2DMP), 98.6 (CHAr(DMP)), 104.1 (CHAr(DMP)), 117.1 (CAr(DMP)), 119.6 (d, 3JC-P = 3.7 Hz, CHArCArOP), 119.7 (d, 3JC-P = 3.9 Hz, CHArCArOP), 121.0 (d, 3JC-P = 10.4 Hz, CArCH2OP), 121.4 (d, 3JC-P = 10.4 Hz, CArCH2OP), 122.8 (CHAr), 123.1 (CHAr), 127. (CHAr), 131.9 (CHAr(DMP)), 159.1 (CArOCH3), 161.5 (CArOCH3); 31P-NMR (121.5 MHz, CDCl3): −10.4, −10.5, −19.7, −19.8; 19F-NMR (282.4 MHz, CDCl3): −63.15, −63.16, −63.20, −63.21; HRMS (EI)+: m/z calculated for C28H25F6NO11P2Na [M + Na]+ 750.0699, found 750.0650.
- N-((2,4-Dimethoxybenzyl)oxy)-N-(6-methoxy-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)-3-((6-methoxy-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (18g). When general procedure D was applied to synthesize compound 16ag from alcohol 10a (175 mg, 0.69 mmol), a byproduct of 18g was obtained. The crude product was purified by automated flash chromatography (EtOAc/petroleum ether, 3:7 → EtOAc) to give 18g as a colorless oil (45 mg, 12%) and as a mixture of two diastereoisomers. Rf = 0.60 (EtOAc/petroleum ether, 7:3); 1H-NMR (300 MHz, CDCl3): 2.86 (2H, t, 3J = 6.3 Hz, CH2CO), 3.75 (6H, s, OCH3), 3.78 (3H, s, OCH3), 3.81 (3H, s, OCH3), 4.32–4.49 (2H, m, POCH2), 4.89–4.98 (2H, m, OCH2DMP), 5.05–5.20 (2H, m, ArCH2OP), 5.24–3.34 (1H, m, ArCH2OP), 5.38–5.46 (1H, m, ArCH2OP), 6.41–6.45 (3H, m, CHAr(DMP) and CHAr), 6.52 (5/10 of 1H, s, CHAr), 6.53 (5/10 of 1H, s, CHAr), 6.74–6.84 (3H, m, CHAr), 6.93–6.98 (1H, m, CHAr), 7.16 (1H, dd, 3J = 8.8 Hz, 4J = 1.5 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 32.9 (d, 3JC-P = 6.9 Hz, CH2CO), 55.6 (OCH3), 55.7 (OCH3), 55.9 (OCH3), 63.6 (POCH2), 63.7 (POCH2), 68.8 (d, 2JC-P = 6.6 Hz, ArCH2OP), 69.4 (d, 2JC-P = 6.8 Hz, ArCH2OP), 69.5 (d, 2JC-P = 6.8 Hz, ArCH2OP), 71.8 (OCH2DMP), 98.5 (CHAr(DMP)), 104.1 (CHAr(DMP)), 110.1 (CHAr), 110.2 (CHAr), 115.0 (CHAr), 115.1 (CHAr), 117.6 (CAr(DMP)), 119.7 (CHArCArOP), 119.8 (CHArCArOP), 121.1 (d, 3JC-P = 9.4 Hz, CArCH2OP), 121.4 (d, 3JC-P = 9.4 Hz, CArCH2OP), 131.5 (CHAr(DMP)), 143.1 (d, 2JC-P = 9.6 Hz, CArOP), 143.2 (d, 2JC-P = 9.6 Hz, CArOP), 143.7 (d, 2JC-P = 7.3 Hz, CArOP), 144.0 (d, 2JC-P = 6.7 Hz, CArOP), 156.1 (CArOCH3), 156.3 (CArOCH3), 158.9 (CArOCH3), 161.2 (CArOCH3), 171.4 (CO); 31P-NMR (121.5 MHz, CDCl3): −9.4, −9.5, −18.5; HRMS (EI)+: m/z calculated for C28H31NO13P2Na [M + Na]+ 674.1163, found 674.1134.
- N-Hydroxy-N-(2-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)formamide (7aa). The general procedure F was applied to synthesize compound 7aa from cycloSal 17aa (93 mg, 0.22 mmol) and 2% TFA in DCM. The product 7aa was obtained without purification as an orange oil (48 mg, 80%) and as a mixture of the three Z, E, and another conformer in a 20:20:60 ratio, respectively. Rf = 0.33 (EtOAc); 1H-NMR (500 MHz, CD3OD): 3.57–3.61 (2/10 of 2H, m, NCH2), 3.76–3.80 (6/10 of 2H, m, NCH2), 3.85–3.89 (2/10 of 2H, m, NCH2), 4.35–4.42 (8/10 of 2H, m, POCH2), 4.48–4.59 (2/10H of 2H, bs, POCH2), 5.39–5.56 (2H, m, ArCH2OP), 7.11 (1H, m, CHAr(cycloSal)), 7.22 (2H, m, CHAr(cycloSal), 7.38 (1H, m, CHAr(cycloSal)), 7.86 (6/10 of 1H, bs, CHO), 7.91 (2/10 of 1H, bs, CHO), 8.19 (2/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz, CD3OD): 47.8 (d, 3JCP = 6.9 Hz, NCH2), 51.6 (d, 3JCP = 7.2 Hz, NCH2), 52.0 (d, 3JCP = 8.1 Hz, NCH2), 62.4 (d, 2JCP = 4.8 Hz, POCH2), 64.9 (d, 2JCP = 5.6 Hz, POCH2), 65.2 (d, 2JCP = 5.6 Hz, POCH2), 70.3 (ArCH2OP), 70.4 (ArCH2OP), 70.5 (ArCH2OP), 119.6 (CHArCArOP), 119.7 (CHArCArOP), 119.8 (CHArCArOP), 122.3 (CArCH2OP), 122.4 (CArCH2OP), 122.5 (CArCH2OP), 125.9 (CHAr), 126.1 (CHAr), 126.2 (CHAr), 126.9 (CHAr), 127.1 (CHAr), 127.2 (CHAr), 131.1 (CHAr), 131.2 (CHAr), 131.4 (CHAr), 151.3 (d, 2JCP = 6.8 Hz, CArOP), 166.3 (CHO), 164.9 (CHO); 31P-NMR (121.5 MHz, CD3OD): −10.4; −10.5; −10.6; HRMS (EI)+: m/z calculated for C10H12NO6PNa [M + Na]+ 296.0294, found 296.0304.
- N-(2-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-hydroxyformamide (7ac). The general procedure F was applied to synthesize compound 7ac from cycloSal 17ac (77 mg, 0.17 mmol) and 2% TFA in DCM. The product 7ac was obtained without purification as an orange oil (38 mg, 73%) and as a mixture of the three Z, E and another conformer in a 30:40:30 ratio, respectively. Rf = 0.33 (EtOAc); 1H-NMR (400 MHz, CDCl3): 3.58 (3/10 of 2H, bs, NCH2), 3.81 (3/10 of 2H, bs, NCH2), 3.91 (4/10 of 2H, m, NCH2), 4.41–4.50 (8/10 of 2H, m, POCH2), 4.58–4.63 (2/10 of 2H, m, POCH2), 5.30–5.42 (2H, m, ArCH2OP), 7.03 (1H, m, CHAr), 7.10 (1H, d, 4J = 2.2 Hz, CHAr), 7.31 (1H, d, 3J = 8.6 Hz, CHAr), 7.93 (3/10 of 1H, bs, CHO), 8.07 (1/10 of 1H, bs, CHO), 8.54 (4/10 of 1H, bs, CHO); 13C-NMR (125.8 MHz CDCl3): 46.4 (NCH2), 63.2 (d, 2J = 6.0 Hz, POCH2), 64.2 (d, 2J = 6.6 Hz, POCH2), 68.6 (d, 2JCP = 6.9 Hz, ArCH2OP), 68.7 (d, 2JCP = 6.9 Hz, ArCH2OP), 69.0 (d, 2JCP = 6.9 Hz, ArCH2OP), 120.5 (d, 3JCP = 9.2 Hz, CHArCArOP), 122.0 (d, 3JCP = 10.1 Hz, CArCH2OP), 125.4 (d, 4J = 14.2 Hz CHAr(cycloSal)), 130.4 (CHAr(cycloSal)), 148.5 (CArOP), 164.8 (CHO); 31P-NMR (162.0 MHz, CDCl3): −6.71, −9.07, −9.63; HRMS (EI)+: m/z calculated for C10H12ClNO6PNa [M + Na]+ 308.0043, found 308.0085.
- N-Hydroxy-N-(2-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)acetamide (7ba). The general procedure F was applied to synthesize compound 7ba from cycloSal 17ba (73 mg, 0.17 mmol) and 2% TFA in DCM. The product 7ba was obtained without purification as an orange oil (41 mg, 84%). Rf = 0.35 (EtOAc); 1H-NMR (500 MHz, CDCl3): 1.99 (3H, s, CH3CO), 3.83–3.94 (2H, m, CH2N), 4.35–4.40 (2H, m, POCH2), 5.39–5.51 (2H, m, ArCH2OP), 7.10 (1H, m, CHAr(cycloSal)), 7.22 (2H, m, CHAr(cycloSal)), 7.37 (1H, t, 3J = 7.6 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.3 (CH3CO), 48.9 (d, 3J = 6.9 Hz, NCH2), 65.6 (d, 2J = 6.9 Hz, POCH2), 70.4 (d, 2JCP = 6.9 Hz, ArCH2OP), 119.7 (d, 3JCP = 9.1 Hz, CHArCArOP), 122.4 (d, 3JCP = 9.9 Hz, CArCH2OP), 124.2 (CHAr), 125.9 (CHAr), 126.6 (CHAr), 127.0 (CHAr), 130.2 (CHAr), 131.1 (CHAr), 151.3 (d, 2JCP = 6.8 Hz, CArOP), 172.3 (CO), 174.7 (CO); 31P-NMR (162.0 MHz, CDCl3): −8.9, −10.5; HRMS (EI)+: m/z calculated for C11H14NO6PNa [M + Na]+ 310.0451, found 310.0441.
- N-(2-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)ethyl)-N-hydroxyacetamide (7bc). The general procedure F was applied to synthesize compound 7bc from cycloSal 17bc (71 mg, 0.15 mmol) 2% TFA in DCM. The product 200c was obtained without purification as an orange oil (33 mg, 68%). Rf = 0.43 (EtOAc); 1H-NMR (400 MHz, CDCl3): 2.12–2.28 (3H, s, CH3CO), 3.58 (3/10 of 2H, bs, NCH2), 3.93–4.01 (2H, m, NCH2), 4.41–4.48 (2H, m, POCH2), 5.30–5.34 (2H, m, ArCH2OP), 7.03 (1H, d, 3J = 8.7 Hz CHAr), 7.10 (1H, d, 4J = 2.2 Hz, CHAr), 7.31 (1H, d, 3J = 8.1 Hz, CHAr); 13C-NMR (125.8 MHz, CDCl3): 20.6 (CH3CO), 20.7 (CH3CO), 47.2 (NCH2), 65.2 (d, 2J = 6.9 Hz, POCH2), 68.7 (d, 2JCP = 6.9 Hz, ArCH2OP), 120.2 (d, 3JCP = 8.0 Hz, CHArCArOP), 120.5 (d, 3JCP = 8.2 Hz, CHArCArOP), 122.7 (CArCH2OP), 125.4 (CHAr(cycloSal)), 125.5 (CHAr(cycloSal)), 128.7 (CArCl), 129.5 (CHAr(cycloSal)), 130.4 (CHAr(cycloSal));31P-NMR (162.0 MHz, CDCl3): −6.71; HRMS (EI)+: m/z calculated for C11H14ClNO6PNa [M + Na]+ 322.0242, found 322.0202.
- N-Hydroxy-3-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (8aa). The general procedure F was applied to synthesize compound 8aa from cycloSal 16aa (80 mg, 0.19 mmol) and 3% TFA in DCM. The crude product was purified by preparative TLC (MeOH/EtOAc, 5:95 v/v) to give 8aa as a colorless oil (31 mg, 60%) and as a sole Z conformer. Rf = 0.39 (EtOAc/methanol, 95:5);1H-NMR (400 MHz, CD3OD): 2.49 (2H, m, CH2CO), 4.37–4.51 (2H, m, POCH2), 5.38–5.48 (ArCH2OP), 7.10 (1H, pseudo-d, 3J = 7.9 Hz, CHAr(cycloSal)), 7.17–7.23 (2H, m, CHAr(cycloSal)), 7.37 (1H, pseudo-t, 3J = 7.6 Hz, CHAr); 13C-NMR (125.8 MHz, CD3OD): 34.8 (d, 3JC-P = 7.5 Hz, CH2CO), 65.9 (d, 2JC-P = 5.3 Hz, POCH2), 70.2 (d, 2JC-P = 6.9 Hz, ArCH2OP), 119.5 (d, 3JC-P = 9.0 Hz, CHArCArOP), 122.4 (d, 3JC-P = 9.8 Hz, CArCH2OP), 125.9 (CHAr(cycloSal)), 127.1 (CHAr(cycloSal)), 131.2 (CHAr(cycloSal)), 151.3 (d, 2JC-P = 6.8 Hz, CArOP), 169.0 (CO); 31P-NMR (162 MHz, CD3OD): −9.3; HRMS (EI)+: m/z calculated for C10H12NO6PNa [M + Na]+ 296.0294, found 296.0266.
- 3-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-hydroxypropanamide (8ac). The general procedure F was applied to synthesize compound 8ac from cycloSal 16ac (93 mg, 0.20 mmol) and 2% TFA in DCM. The product 201c was obtained without purification as a colorless oil (50 mg, 81%) and as a mixture of two Z and E conformers in a 60:40 ratio. Rf = 0.18 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.61 (6/10 of 2H, bs, CH2CO), 2.93 (4/10 of 2H, bs, CH2CO), 4.42–4.47 (2H, m, POCH2), 5.24–5.35 (2H, m, ArCH2OP), 6.97 (1H, pseudo-d, 3J = 8.4 Hz, CHAr(cycloSal)), 7.05 (1H, pseudo-s, CHAr(cycloSal)), 7.24 (1H, pseudo-s, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 29.9 (CH2CO), 64.9 (POCH2), 68.5 (d, 2JC-P = 6.5 Hz, ArCH2OP), 123.5 (d, 3JC-P = 9.4 Hz, ClCArCArOP), 124.1 (CHAr(cycloSal)), 124.2 (CHAr(cycloSal)), 124.5 (CArCH2OP), 125.6 (CHAr(cycloSal)), 130.0 (CArCl), 130.3 (CHAr(cycloSal)), 148.5 (d, 2JC-P = 5.2 Hz, CArOP); 31P-NMR (162.0 MHz, CDCl3): −10.3; HRMS (EI)+: m/z calculated for C11H12ClNO6P [M + H]+ 308.0085, found 308.0110.
- N-Hydroxy-N-methyl-3-((2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (8ba). The general procedure F was applied to synthesize the compound 8ba from cycloSal 16ba (20 mg, 0.05 mmol) and 2% TFA in DCM. The product 8ba was obtained without purification as a colorless oil (11 mg, 80%) and as a mixture of two Z and E conformers in a 30:70 ratio. Rf = 0.30 (EtOAc); 1H-NMR (400 MHz, CDCl3): 2.76 (30/100H of 2H, bs, CH2CO), 2.87–3.06 (70/100H of 2H, m, CH2CO), 3.24 (70/100H of 3H, s, NCH3), 3.33 (30/100H of 3H, s, NCH3), 4.43–4.53 (2H, m, POCH2), 5.30–5.44 (2H, m, ArCH2OP), 6.84 (1H, bs, OH), 7.05–7.09 (2H, m, CHAr(cycloSal)), 7.15 (1H, t, 3J = 7.6 Hz, CHAr(cycloSal)), 7.32 (1H, t, 3J = 7.8 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 32.3 (CH2CO), 33.3 (CH2CO), 36.3 (NCH3), 64.4 (POCH2), 65.3 (d, 2JC-P = 5.3 Hz, POCH2), 69.0 (d, 2JC-P = 6.8 Hz, ArCH2OP), 118.9 (d, 3JC-P = 9.1 Hz, CHArCArOP), 120.6 (d, 3JC-P = 10.1 Hz, CArCH2OP), 124.8 (CHAr(cycloSal)), 125.6 (CHAr(cycloSal)), 130.2 (CHAr(cycloSal)), 150.0 (d, 2JC-P = 6.1 Hz, CArOP), 170.4 (CO); 31P-NMR (162.0 MHz, CDCl3): −9.7; MS (EI)+: m/z calculated for C11H14NO6PNa [M + Na]+ 310.043, found 310.043.
- N-Hydroxy-N-methyl-3-((6-methyl-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (8bb). The general procedure F was applied to synthesize the compound 8bb from cycloSal 16bb (20 mg, 44 μmol) in 2% TFA in DCM. The product 202b was obtained without purification as a colorless oil (11 mg, 83%) and as a mixture of two Z and E conformers in a 30:70 ratio, respectively. Rf = 0.24 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.31 (3H, s, CH3), 2.77–3.07 (3H, m, CH2CO and OH), 3.26 (7/10 of 3H, s, NCH3), 3.34 (3/10 of 3H, s, NCH3), 4.47 (2H, bs, POCH2), 5.27–5.39 (2H, m, ArCH2OP), 6.86 (1H, s, CHAr(cycloSal)), 6.93–6.96 (1H, m, CHAr(cycloSal)), 7.10 (1H, d, 3J = 8.3 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 20.9 (CH3), 33.3 (CH2CO), 36.2 (NCH3), 65.6 (d, 2JC-P = 6.4 Hz, POCH2), 69.3 (d, 2JC-P = 6.6 Hz, ArCH2OP), 118.6 (d, 3JC-P = 8.9 Hz, CHArCArOP), 120.1 (d, 3JC-P = 9.7 Hz, CArCH2OP), 125.8 (CHAr(cycloSal)), 130.6 (CHAr(cycloSal)), 134.6 (CArCH3), 147.8 (d, 2JC-P = 7.1 Hz, CArOP); 31P-NMR (162.0 MHz, CDCl3): −9.2; MS (EI)+: m/z calculated for C12H16NO6P [M + H]+ 302.0788, found 302.077.
- 3-((6-Chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)-N-hydroxy-N-methylpropanamide (8bc). The general procedure F was applied to synthesize compound 8bc from cycloSal 16bc (87 mg, 0.18 mmol) and 2% TFA in DCM. The product 8bcc was obtained without purification as a colorless oil (25 mg, 44%) and as a mixture of two Z and E conformers in a 30:70 ratio. Rf = 0.31 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.80 (3/10 of 2H, bs, CH2CO), 2.94–3.09 (7/10 of 2H, bs, CH2CO), 3.29 (7/10 of 3H, bs, NCH3), 3.37 (3/10 of 3H, bs, NCH3), 4.51 (2H, bs, POCH2), 5.28–5.41 (2H, m, ArCH2OP), 7.02 (1H, pseudo-d, 3J = 8.7 Hz, CHAr(cycloSal)), 7.09 (1H, pseudo-d, 3J = 2.3 Hz, CHAr(cycloSal)), 7.30 (1H, pseudo-d, 3J = 8.7 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 29.9 (CH2CO), 32.3 (CH2CO), 36.2 (NCH3), 36.4 (NCH3), 64.5 (POCH2), 65.6 (d, 2JC-P = 5.3 Hz, POCH2) 68.4 (ArCH2OP), 68.6 (d, 2JC-P = 6.1 Hz, ArCH2OP), 120.3 (d, 3JC-P = 8.8 Hz, CHArCArOP), 121.9 (CArCH2OP), 125.5 (CHAr(cycloSal)), 130.3 (CHAr(cycloSal)), 148.5 (CArOP); 31P-NMR (162.0 MHz, CDCl3): −10.2; HRMS (EI)+: m/z calculated for C11H13ClNO6PNa [M + Na]+ 344.0061, found 344.0030.
- N-Hydroxy-N-methyl-3-((2-oxido-6-(trifluoromethyl)-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)propanamide (8bf). The general procedure F was applied to synthesize compound 8bf from cycloSal 16bf (20 mg, 40 μmol) and 2% TFA in DCM. The product 8bf was obtained without purification as a colorless oil (11 mg, 80%) and as a mixture of two Z and E conformers in a 40:60 ratio. Rf = 0.57 (EtOAc); 1H-NMR (500 MHz, CDCl3): 2.77–3.06 (3H, m, CH2CO and OH), 3.26 (6/10 of 3H, s, NCH3), 3.36 (4/10 of 3H, s, NCH3), 4.63 (2H, m, POCH2), 5.35–5.49 (2H, m, ArCH2OP), 7.16–7.20 (1H, m, CHAr(cycloSal)), 7.38 (1H, s, CHAr(cycloSal)), 7.61 (1H, d, 3J = 8.5 Hz, CHAr(cycloSal)); 13C-NMR (125.8 MHz, CDCl3): 31.9 (CH2CO), 33.3 (CH2CO), 36.0 (NCH3), 36.3 (NCH3), 64.5 (POCH2), 65.8 (d, 2JC-P = 6.2 Hz, POCH2), 68.7 (d, 2JC-P = 7.0 Hz, ArCH2OP), 119.7 (d, 3JC-P = 9.2 Hz, CHArCArOP), 121.2 (d, 3JC-P = 9.8 Hz, CArCH2OP), 123.2 (CHAr(cycloSal)), 127.5 (CHAr(cycloSal)), 152.4 (d, 2JC-P = 7.3 Hz, CArOP), 170.3 (CO); 31P-NMR (162.0 MHz, CDCl3): −10.3; 19F-NMR (282.4 MHz, CDCl3): −63.3; HRMS (EI)+: m/z calculated for C12H13F3NO6PNa [M + Na]+ 378.0325, found 378.0331.
3.2. Bacterial Growth Inhibition
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Word Health Organization. Antimicrobial Resistance. Key Facts 17 November 2017. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 12 July 2023).
- Word Health Organization. Tuberculosis. Key Facts 21 April 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 12 July 2023).
- Sulis, G.; Pa, M. Isoniazid-resistant tuberculosis: A problem we can no longer ignore. PLoS Med. 2020, 17, e1003023. [Google Scholar] [CrossRef] [PubMed]
- Mori, G.; Roberto Chiarelli, L.; Riccardi, G.; Pasca, M.R. New prodrugs against tuberculosis. Drug Discov. Today 2017, 22, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Rohmer, M.; Grosdemange-Billiard, C.; Seemann, M.; Tritsch, D. Isoprenoid biosynthesis as a novel target for antibacterial and antiparasitic drugs. Curr. Opin. Investig. Drugs 2004, 5, 154–162. [Google Scholar] [PubMed]
- Katayama, N.; Nozaki, Y.; Harada, S.; Ono, H.; Industries, T.C.J. Fosfadecyn and fosfocytocin, new nucleotide antibiotics produced by bacteria. Antibiotics 1990, 43, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Masini, T.; Hirsch, A.K.H. Development of inhibitors of the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents. J. Med. Chem. 2014, 57, 9740–9763. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Sakamoto, H.; Fukada, S.; Konishi, T.; Nishida, M. Pharmacokinetics of fosmidomycin, a new phosphonic acid antibiotic. Antimicrob. Agents Chemother. 1982, 21, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, L.; Tritsch, D.; Grosdemange-Billiard, C.; Hemmerlin, A.; Willem, A.; Bach, T.J.; Rohmer, M. Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: Phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reducto-isomerase. Biochem. J. 2005, 386, 127–135. [Google Scholar] [CrossRef]
- Munier, M.; Tritsch, D.; Krebs, F.; Esque, J.; Hemmerlin, A.; Rohmer, M.; Stote, R.H.; Grosdemange-Billiard, C. Synthesis and biological evaluation of phosphate isosters of fosmidomycin and analogs as inhibitors of Escherichia coli and Mycobacterium smegmatis 1-deoxyxylulose 5-phosphate reductoisomerases. Bioorg. Med. Chem. 2017, 25, 684–689. [Google Scholar] [CrossRef]
- Brown, A.C.; Parish, T. Dxr is essential in Mycobacterium tuberculosis and fosmidomycin resistance is due to a lack of uptake. BMC Microbiol. 2008, 8, 78. [Google Scholar] [CrossRef]
- Albert, A. Chemical aspects of selective toxicity. Nature 1958, 182, 421–422. [Google Scholar] [CrossRef]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Cos, P.; Van Calenberghc, S. Acyloxybenzyl and Alkoxyalkyl Prodrugs of a Fosmidomycin Surrogate as Antimalarial and Antitubercular Agents. ACS Med. Chem. Lett. 2018, 9, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Courtens, C.; van Charante, F.; Quennesson, T.; Risseeuw, M.; Cos, P.; Caljon, G.; Coenye, T.; Van Calenbergh, S. Acyloxymethyl and alkoxycarbonyloxymethyl prodrugs of a fosmidomycin surrogate as antimalarial and antibacterial agents. Eur. J. Med. Chem. 2023, 245, 114924. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Ferla, S.; Brancale, A.; McGuigan, C. Synthesis and biological evaluation of 6-substituted-5-fluorouridine ProTides. Bioorg. Med. Chem. 2018, 26, 551–565. [Google Scholar] [CrossRef]
- Munier, M.; Tritsch, D.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. Synthesis and biological evaluation of aryl phosphoramidate prodrugs of fosfoxacin and its derivatives. Bioorg. Chem. 2019, 89, 103012. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Lorey, M.; De Clercq, E.; Balzani, J. Cyclic saligenyl phosphotriesters of 2’,3’-dideoxy-2’,3’-didehydrothymidine (d4T)—A new pro-nucleotide approach. Bioorg. Med. Chem. Lett. 1997, 7, 99–104. [Google Scholar] [CrossRef]
- Hecker, S.J.; Erion, M.D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef]
- Meier, C.; Balzarini, J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antiviral Res. 2006, 71, 282–292. [Google Scholar] [CrossRef]
- Meier, C. cycloSal Phosphates as chemical trojan horses for intracellular nucleotide and glycosylmonophosphate delivery—Chemistry meets biology. Eur. J. Org. Chem 2006, 5, 1081–1102. [Google Scholar] [CrossRef]
- Ruda, G.F.; Wong, P.E.; Alibu, V.P.; Norval, S.; Read, K.D.; Barrett, M.P.; Gilbert, I.H. Aryl phosphoramidates of 5-phospho erythronohydroxamic acid, a new class of potent trypanocidal compounds. J. Med. Chem. 2010, 53, 6071–6078. [Google Scholar] [CrossRef]
- Zuo, L.; Yao, S.; Wang, W.; Duan, W. An efficient method for demethylation of aryl methyl ethers. Tetrahedron Lett. 2008, 49, 4054–4056. [Google Scholar] [CrossRef]
- Cho, S.-D.; Park, Y.-D.; Kim, J.-J.; Falck, J.R.; Yoon, Y.-J. Facile reduction of carboxylic acids, esters, acid chlorides, amides and nitriles to alcohols or amines using NaBH4/BF3.Et2O. Bull. Korean Chem. Soc. 2004, 25, 407–409. [Google Scholar] [CrossRef]
- Barlaam, B.; Hamon, A.; Maudet, M. New hydroxylamines for the synthesis of hydroxamic acids. Tetrahedron Lett. 1998, 39, 7865–7868. [Google Scholar] [CrossRef]
- San Jose, G.; Jackson, E.R.; Haymond, A.; Johny, C.; Edwards, R.L.; Wang, X.; Brothers, R.C.; Edelstein, E.K.; Odom, A.R.; Boshoff, H.I.; et al. Structure—activity relationships of the MEPicides: N-Acyl and O-linked analogs of FR900098 as inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis. ACS Infect. Dis. 2016, 2, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Courtens, C.; Risseeuwa, M.; Caljonb, G.; Maesb, L.; Cosb, P.; Martin, A.; Van Calenbergh, S. Double prodrugs of a fosmidomycin surrogate as antimalarial and antitubercular agents. Bioorg. Med. Chem. Lett. 2019, 29, 1232–1235. [Google Scholar] [CrossRef]
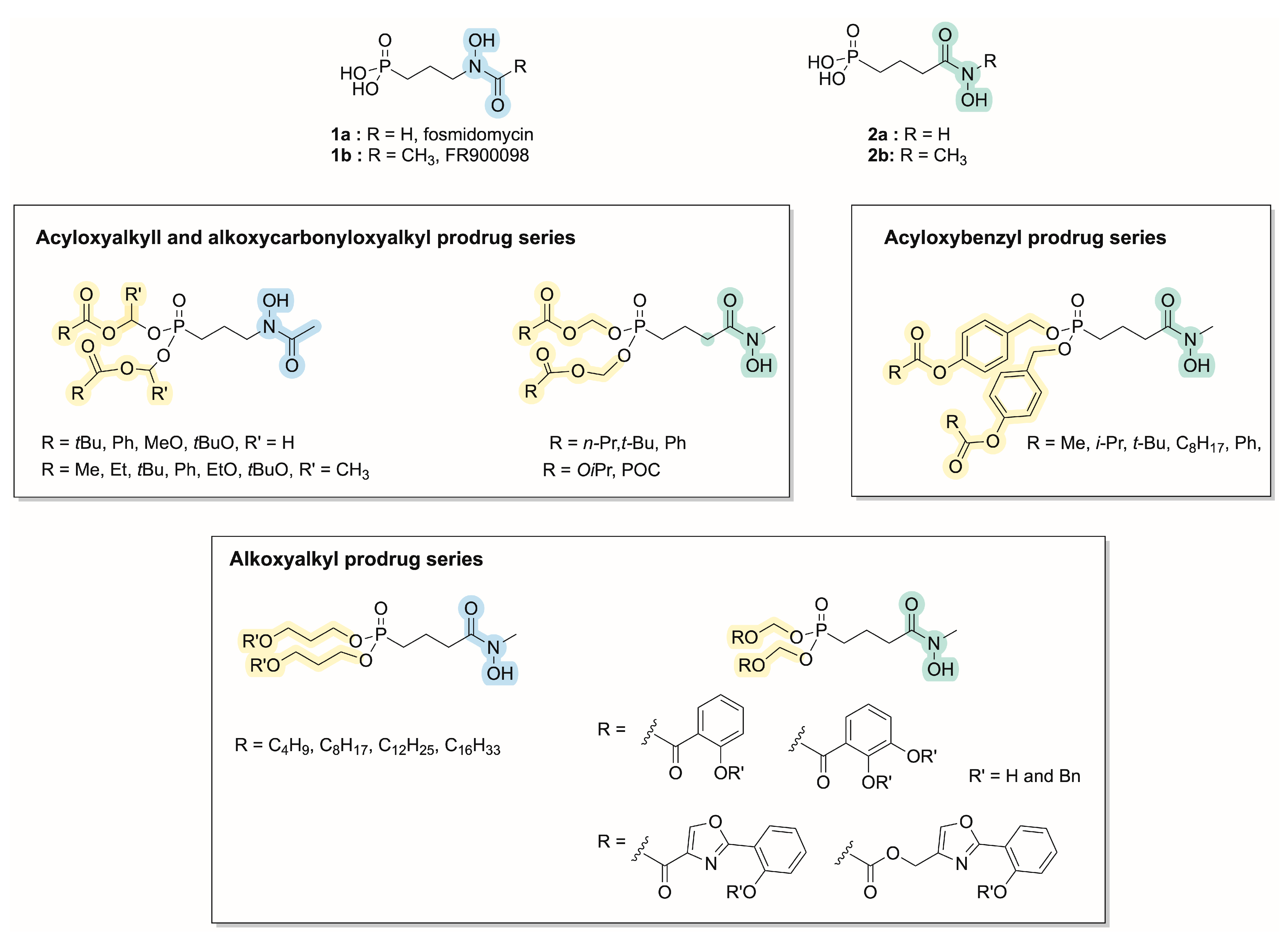
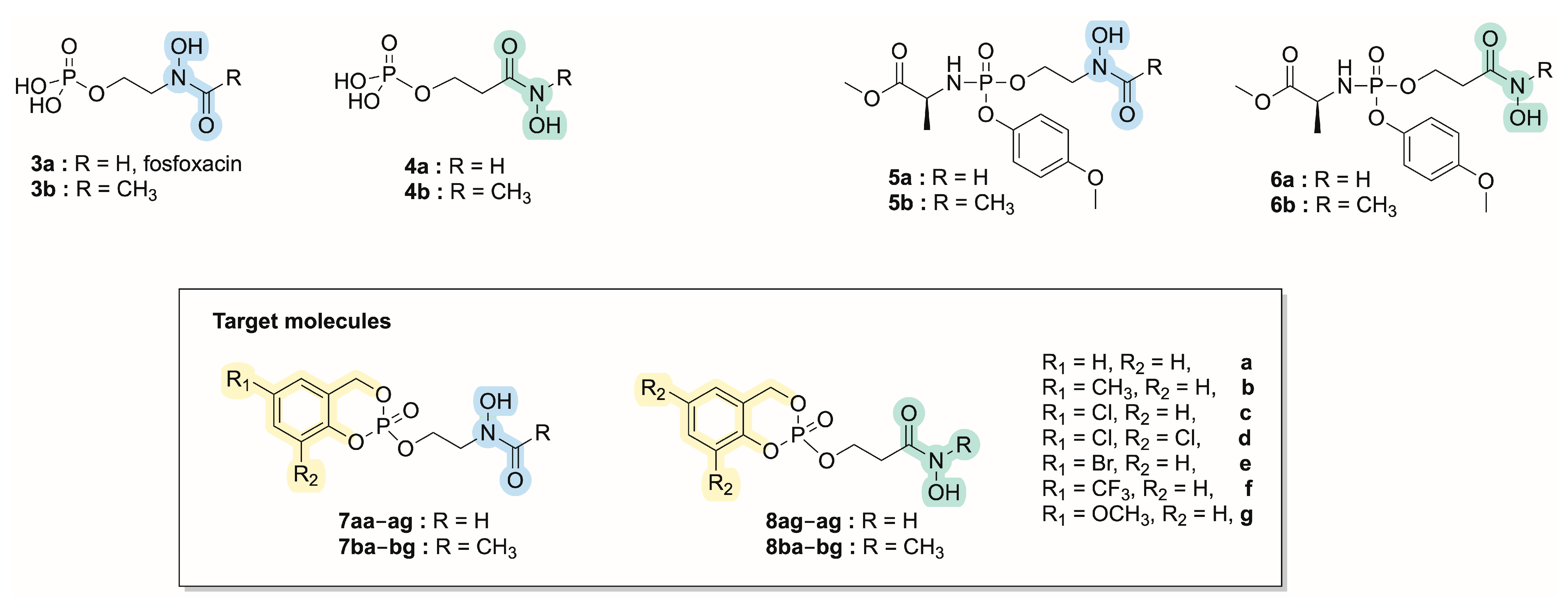

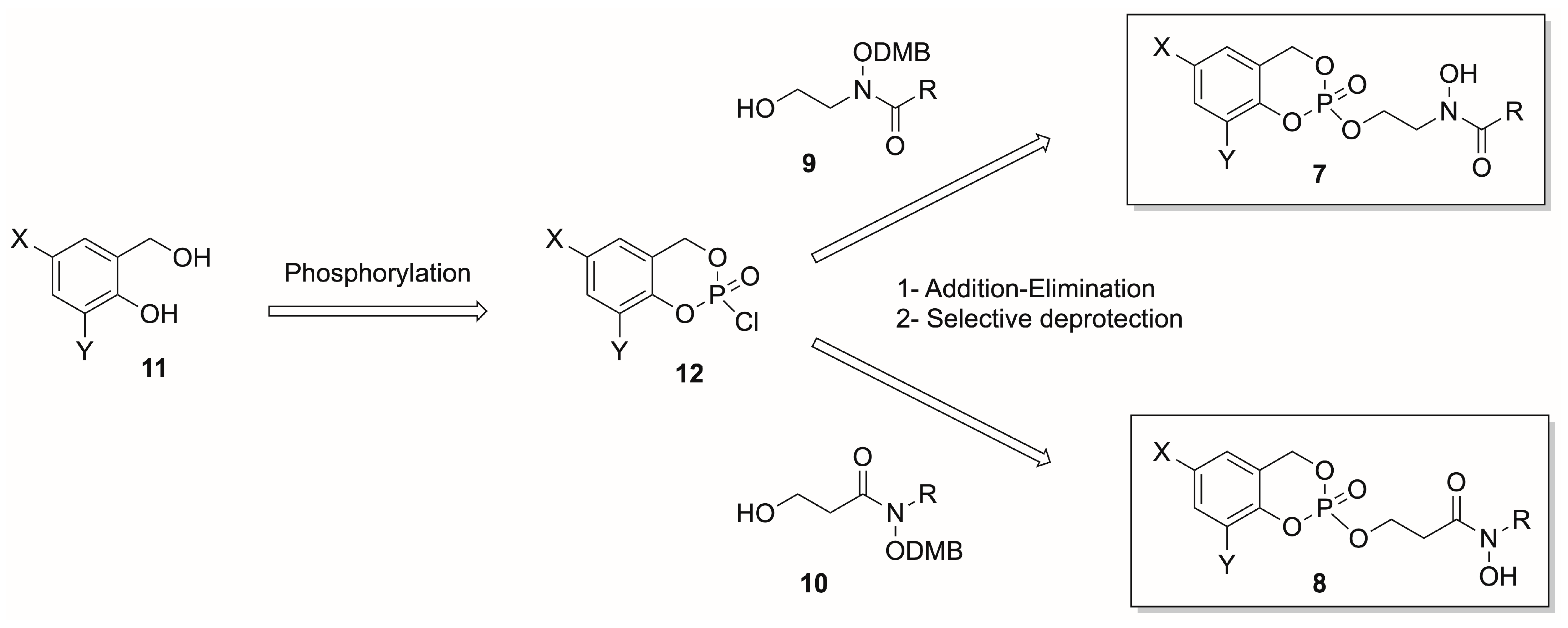
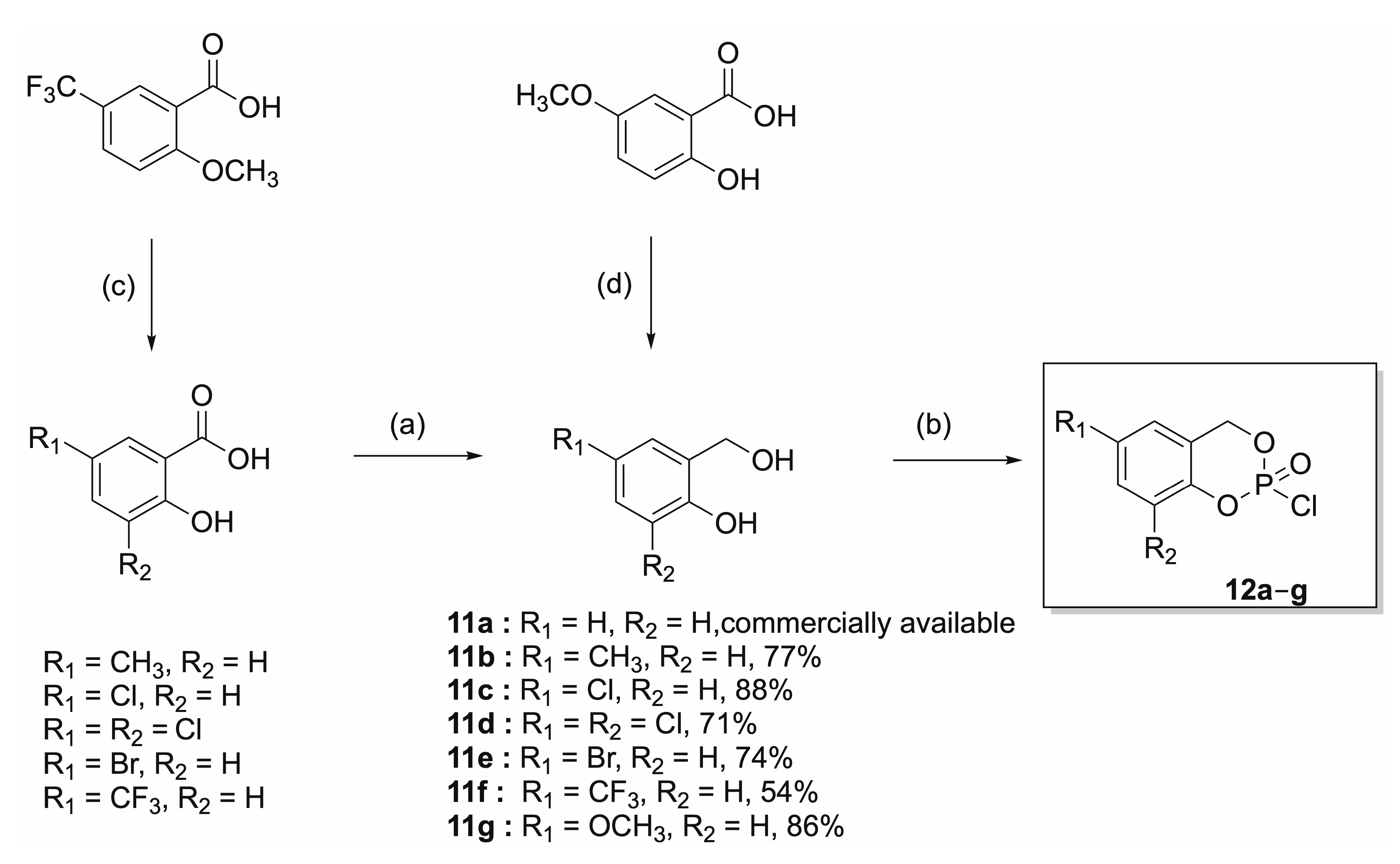
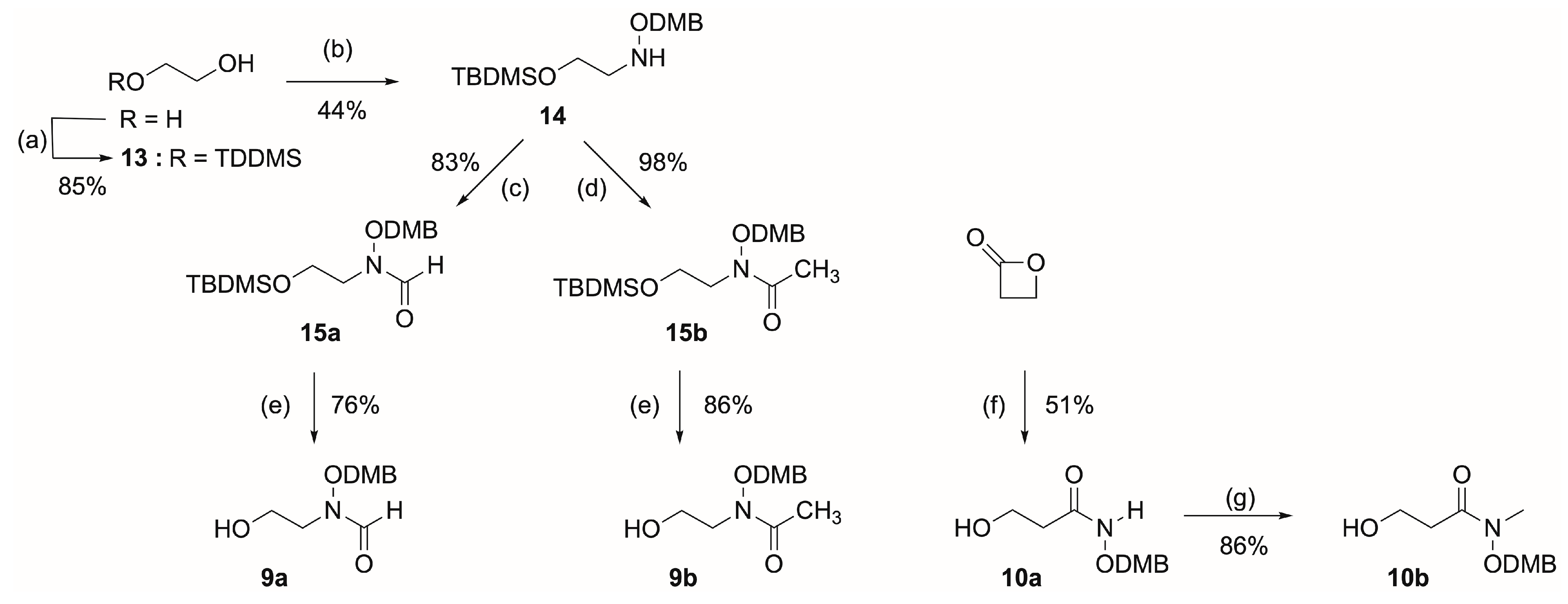
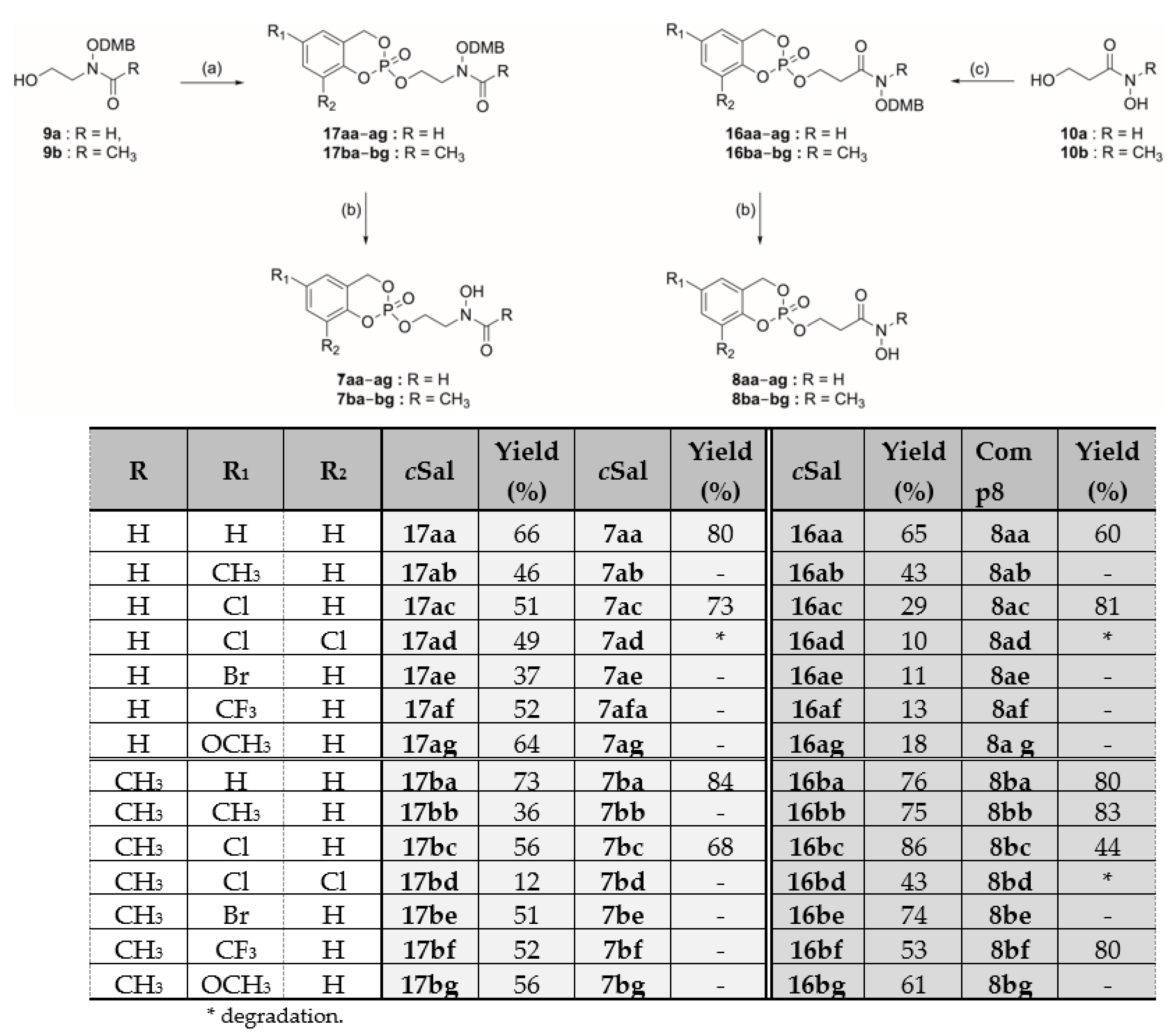
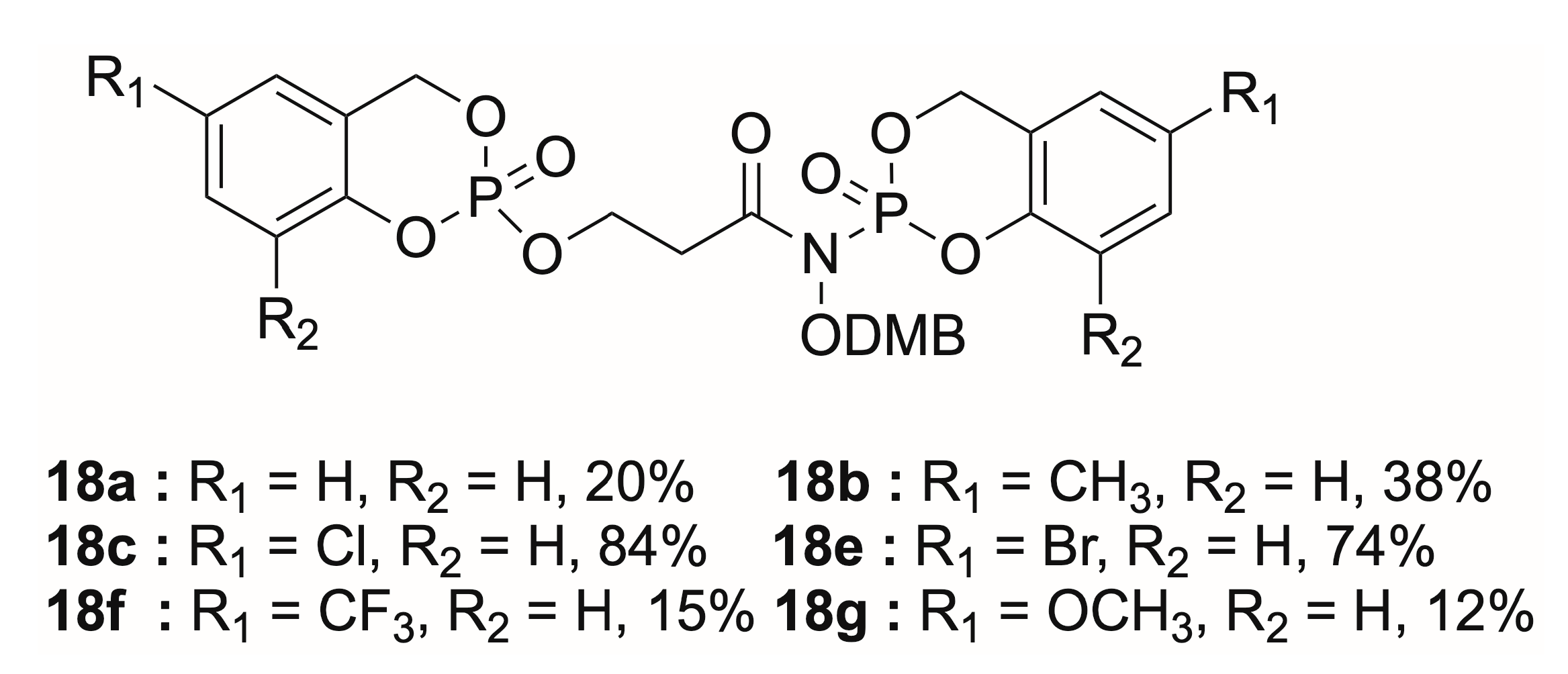

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli | M. smegmatis | ||
|---|---|---|---|
| Compound | Diameter (mm) | Compound | Diameter (mm) |
| Fosmidomycin | 40–45 | Isoniazide | 30–35 |
| 7aa | - | 7aa | - |
| 7ba | - | 7ba | - |
| 8aa | - | 8aa | - |
| 8ba | - | 8ba | - |
| 8bb | - | 8bb | - |
| 7ac | 8 | 7ac | 8 |
| 7bc | 23 | 7bc | - |
| 8ac | - | 8ac | 10 |
| 8bc | - | 8bc | 8 |
| 8bf | - | 8bf | - |
| Entry | R1 | R2 | Prodrugs | Prodrugs Inhibition Zone (mm) | Isoniazide Inhibition Zone (mm) |
|---|---|---|---|---|---|
| 1 | H | H | 16aa | - | 33 |
| 2 | 16ba | 26 | |||
| 3 | 17aa | 19 | 31 | ||
| 4 | 17ba | - | |||
| 5 | CH3 | H | 16ab | - | 28 |
| 6 | 16bb | - | |||
| 7 | 17ab | - | |||
| 8 | 17bb | - | |||
| 9 | Cl | H | 16ac | - | 36 |
| 10 | 16bc | 33 | |||
| 11 | 17ac | 25 | 35 | ||
| 12 | 17bc | 22 | |||
| 13 | Cl | Cl | 16ad | - | 35 |
| 14 | 16bd | 16 | |||
| 15 | 17ad | 11 | |||
| 16 | 17bd | 12 | |||
| 17 | Br | H | 16ae | 8 | 42 |
| 18 | 16be | 31 | |||
| 19 | 17ae | 20 | |||
| 20 | 17be | 21 | |||
| 21 | CF3 | H | 16af | - | 24 |
| 22 | 16bf | 25 | |||
| 23 | 17af | 19 | |||
| 24 | 17bf | 17 | |||
| 25 | OCH3 | H | 16ag | - | 27 |
| 26 | 16bg | - | |||
| 27 | 17ag | - | |||
| 28 | 17bg | - |
| Entry | Bis(cyclosal) | Bis(cyclosal) Inhibition Zone (mm) | Isoniazide Inhibition Zone (mm) |
|---|---|---|---|
| 1 | 18a | - | 35 |
| 2 | 18b | - | 35 |
| 3 | 18c | 31 | 35 |
| 4 | 18e | 10 | 35 |
| 5 | 18f | 8 | 24 |
| 6 | 18g | - | 24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munier, M.; Tritsch, D.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. New Application of cycloSaligenyl Prodrugs Approach for the Delivery of Fosfoxacin Derivatives in Mycobacteria. Molecules 2023, 28, 7713. https://doi.org/10.3390/molecules28237713
Munier M, Tritsch D, Lièvremont D, Rohmer M, Grosdemange-Billiard C. New Application of cycloSaligenyl Prodrugs Approach for the Delivery of Fosfoxacin Derivatives in Mycobacteria. Molecules. 2023; 28(23):7713. https://doi.org/10.3390/molecules28237713
Chicago/Turabian StyleMunier, Mathilde, Denis Tritsch, Didier Lièvremont, Michel Rohmer, and Catherine Grosdemange-Billiard. 2023. "New Application of cycloSaligenyl Prodrugs Approach for the Delivery of Fosfoxacin Derivatives in Mycobacteria" Molecules 28, no. 23: 7713. https://doi.org/10.3390/molecules28237713
APA StyleMunier, M., Tritsch, D., Lièvremont, D., Rohmer, M., & Grosdemange-Billiard, C. (2023). New Application of cycloSaligenyl Prodrugs Approach for the Delivery of Fosfoxacin Derivatives in Mycobacteria. Molecules, 28(23), 7713. https://doi.org/10.3390/molecules28237713