
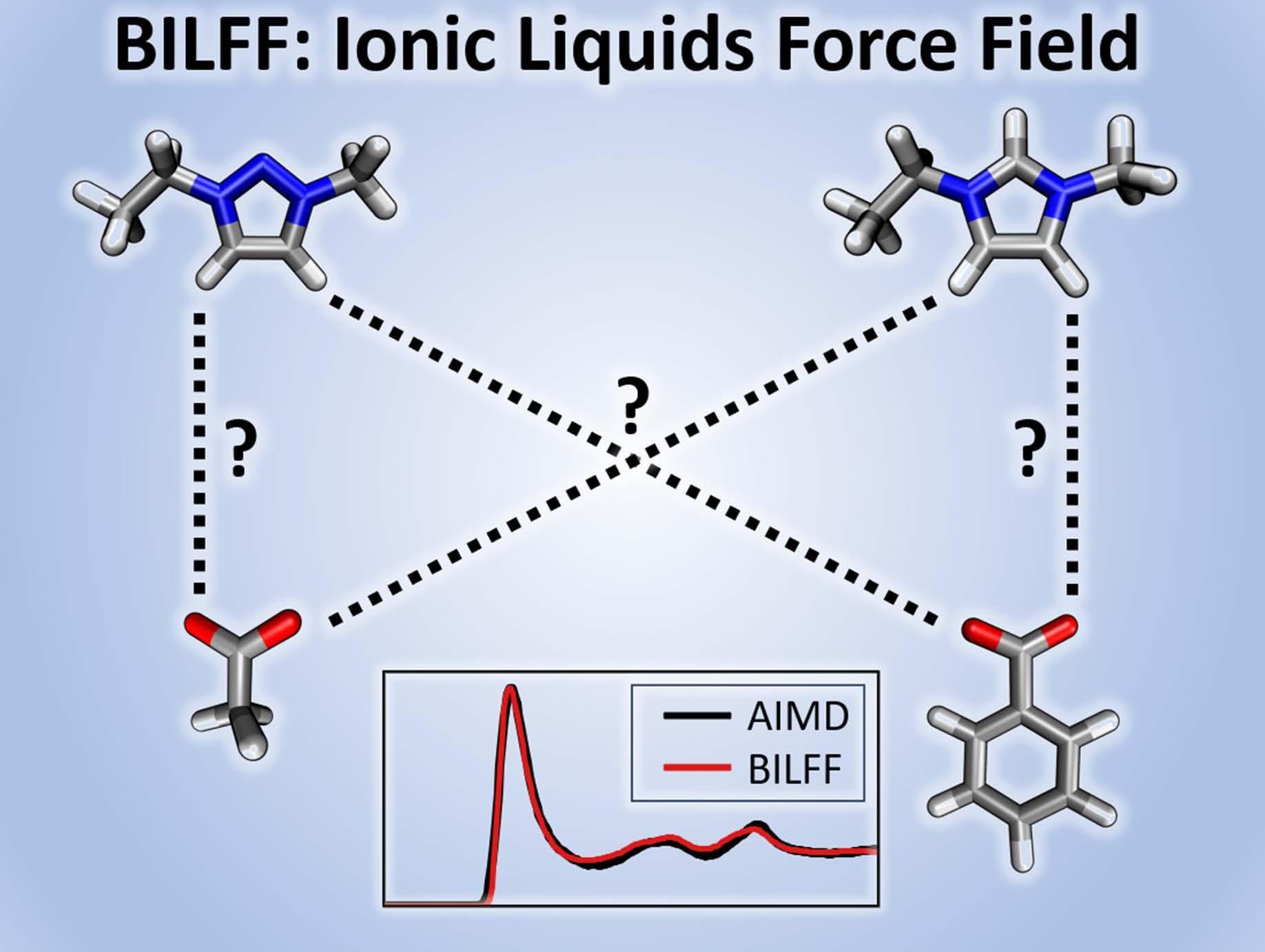
BILFF: All-Atom Force Field for Modeling Triazolium- and Benzoate-Based Ionic Liquids



Abstract
:
1. Introduction
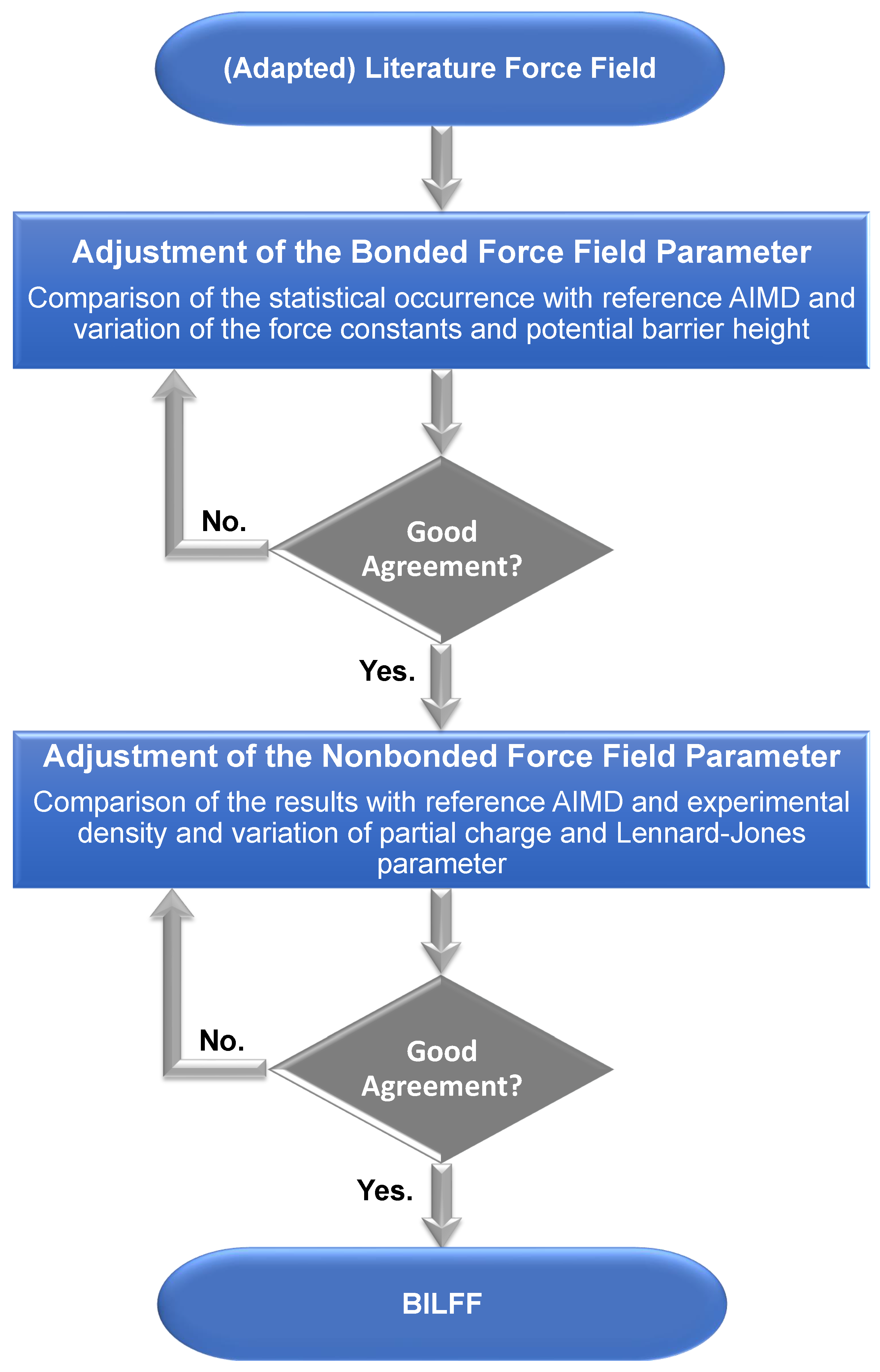
2. Optimization Procedure
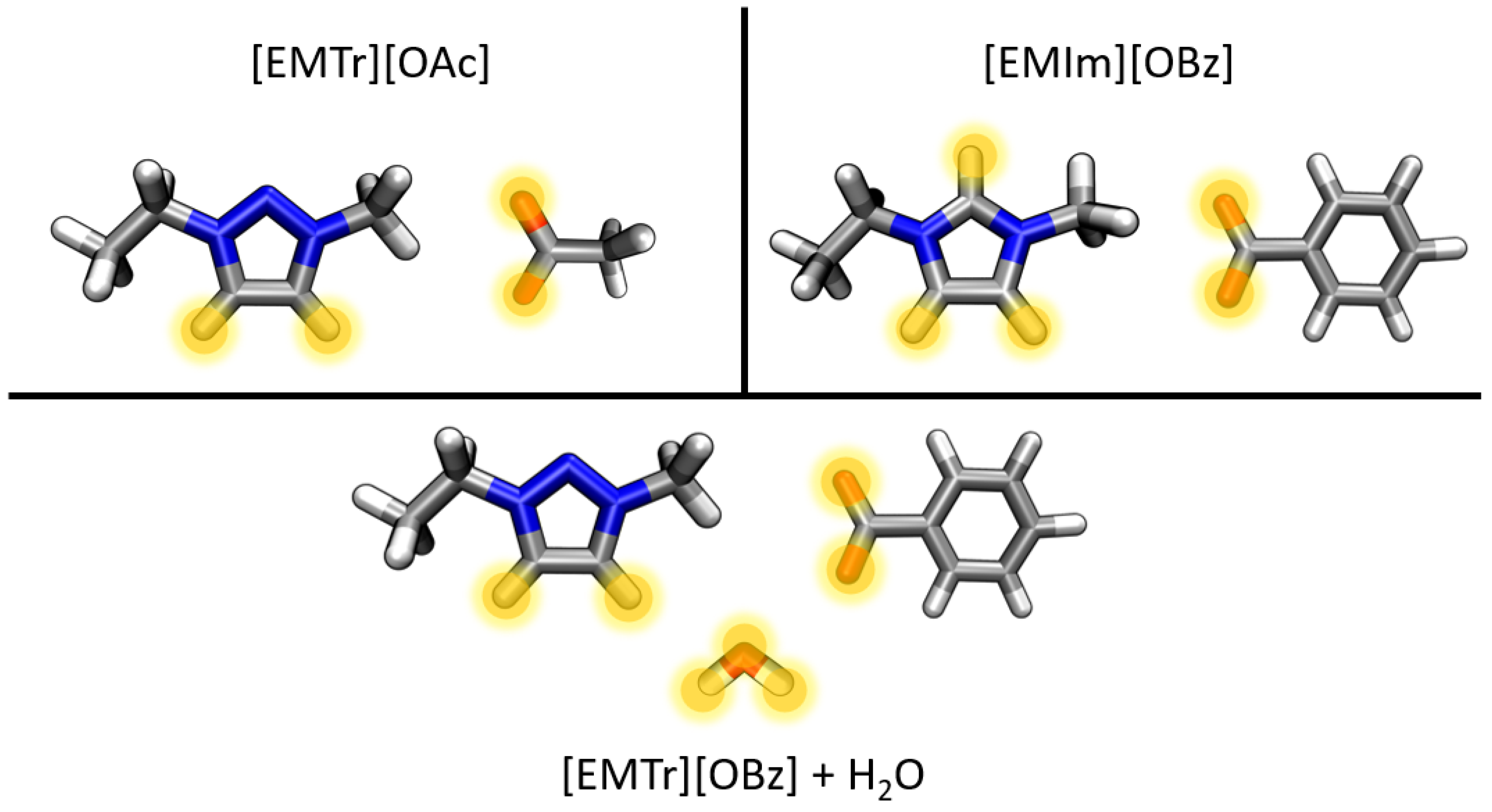
2.1. Microstructure of the Systems
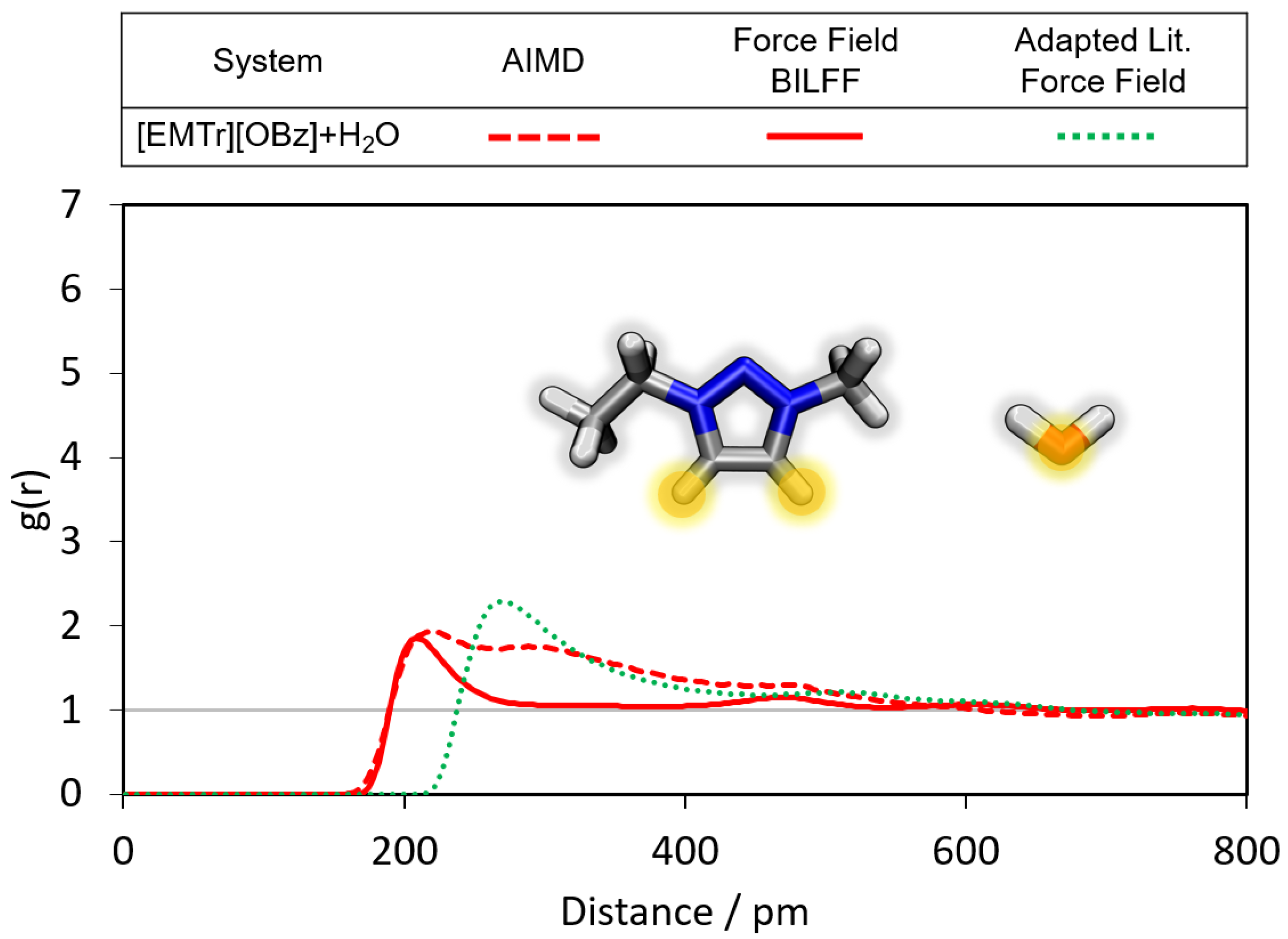
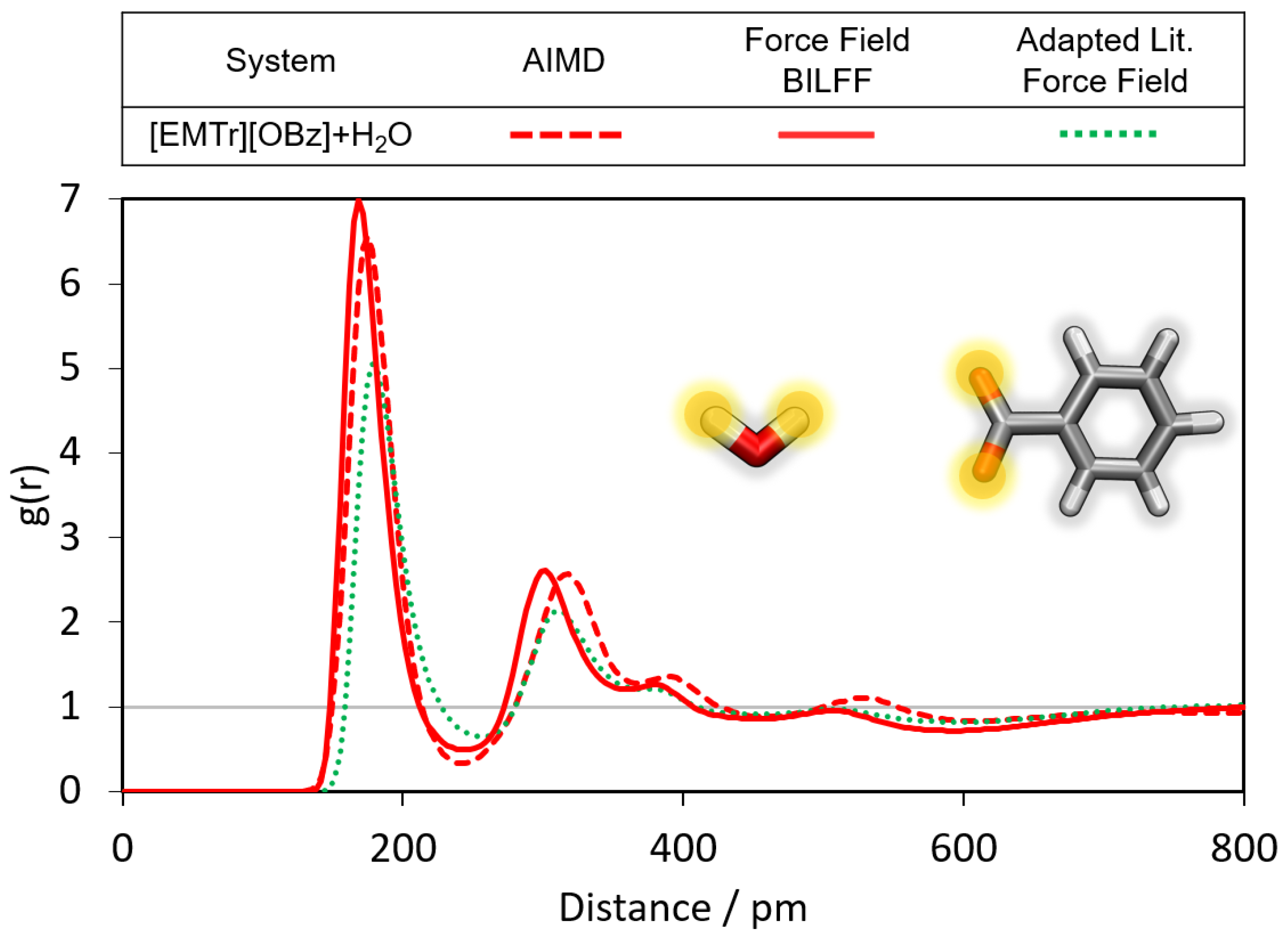
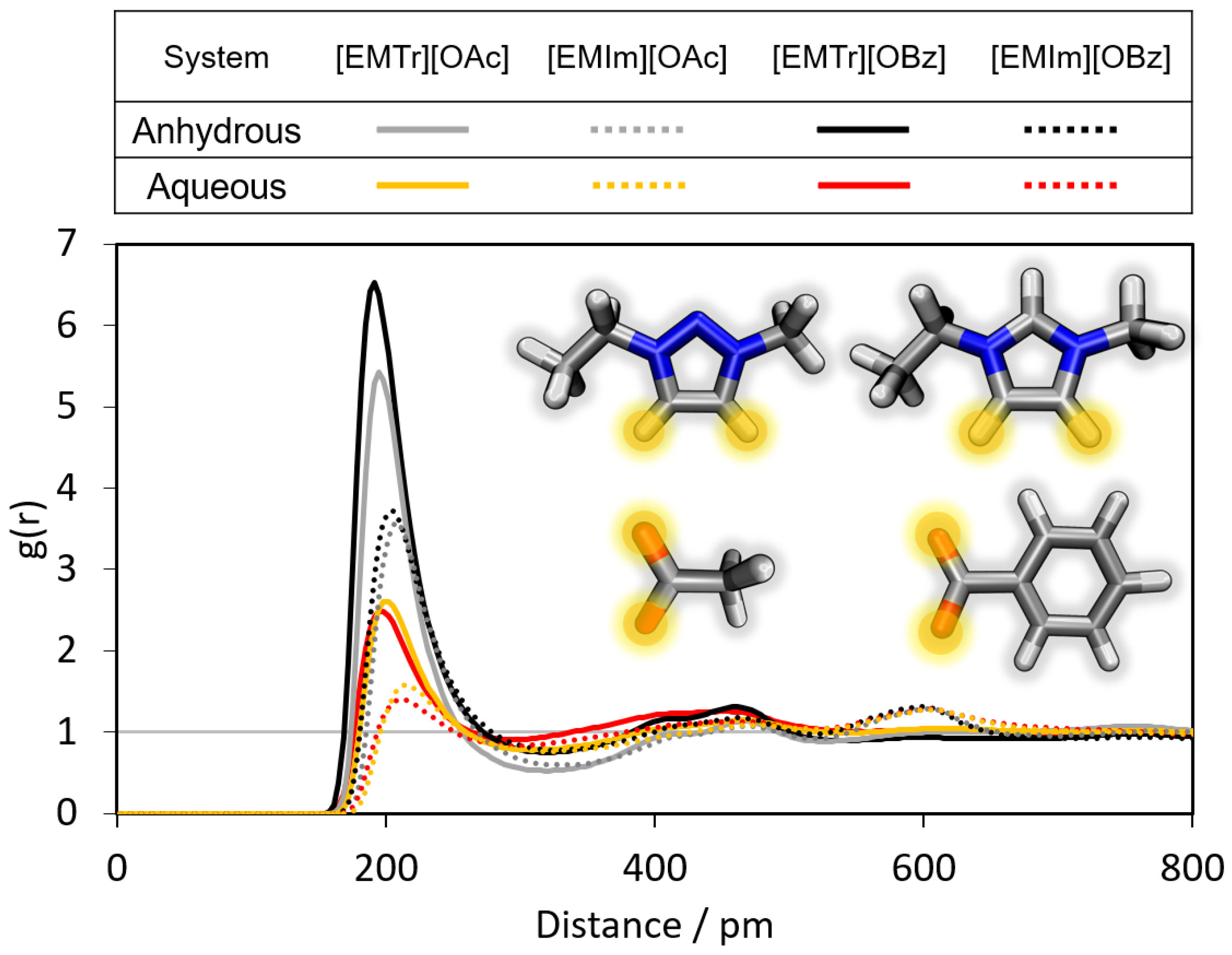
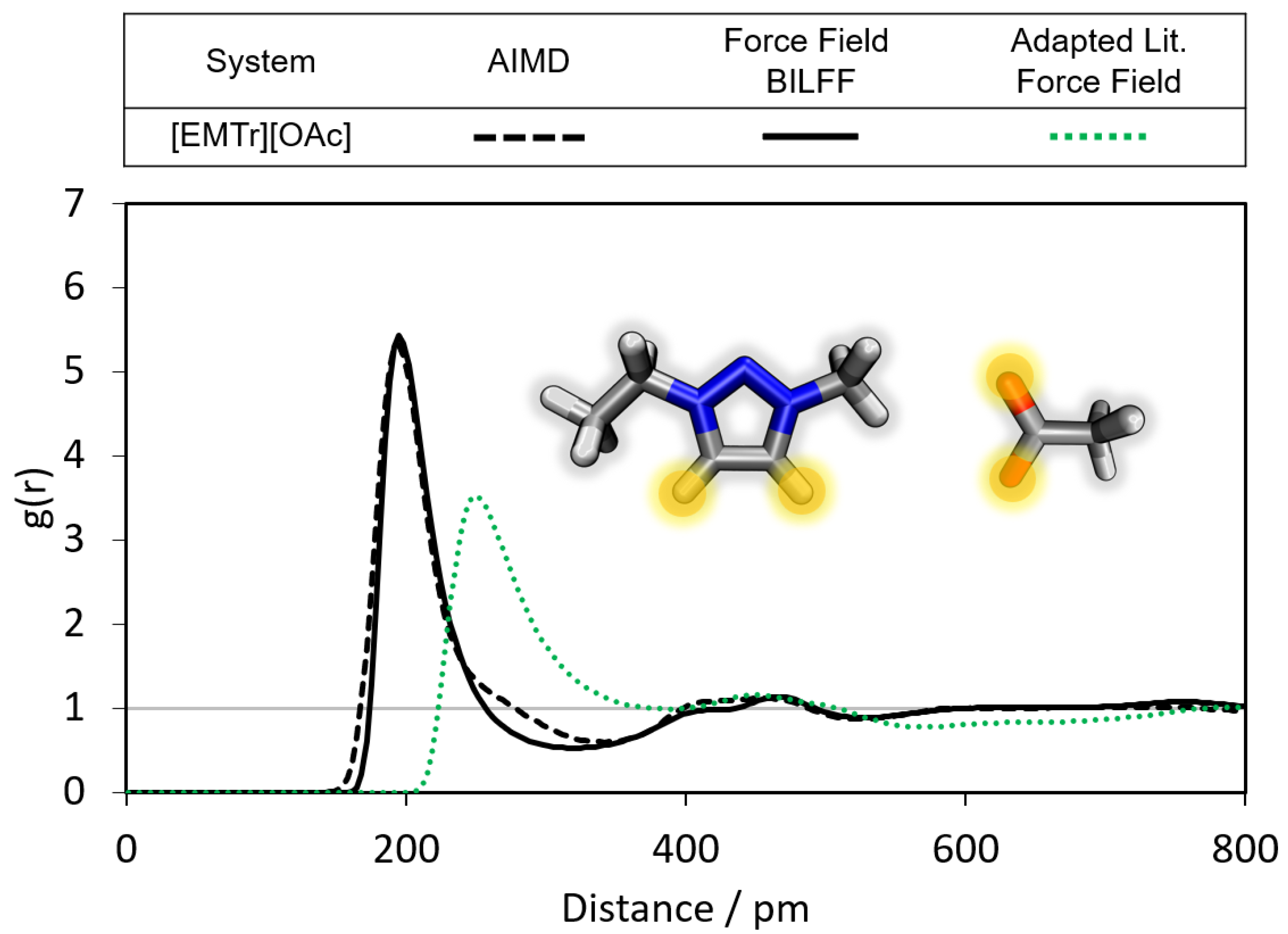
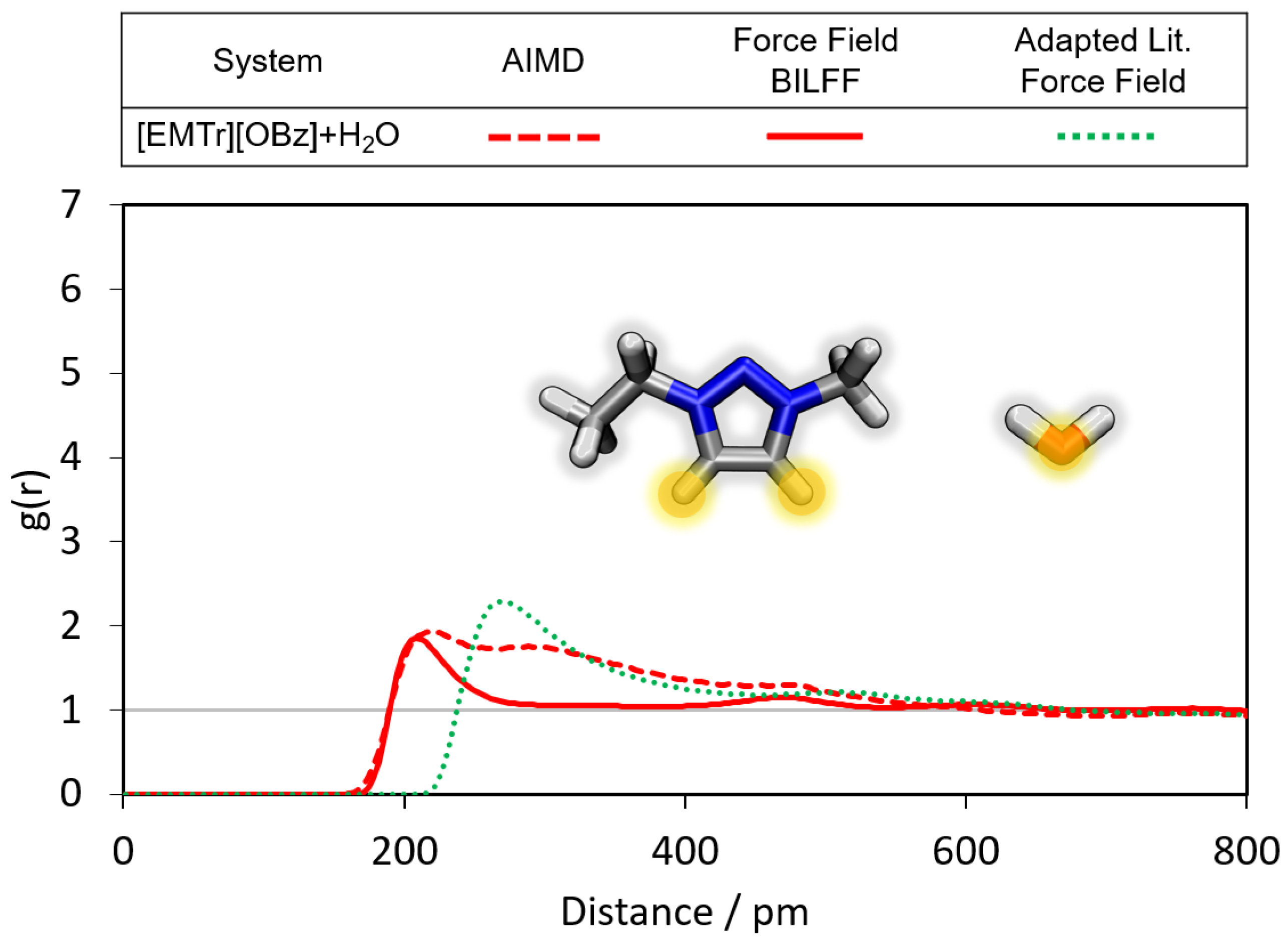
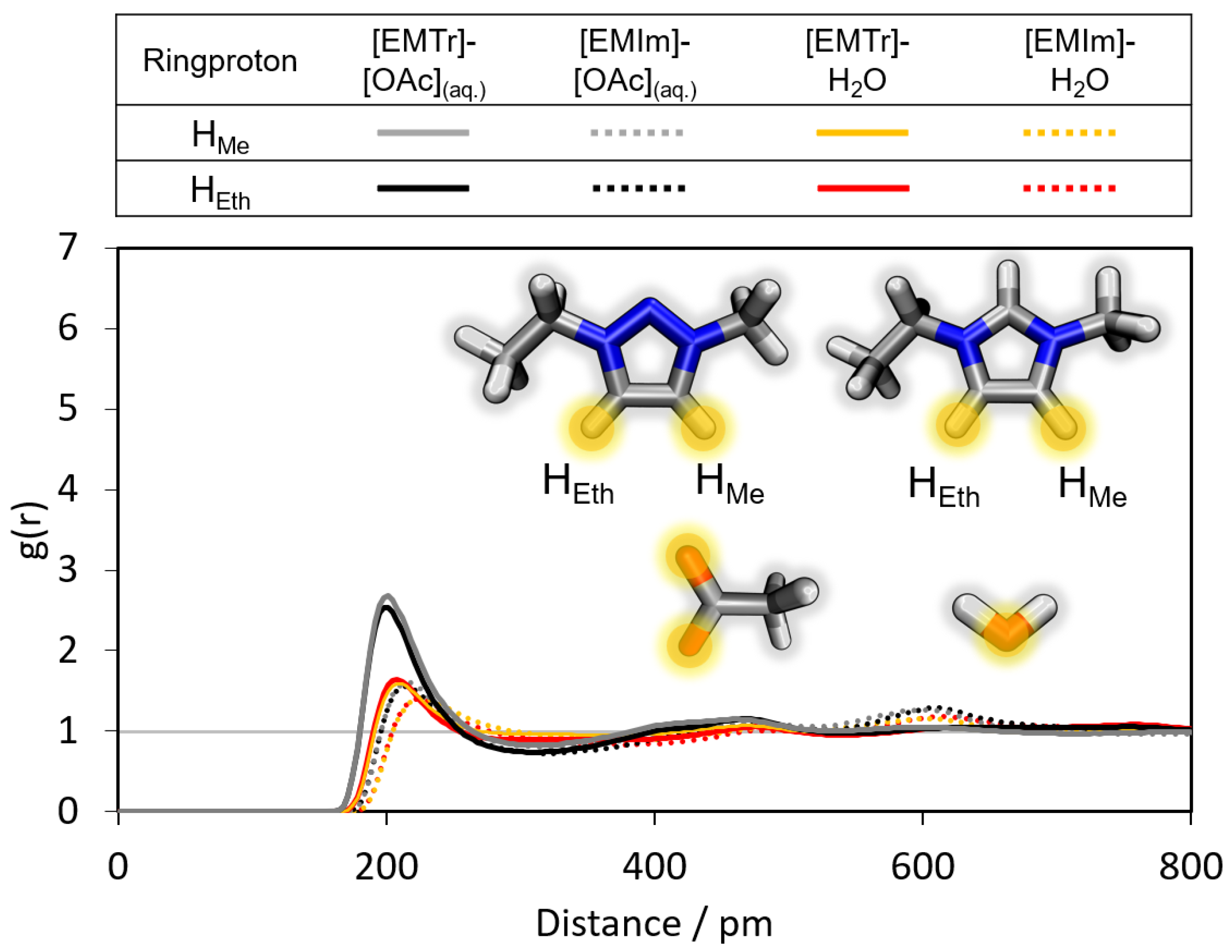
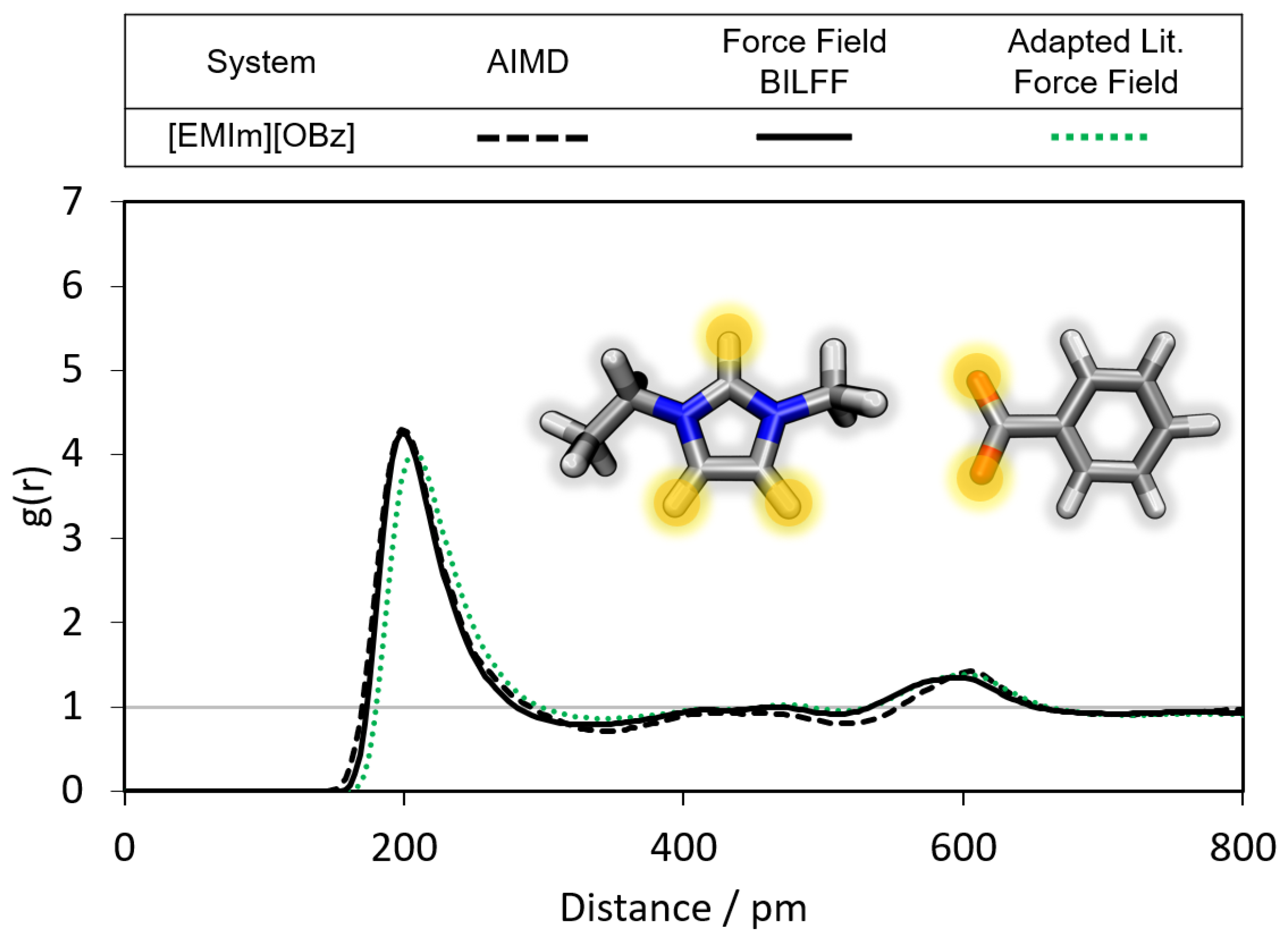
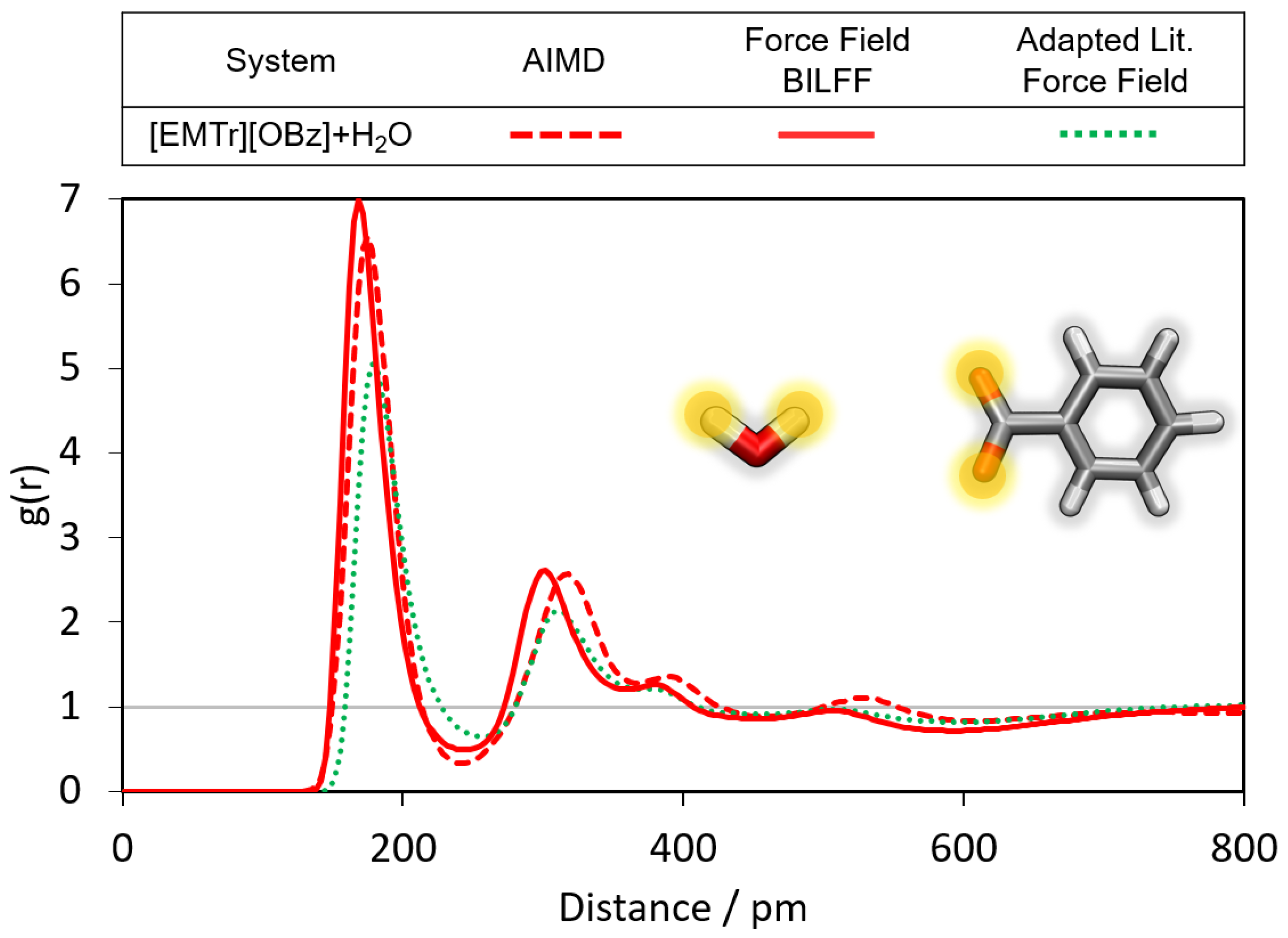
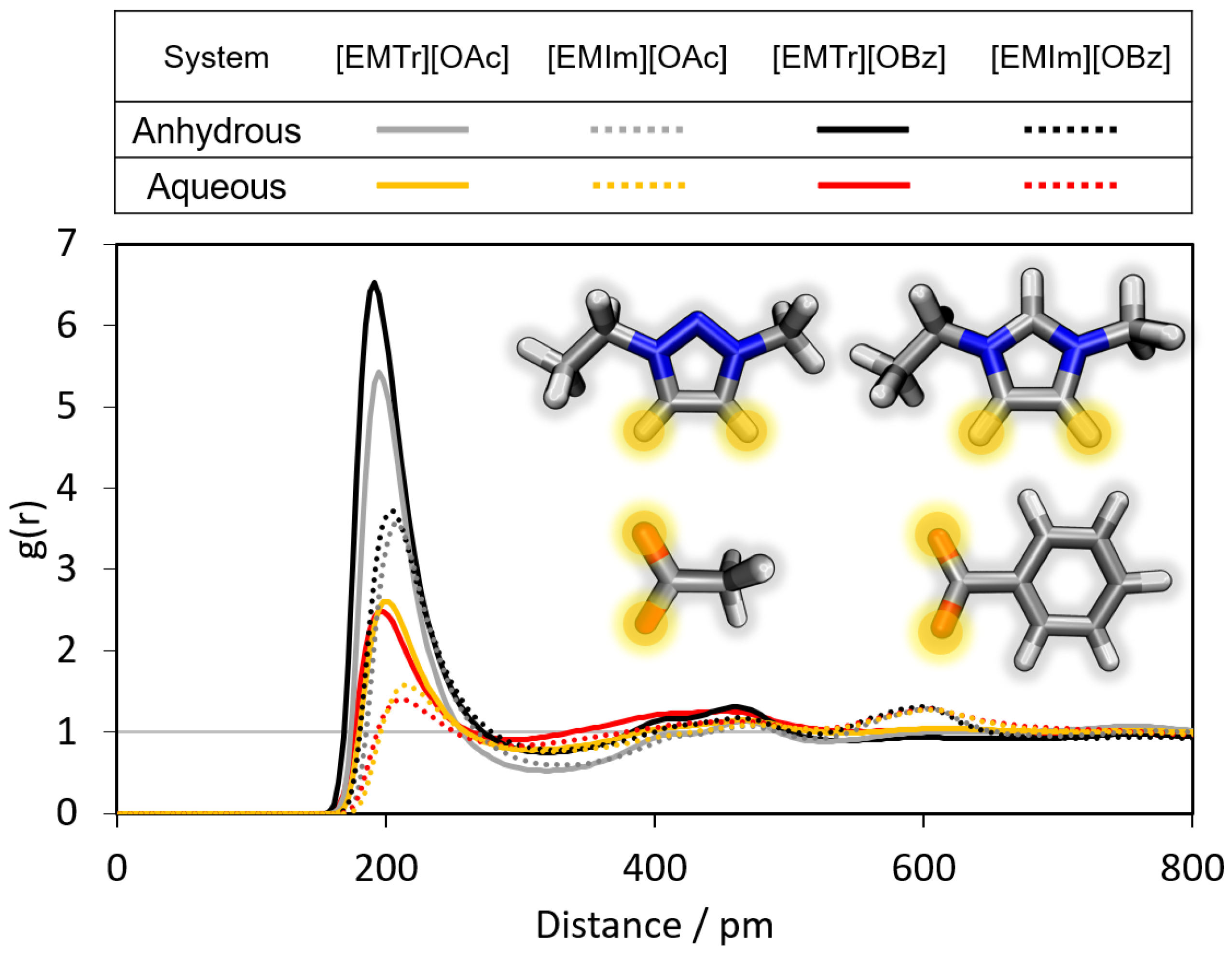
2.1.1. Radial Distribution Functions
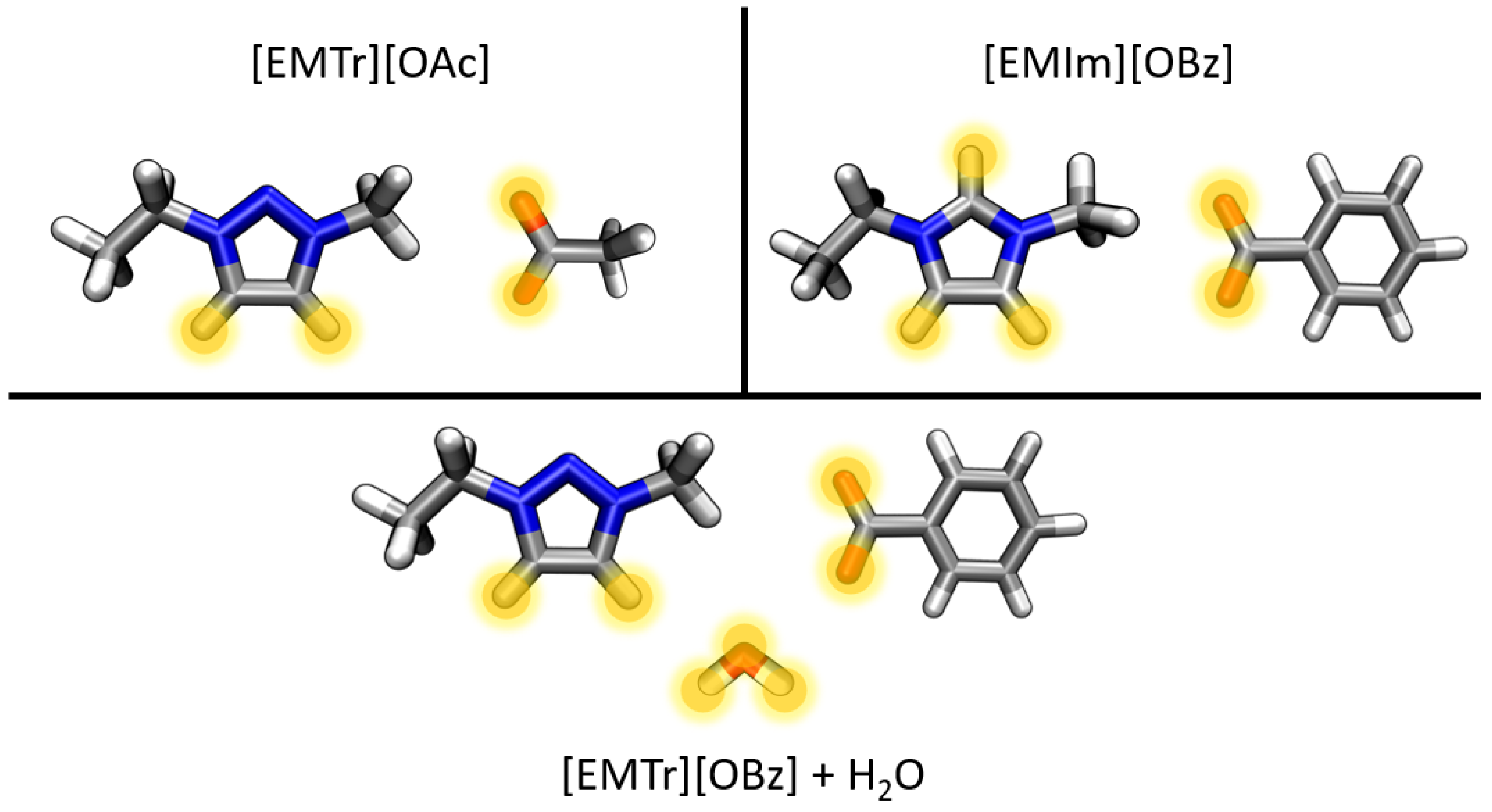
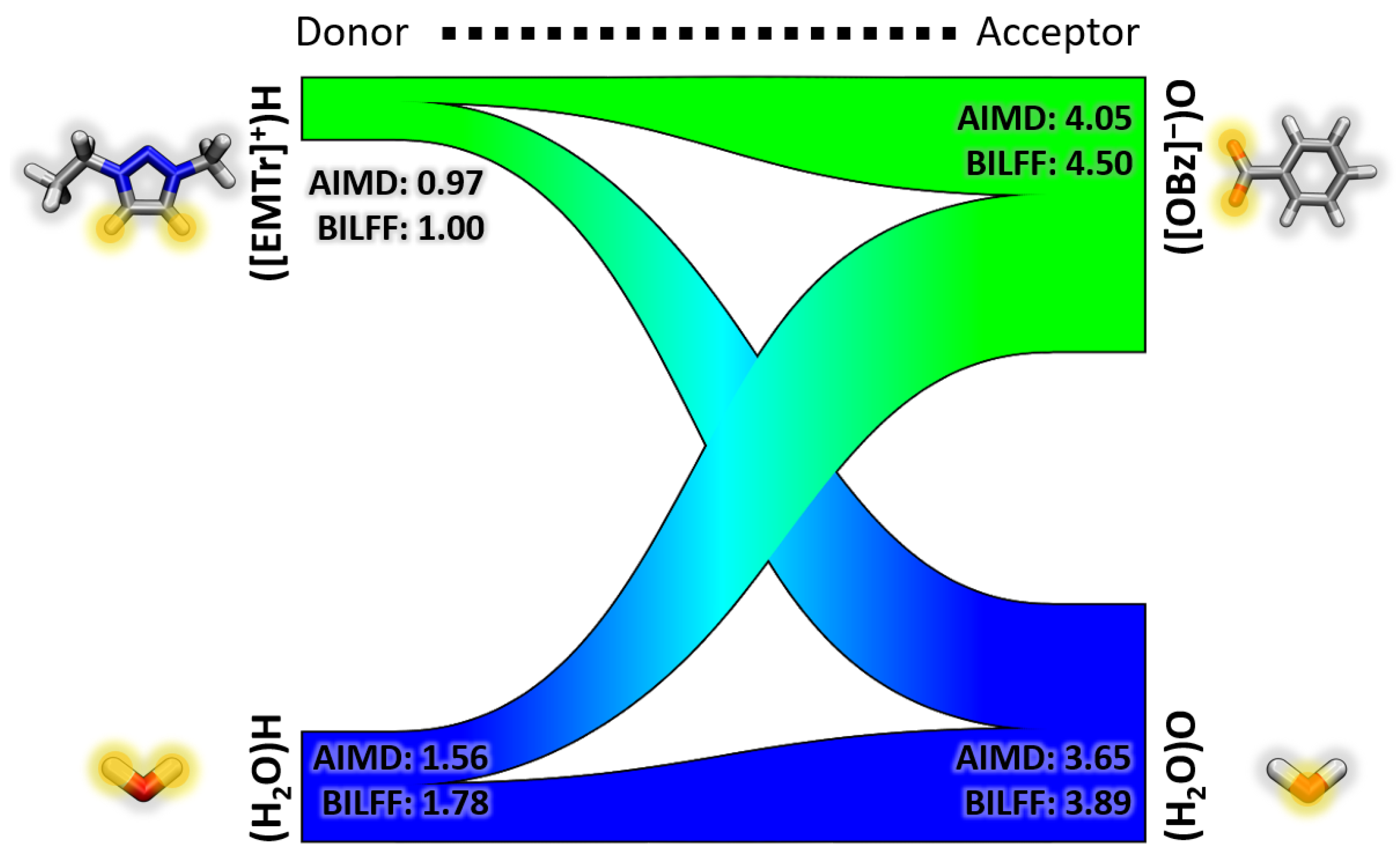
- Triazolium forms stronger hydrogen bonds with both the anion and water than imidazolium.
- Benzoate forms stronger hydrogen bonds with the cation than acetate in the anhydrous system.
- Benzoate forms stronger hydrogen bonds with water than acetate, resulting in a greater attenuation of the cation–anion interaction in the aqueous system.
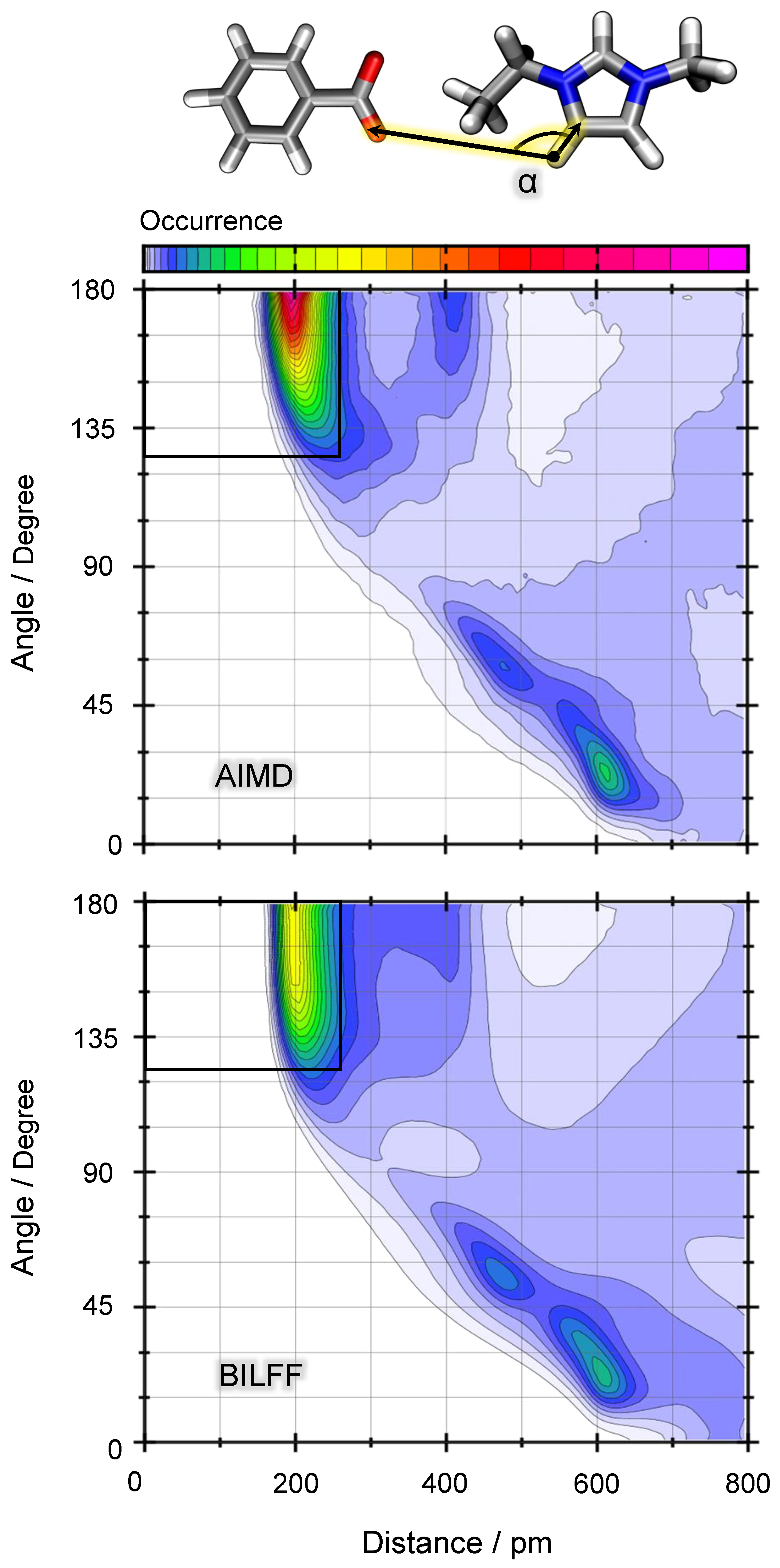
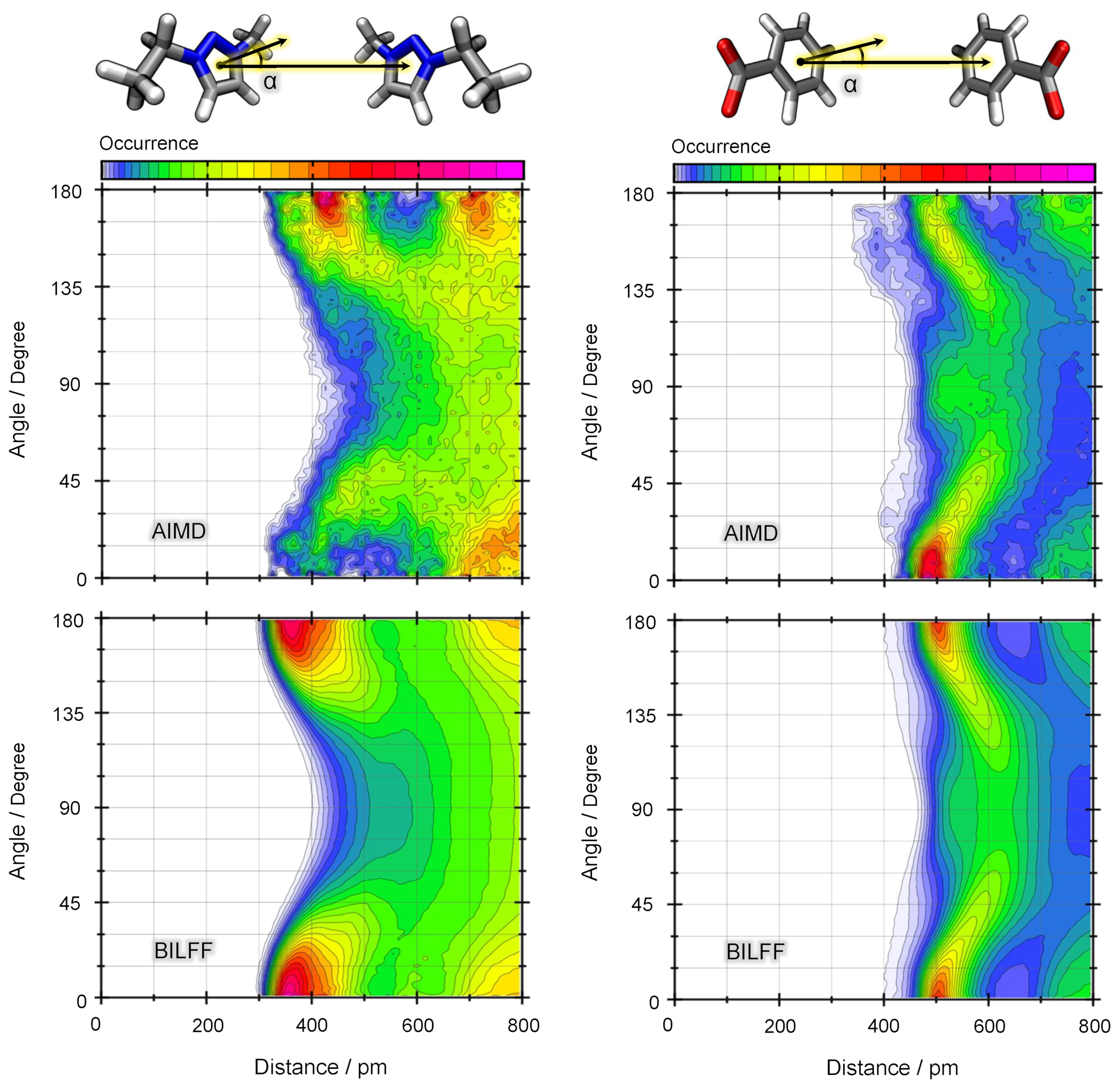
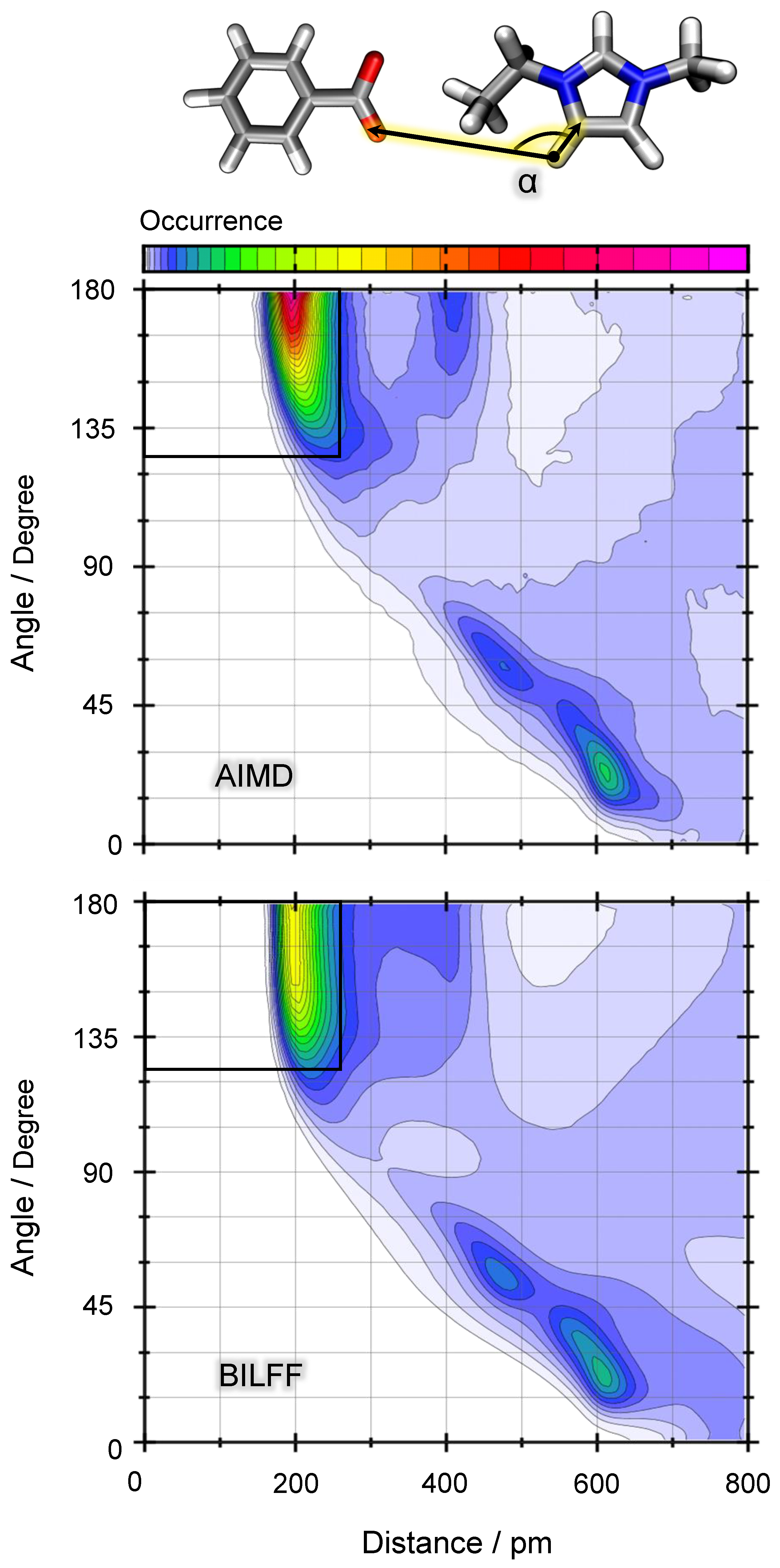
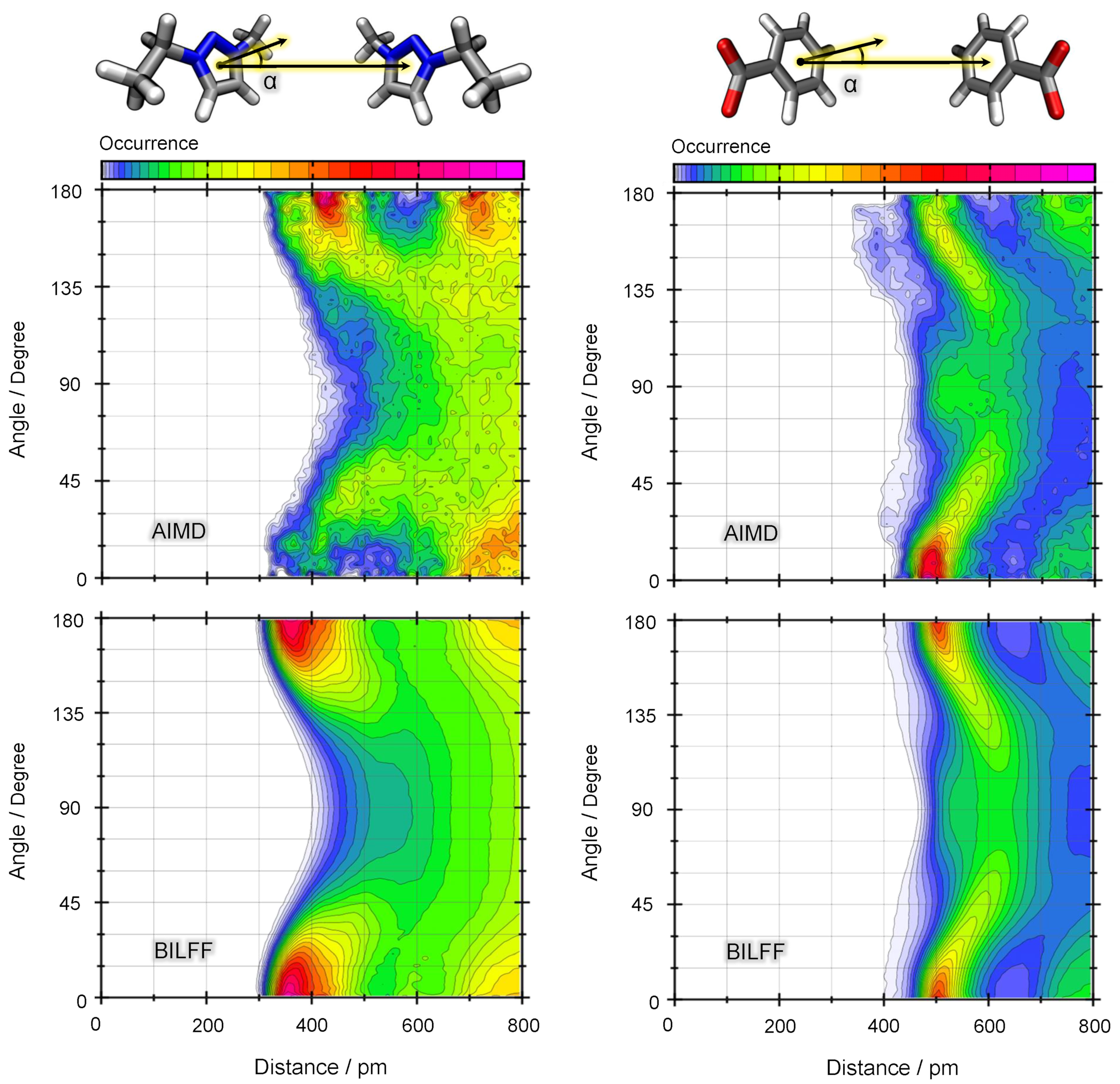
2.1.2. Combined Distance–Angle Distribution Functions
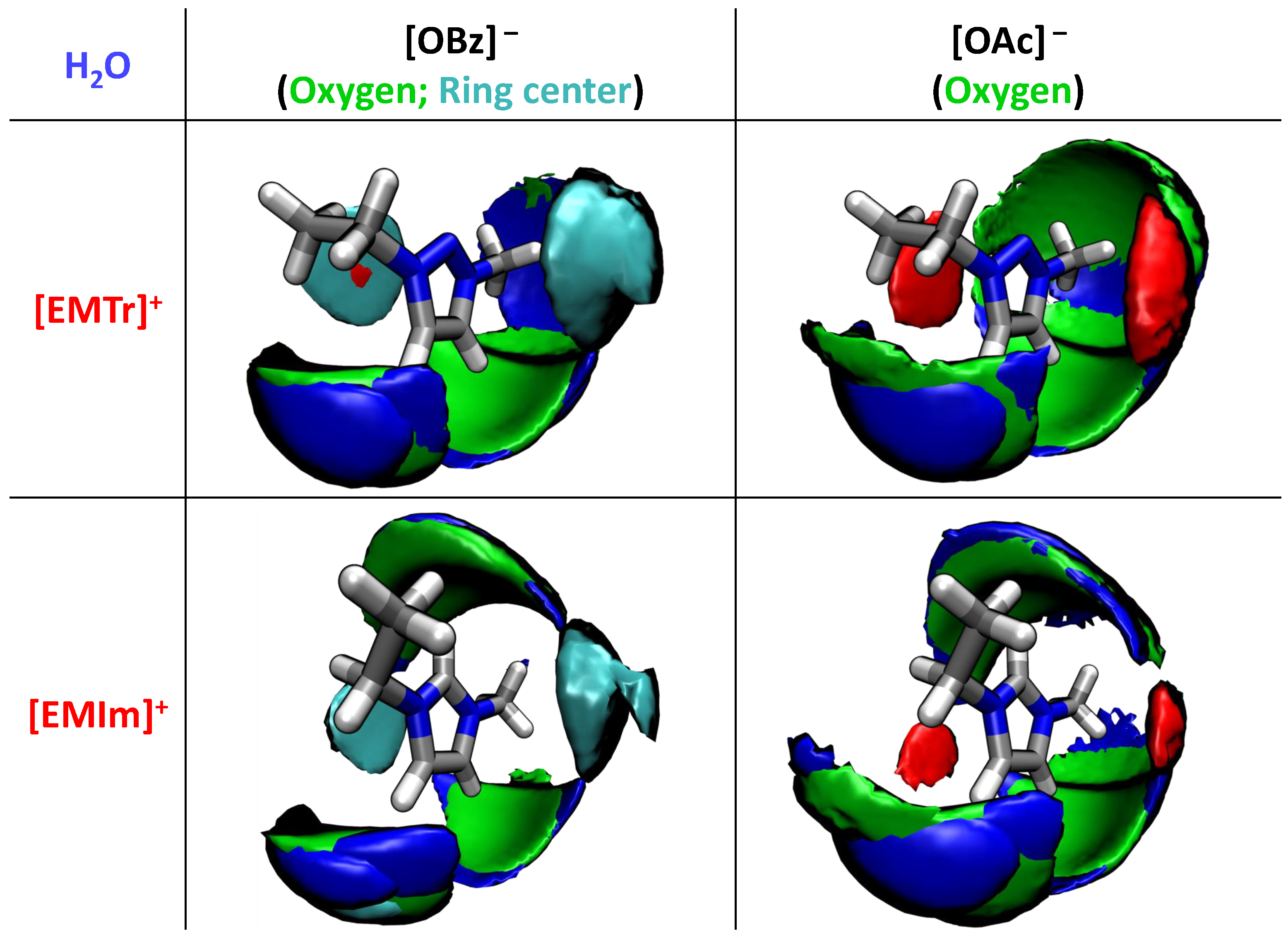
2.1.3. Spatial Distribution Functions
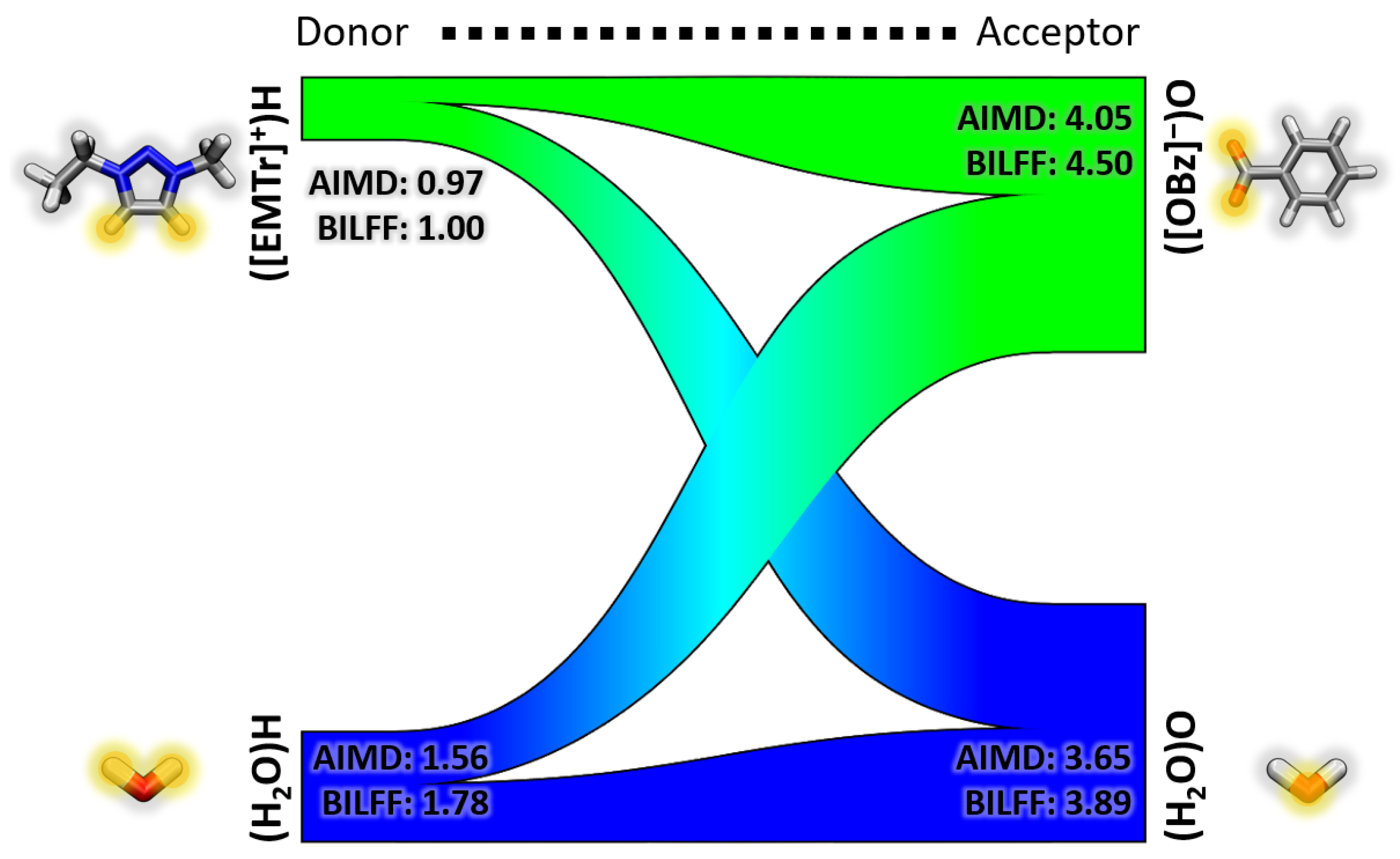
2.2. Competing Hydrogen Bonds
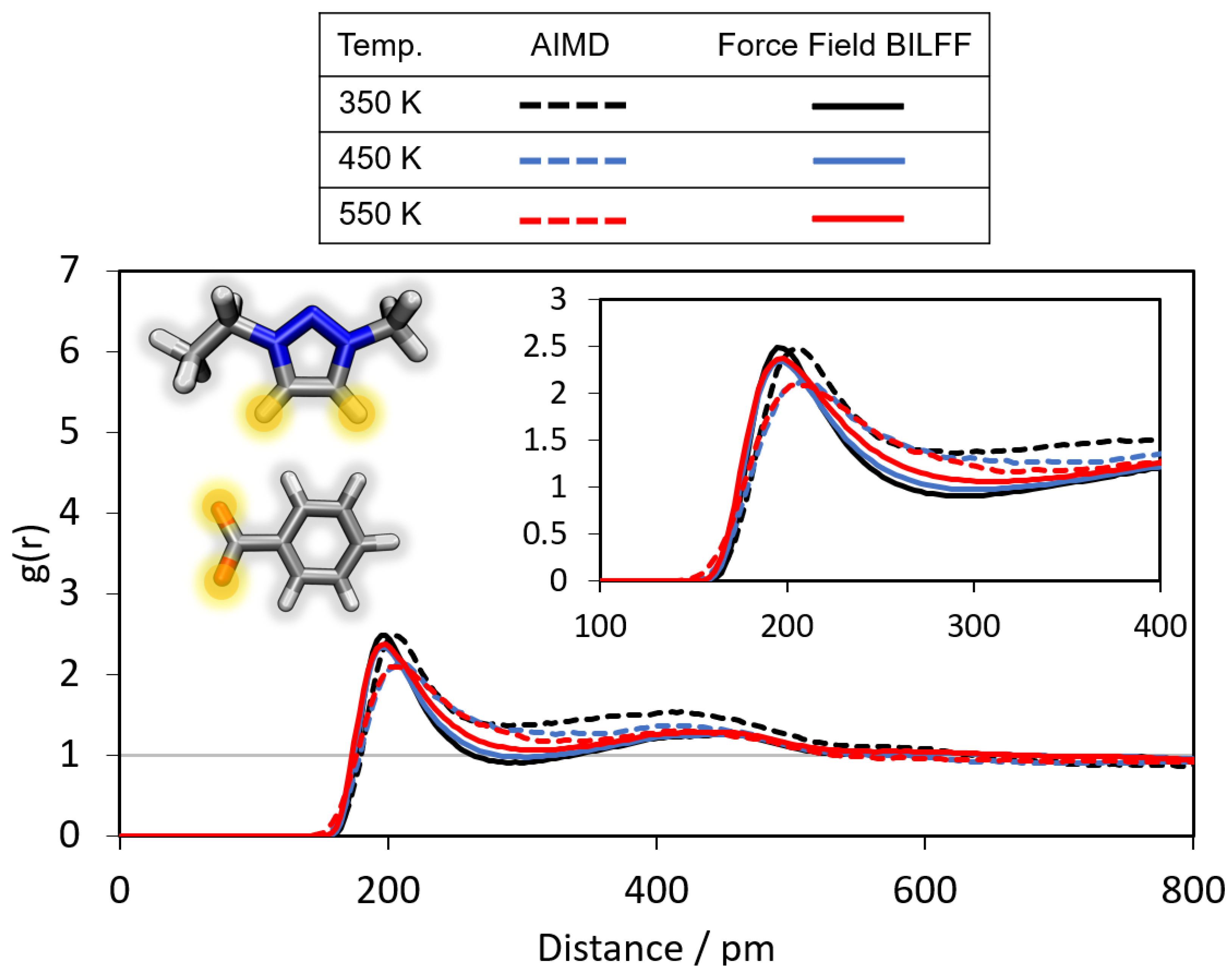
2.3. Temperature Dependence
2.3.1. Radial Distribution Functions at Higher Temperatures
2.3.2. Hydrogen Bond Lifetime
2.3.3. Validation of the Density and Diffusion
- The diffusion coefficient is higher at elevated temperatures.
- In the presence of water, the diffusion coefficient of all ionic liquids increases significantly.
- The diffusion coefficient is influenced by both the cations and anions in the IL. When comparing the diffusion of the cations in the different ILs containing either [EMTr] or [EMIm], it is observed that the triazolium cations exhibit a slower diffusion compared to imidazolium. Furthermore, when comparing the diffusion rates of the anions, it is found that benzoate diffuses slower than acetate in both pure and aqueous systems. This is due to the different occupied volumes and thus the bulkiness of the molecules as well as their more pronounced hydrogen bond formation.
- The lowest diffusion rate, and, at the same time, the highest density, can be found in pure [EMTr][OBz].
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIMD | Ab initio Molecular Dynamics |
| BILFF | Bio-polymers in Ionic Liquids Force Field |
| CDF | Combined Distribution Function |
| FFMD | Force Field Molecular Dynamics |
| IL | Ionic Liquid |
| MD | Molecular Dynamics |
| RDF | Radial Distribution Function |
| SDF | Spatial Distribution Function |
References
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, N.; He, X.; Lu, X.; Zhang, X. Physical properties of ionic liquids: Database and evaluation. J. Phys. Chem. Ref. Data 2006, 35, 1475–1517. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef]
- Zhao, D.; Wu, M.; Kou, Y.; Min, E. Ionic liquids: Applications in catalysis. Catal. Today 2002, 74, 157–189. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Forsyth, M.; Howlett, P.C.; Pringle, J.M.; Sun, J.; Annat, G.; Neil, W.; Izgorodina, E.I. Ionic liquids in electrochemical devices and processes: Managing interfacial electrochemistry. Acc. Chem. Res. 2007, 40, 1165–1173. [Google Scholar] [CrossRef]
- Obadia, M.M.; Drockenmuller, E. Poly(1,2,3-triazolium)s: A new class of functional polymer electrolytes. Chem. Commun. 2016, 52, 2433–2450. [Google Scholar] [CrossRef]
- Guo, N.; Li, M.; Sun, X.; Wang, F.; Yang, R. Enzymatic hydrolysis lignin derived hierarchical porous carbon for supercapacitors in ionic liquids with high power and energy densities. Green Chem. 2017, 19, 2595–2602. [Google Scholar] [CrossRef]
- Elia, G.A.; Ulissi, U.; Mueller, F.; Reiter, J.; Tsiouvaras, N.; Sun, Y.K.; Scrosati, B.; Passerini, S.; Hassoun, J. A long-life lithium ion battery with enhanced electrode/electrolyte interface by using an ionic liquid solution. Chemistry 2016, 22, 6808–6814. [Google Scholar] [CrossRef]
- Fletcher, J.T.; Sobczyk, J.M.; Gwazdacz, S.C.; Blanck, A.J. Antimicrobial 1,3,4-trisubstituted-1,2,3-triazolium salts. Bioorg. Med. Chem. 2018, 28, 3320–3323. [Google Scholar] [CrossRef]
- Tan, W.; Li, Q.; Dong, F.; Zhang, J.; Luan, F.; Wei, L.; Chen, Y.; Guo, Z. Novel cationic chitosan derivative bearing 1,2,3-triazolium and pyridinium: Synthesis, characterization, and antifungal property. Carbohydr. Polym. 2018, 182, 180–187. [Google Scholar] [CrossRef]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of cellulose correction of cellose with ionic liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef]
- Brandt, A.; Ray, M.J.; To, T.Q.; Leak, D.J.; Murphy, R.J.; Welton, T. Ionic liquid pretreatment of lignocellulosic biomass with ionic liquid–water mixtures. Green Chem. 2011, 13, 2489. [Google Scholar] [CrossRef]
- Gupta, K.M.; Jiang, J. Cellulose dissolution and regeneration in ionic liquids: A computational perspective. Chem. Eng. Sci. 2015, 121, 180–189. [Google Scholar] [CrossRef]
- Brehm, M.; Pulst, M.; Kressler, J.; Sebastiani, D. Triazolium-based ionic liquids: A novel class of cellulose solvents. J. Phys. Chem. B 2019, 123, 3994–4003. [Google Scholar] [CrossRef]
- Brehm, M.; Radicke, J.; Pulst, M.; Shaabani, F.; Sebastiani, D.; Kressler, J. Dissolving cellulose in 1,2,3-triazolium- and imidazolium-based ionic liquids with aromatic anions. Molecules 2020, 25, 3539. [Google Scholar] [CrossRef]
- Radicke, J.; Roos, E.; Sebastiani, D.; Brehm, M.; Kressler, J. Lactate–based ionic liquids as chiral solvents for cellulose. J. Polym. Sci. 2023, 61, 372–384. [Google Scholar] [CrossRef]
- Zhang, L.; Ruan, D.; Gao, S. Dissolution and regeneration of cellulose in NaOH/thiourea aqueous solution. J. Polym. Sci. B Polym. Phys. 2002, 40, 1521–1529. [Google Scholar] [CrossRef]
- Heinze, T.; Schwikal, K.; Barthel, S. Ionic liquids as reaction medium in cellulose functionalization. Macromol. Biosci. 2005, 5, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Rahman, M.; Qin, Y.; Maxim, M.L.; Rodríguez, H.; Rogers, R.D. Complete dissolution and partial delignification of wood in the ionic liquid 1-ethyl-3-methylimidazolium acetate. Green Chem. 2009, 11, 646. [Google Scholar] [CrossRef]
- Fort, D.A.; Remsing, R.C.; Swatloski, R.P.; Moyna, P.; Moyna, G.; Rogers, R.D. Can ionic liquids dissolve wood? Processing and analysis of lignocellulosic materials with 1-n-butyl-3-methylimidazolium chloride. Green Chem. 2007, 9, 63–69. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose Ibeta from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef]
- Olsson, C.; Westm, G. Direct dissolution of cellulose: Background, means and applications. Cellul. Fundam. Asp. 2013. [Google Scholar] [CrossRef]
- Ren, F.; Wang, J.; Yu, J.; Zhong, C.; Xie, F.; Wang, S. Dissolution of cellulose in ionic liquid-DMSO mixtures: Roles of DMSO/IL ratio and the cation alkyl chain length. ACS Omega 2021, 6, 27225–27232. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, N.; Stoner, B.; Jiang, X.; Lu, X.; Rogers, R.D. Rapid dissolution of lignocellulosic biomass in ionic liquids using temperatures above the glass transition of lignin. Green Chem. 2011, 13, 2038. [Google Scholar] [CrossRef]
- Liebert, T.F.; Heinze, T.J.; Edgar, K.J. Cellulose Solvents: For Analysis, Shaping and Chemical Modification; American Chemical Society: Washington, DC, USA, 2010; Volume 1033. [Google Scholar] [CrossRef]
- Roos, E.; Brehm, M. A force field for bio-polymers in ionic liquids (BILFF)—Part 1: EMImOAc/water mixtures. Phys. Chem. Chem. Phys. 2021, 23, 1242–1253. [Google Scholar] [CrossRef]
- Roos, E.; Sebastiani, D.; Brehm, M. A force field for bio-polymers in ionic liquids (BILFF)—Part 2: Cellulose in EMImOAc/water mixtures. Phys. Chem. Chem. Phys. 2023, 25, 8755–8766. [Google Scholar] [CrossRef]
- Olsson, C.; Idström, A.; Nordstierna, L.; Westman, G. Influence of water on swelling and dissolution of cellulose in 1-ethyl-3-methylimidazolium acetate. Carbohydr. Polym. 2014, 99, 438–446. [Google Scholar] [CrossRef]
- Le, K.A.; Sescousse, R.; Budtova, T. Influence of water on cellulose-EMIMAc solution properties: A viscometric study. Cellulose 2012, 19, 45–54. [Google Scholar] [CrossRef]
- Froschauer, C.; Hummel, M.; Iakovlev, M.; Roselli, A.; Schottenberger, H.; Sixta, H. Separation of hemicellulose and cellulose from wood pulp by means of ionic liquid/cosolvent systems. Biomacromolecules 2013, 14, 1741–1750. [Google Scholar] [CrossRef]
- Bowron, D.T.; D’Agostino, C.; Gladden, L.F.; Hardacre, C.; Holbrey, J.D.; Lagunas, M.C.; McGregor, J.; Mantle, M.D.; Mullan, C.L.; Youngs, T.G.A. Structure and dynamics of 1-ethyl-3-methylimidazolium acetate via molecular dynamics and neutron diffraction. J. Phys. Chem. B 2010, 114, 7760–7768. [Google Scholar] [CrossRef] [PubMed]
- Yaghini, N.; Pitawala, J.; Matic, A.; Martinelli, A. Effect of water on the local structure and phase behavior of imidazolium-based protic ionic liquids. J. Phys. Chem. B 2015, 119, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [CrossRef] [PubMed]
- Sambasivarao, S.V.; Acevedo, O. Development of OPLS-AA force field parameters for 68 unique ionic liquids. J. Chem. Theory Comput. 2009, 5, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.; Deschamps, J.; Pádua, A.A.H. Modeling ionic liquids using a systematic all-atom force field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A.H. Molecular force field for ionic liquids III: Imidazolium, pyridinium, and phosphonium cations; chloride, bromide, and dicyanamide anions. J. Phys. Chem. B 2006, 110, 19586–19592. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A.H. A generic and systematic force field for ionic liquids modeling. Theor. Chem. Acc. 2012, 131, 3330. [Google Scholar] [CrossRef]
- Pike, S.J.; Hutchinson, J.J.; Hunter, C.A. H-Bond Acceptor Parameters for Anions. J. Am. Chem. Soc. 2017, 139, 6700–6706. [Google Scholar] [CrossRef]
- Quijada-Maldonado, E.; van der Boogaart, S.; Lijbers, J.H.; Meindersma, G.W.; de Haan, A.B. Experimental densities, dynamic viscosities and surface tensions of the ionic liquids series 1-ethyl-3-methylimidazolium acetate and dicyanamide and their binary and ternary mixtures with water and ethanol at T = (298.15 to 343.15K). J. Chem. Thermodyn. 2012, 51, 51–58. [Google Scholar] [CrossRef]
- Urahata, S.M.; Ribeiro, M.C.C. Single particle dynamics in ionic liquids of 1-alkyl-3-methylimidazolium cations. Chem. Phys. 2005, 122, 024511. [Google Scholar] [CrossRef]
- Hall, C.A.; Le, K.A.; Rudaz, C.; Radhi, A.; Lovell, C.S.; Damion, R.A.; Budtova, T.; Ries, M.E. Macroscopic and microscopic study of 1-ethyl-3-methyl-imidazolium acetate-water mixtures. J. Phys. Chem. B 2012, 116, 12810–12818. [Google Scholar] [CrossRef] [PubMed]
- Green, S.M.; Ries, M.E.; Moffat, J.; Budtova, T. NMR and rheological study of anion size influence on the properties of two imidazolium-based ionic liquids. Sci. Rep. 2017, 7, 8968. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- The CP2K Developers Group. 2023. Available online: http://www.cp2k.org/ (accessed on 18 June 2023).
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package—Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. An efficient orbital transformation method for electronic structure calculations. J. Chem. Phys. 2003, 118, 4365–4369. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133. [Google Scholar] [CrossRef]
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.A.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised damping parameters for the D3 dispersion correction to density functional theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Nose, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Martyna, G.; Klein, M.; Tuckerman, M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1192, 97, 2535–2643. [Google Scholar] [CrossRef]
- Schneider, T.; Stoll, E. Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions. Phys. Rev. B 1978, 17, 1302–1322. [Google Scholar] [CrossRef]
- Dünweg, B.; Paul, W. Brownian dynamics simulations without Gaussian random numbers. Int. J. Mod. Phys. C 1991, 2, 817–827. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Bhargava, B.L.; Balasubramanian, S. Refined potential model for atomistic simulations of ionic liquid [bmim][PF6]. J. Chem. Phys. 2007, 127, 114510. [Google Scholar] [CrossRef]
- Liu, H.; Maginn, E. A molecular dynamics investigation of the structural and dynamic properties of the ionic liquid 1-n-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide. J. Chem. Phys. 2011, 135, 124507. [Google Scholar] [CrossRef]
- Doherty, B.; Zhong, X.; Gathiaka, S.; Li, B.; Acevedo, O. Revisiting OPLS force field parameters for ionic liquid simulations. J. Chem. Theory Comput. 2017, 13, 6131–6145. [Google Scholar] [CrossRef] [PubMed]
- Doherty, B.; Acevedo, O. OPLS force field for choline chloride-based deep eutectic solvents. J. Phys. Chem. B 2018, 122, 9982–9993. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L. Competitive microstructures versus cooperative dynamics of hydrogen bonding and Pi-type stacking interactions in imidazolium bis(oxalato)borate ionic liquids. J. Phys. Chem. B 2018, 122, 6570–6585. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Thomas, M.; Gehrke, S.; Kirchner, B. TRAVIS – A free analyzer for trajectories from molecular simulation. J. Chem. Phys. 2020, 152, 164105. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Kirchner, B. TRAVIS—A free analyzer and visualizer for Monte Carlo and molecular dynamics trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef]
- Grace, 1996–2023 Grace Development Team. Available online: http://plasma-gate.weizmann.ac.il/Grace (accessed on 18 June 2023).
- Wolfram Research, Inc. Mathematica, Version 8.0; Wolfram Research, Inc.: Champaign, IL, USA, 2010.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J. An Efficient Library for Parallel Ray Tracing and Animation. Ph.D. Thesis, Computer Science Department, University of Missouri-Rolla, Rolla, MO, USA, 1998. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
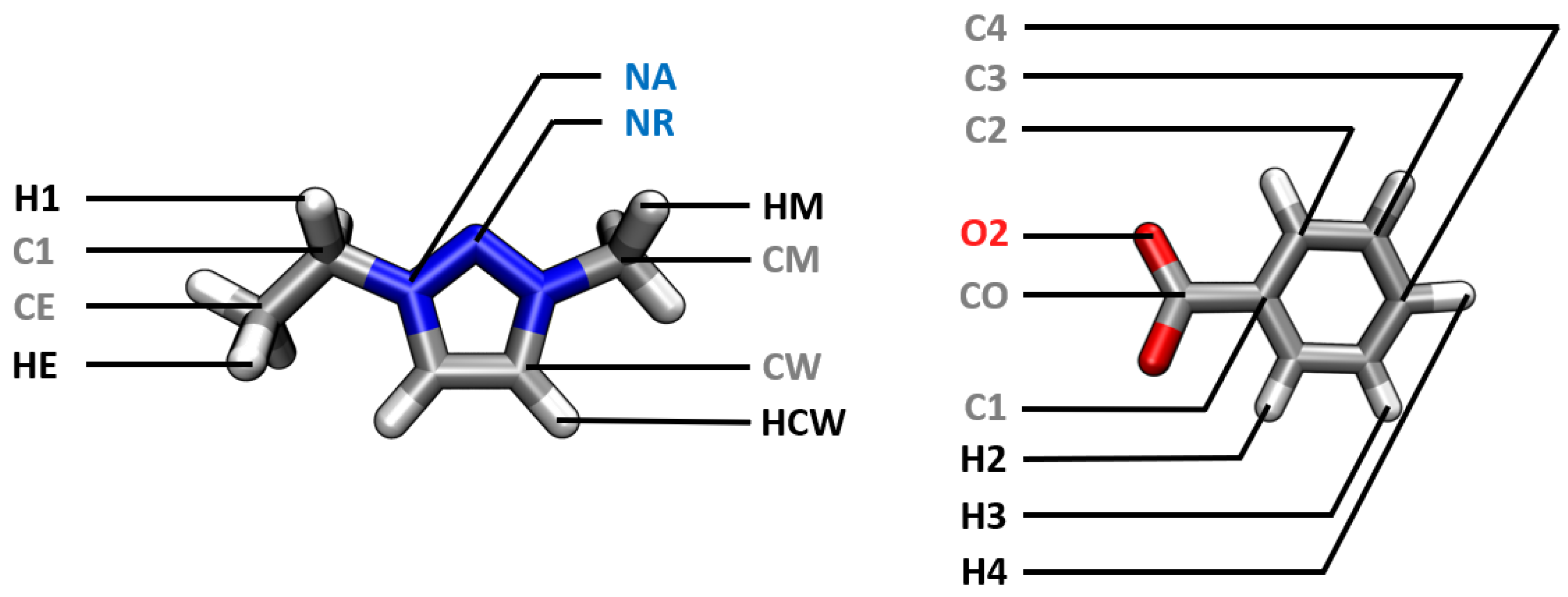
| Atom Type | Atom Class |
|---|---|
| [EMTr] | |
| C1 | CT |
| CE | CT |
| CM | CT |
| CW | CW |
| HCW | HA |
| H1 | HC |
| HE | HC |
| HM | HC |
| NR | NR |
| NA | NA |
| [OBz] | |
| C1 | CA |
| C2 | CA |
| C3 | CA |
| C4 | CA |
| CO | CO |
| H2 | HA |
| H3 | HA |
| H4 | HA |
| O2 | O2 |
| Atom Type | q | ||
|---|---|---|---|
| / | / Å | / kJ mol | |
| [EMTr] | |||
| C1 | −0.187 | 3.34 | 0.2760 |
| CE | −0.054 | 3.34 | 0.2760 |
| CW | −0.144 | 3.38 | 0.2930 |
| HCW | 0.191 | 1.48 | 0.1260 |
| HC | 0.070 | 2.38 | 0.1260 |
| H1 | 0.148 | 2.38 | 0.1260 |
| NR | −0.204 | 3.10 | 0.7110 |
| NA | 0.204 | 3.10 | 0.7110 |
| [OBz] | |||
| C1 | 0.005 | 3.70 | 0.2929 |
| C2 | −0.118 | 3.70 | 0.2929 |
| C3 | −0.121 | 3.70 | 0.2929 |
| C4 | −0.299 | 3.70 | 0.2929 |
| CO | 0.398 | 3.90 | 0.4393 |
| H2 | 0.070 | 2.42 | 0.1255 |
| H3 | 0.157 | 2.42 | 0.1255 |
| H4 | 0.200 | 2.42 | 0.1255 |
| O2 | −0.550 | 2.80 | 0.8786 |
| Bond | ||
|---|---|---|
| / Å | / kJ mol | |
| [EMTr] | ||
| NA–NR | 1.344 | 3199.2 |
| CW–HA | 1.088 | 2633.8 |
| CW–NA | 1.375 | 3108.7 |
| CW–CW | 1.386 | 3773.2 |
| NA–CT | 1.488 | 2046.3 |
| HC–CT | 1.099 | 2679.4 |
| CT–CT | 1.533 | 2125.5 |
| [OBz] | ||
| CA–CA | 1.387 | 3274.1 |
| CA–HA | 1.088 | 2707.4 |
| CA–CO | 1.504 | 1906.9 |
| CO–O2 | 1.282 | 4273.1 |
| Angle | ||
|---|---|---|
| / Deg | / kJ mol rad | |
| [EMTr] | ||
| CW–NA–NR | 112.1 | 568.7 |
| NR–NA–CT | 118.6 | 396.5 |
| NA–NR–NA | 104.4 | 610.1 |
| NA–CT–CT | 110.9 | 361.2 |
| NA–CW–CW | 107.0 | 579.7 |
| NA–CW–HA | 120.8 | 200.9 |
| CW–CW–HA | 131.7 | 190.5 |
| NA–CT–HC | 107.2 | 375.9 |
| CT–CT–HC | 111.4 | 296.2 |
| HC–CT–HC | 109.2 | 226.5 |
| CW–NA–CT | 125.2 | 242.9 |
| [OBz] | ||
| CA–CA–CA | 120.0 | 446.0 |
| CA–CA–HA | 120.0 | 258.1 |
| CA–CA–CO | 120.0 | 397.6 |
| CA–CO–O2 | 117.0 | 550.2 |
| O2–CO–O2 | 126.0 | 735.9 |
| Torsion Angle | ||||
|---|---|---|---|---|
| / kJ mol | / kJ mol | / kJ mol | / kJ mol | |
| [EMTr] | ||||
| CW–NA–NR–NA | 0.0000 | 19.4600 | 0.0000 | 0.0000 |
| CT–NA–NR–NA | 0.0000 | 19.4600 | 0.0000 | 0.0000 |
| NR–NA–CW–CW | 0.0000 | 12.5500 | 0.0000 | 0.0000 |
| NR–NA–CW–HA | 0.0000 | 12.5500 | 0.0000 | 0.0000 |
| NR–NA–CT–HC | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| NR–NA–CT–CT | 0.1000 | 1.0000 | 0.1000 | −0.3000 |
| CT–NA–CW–CW | 0.0000 | 12.5500 | 0.0000 | 0.0000 |
| CT–NA–CW–HA | 0.0000 | 12.5500 | 0.0000 | 0.0000 |
| NA–CW–CW–NA | 0.0000 | 65.0000 | 0.0000 | 0.0000 |
| NA–CW–CW–HA | 0.0000 | 44.9800 | 0.0000 | 0.0000 |
| HA–CW–CW–HA | 0.0000 | 30.0000 | 0.0000 | 0.0000 |
| CW–NA–CT–HC | 0.1000 | 0.2000 | 0.0000 | 0.0000 |
| CW–NA–CT–CT | 0.4000 | 1.0000 | 0.0000 | 0.2000 |
| NA–CT–CT–HC | 0.0000 | 0.0000 | 0.3670 | 0.0000 |
| HC–CT–CT–HC | 0.0000 | 0.0000 | 1.2552 | 0.0000 |
| [OBz] | ||||
| CA–CA–CA–CA | 0.0000 | 30.334 | 0.0000 | 0.0000 |
| HA–CA–CA–CA | 0.0000 | 30.334 | 0.0000 | 0.0000 |
| HA–CA–CA–CO | 0.0000 | 30.334 | 0.0000 | 0.0000 |
| HA–CA–CA–HA | 0.0000 | 30.334 | 0.0000 | 0.0000 |
| CA–CA–CA–CO | 0.0000 | 30.334 | 0.0000 | 0.0000 |
| CA–CA–CO–O2 | 0.0000 | 8.000 | 0.0000 | 0.0000 |
| Temp. | Intermittent | Continuous | ||
|---|---|---|---|---|
| (AIMD) | (FFMD) | (AIMD) | (FFMD) | |
| / K | / ps | / ps | / ps | / ps |
| [EMTr][OAc] | ||||
| (C)H⋯O(A) | ||||
| 350 | 627.1 | 855.3 | 4.0 | 4.1 |
| [EMTr][OAc]/HO | ||||
| (C)H⋯O(A) | ||||
| 350 | – | 117.6 | – | 1.9 |
| (C)H⋯O(HO) | ||||
| 350 | – | 32.9 | – | 1.0 |
| (HO)H⋯O(A) | ||||
| 350 | – | 58.3 | – | 0.2 |
| [EMIm][OAc] | ||||
| (C)H⋯O(A) | ||||
| 350 | 472.0 | 779.7 | 3.0 | 4.5 |
| [EMIm][OAc]/HO | ||||
| (C)H⋯O(A) | ||||
| 350 | 73.2 | 146.0 | 1.3 | 1.8 |
| (C)H⋯O(HO) | ||||
| 350 | 31.2 | 40.4 | 0.6 | 0.8 |
| (HO)H⋯O(A) | ||||
| 350 | 153.2 | 165.7 | 0.2 | 0.8 |
| [EMIm][OBz] | ||||
| (C)H⋯O(A) | ||||
| 350 | 242.2 | 1841.6 | 1.0 | 2.0 |
| [EMIm][OBz]/HO | ||||
| (C)H⋯O(A) | ||||
| 350 | – | 207.3 | – | 1.2 |
| (C)H⋯O(HO) | ||||
| 350 | – | 236.0 | – | 1.7 |
| (HO)H⋯O(A) | ||||
| 350 | – | 299.7 | – | 6.6 |
| [EMTr][OBz] | ||||
| (C)H⋯O(A) | ||||
| 350 | – | 2821.1 | – | 6.3 |
| [EMTr][OBz]/HO | ||||
| (C)H⋯O(A) | ||||
| 350 | 105.0 | 203.8 | 1.6 | 2.5 |
| 450 | 25.5 | 27.8 | 0.8 | 1.4 |
| 550 | 15.5 | 11.2 | 0.6 | 1.0 |
| (C)H⋯O(HO) | ||||
| 350 | 38.1 | 48.3 | 0.7 | 1.0 |
| 450 | 5.7 | 6.4 | 0.4 | 0.6 |
| 550 | – | 2.3 | 0.3 | 0.5 |
| (HO)H⋯O(A) | ||||
| 350 | 466.6 | 273.1 | 4.4 | 8.8 |
| 450 | 36.7 | 34.8 | 1.0 | 2.2 |
| 550 | 15.6 | 11.4 | 0.5 | 1.3 |
| System | Temp. | Box Size | (FFMD) | (Lit.) |
|---|---|---|---|---|
| / K | / pm | / g cm | / g cm | |
| [EMTr][OAc] | 350 | 3198 | 1.11 | 1.12 |
| [EMTr][OAc]/HO | 350 | 3027 | 1.09 | – |
| [EMIm][OAc] | 350 | 3225 | 1.08 | 1.07 |
| [EMIm][OAc]/HO | 350 | 3043 | 1.07 | 1.07 |
| [EMIm][OBz] | 350 | 3550 | 1.10 | 1.10 |
| [EMIm][OBz]/HO | 350 | 3286 | 1.09 | – |
| [EMTr][OBz] | 350 | 3529 | 1.13 | 1.14 |
| [EMTr][OBz]/HO | 350 | 3271 | 1.10 | – |
| 450 | 3369 | 1.01 | – | |
| 550 | 3502 | 0.90 | – |
| Molecule | Temp. | D(AIMD) | D(FFMD) |
|---|---|---|---|
| / K | / m s | / m s | |
| [EMTr][OAc] | |||
| [EMTr] | 350 | 4.02 | 5.94 |
| [OAc] | 4.79 | 5.35 | |
| [EMTr][OAc]/HO | |||
| [EMTr] | 350 | – | 24.42 |
| [OAc] | – | 22.15 | |
| HO | – | 54.88 | |
| [EMIm][OAc] | |||
| [EMIm] | 350 | 7.09 | 9.26 |
| [OAc] | 6.61 | 6.72 | |
| [EMIm][OAc]/HO | |||
| [EMIm] | 350 | 24.95 | 21.77 |
| [OAc] | 28.10 | 22.42 | |
| HO | 46.88 | 55.97 | |
| [EMIm][OBz] | |||
| [EMIm] | 350 | 10.33 | 3.72 |
| [OBz] | 8.92 | 1.89 | |
| [EMIm][OBz]/HO | |||
| [EMIm] | 350 | – | 16.01 |
| [OBz] | – | 11.42 | |
| HO | – | 42.83 | |
| [EMTr][OBz] | |||
| [EMTr] | 350 | – | 1.77 |
| [OBz] | – | 1.13 | |
| [EMTr][OBz]/HO | |||
| [EMTr] | 350 | 9.41 | 15.30 |
| 450 | 96.73 | 123.40 | |
| 550 | 244.91 | 370.79 | |
| [OBz] | 350 | 11.75 | 10.54 |
| 450 | 97.40 | 102.42 | |
| 550 | 186.37 | 310.49 | |
| HO | 350 | 25.56 | 48.66 |
| 450 | 252.32 | 358.21 | |
| 550 | 562.49 | 102.76 | |
| System | Composition | Sim. Time | Box Size | Density |
|---|---|---|---|---|
| / ps | / pm | / g cm | ||
| AIMD | ||||
| [EMTr][OAc] | 36 [EMTr][OAc] | 30 + 223 | 2121 | 1.072 |
| [EMIm][OBz] | 36 [EMIm][OBz] | 50 + 46 | 2319 | 1.114 |
| [EMTr][OBz] | 27 [EMTr][OBz] | 50 + 103 | 2319 | 1.033 |
| /HO | 81 Water | |||
| FFMD | ||||
| [EMTr][OAc] | 128 [EMTr][OAc] | 10,000 | 3198 | 1.113 |
| [EMTr][OAc] | 81 [EMTr][OBz] | 10,000 | 3027 | 1.092 |
| /HO | 243 Water | |||
| [EMIm][OBz] | 128 [EMIm][OBz] | 10,000 | 3550 | 1.103 |
| [EMIm][OBz] | 81 [EMTr][OBz] | 10,000 | 3286 | 1.086 |
| /HO | 243 Water | |||
| [EMTr][OBz] | 128 [EMTr][OBz] | 10,000 | 3529 | 1.128 |
| [EMTr][OBz] | 81 [EMTr][OBz] | 10,000 | 3271 | 1.105 |
| /HO | 243 Water |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roos, E.; Sebastiani, D.; Brehm, M. BILFF: All-Atom Force Field for Modeling Triazolium- and Benzoate-Based Ionic Liquids. Molecules 2023, 28, 7592. https://doi.org/10.3390/molecules28227592
Roos E, Sebastiani D, Brehm M. BILFF: All-Atom Force Field for Modeling Triazolium- and Benzoate-Based Ionic Liquids. Molecules. 2023; 28(22):7592. https://doi.org/10.3390/molecules28227592
Chicago/Turabian StyleRoos, Eliane, Daniel Sebastiani, and Martin Brehm. 2023. "BILFF: All-Atom Force Field for Modeling Triazolium- and Benzoate-Based Ionic Liquids" Molecules 28, no. 22: 7592. https://doi.org/10.3390/molecules28227592
APA StyleRoos, E., Sebastiani, D., & Brehm, M. (2023). BILFF: All-Atom Force Field for Modeling Triazolium- and Benzoate-Based Ionic Liquids. Molecules, 28(22), 7592. https://doi.org/10.3390/molecules28227592