
Computational Design of Ni6@Pt1M31 Clusters for Multifunctional Electrocatalysts

Abstract
:
1. Introduction
2. Results and Discussion
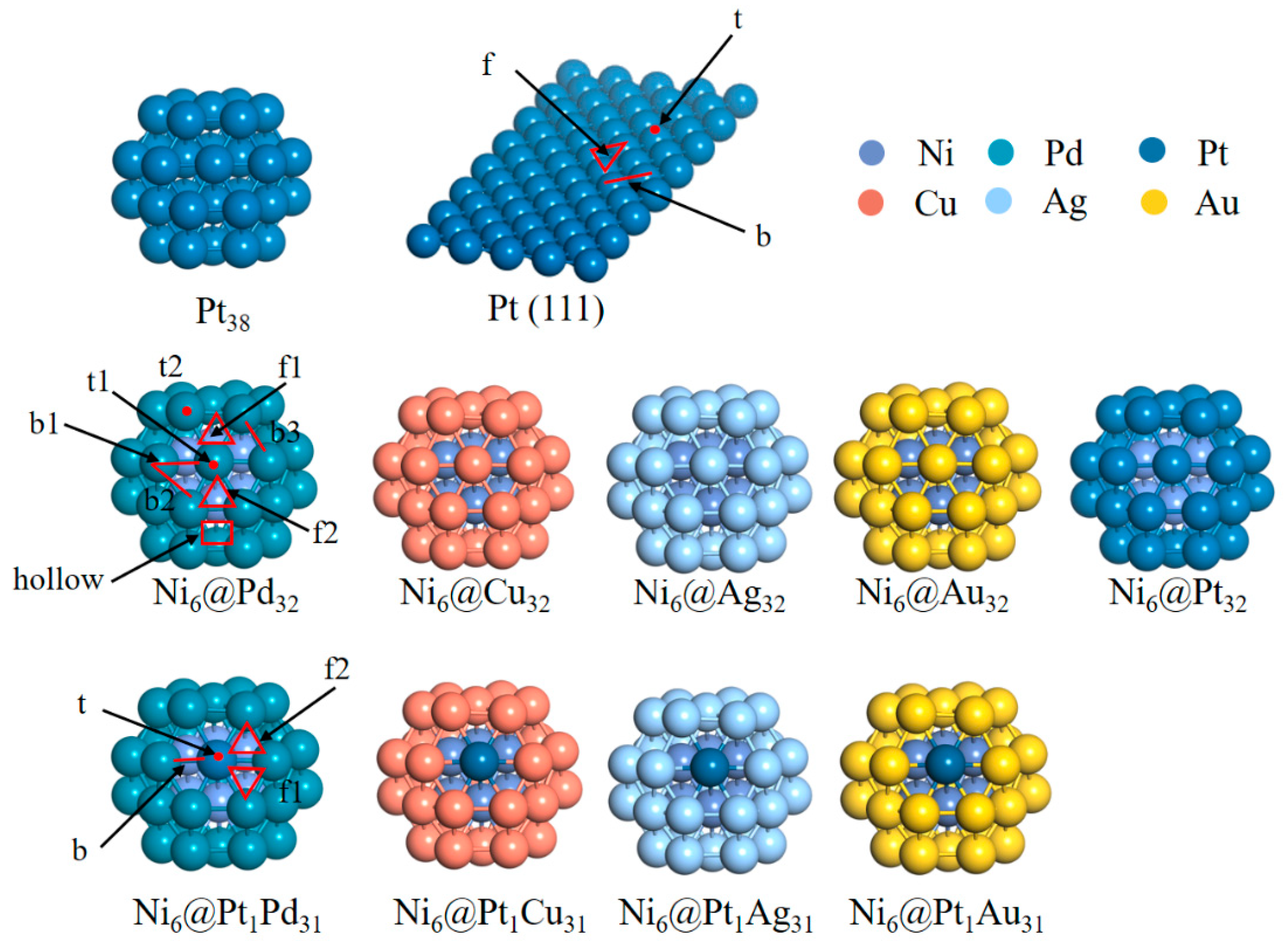
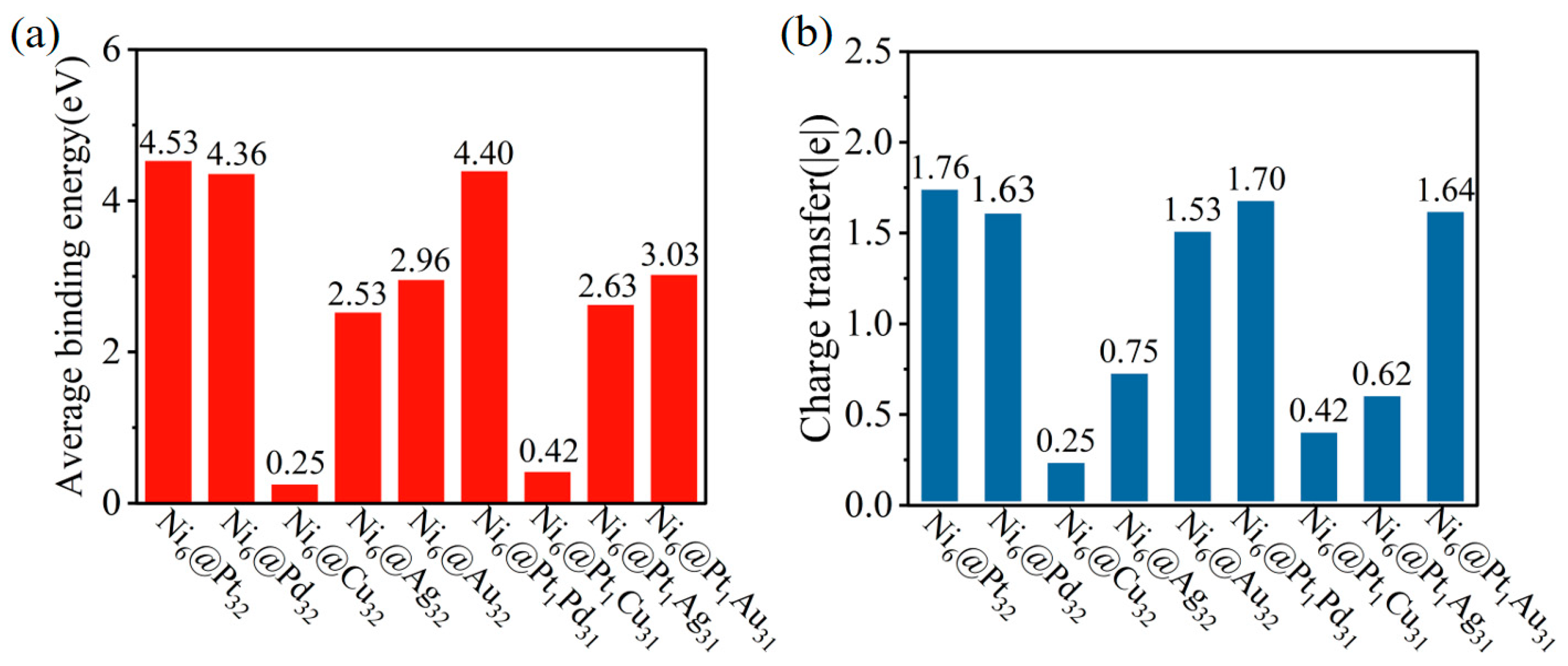
2.1. Structure and Stability Analysis
2.2. Hydrogen Evolution Reaction (HER) Catalytic Activity
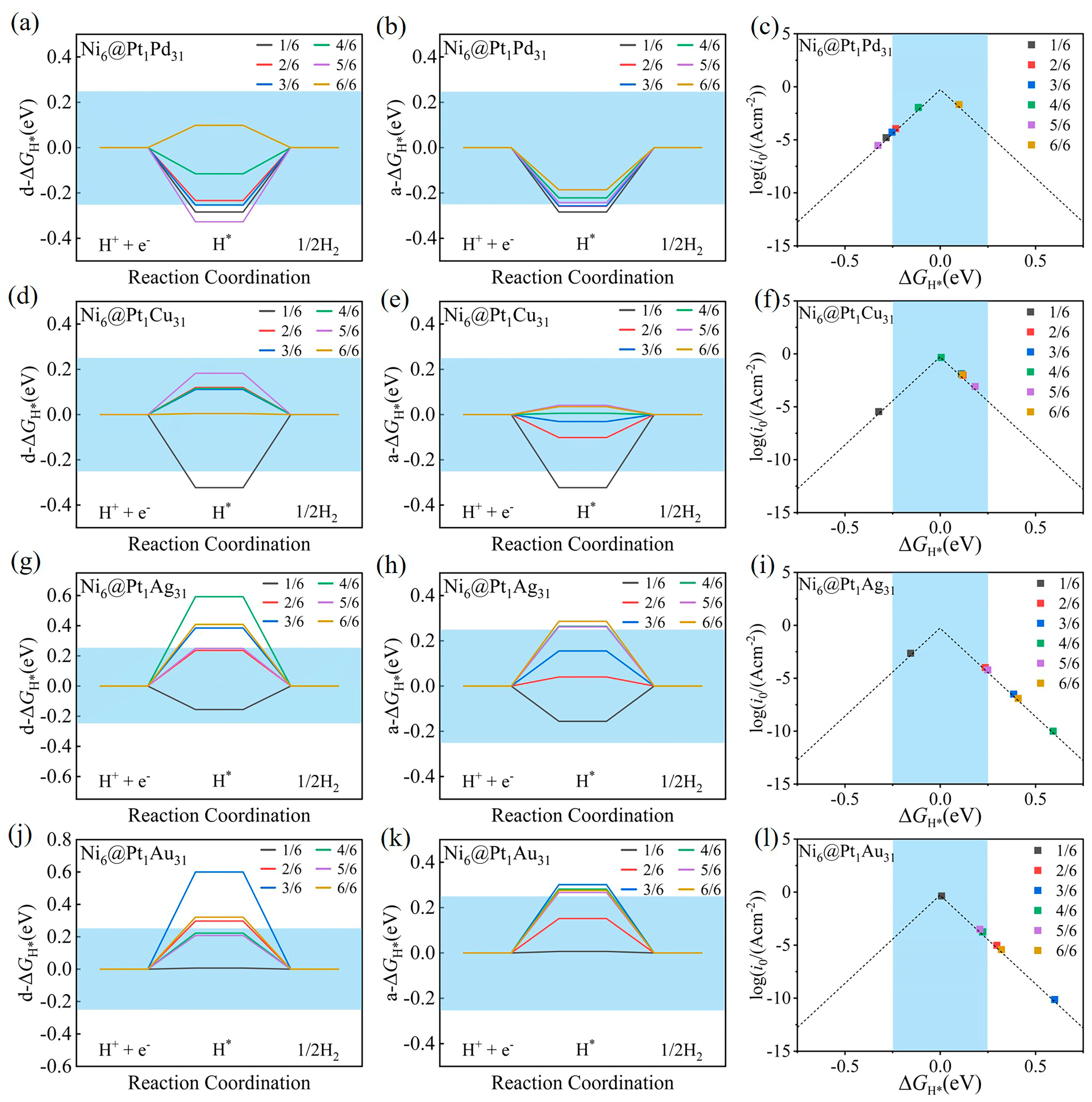
2.2.1. The Hydrogen-Adsorption Gibbs Free Energies of Ni6@M32 and Ni6@Pt1M31
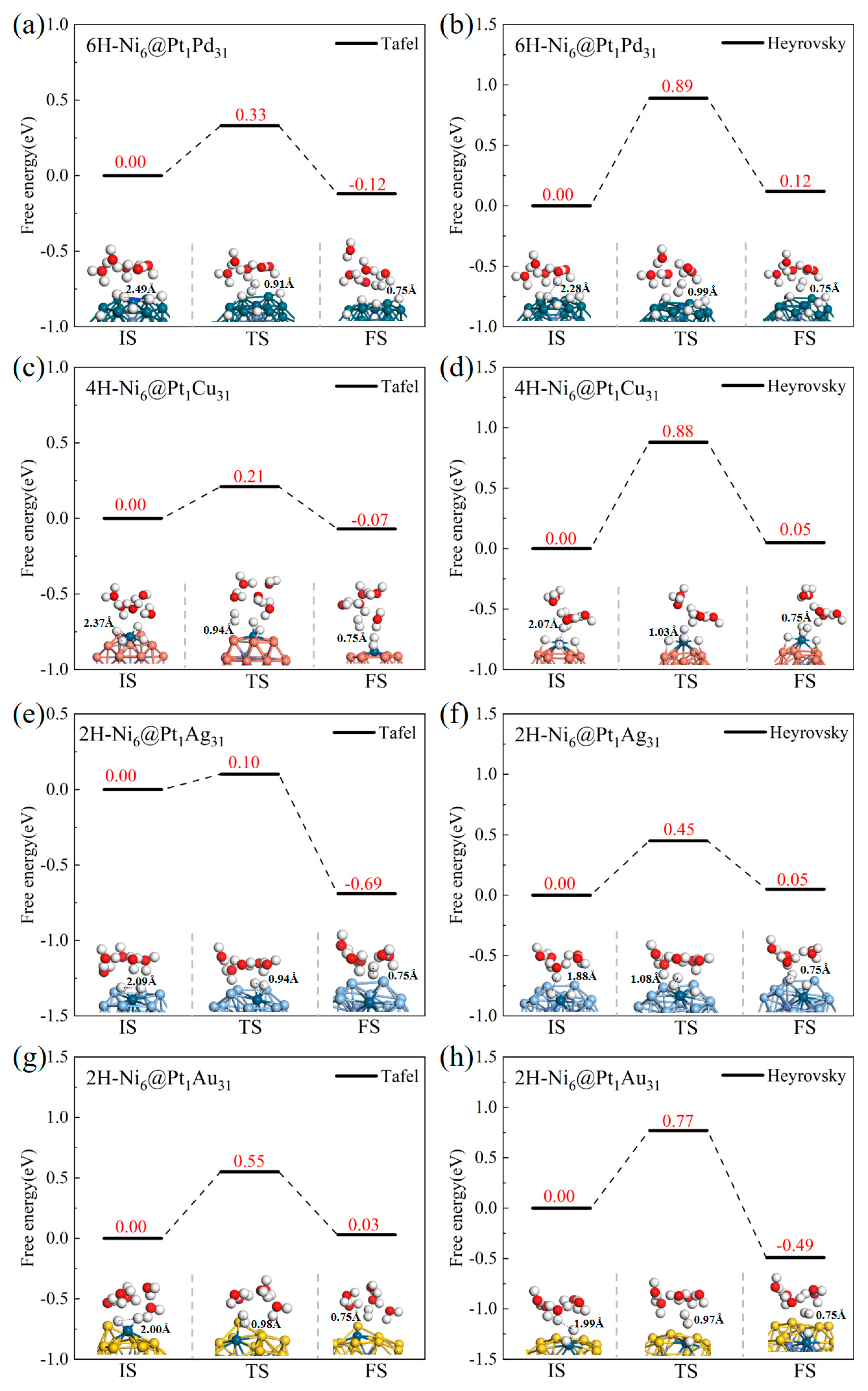
2.2.2. The Energy Barriers of the Tafel and Heyrovsky Processes
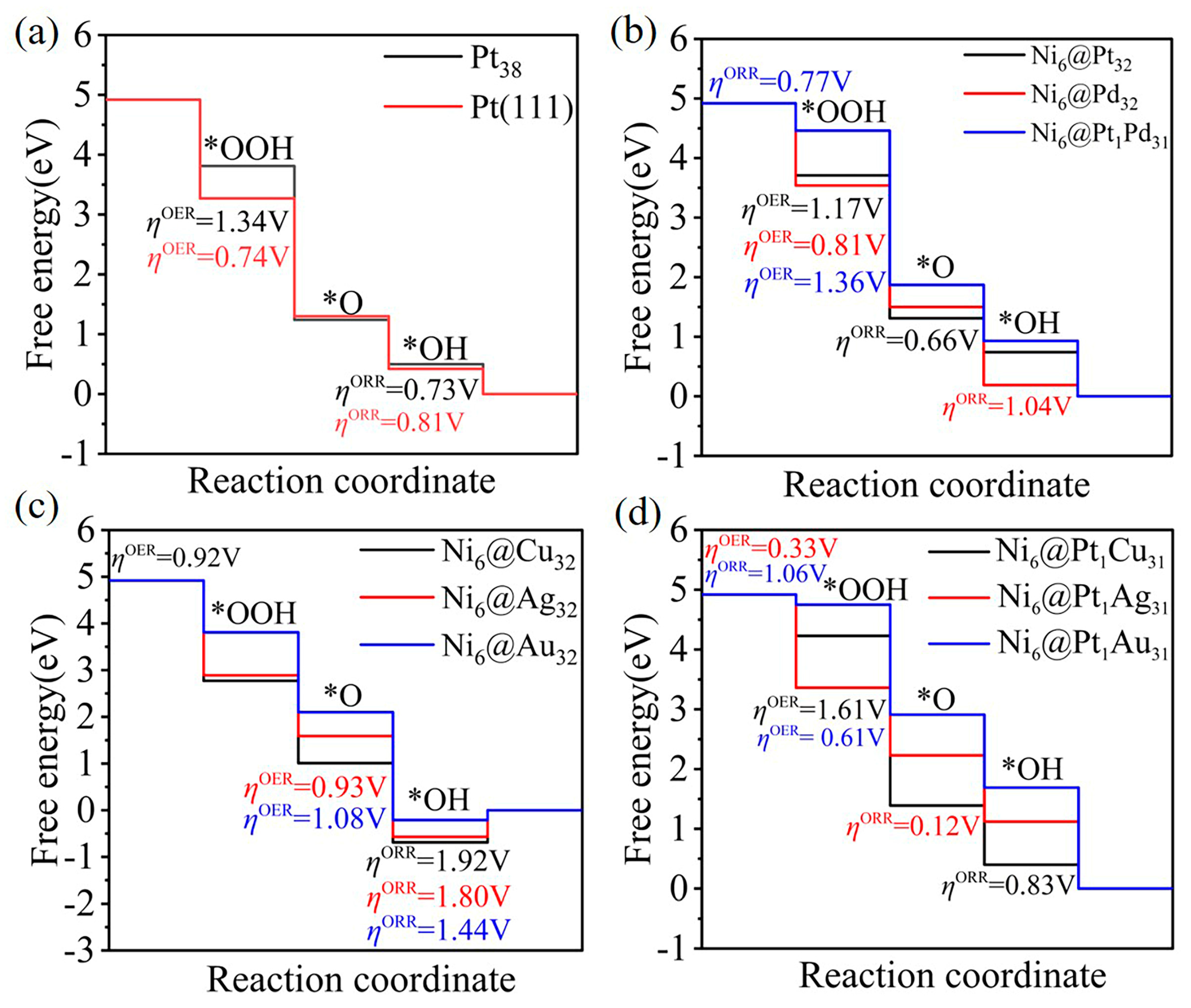
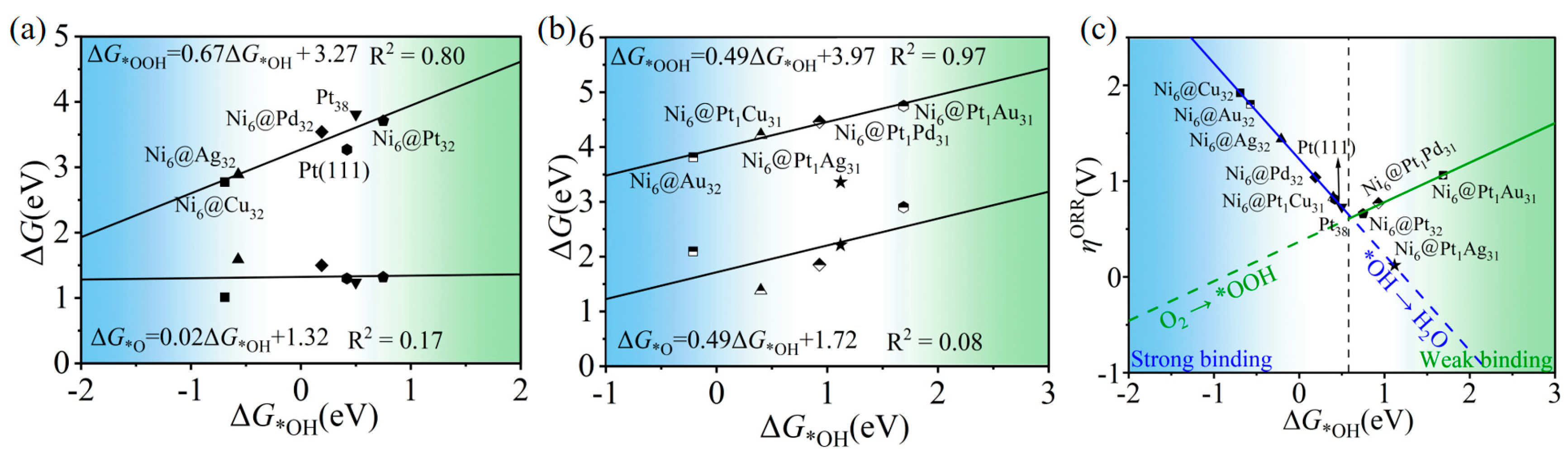
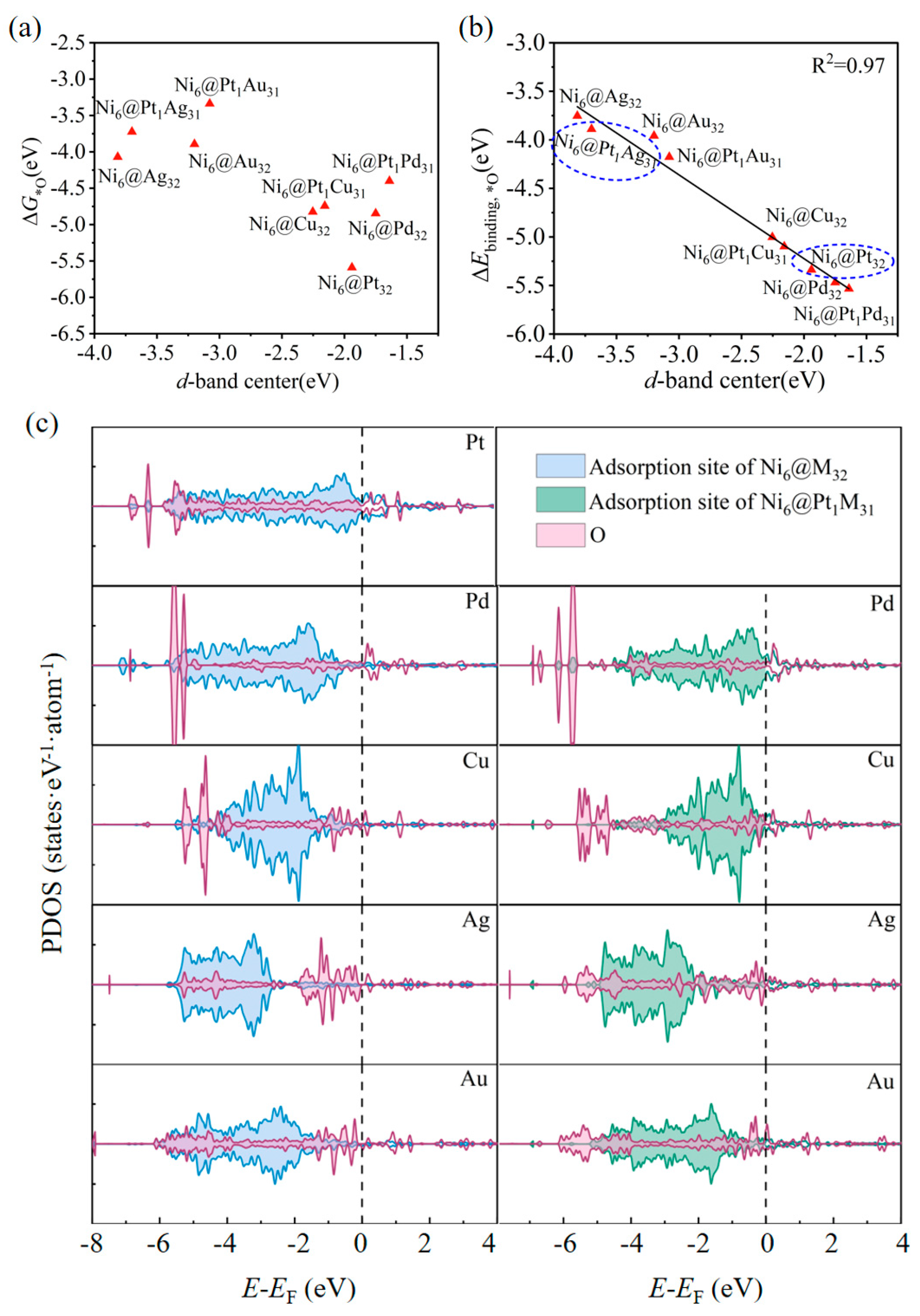
2.3. The Oxygen Reduction Reaction (ORR)/Oxygen Evolution Reaction (OER): Catalytic Activity
3. Computational Details
3.1. Computational Methods
3.2. Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gabbasa, M.; Sopian, K.; Fudholi, A.; Asim, N. A Review of Unitized Regenerative Fuel Cell Stack: Material, Design and Research Achievements. Int. J. Hydrogen Energy 2014, 39, 17765–17778. [Google Scholar] [CrossRef]
- Gewirth, A.A.; Varnell, J.A.; DiAscro, A.M. Nonprecious Metal Catalysts for Oxygen Reduction in Heterogeneous Aqueous Systems. Chem. Rev. 2018, 118, 2313–2339. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, H.X.; Wang, Z.L.; Meng, F.L.; Zhang, X.B. Integrated Three-Dimensional Carbon Paper/Carbon Tubes/Cobalt-Sulfide Sheets as an Efficient Electrode for Overall Water Splitting. ACS Nano 2016, 10, 2342–2348. [Google Scholar] [CrossRef] [PubMed]
- Reier, T.; Oezaslan, M.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials. ACS Catal. 2012, 2, 1765–1772. [Google Scholar] [CrossRef]
- Suen, N.T.; Hung, S.F.; Quan, Q.; Zhang, N.; Xu, Y.J.; Chen, H.M. Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Stoerzinger, K.A.; Diaz-Morales, O.; Kolb, M.; Rao, R.R.; Frydendal, R.; Qiao, L.; Wang, X.R.; Halck, N.B.; Rossmeisl, J.; Hansen, H.A.; et al. Orientation-Dependent Oxygen Evolution on RuO2 without Lattice Exchange. ACS Energy Lett. 2017, 2, 876–881. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, X.; Su, Y.Q.; Wang, L.; Wang, H.; Dang, D.; Chi, B.; Liu, H.; Hensen, E.J.M.; Lou, X.W.D.; et al. Engineering Bunched Pt-Ni Alloy Nanocages for Efficient Oxygen Reduction in Practical Fuel Cells. Science 2019, 366, 850–856. [Google Scholar] [CrossRef]
- Sealy, C. The Problem with Platinum. Mater. Today 2008, 11, 65–68. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of Electrocatalysts for Oxygen- and Hydrogen-Involving Energy Conversion Reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Norskov, J.K. Materials for Solar Fuels and Chemicals. Nat. Mater. 2016, 16, 70–81. [Google Scholar] [CrossRef]
- Anantharaj, S.; Ede, S.R.; Sakthikumar, K.; Karthick, K.; Mishra, S.; Kundu, S. Recent Trends and Perspectives in Electrochemical Water Splitting with an Emphasis on Sulfide, Selenide, and Phosphide Catalysts of Fe, Co, and Ni: A Review. ACS Catal. 2016, 6, 8069–8097. [Google Scholar] [CrossRef]
- Reier, T.; Nong, H.N.; Teschner, D.; Schlögl, R.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction in Acidic Environments—Reaction Mechanisms and Catalysts. Adv. Energy Mater. 2017, 7, 1601275. [Google Scholar] [CrossRef]
- Yu, F.Y.; Lang, Z.L.; Yin, L.Y.; Feng, K.; Xia, Y.J.; Tan, H.Q.; Zhu, H.T.; Zhong, J.; Kang, Z.H.; Li, Y.G. Pt-O Bond as an Active Site Superior to Pt0 in Hydrogen Evolution Reaction. Nat. Commun. 2020, 11, 490. [Google Scholar] [CrossRef]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Norskov, J.K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- Yang, H. Platinum-Based Electrocatalysts with Core-Shell Nanostructures. Angew. Chem. Int. Ed. Engl. 2011, 50, 2674–2676. [Google Scholar] [CrossRef]
- Zhu, B.; Lu, J.; Sakaki, S. Catalysis of Core-Shell Nanoparticle M@Pt (M = Co and Ni) for Oxygen Reduction Reaction and Its Electronic Structure in Comparison to Pt Nanoparticle. J. Catal. 2021, 397, 13–26. [Google Scholar] [CrossRef]
- Park, H.U.; Park, A.H.; Shi, W.; Park, G.G.; Kwon, Y.U. Ternary Core-Shell PdM@Pt (M = Mn and Fe) Nanoparticle Electrocatalysts with Enhanced ORR Catalytic Properties. Ultrason. Sonochem. 2019, 58, 104673. [Google Scholar] [CrossRef]
- Xiong, Y.; Xiao, L.; Yang, Y.; DiSalvo, F.J.; Abruña, H.D. High-Loading Intermetallic Pt3Co/C Core–Shell Nanoparticles as Enhanced Activity Electrocatalysts toward the Oxygen Reduction Reaction (ORR). Chem. Materials 2018, 30, 1532–1539. [Google Scholar]
- Ball, S.C.; Burton, S.L.; Fisher, J.; O’Malley, R.; Tessier, B.C.; Theobald, B.; Thompsett, D.; Zhou, W.-P.; Su, D.; Zhu, Y.; et al. Structure and Activity of Novel Pt Core-Shell Catalysts for the Oxygen Reduction Reaction. ECS Trans. 2009, 25, 1023–1036. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Liang, Z.-X.; Chen, S.-L. Synthesis and Electrocatalytic Properties of Ni-Pd/C Catalysts with Pd-Enriched Surface. J. Electrochem. 2009, 15, 371–376. [Google Scholar] [CrossRef]
- Lin, Y.; Pan, Y.; Liu, S.; Sun, K.; Cheng, Y.; Liu, M.; Wang, Z.; Li, X.; Zhang, J. Construction of Multi-Dimensional Core/Shell Ni/Nicop Nano-Heterojunction for Efficient Electrocatalytic Water Splitting. Appl. Catal. B 2019, 259, 118039. [Google Scholar] [CrossRef]
- Ma, W.; Xie, M.; Xie, S.; Wei, L.; Cai, Y.; Zhang, Q.; Wang, Y. Nickel and Indium Core-Shell co-Catalysts Loaded Silicon Nanowire Arrays for Efficient Photoelectrocatalytic Reduction of CO2 to Formate. J. Energy Chem. 2021, 54, 422–428. [Google Scholar] [CrossRef]
- Seok, S.; Choi, M.; Lee, Y.; Jang, D.; Shin, Y.; Kim, Y.-H.; Jo, C.; Park, S. Ni Nanoparticles on Ni Core/N-Doped Carbon Shell Heterostructures for Electrocatalytic Oxygen Evolution. ACS Appl. Nano Mater. 2021, 4, 9418–9429. [Google Scholar] [CrossRef]
- Jiang, J.; Gao, H.; Lu, S.; Zhang, X.; Wang, C.-Y.; Wang, W.-K.; Yu, H.-Q. Ni–Pd Core–Shell Nanoparticles with Pt-Like Oxygen Reduction Electrocatalytic Performance in Both Acidic and Alkaline Electrolytes. J. Mater. Chem. A 2017, 5, 9233–9240. [Google Scholar] [CrossRef]
- Leteba, G.M.; Mitchell, D.R.G.; Levecque, P.B.J.; van Steen, E.; Lang, C.I. Topographical and Compositional Engineering of Core-Shell Ni@Pt ORR Electro-Catalysts. RSC Adv. 2020, 10, 29268–29277. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.S.; Pathak, B. Computational Screening for ORR Activity of 3d Transition Metal Based M@Pt Core–Shell Clusters. J. Phys. Chem. C 2019, 123, 3634–3644. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-Atom Catalysis of Co Oxidation Using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Javed, M.S.; Zhang, X.; Ali, S.; Mateen, A.; Idrees, M.; Sajjad, M.; Batool, S.; Ahmad, A.; Imran, M.; Najam, T.; et al. Heterostructured bimetallic–sulfide@ layered Ti3C2Tx–MXene as a synergistic electrode to realize high-energy-density aqueous hybrid-supercapacitor. Nano Energy 2022, 101, 107624. [Google Scholar] [CrossRef]
- Shah, S.S.A.; Najam, T.; Bashir, M.S.; Peng, L.; Nazir, M.A.; Javed, M.S. Single-atom catalysts for next-generation rechargeable batteries and fuel cells. Energy Storage Mater. 2022, 45, 301–322. [Google Scholar] [CrossRef]
- Najam, T.; Shah SS, A.; Javed, M.S.; Chen, P.T.; Chuang, C.; Saad, A.; Cai, X. Modulating the electronic structure of zinc single atom catalyst by P/N coordination and Co2P supports for efficient oxygen reduction in Zn-Air battery. Chem. Eng. J. 2022, 440, 135928. [Google Scholar] [CrossRef]
- Talib, S.H.; Lu, Z.; Yu, X.; Ahmad, K.; Bashir, B.; Yang, Z.; Li, J. Theoretical Inspection of M1/PMA Single-Atom Electrocatalyst: Ultra-High Performance for Water Splitting (HER/OER) and Oxygen Reduction Reactions (OER). ACS Catal. 2021, 11, 8929–8941. [Google Scholar] [CrossRef]
- Zhao, C.; Tang, Y.; Yu, C.; Tan, X.; Banis, M.N.; Li, S.; Wan, G.; Huang, H.; Zhang, L.; Yang, H.; et al. Insights into the Electronic Origin of Enhancing the Catalytic Activity of Co3o4 for Oxygen Evolution by Single Atom Ruthenium. Nano Today 2020, 34, 100955. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Xi, L.; Yu, Y.; Chen, N.; Sun, S.; Wang, W.; Lange, K.M.; Zhang, B. Single-Atom Au/NiFe Layered Double Hydroxide Electrocatalyst: Probing the Origin of Activity for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2018, 140, 3876–3879. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, P. Hydrogénations et déshydrogénations par catalyse. Berichte Dtsch. Chem. Ges. 1911, 44, 1984–2001. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of Water on Oxide Surfaces. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Wigner, E.P. On the Quantum Correction for Thermodynamic Equilibrium. Phys. Rev. 1932, 40, 749–759. [Google Scholar] [CrossRef]
- Nilsson, A.; Pettersson, L.G.M.; Hammer, B.; Bligaard, T.; Christensen, C.H.; Norskov, J.K. The Electronic Structure Effect in Heterogeneous Catalysis. Catal. Lett. 2005, 100, 111–114. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B Condens. Matter. 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys. Rev. B Condens. Matter. 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 1986, 33, 8800–8802. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Gennero de Chialvo, M.R.; Chialvo, A.C. Kinetics of Hydrogen Evolution Reaction with Frumkin Adsorption: Re-Examination of the Volmer–Heyrovsky and Volmer–Tafel Routes. Electrochim. Acta 1998, 44, 841–851. [Google Scholar] [CrossRef]
- de Chialvo, M.R.G.; Chialvo, A.C. Hydrogen Evolution Reaction: Analysis of the Volmer-Heyrovsky-Tafel Mechanism with a Generalized Adsorption Model. Eelectroanal. Chem. 1994, 372, 209–223. [Google Scholar] [CrossRef]
- Gopalakrishnan, D.; Lee, A.; Thangavel, N.K.; Reddy Arava, L.M. Facile Synthesis of Electrocatalytically Active NbS2 Nanoflakes for an Enhanced Hydrogen Evolution Reaction (HER). Sustain. Energ. Fuels 2018, 2, 96–102. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Coordination | Ef/eV |
|---|---|
| Ni6@Pd32 + Pt1 → Ni6@Pt1Pd31 + Pd1 | −0.56 |
| Ni6@Cu32 + Pt1 → Ni6@Pt1Cu31 + Cu1 | −0.08 |
| Ni6@Ag32 + Pt1 → Ni6@Pt1Ag31 + Ag1 | −0.51 |
| Ni6@Au32 + Pt1 → Ni6@Pt1Au31 + Au1 | 0.33 |
| System | ΔGTafel≠/eV | Tafel (k/s) | ΔGHeyrovsky≠/eV | Heyrovsky (k/s) |
|---|---|---|---|---|
| 6H-Ni6@Pt32 | 0.55 | 4.35 × 103 | 0.43 | 3.92 × 105 |
| 6H-Ni6@Pd32 | 0.66 | 54.4 | 0.54 | 4.78 × 103 |
| 2H-Ni6@Cu32 | 0.78 | 0.72 | 0.14 | 3.17 × 1010 |
| 3H-Ni6@Ag32 | 1.06 | 1.84 × 10−5 | 0.70 | 9.84 |
| 6H-Ni6@Pt1Pd31 | 0.33 | 1.76 × 107 | 0.89 | 6.17 × 103 |
| 4H-Ni6@Pt1Cu31 | 0.21 | 1.84 × 109 | 0.88 | 9.25 × 10−3 |
| 2H-Ni6@Pt1Ag31 | 0.10 | 1.83 × 1011 | 0.45 | 1.69 × 105 |
| 2H-Ni6@Pt1Au31 | 0.55 | 3.27 × 103 | 0.77 | 0.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, J.; Tian, D. Computational Design of Ni6@Pt1M31 Clusters for Multifunctional Electrocatalysts. Molecules 2023, 28, 7563. https://doi.org/10.3390/molecules28227563
Jia J, Tian D. Computational Design of Ni6@Pt1M31 Clusters for Multifunctional Electrocatalysts. Molecules. 2023; 28(22):7563. https://doi.org/10.3390/molecules28227563
Chicago/Turabian StyleJia, Jiaojiao, and Dongxu Tian. 2023. "Computational Design of Ni6@Pt1M31 Clusters for Multifunctional Electrocatalysts" Molecules 28, no. 22: 7563. https://doi.org/10.3390/molecules28227563
APA StyleJia, J., & Tian, D. (2023). Computational Design of Ni6@Pt1M31 Clusters for Multifunctional Electrocatalysts. Molecules, 28(22), 7563. https://doi.org/10.3390/molecules28227563