
New Spectroelectrochemical Insights into Manganese and Rhenium Bipyridine Complexes as Catalysts for the Electrochemical Reduction of Carbon Dioxide

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
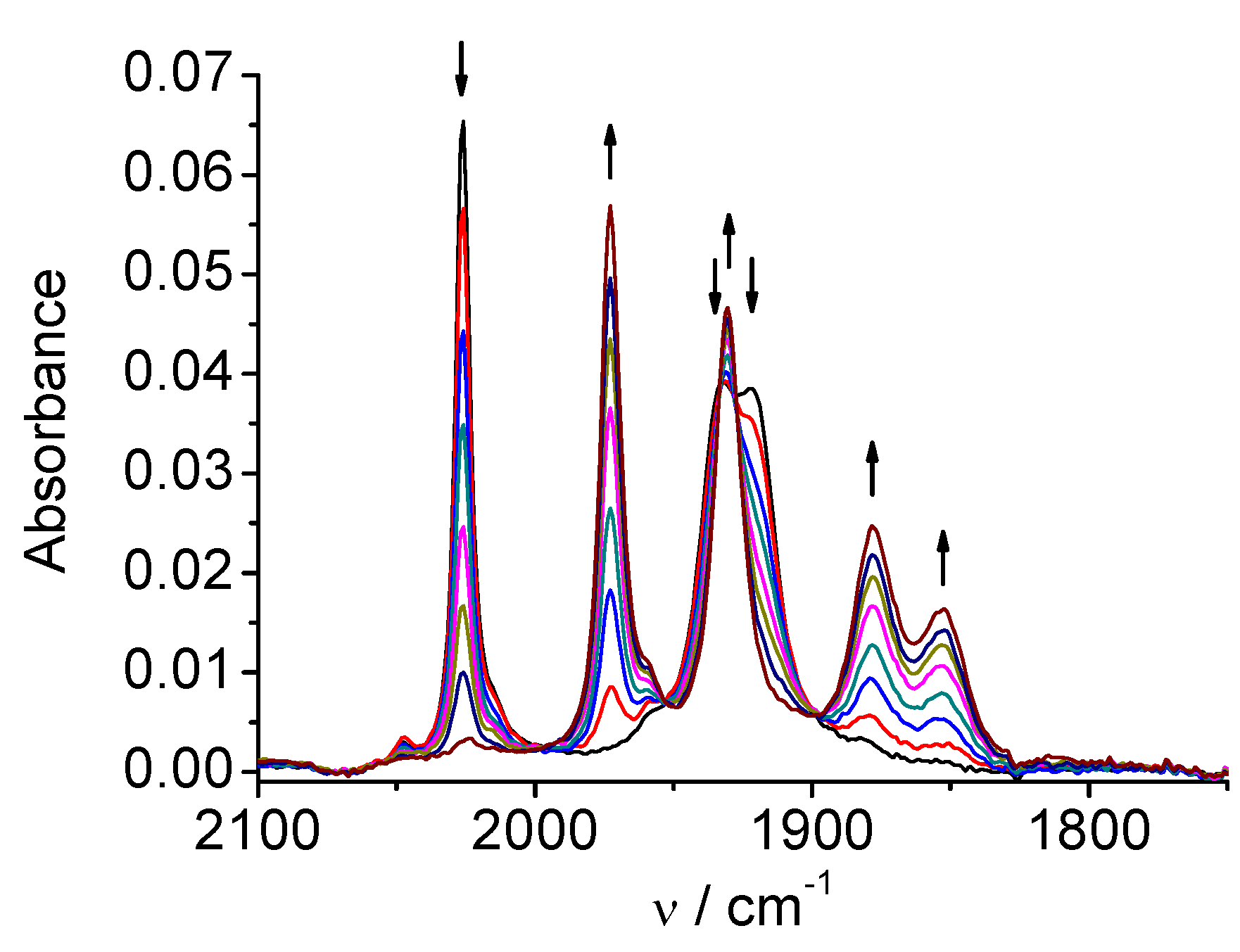
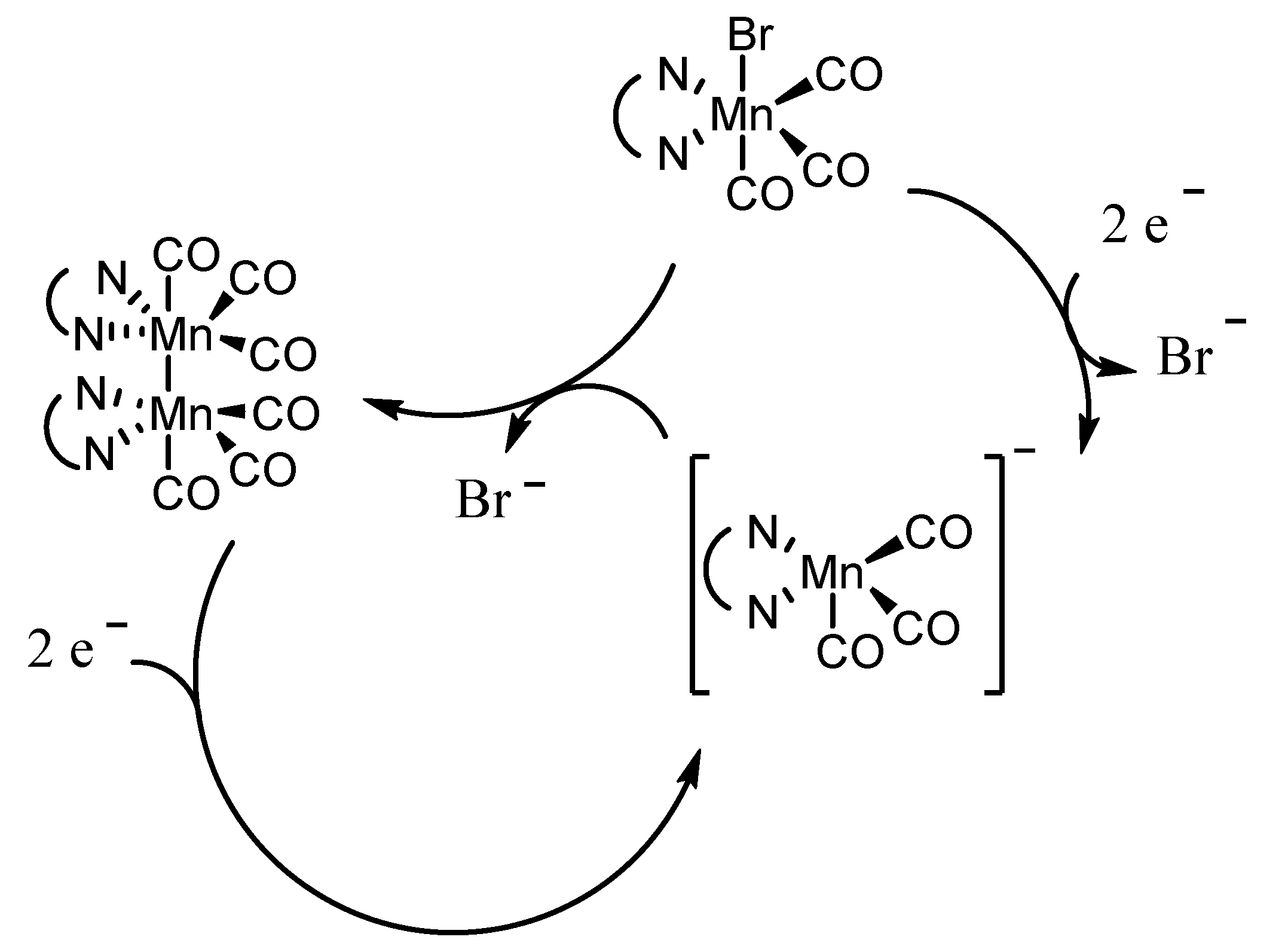
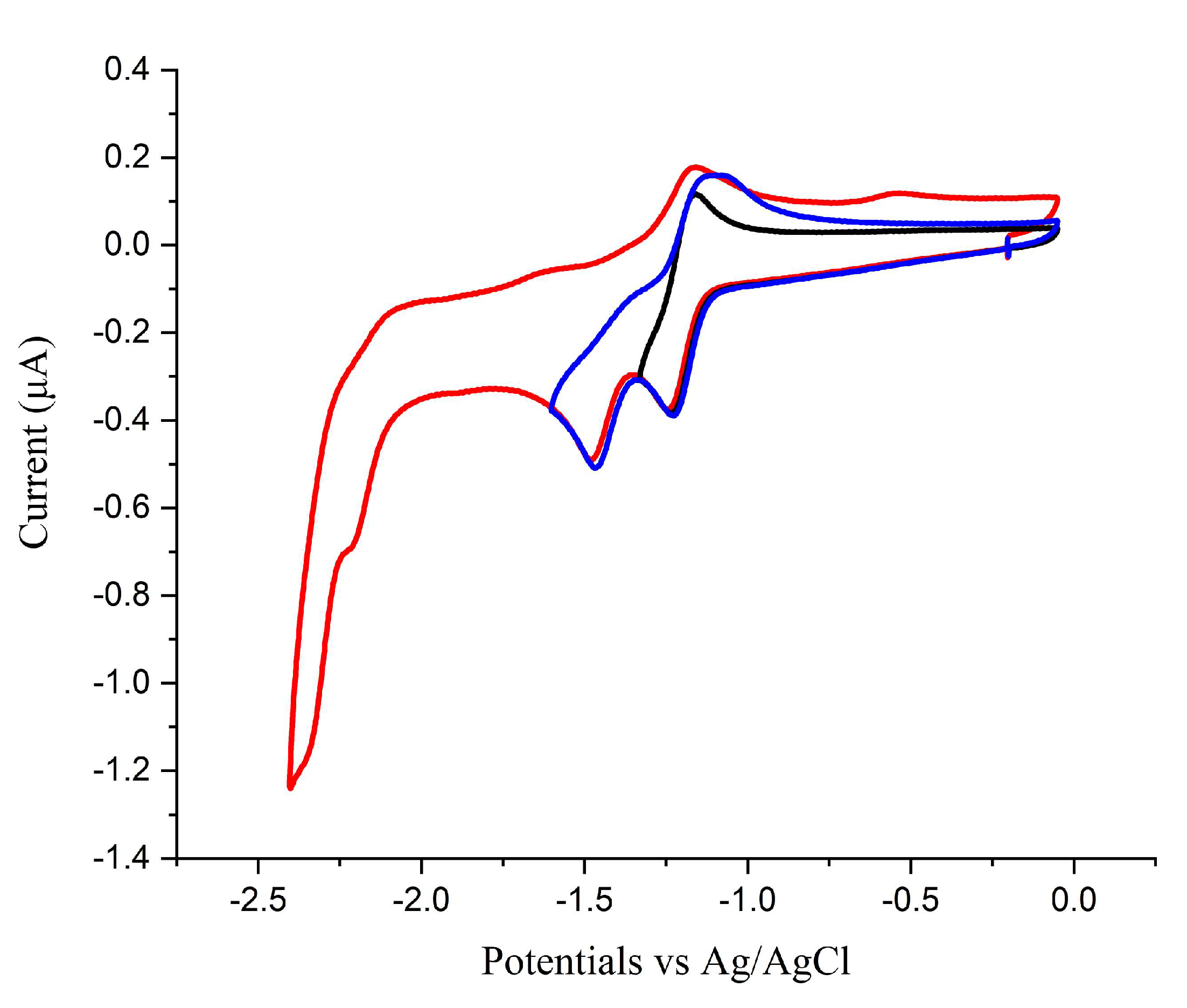
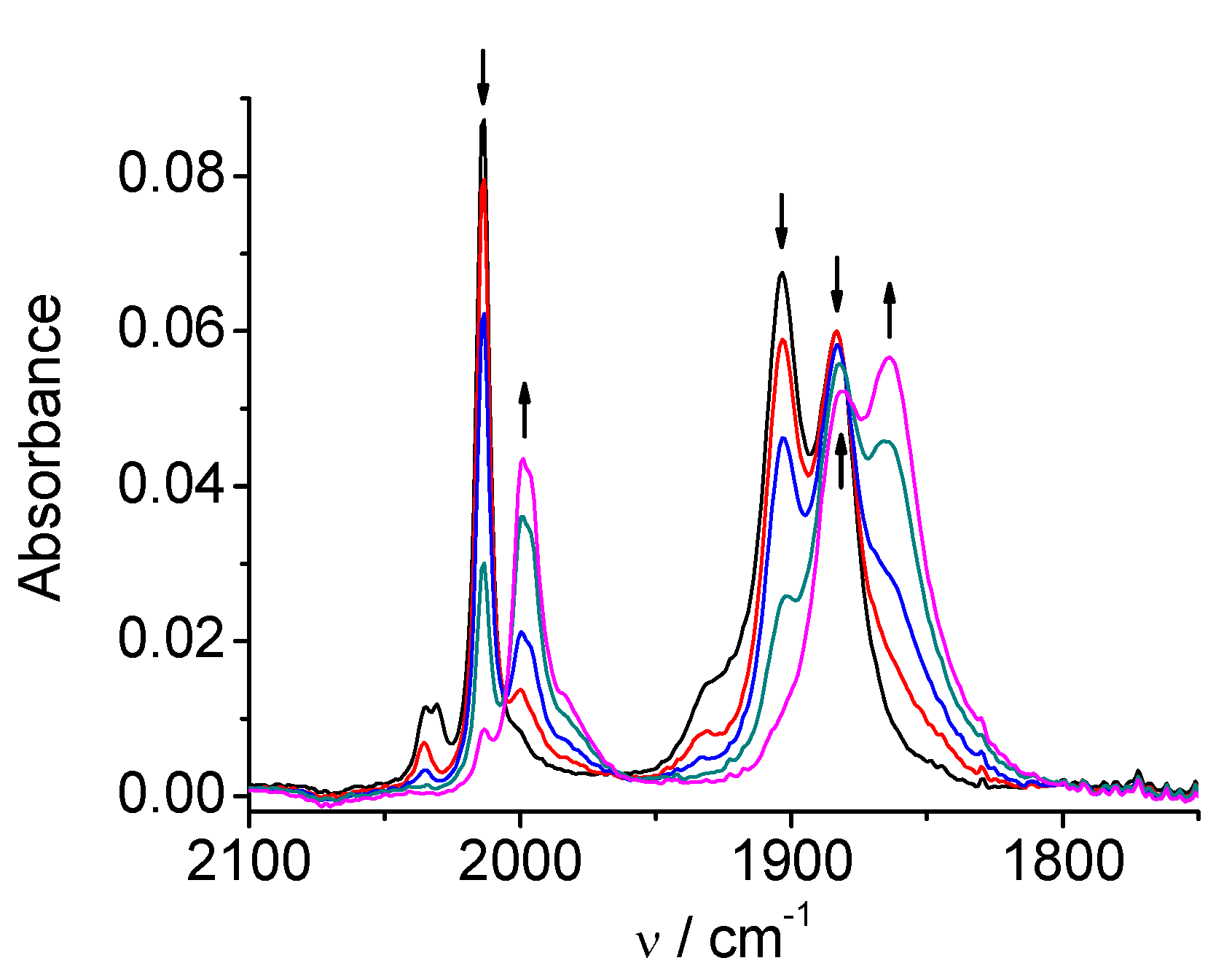
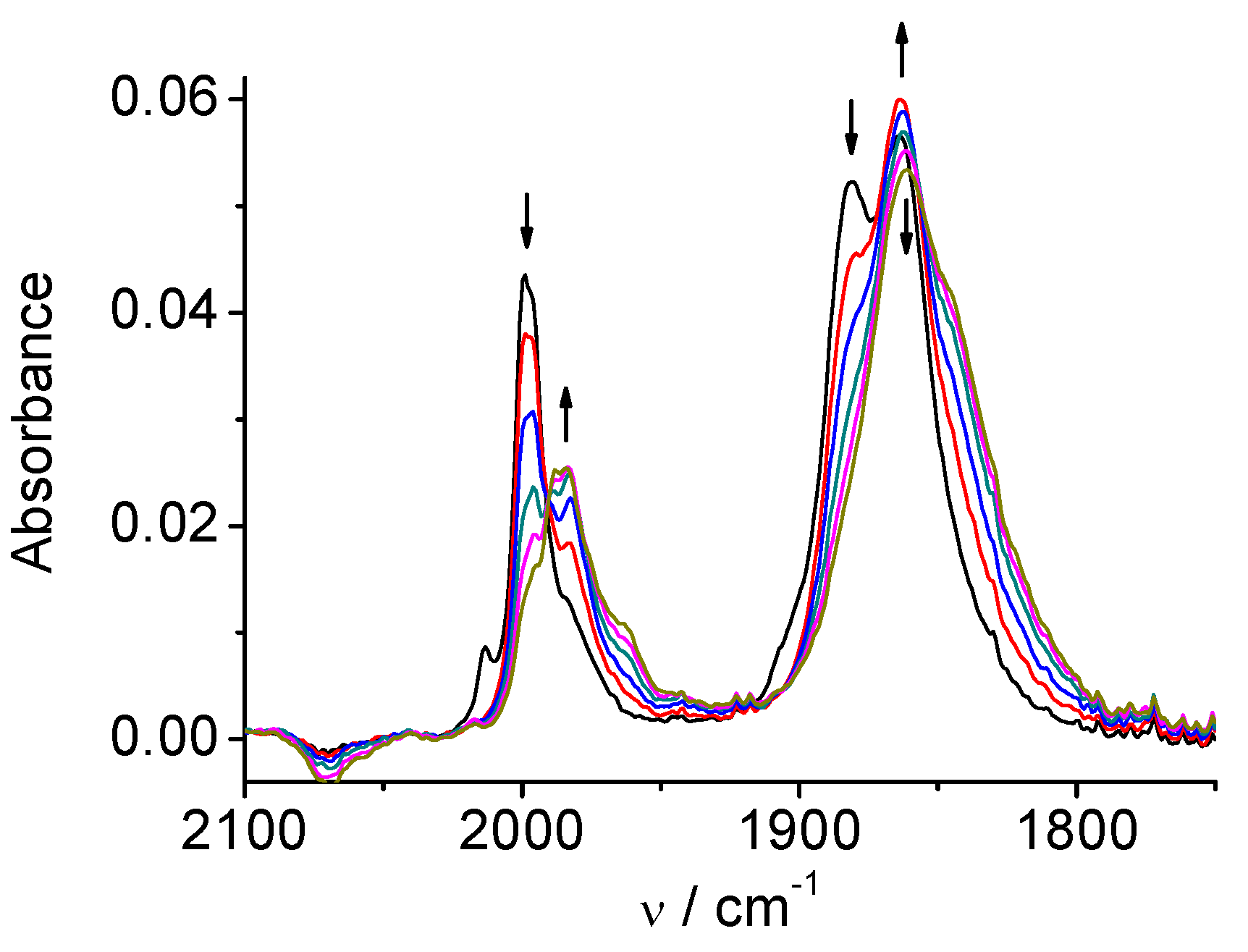
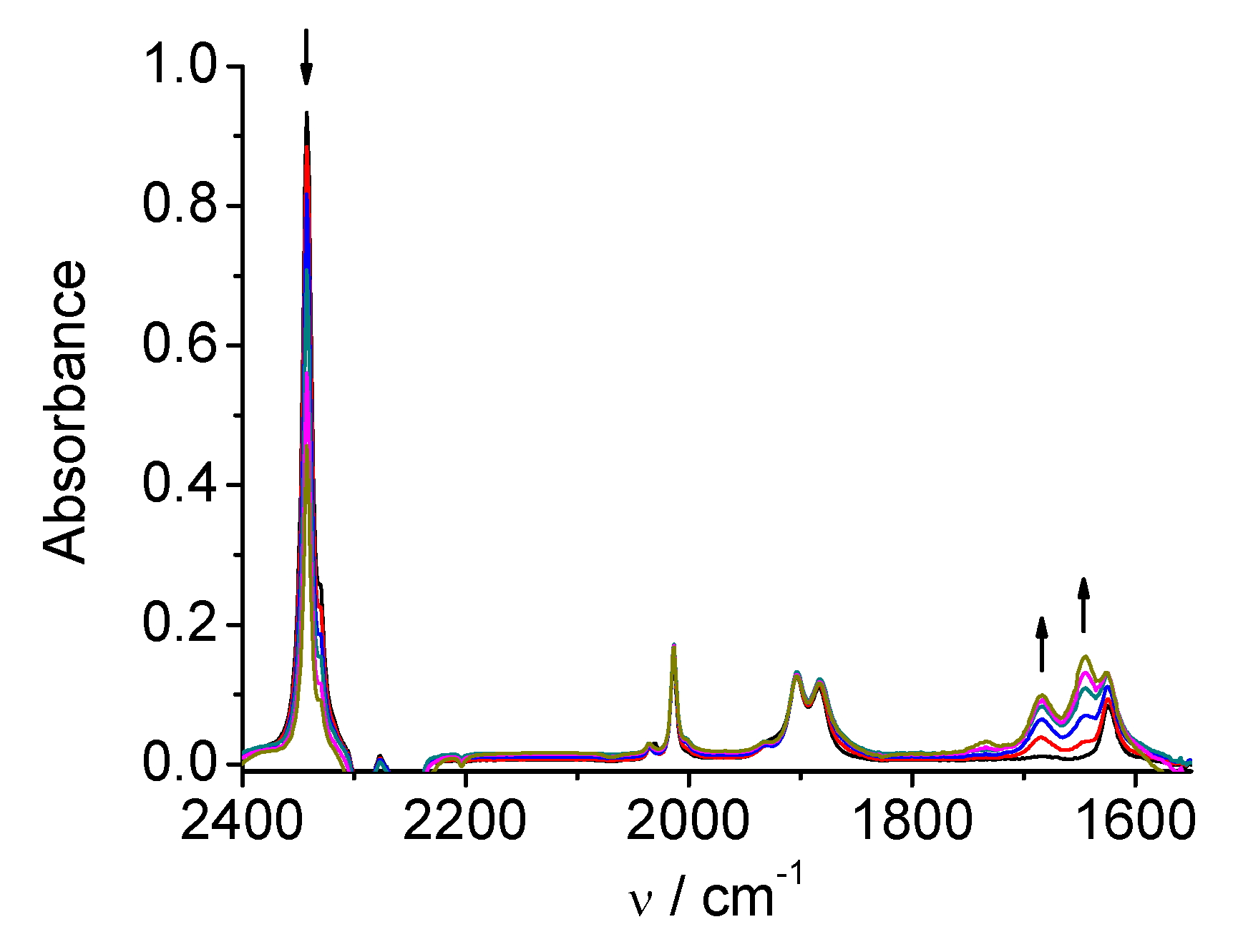
2.1. Mn(apbpy)(CO)3Br
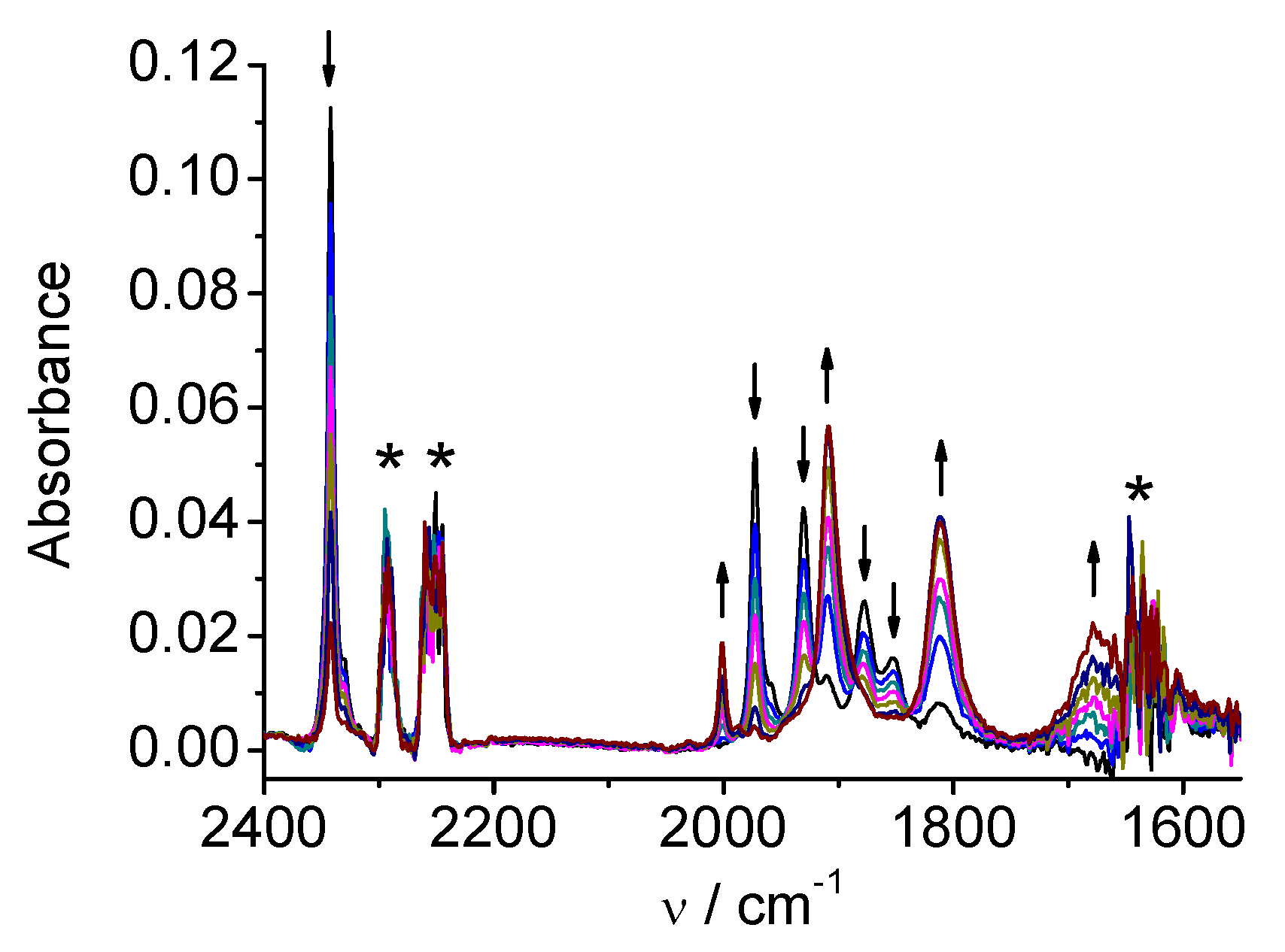
2.2. Re(apbpy)(CO)3Cl
2.3. Mn(bpy-4,-4′-CF3-NMe2)(CO)3Br
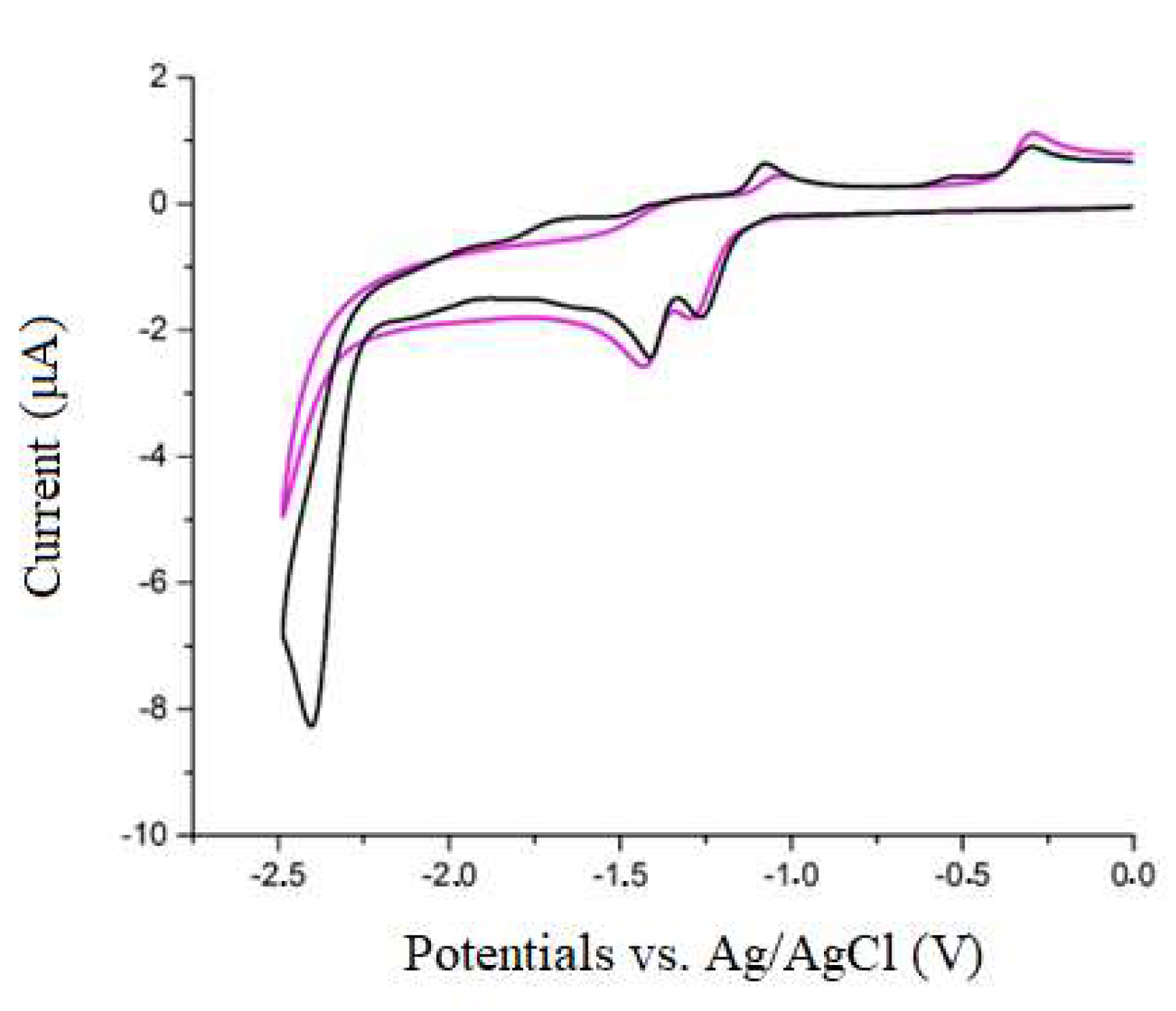
2.4. Re(bpy-4,4′-NMe2)(CO)3Cl
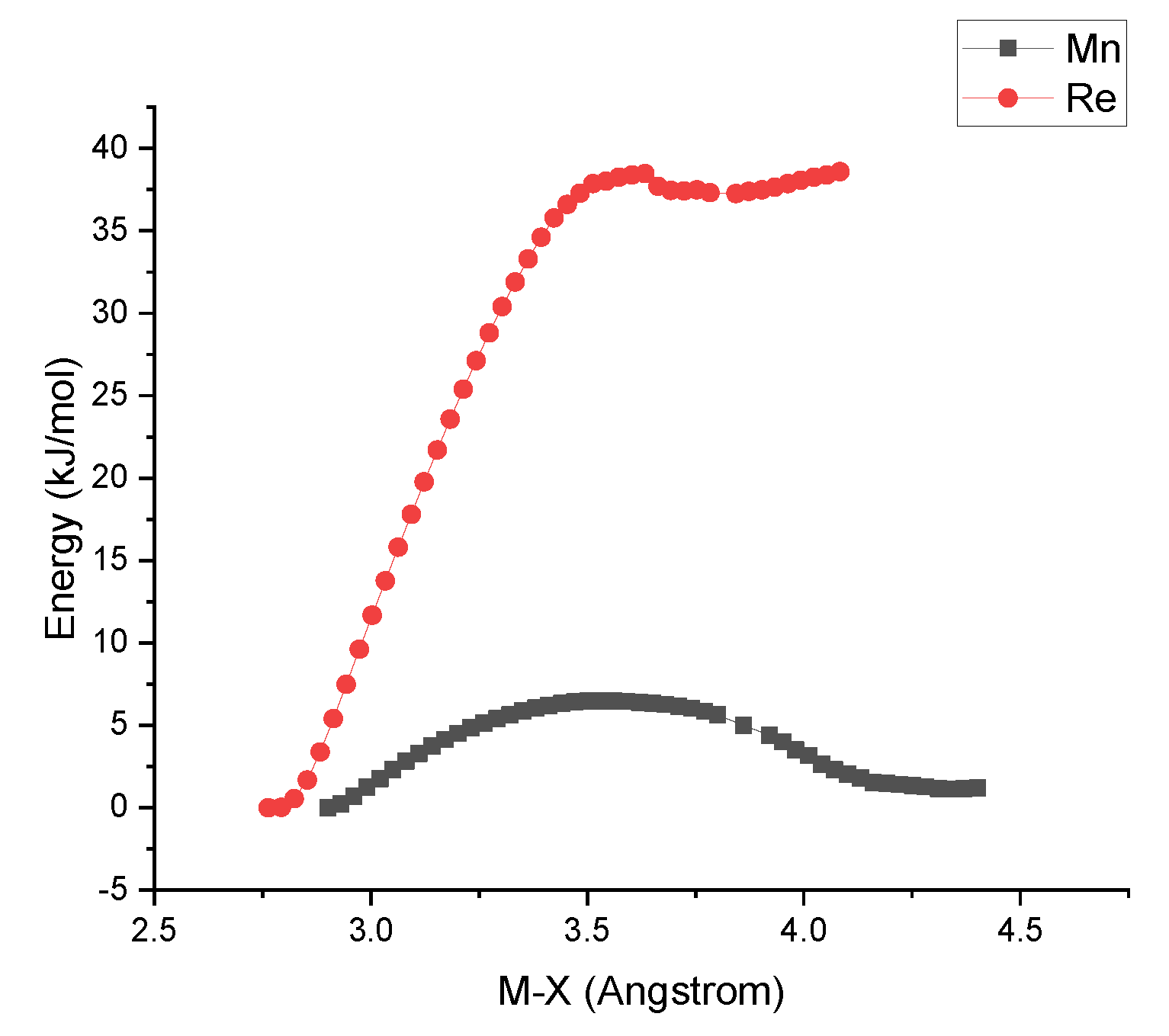
3. Discussion and Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rotundo, L.; Gobetto, R.; Nervi, C. Electrochemical CO2 reduction with earth-abundant metal catalysts. Curr. Opin. Green Sust. Chem. 2021, 31, 100509. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xiao, F.-X. An overview of solar-driven photoelectrochemical CO2 conversion to chemical fuels. ACS Catal. 2022, 12, 9023–9057. [Google Scholar] [CrossRef]
- Cheon, J.; Yang, J.Y.; Koper, M.; Ishitani, O. From Pollutant to Chemical Feedstock: Valorizing Carbon Dioxide through Photo-and Electrochemical Processes. Acc. Chem. Res. 2022, 55, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Jin, B.-K. FTIR Spectroelectrochemistry Study on the Reduction of CO2 at a Gold Electrode Interface. Chin. J. Inorg. Chem. 2012, 28, 541–545. [Google Scholar]
- Hartl, F.; Aarnts, M.P.; Peelen, K. Infrared Spectroelectrochemical Investigation of Carbon Dioxide Reduction Mediated by the Anion [Ru (SnPh3)(Co)2(iPr-DAB)]-(iPr-DAB= N,N′-Diisopropyl-1,4-diaza-1,3-butadiene). Coll. Czech. Chem. Comm. 1996, 61, 1342–1352. [Google Scholar] [CrossRef]
- Breelove, B.K.; Takayama, D.; Ito, T. Synthesis of an electron pooling ligand based on triruthenium clusters and electrochemical reduction of CO2 by its zinc(II)complex. Chem. Lett. 2004, 33, 1624–1625. [Google Scholar] [CrossRef]
- Behnke, S.L.; Manesis, A.C.; Shafaat, H.S. Spectroelectrochemical investigations of nickel cyclam indicate different reaction mechanisms for electrocatalytic CO2 and H+ reduction. Dalton Trans. 2018, 47, 15206–15216. [Google Scholar] [CrossRef]
- Polyansky, D.E.; Grills, D.C.; Ertem, M.Z.; Ngo, K.T.; Fujita, E. Role of Bimetallic Interactions in the Enhancement of Catalytic CO2 Reduction by a Macrocyclic Cobalt Catalyst. ACS Catal. 2022, 12, 1706–1717. [Google Scholar] [CrossRef]
- Johnson, F.P.A.; George, M.W.; Hartl, F.; Turner, J.J. Electrocatalytic Reduction of CO2 Using the Complexes [Re (bpy)(CO)3L] n (n=+ 1, L= P (OEt)3, CH3CN; n= 0, L= Cl-, Otf-; bpy= 2,2‘-Bipyridine; Otf-= CF3SO3) as Catalyst Precursors: Infrared Spectroelectrochemical Investigation. Organometallics 1996, 15, 3374–3387. [Google Scholar] [CrossRef]
- Neri, G.; Donaldsonb, P.M.; Cowan, A.J. In situ study of the low overpotential “dimer pathway” for electrocatalytic carbon dioxide reduction by manganese carbonyl complexes. Phys. Chem. Chem. Phys. 2019, 21, 7389–7397. [Google Scholar] [CrossRef] [PubMed]
- Filippi, J.; Rotundo, L.; Gobetto, R.; Miller, H.A.; Nervi, C.; Lavacchi, A.; Vizza, F. Turning manganese into gold: Efficient electrochemical CO2 reduction by a fac-Mn (apbpy)(CO)3Br complex in a gas–liquid interface flow cell. Chem. Eng. J. 2021, 416, 129050. [Google Scholar] [CrossRef]
- Rotundo, L.; Filippi, J.; Gobetto, R.; Miller, H.A.; Rocca, R.; Nervi, C.; Vizza, F. Electrochemical CO2 reduction in water at carbon cloth electrodes functionalized with a fac-Mn (apbpy)(CO)3 Br complex. Chem. Commun. 2019, 55, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Garino, C.; Priola, E.; Sassone, D.; Rao, H.; Ma, B.; Robert, M.; Fiedler, J.; Gobetto, R.; Nervi, C. Electrochemical and photochemical reduction of CO2 catalyzed by Re(I)complexes carrying local proton sources. Organometallics 2019, 38, 1351–1360. [Google Scholar] [CrossRef]
- Sun, C.; Rotundo, L.; Garino, C.; Nencini, L.; Yoon, S.S.; Gobetto, R.; Nervi, C. Electrochemical CO2 reduction at glassy carbon electrodes functionalized by MnI and ReI organometallic complexes. ChemPhysChem 2017, 18, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Azzi, E.; Deagostino, A.; Garino, C.; Nencini, L.; Priola, E.; Quagliotto, P.; Rocca, R.; Gobetto, R.; Nervi, C. Electronic effects of substituents on fac-M (bpy-R)(CO)3(M = Mn, Re) complexes for homogeneous CO2 electroreduction. Front. Chem. 2019, 7, 417. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn (bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Garino, C.; Minero, C.; Sordello, F.; Nervi, C.; Gobetto, R. Photo-and Electrocatalytic Reduction of CO2 by [Re (CO)3{α,α′-Diimine-(4-piperidinyl-1,8-naphthalimide)}Cl] Complexes. Eur. J. Inorg. Chem. 2015, 2015, 296–304. [Google Scholar] [CrossRef]
- Taylor, J.O.; Neri, G.; Banerji, L.; Cowan, A.J.; Hartl, F. Strong impact of intramolecular hydrogen bonding on the cathodic path of [Re (3,3′-dihydroxy-2, 2′-bipyridine)(CO)3Cl] and catalytic reduction of carbon dioxide. Inorg. Chem. 2020, 59, 5564–5578. [Google Scholar] [CrossRef]
- Madsen, M.R.; Rønne, M.H.; Heuschen, M.; Golo, D.; Ahlquist, M.S.G.; Skrydstrup, T.; Pedersen, S.U.; Daasbjerg, K. Promoting selective generation of formic acid from CO2 using Mn (bpy)(CO)3Br as electrocatalyst and triethylamine/isopropanol as additives. J. Am. Chem. Soc. 2021, 143, 20491–20500. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local proton source in electrocatalytic CO2 reduction with [Mn (bpy–R)(CO)3Br] complexes. Chem. Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef] [PubMed]
- Rønne, M.H.; Cho, D.; Madsen, M.R.; Jakobsen, J.B.; Eom, S.; Escoudé, É.; Hammershøj, H.C.D.; Nielsen, D.U.; Pedersen, S.U.; Baik, M.-H.; et al. Ligand-controlled product selectivity in electrochemical carbon dioxide reduction using manganese bipyridine catalysts. J. Am. Chem. Soc. 2020, 142, 4265–4275. [Google Scholar] [CrossRef] [PubMed]
- Rønne, M.H.; Madsen, M.R.; Skrydstrup, T.; Pedersen, S.U.; Daasbjerg, K. Mechanistic Elucidation of Dimer Formation and Strategies for Its Suppression in Electrochemical Reduction of Fac-Mn (bpy)(CO)3Br. ChemElectroChem 2021, 8, 2108–2114. [Google Scholar] [CrossRef]
- Madsen, M.R.; Jakobsen, J.B.; Rønne, M.H.; Liang, H.; Hammershøj, H.C.D.; Nørby, P.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Evaluation of the electrocatalytic reduction of carbon dioxide using rhenium and ruthenium bipyridine catalysts bearing pendant amines in the secondary coordination sphere. Organometallics 2020, 39, 1480–1490. [Google Scholar] [CrossRef]
- Stor, G.J.; Hartl, F.; van Outersterp, J.W.M.; Stufkens, D.J. Spectroelectrochemical (IR, UV/Vis) Determination of the Reduction Pathways for a Series of [Re(CO)3(.alpha.-diimine)L′]0/+ (L′ = Halide, OTf-, THF, MeCN, n-PrCN, PPh3, P(OMe)3) Complexes. Organometallics 1995, 14, 1115–1131. [Google Scholar] [CrossRef]
- Sampson, M.D.; Nguyen, A.D.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: Eliminating dimerization and altering catalysis. J. Am. Chem. Soc. 2014, 136, 5460–5471. [Google Scholar] [CrossRef] [PubMed]
- Machan, C.W.; Sampson, M.D.; Chabolla, S.A.; Dang, T.; Kubiak, C.P. Developing a Mechanistic Understanding of Molecular Electrocatalysts for CO2 Reduction using Infrared Spectroelectrochemistry. Organometallics 2014, 33, 4550–4559. [Google Scholar] [CrossRef]
- Clark, M.L.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Reduction by M (bpy-R)(CO)4 (M = Mo, W; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts. Chem. Sci. 2014, 5, 1894–1900. [Google Scholar] [CrossRef]
- Cheng, S.C.; Blaine, C.A.; Hill, M.G.; Mann, K.R. Electrochemical and IR spectroelectrochemical studies of the electrocatalytic reduction of carbon dioxide by [Ir2(dimen)4]2+ (dimen = 1,8-Diisocyanomenthane). Inorg. Chem. 1996, 35, 7704–7708. [Google Scholar] [CrossRef]
- Johansson, O.; Borgström, M.; Lomoth, R.; Palmblad, M.; Bergquist, J.; Hammarström, L.; Sun, L.; Åkermark, B. Electron Donor–Acceptor Dyads Based on Ruthenium (II) Bipyridine and Terpyridine Complexes Bound to Naphthalenediimide. Inorg. Chem. 2003, 42, 2908–2918. [Google Scholar] [CrossRef]
- Krejčik, M.; Daněk, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer electrochemical cell: Applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholate)−. J. Electroanal. Chem. Interfacial Electrochem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: The best of two worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01 ed.; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
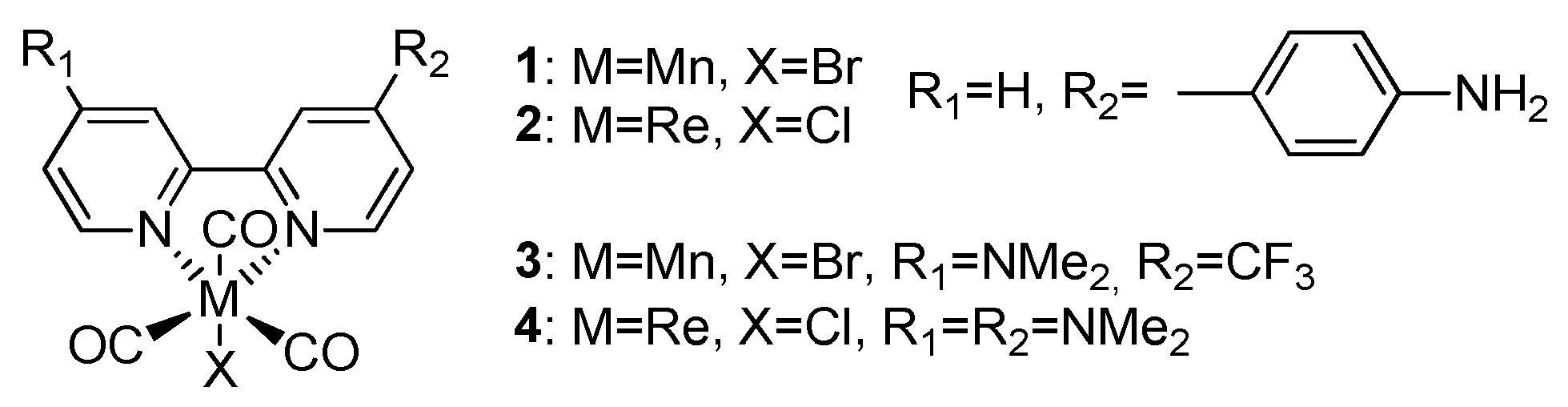







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | CPE (V) | Ep1 (V) | Mixture | Time (min) | Ep2 (V) | TONCO | FECO% |
|---|---|---|---|---|---|---|---|
| 1 | −1.73 | −1.51 | 4% water | 90 | −1.76 | 12 | 93 |
| 2 | −2.00 | −1.70 | 8% MeOH | 120 | −2.12 | 12 | 96.4 |
| 3 | −1.50 | −1.31 | 5% water | 420 | −1.42 | 26 | 84 |
| 4 | −2.00 | −1.82 | 5% MeOH | 600 | −1.82 | 31.5 | 100 |
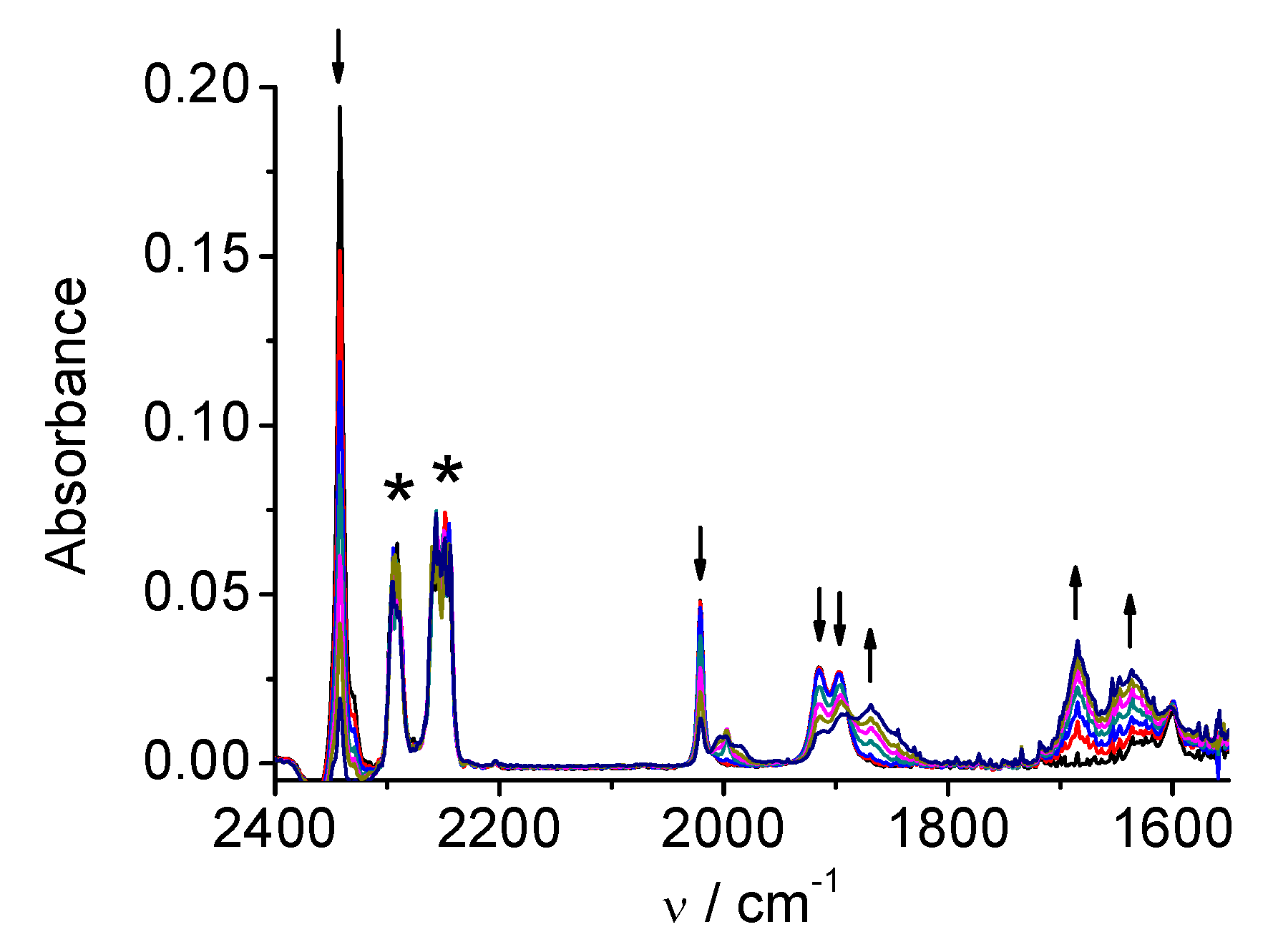
| Complex | Solvent | Native State | 1e Reduction State | 2e Reduction State |
|---|---|---|---|---|
| 1 | MeCN | 2026, 1933, 1922 | 1973, 1931, 1878, 1852 | 1909, 1811 |
| DMA | 2021, 1930, 1912 | 1969, 1927, 1875, 1853 | 1907, 1810 | |
| 2 | MeCN | 2021, 1915, 1897 | 1997, 1886, 1866 | 1986, 1850 |
| DMA | 2016, 1912, 1890 | 1992, 1879, 1860 | 1981, 1860 | |
| 3 | MeCN | 2026, 1932, 1921 | 1974, 1932, 1878, 1854 | 1917, 1816 |
| DMA | 2021, 1930, 1912 | 1972, 1929, 1877, 1855 | 1916, 1815 | |
| 4 | MeCN | 2014, 1903, 1883 | 1998, 1882, 1864 | 1986, 1862 |
| DMA | 2009, 1899, 1877 | not observed | 1980, 1859 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbero, A.; Rotundo, L.; Reviglio, C.; Gobetto, R.; Sokolova, R.; Fiedler, J.; Nervi, C. New Spectroelectrochemical Insights into Manganese and Rhenium Bipyridine Complexes as Catalysts for the Electrochemical Reduction of Carbon Dioxide. Molecules 2023, 28, 7535. https://doi.org/10.3390/molecules28227535
Barbero A, Rotundo L, Reviglio C, Gobetto R, Sokolova R, Fiedler J, Nervi C. New Spectroelectrochemical Insights into Manganese and Rhenium Bipyridine Complexes as Catalysts for the Electrochemical Reduction of Carbon Dioxide. Molecules. 2023; 28(22):7535. https://doi.org/10.3390/molecules28227535
Chicago/Turabian StyleBarbero, Alice, Laura Rotundo, Chiara Reviglio, Roberto Gobetto, Romana Sokolova, Jan Fiedler, and Carlo Nervi. 2023. "New Spectroelectrochemical Insights into Manganese and Rhenium Bipyridine Complexes as Catalysts for the Electrochemical Reduction of Carbon Dioxide" Molecules 28, no. 22: 7535. https://doi.org/10.3390/molecules28227535
APA StyleBarbero, A., Rotundo, L., Reviglio, C., Gobetto, R., Sokolova, R., Fiedler, J., & Nervi, C. (2023). New Spectroelectrochemical Insights into Manganese and Rhenium Bipyridine Complexes as Catalysts for the Electrochemical Reduction of Carbon Dioxide. Molecules, 28(22), 7535. https://doi.org/10.3390/molecules28227535