
A Holistic View of the Interactions between Electron-Deficient Systems: Clustering of Beryllium and Magnesium Hydrides and Halides



Abstract
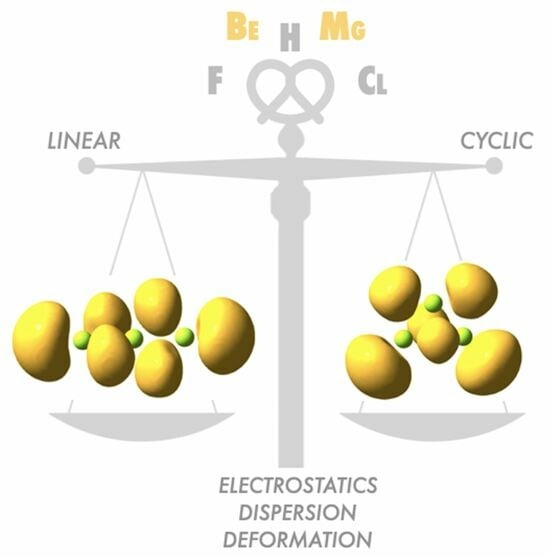
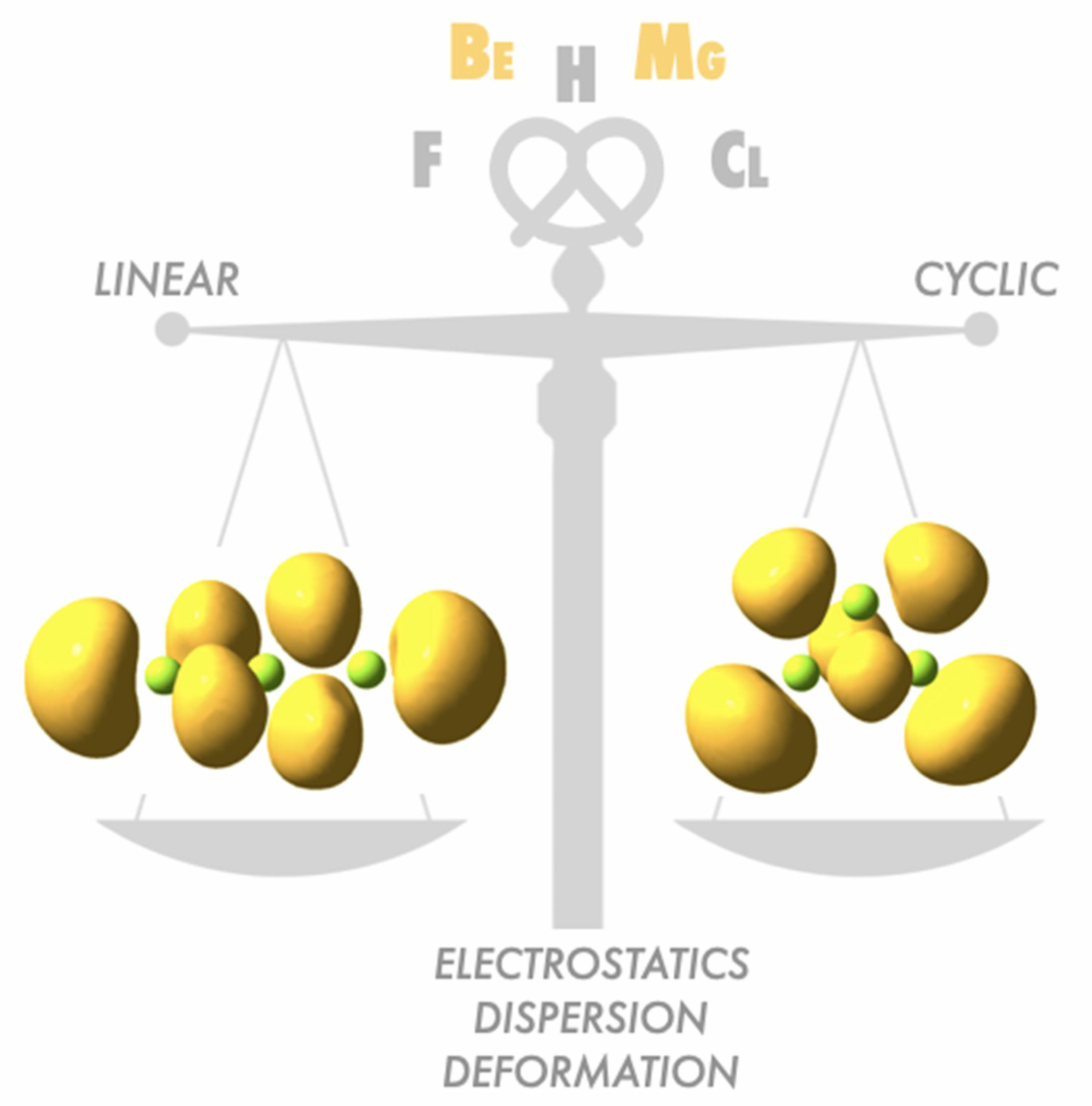
1. Introduction
2. Results and Discussion
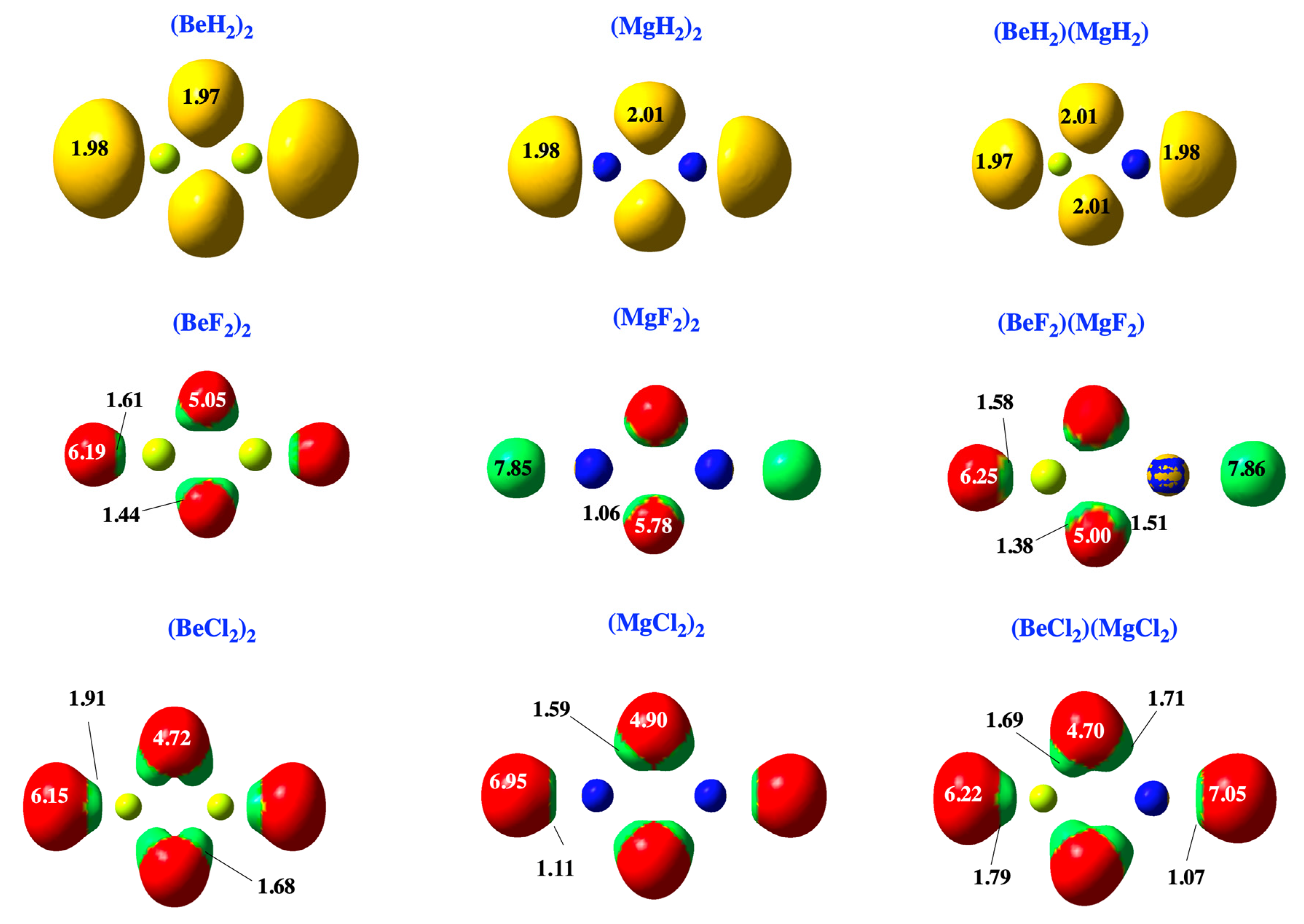
2.1. Dimers
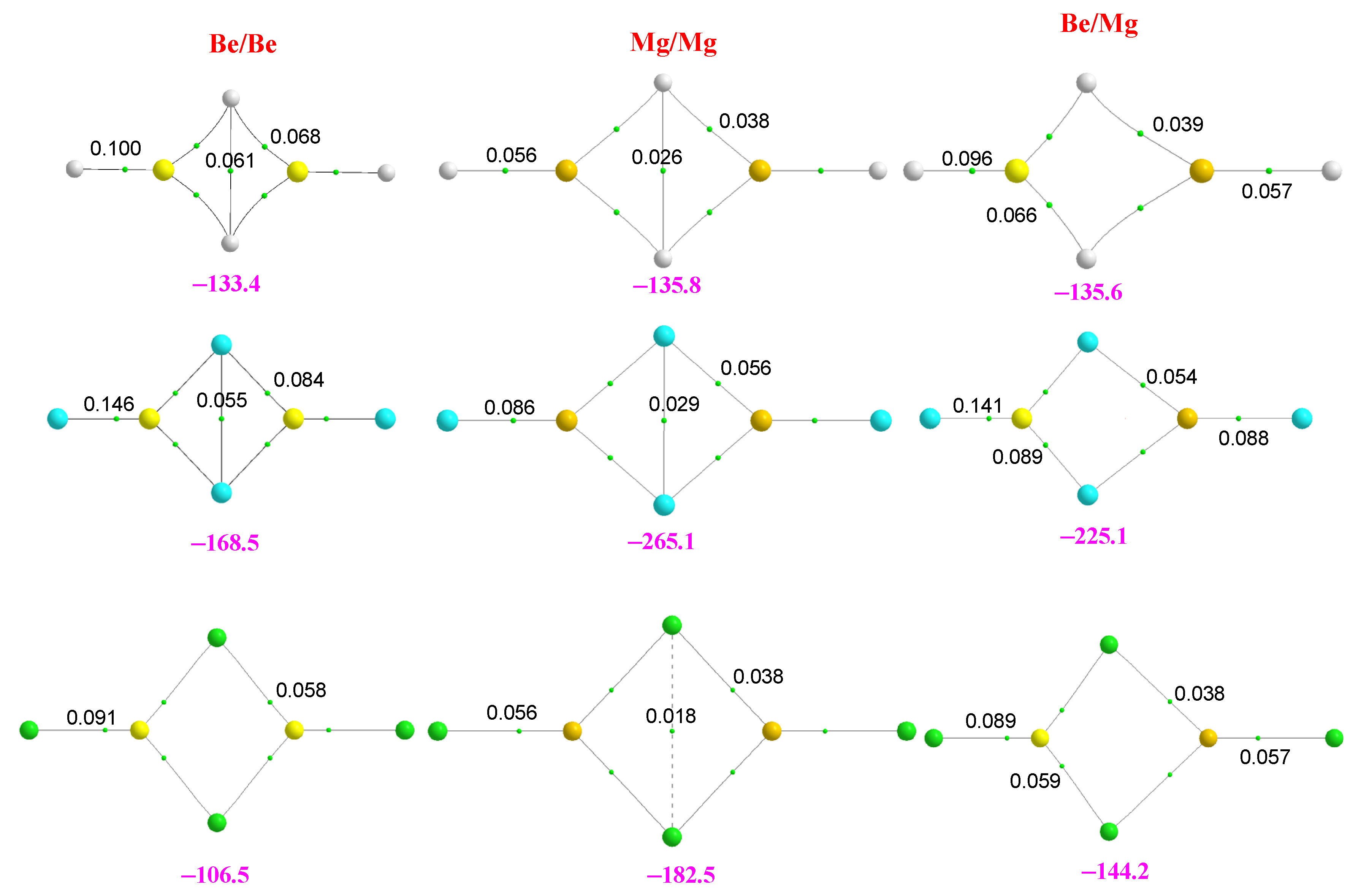
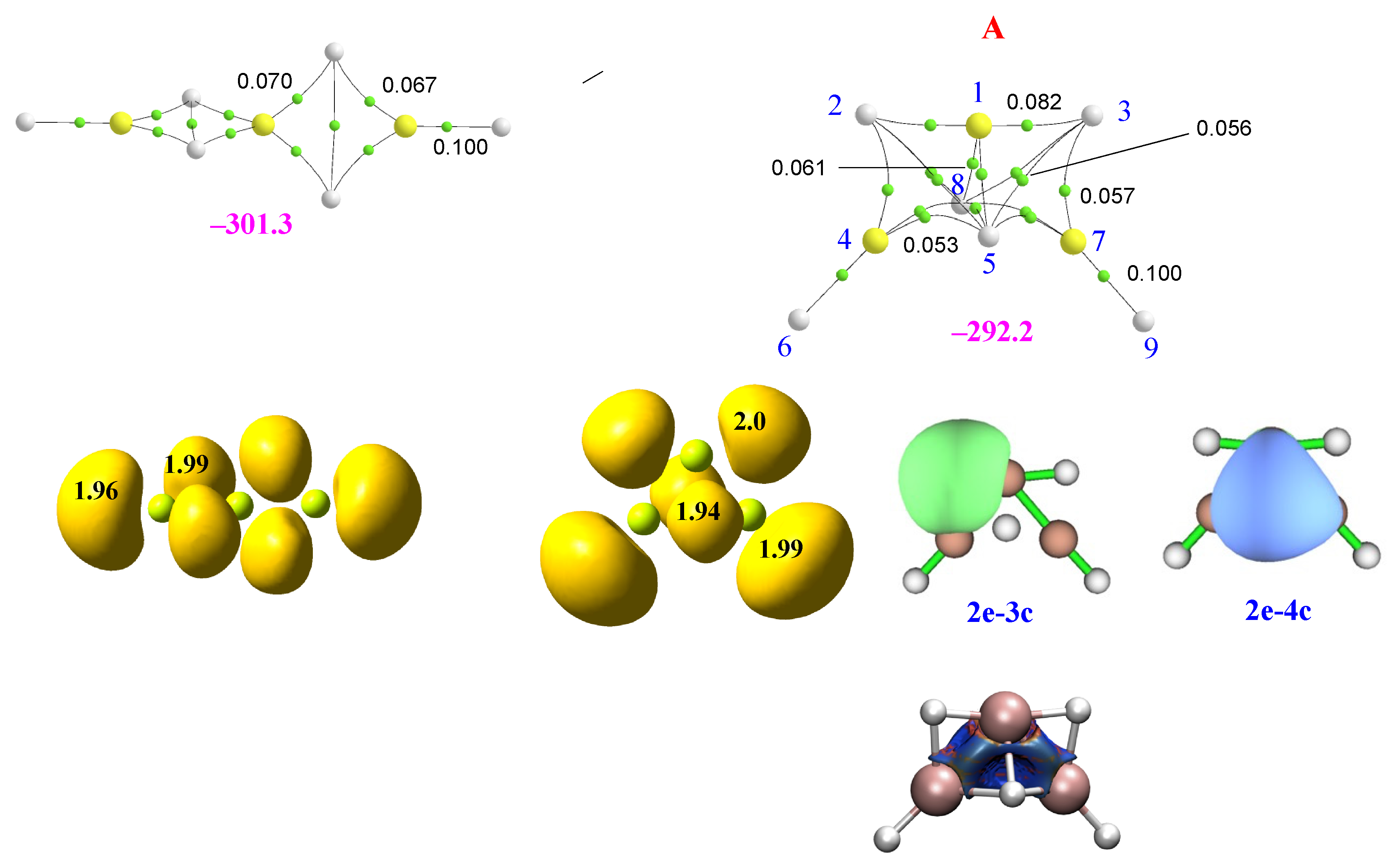
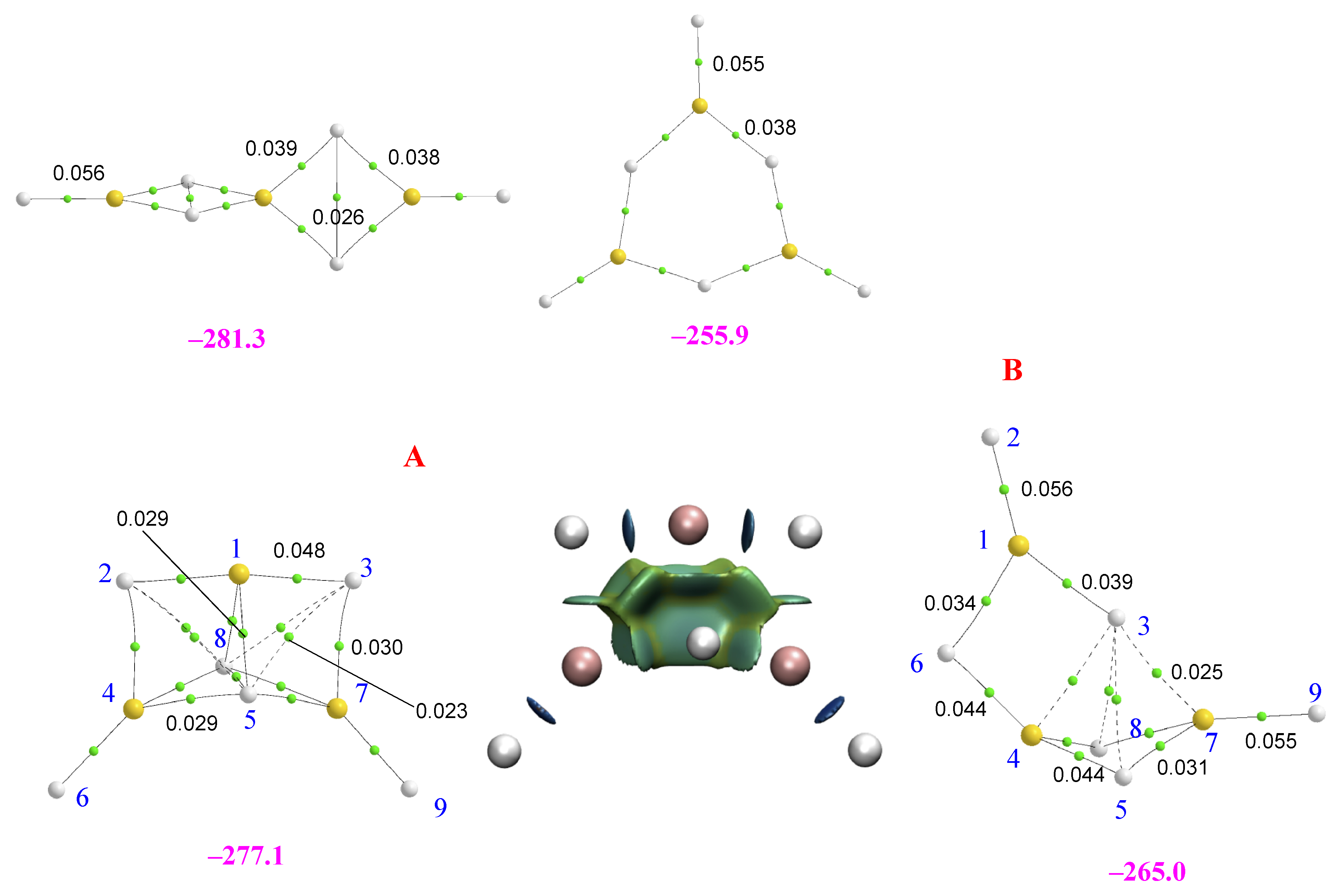
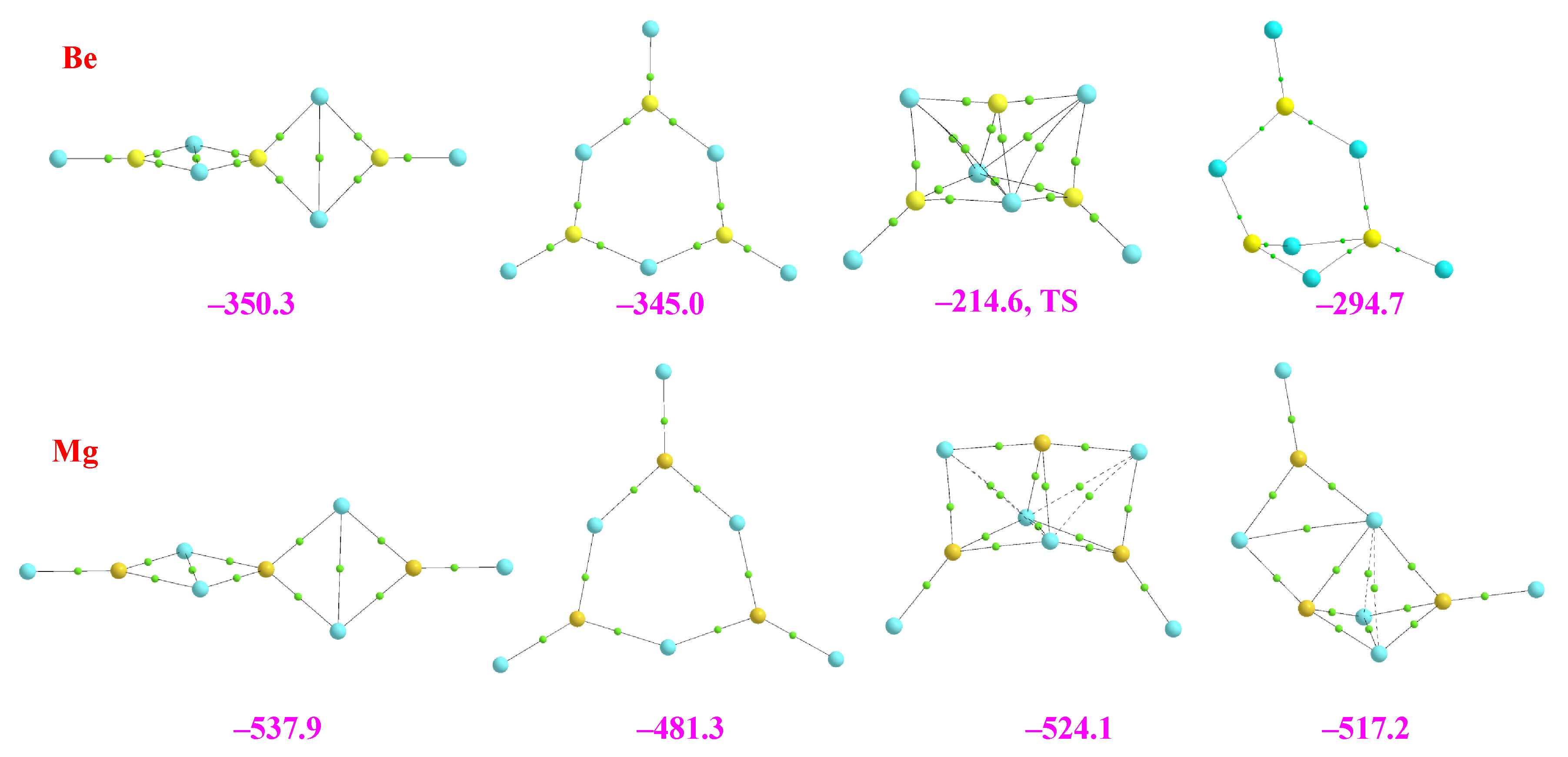
2.2. Homotrimers
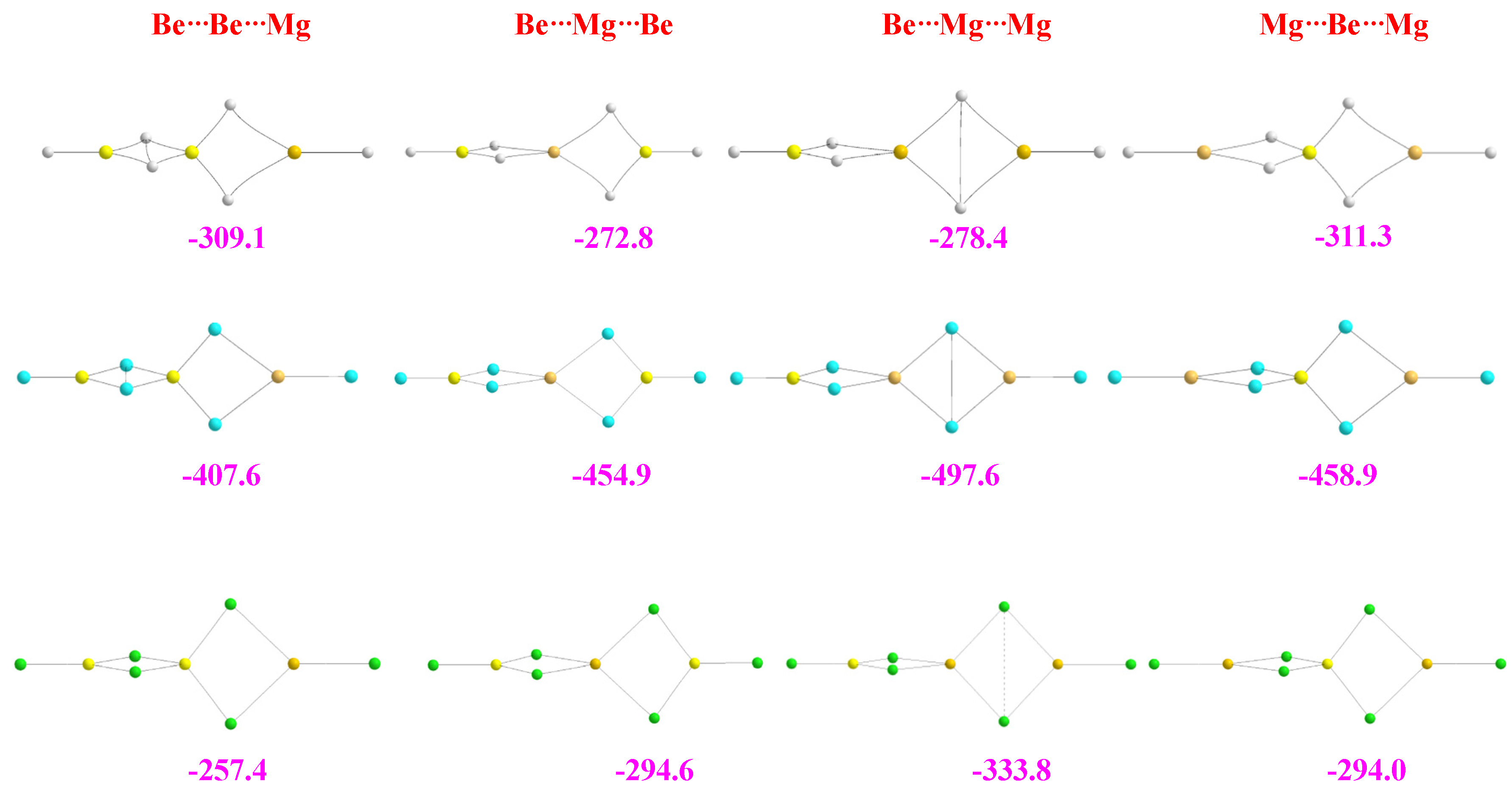
2.3. Heterotrimers
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Lipscomb, W.N. Advances in Theoretical Studies of Boron Hydrides and Carboranes. In Boron Hydride Chemistry; Muetterties, E.L., Ed.; Academic Press: Itaca, NY, USA, 1975; pp. 39–78. [Google Scholar]
- Mayer, I. Bond orders in 3-center bonds—An analytical investigation into the electronic-structure of diborane and the 3-center 4-electron bonds of hypervalent sulfur. J. Mol. Struct. Theochem 1989, 55, 43–52. [Google Scholar] [CrossRef]
- Liang, C.X.; Davy, R.D.; Schaefer, H.F. Infrared-spectra of the unknown dialane (Al2H6) and recently observed digallane (Ga2H6) molecules. Chem. Phys. Lett. 1989, 159, 393–398. [Google Scholar] [CrossRef]
- Lammertsma, K.; Leszczynski, J. Ab initio study on dialane(6) and digallane(6). J. Phys. Chem. 1990, 94, 2806–2809. [Google Scholar] [CrossRef]
- Andrews, L.; Wang, X.F. The infrared spectrum of Al2H6 in solid hydrogen. Science 2003, 299, 2049–2052. [Google Scholar] [CrossRef]
- Nori-Shargh, D.; Yahyaei, H.; Mousavi, S.N.; Maasoomi, A.; Kayi, H. Natural bond orbital, nuclear magnetic resonance analysis and hybrid-density functional theory study of sigma-aromaticity in Al2F6, Al2Cl6, Al2Br6 and Al2I6. J. Mol. Model. 2013, 19, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Mó, O.; Montero-Campillo, M.M.; Yáñez, M.; Alkorta, I.; Elguero, J. Dispersion, Rehybridization, and Pentacoordination: Keys to Understand Clustering of Boron and Aluminum Hydrides and Halides. J. Phys. Chem. A 2023, 127, 5860–5871. [Google Scholar] [CrossRef]
- Ahlrichs, R. Ab initio Calculations on Small Hydrides Including Electron Correlation. Theoret. Chim. Acta 1970, 17, 348–361. [Google Scholar] [CrossRef]
- Kirillov, Y.B.; Boldyrev, A.I.; Klimenko, N.M.; Charkin, O.P. An ab initio calculation of the structure and stability of the complex hydrides MgBeH- and Mg2H-. J. Struct. Chem. 1983, 24, 134–136. [Google Scholar] [CrossRef]
- Chen, Y.L.; Huang, C.H.; Hu, W.P. Theoretical study on the small clusters of LiH, NaH, BeH2, and MgH2. J. Phys. Chem. A 2005, 109, 9627–9636. [Google Scholar] [CrossRef] [PubMed]
- Lingam, C.B.; Babu, K.R.; Tewari, S.P.; Vaitheeswaran, G. Quantum chemical studies on beryllium hydride oligomers. Comput. Theor. Chem. 2011, 963, 371–377. [Google Scholar] [CrossRef][Green Version]
- Alkorta, I.; Elguero, J.; Solimannejad, M.; Grabowski, S.J. Dihydrogen Bonding vs. Metal-σ Interaction in Complexes between H2 and Metal Hydride. J. Phys. Chem. A 2011, 115, 201–210. [Google Scholar] [CrossRef]
- Erfort, S.; Tschope, M.; Rauhut, G. Efficient and automated quantum chemical calculation of rovibrational nonresonant Raman spectra. J. Chem. Phys. 2022, 156, 124102. [Google Scholar] [CrossRef]
- Erfort, S.; Tschoepe, M.; Rauhut, G. Toward a fully automated calculation of rovibrational infrared intensities for semi-rigid polyatomic molecules. J. Chem. Phys. 2020, 152, 244104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Andrews, L. Infrared spectra of magnesium hydride molecules, complexes, and solid magnesium dihydride. J. Phys. Chem. A 2004, 108, 11511–11520. [Google Scholar] [CrossRef]
- Abdurahman, A. Ab initio studies of static dipole polarizability of the polymeric beryllium hydride chain. J. Phys. Chem. A 2003, 107, 11547–11552. [Google Scholar] [CrossRef]
- Wang, X.F.; Andrews, L. One-dimensional BeH2 polymers: Infrared spectra and theoretical calculations. Inorg. Chem. 2005, 44, 610–614. [Google Scholar] [CrossRef]
- Ramondo, F.; Bencivenni, L.; Spoliti, M. Ab initio study on the Be2F4, Mg2F4 dimers, on the mixed dimers BeMgF4 and LiNaF2 and on the Li2BeF4, LiBCl4 and LiAlCl4 ion-pairs. J. Mol. Struct. Theochem 1992, 96, 171–184. [Google Scholar] [CrossRef]
- Gigli, G. On the structure of the alkaline earth dihalides dimers. J. Chem. Phys. 1990, 93, 5224–5233. [Google Scholar] [CrossRef]
- Ystenes, B.K. Quantum chemical studies of molecular difluorides and dichlorides of beryllium and magnesium. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1998, 54, 855–868. [Google Scholar] [CrossRef]
- Molnar, J.; Marsden, C.J.; Hargittai, M. Molecular-structures and force-fields of monomeric and dimeric magnesium dichloride from electron-diffraction and quantum-chemical calculations. J. Phys. Chem. 1995, 99, 9062–9071. [Google Scholar] [CrossRef]
- Girichev, A.G.; Giricheva, N.I.; Vogt, N.; Girichev, G.V.; Vogt, J. Structural investigation of molecules in the vapour over beryllium dichloride using electron diffraction and mass spectrometric data. J. Mol. Struct. 1996, 384, 175–182. [Google Scholar] [CrossRef]
- Buchner, M.R.; Spang, N.; Ivlev, S.I. Hydrolysis and oxidation products of phosphine adducts to beryllium chloride. Z. Naturforsch. B J. Chem. Sci. 2022, 77, 381–390. [Google Scholar] [CrossRef]
- Eichkorn, K.; Schneider, U.; Ahlrichs, R. An ab-initio investigation of structure and energetics of clusters MgnCl2n. J. Chem. Phys. 1995, 102, 7557–7563. [Google Scholar] [CrossRef]
- Neogi, S.G.; Chaudhury, P. Structure, electronic properties and vibrational spectra of (MgF2)n clusters through a combination of genetic algorithm and DFT-based approach. Mol. Phys. 2015, 113, 3729–3739. [Google Scholar] [CrossRef]
- Luhtanen, T.N.P.; Linnolahti, M.; Laine, A.; Pakkanen, T.A. Structural characteristics of small magnesium dichloride clusters: A systematic theoretical study. J. Phys. Chem. B 2004, 108, 3989–3995. [Google Scholar] [CrossRef]
- Values calculated at the M06-2X/aug-cc-pVTZ level of theory (this work).
- Wiberg, K.B. Application of pople-santry-segal cndo method to cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1088. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Walker, M.; Harvey, A.J.A.; Sen, A.; Dessent, C.E.H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J. Phys. Chem. A 2013, 117, 12590–12600. [Google Scholar] [CrossRef]
- Castro-Alvarez, A.; Cameros, H.; Sanchez, D.; Vilarrasa, J. Importance of the Electron Correlation and Dispersion Corrections in Calculations Involving Enamines, Hemiaminals, and Aminals. Comparison of B3LYP, M06-2X, MP2, and CCSD Results with Experimental Data. J. Org. Chem. 2015, 80, 11977–11985. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, J.A.; Ayala, R. Assessment of the performance of commonly used DFT functionals vs. MP2 in the study of IL-Water, IL-Ethanol and IL-(H2O)(3) clusters. J. Mol. Liq. 2016, 220, 970–982. [Google Scholar] [CrossRef]
- Stortz, C.A.; Sarotti, A.M. Exhaustive exploration of the conformational landscape of mono- and disubstituted five-membered rings by DFT and MP2 calculations. RSC Adv. 2019, 9, 24134–24145. [Google Scholar] [CrossRef]
- Hankins, D.; Moskowitz, J.W.; Stillinger, F.H. Water molecule interactions. J. Chem. Phys. 1970, 53, 4544–4554. [Google Scholar] [CrossRef]
- Xantheas, S.S. Ab-initio studies of cyclic water clusters (H2O)n, n=1-6. 2. Analysis of many-body interactions. J. Chem. Phys. 1994, 100, 7523–7534. [Google Scholar] [CrossRef]
- Su, P.F.; Jiang, Z.; Chen, Z.C.; Wu, W. Energy Decomposition Scheme Based on the Generalized Kohn-Sham Scheme. J. Phys. Chem. A 2014, 118, 2531–2542. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General atomic and molecular electronic-structure system. J. Comp. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990; pp. 1–456. [Google Scholar]
- Keith, T.A. AIMAll, version 19.10.12; TK Gristmill Software: Overland Parks, KS, USA, 2019. Available online: aim.tkgristmill.com (accessed on 1 January 2023).
- Reed, A.E.; Weinhold, F. Natural localized molecular-orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.G; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2004. [Google Scholar]
- Tkachenko, N.V.; Boldyrev, A.I. Chemical bonding analysis of excited states using the adaptive natural density partitioning method. Phys. Chem. Chem. Phys. 2019, 21, 9590–9596. [Google Scholar] [CrossRef] [PubMed]
- Boto, R.A.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.P.; Maday, Y.; Contreras-García, J. NCIPLOT4: Fast, robust and quantitative analysis of non-covalent interactions. J. Chem. Theory Comp. 2020, 16, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Savin, A.; Nesper, R.; Wengert, S.; Fassler, T.F. ELF: The electron localization function. Angew. Chem. Int. Edit. 1997, 36, 1809–1832. [Google Scholar] [CrossRef]
- Santos, L.D.; Ramalho, T.C.; Hamlin, T.A.; Bickelhaupt, F.M. Intermolecular Covalent Interactions: Nature and Directionality. Chem. Eur. J. 2023, 29, e202203791. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binary Complex | ER(A) | ER(B) | Δ2E(AB) | Etotal |
|---|---|---|---|---|
| Hydrides | ||||
| BeBeH4 | 58.7 | 58.7 | −261.4 | −144.1 |
| MgMgH4 | 36.3 | 36.3 | −216.6 | −143.9 |
| BeMgH4 | 66.3 | 31.6 | −243.2 | −45.2 |
| Fluorides | ||||
| BeBeF4 | 84.1 | 84.1 | −340.8 | −172.6 |
| MgMgF4 | 42.3 | 42.3 | −353.1 | −268.5 |
| BeMgF4 | 85.8 | 42.6 | −357.5 | −229.1 |
| Chlorides | ||||
| BeBeCl4 | 80.5 | 80.5 | −269.8 | −108.9 |
| MgMgCl4 | 44.2 | 44.2 | −273.6 | −185.3 |
| BeMgCl4 | 88.0 | 40.0 | −274.8 | −146.8 |
| Ternary Complex | ER(A) | ER(B) | ER(C) | Δ2E(AB) | Δ2E(AC) | Δ2E(BC) | Δ3E(ABC) | Etotal |
|---|---|---|---|---|---|---|---|---|
| BeBeBeH6 (linear) | 59.2 | 102.6 | 59.2 | −262.0 | 7.8 | −262.0 | −30.9 | −326.2 |
| BeBeBeH6 (cyclic, A) | 87.4 | 8.0 | 86.7 | −202.1 | −115.3 | −116.0 | −66.8 | −318.1 |
| BeBeBeH6 (hexagonal) | 82.9 | 82.9 | 82.9 | −140.1 | −140.1 | −140.1 | −57.0 | −228.7 |
| MgMgMgH6 (linear) | 37.2 | 66.8 | 37.2 | −215.2 | 0.3 | −215.2 | −10.4 | −299.3 |
| MgMgMgH6 (cyclic, A) | 58.4 | 4.2 | 58.2 | −185.5 | −99.6 | −99.9 | −30.6 | −294.8 |
| MgMgMgH6 (cyclic, B) | 28.1 | 44.5 | 54.7 | −124.6 | −54.0 | −188.6 | −42.0 | −281.9 |
| MgMgMgH6 (hexagonal) | 42.9 | 42.9 | 42.9 | −100.1 | −100.1 | −100.1 | −99. 8 | −271.4 |
| MgMgMgH6 (hexagonal, non-planar) | 51.6 | 46.7 | 28.5 | −177.8 | −77.2 | −67.8 | −69.3 | −265.3 |
| BeBeBeF6 (linear) | 84.0 | 167.3 | 84.0 | −345.2 | −2.1 | −345.2 | −2.0 | −359.3 |
| BeBeBeF6 (hexagonal) | 95.1 | 95.1 | 95.1 | −175.1 | −175.1 | −175.1 | −114.4 | −354.4 |
| BeBeBeF6 (cyclic, A) | 162.2 | 29.1 | 162.4 | −358.3 | −146.2 | −146.5 | 80.1 | −217.3 |
| BeBeBeF6 (cyclic, B) | 78.2 | 124.8 | 181.4 | −139.7 | −168.9 | −338.5 | −40.3 | −303.0 |
| MgMgMgF6 (linear) | 42.4 | 82.1 | 42.5 | −356.3 | 0.3 | −356.3 | 1.2 | −546.1 |
| MgMgMgF6 (cyclic, A) | 92.3 | 16.0 | 92.4 | −351.9 | −214.4 | −214.4 | 48.2 | −531.6 |
| MgMgMgF6 (cyclic, B) | 39.5 | 56.2 | 63.1 | −250.2 | −133.0 | −307.4 | 6.8 | −525.0 |
| MgMgMgF6 (hexagonal) | 47.5 | 47.6 | 47.6 | −188.5 | −188.5 | −188.5 | −65.8 | −488.8 |
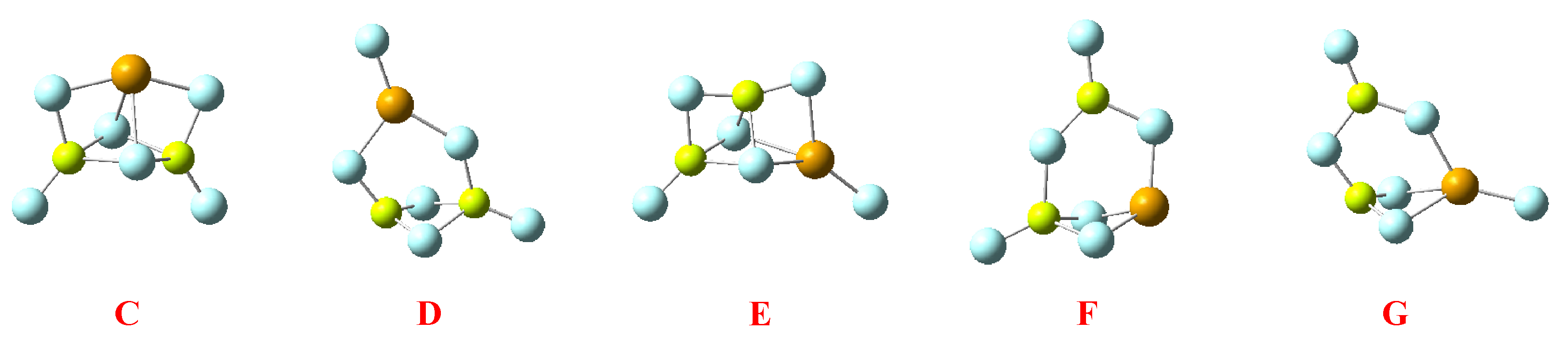
| BeBeMgH6 | BeMgMgH6 | |||
|---|---|---|---|---|
| Molecular graph | Stab. enth./ Rel. Stab. | Molecular graph | Stab. enth./ Rel. Stab. | |
| Trimers | BeBeMg | BeMgMg | ||
| C |  | −253.6 55.5 |  | −319.5 −8.2 |
| D |  | −299.8 9.3 |  | −248.4 62.9 |
| E |  | −313.7 −4.6 |  | −271.3 40.0 |
| F |  | −221.7 87.4 |  | −208.1 103.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mó, O.; Montero-Campillo, M.M.; Yáñez, M.; Alkorta, I.; Elguero, J. A Holistic View of the Interactions between Electron-Deficient Systems: Clustering of Beryllium and Magnesium Hydrides and Halides. Molecules 2023, 28, 7507. https://doi.org/10.3390/molecules28227507
Mó O, Montero-Campillo MM, Yáñez M, Alkorta I, Elguero J. A Holistic View of the Interactions between Electron-Deficient Systems: Clustering of Beryllium and Magnesium Hydrides and Halides. Molecules. 2023; 28(22):7507. https://doi.org/10.3390/molecules28227507
Chicago/Turabian StyleMó, Otilia, M. Merced Montero-Campillo, Manuel Yáñez, Ibon Alkorta, and José Elguero. 2023. "A Holistic View of the Interactions between Electron-Deficient Systems: Clustering of Beryllium and Magnesium Hydrides and Halides" Molecules 28, no. 22: 7507. https://doi.org/10.3390/molecules28227507
APA StyleMó, O., Montero-Campillo, M. M., Yáñez, M., Alkorta, I., & Elguero, J. (2023). A Holistic View of the Interactions between Electron-Deficient Systems: Clustering of Beryllium and Magnesium Hydrides and Halides. Molecules, 28(22), 7507. https://doi.org/10.3390/molecules28227507