
Advancing Our Understanding of Pyranopterin-Dithiolene Contributions to Moco Enzyme Catalysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. What Is Currently Known about Moco in the Enzymes?
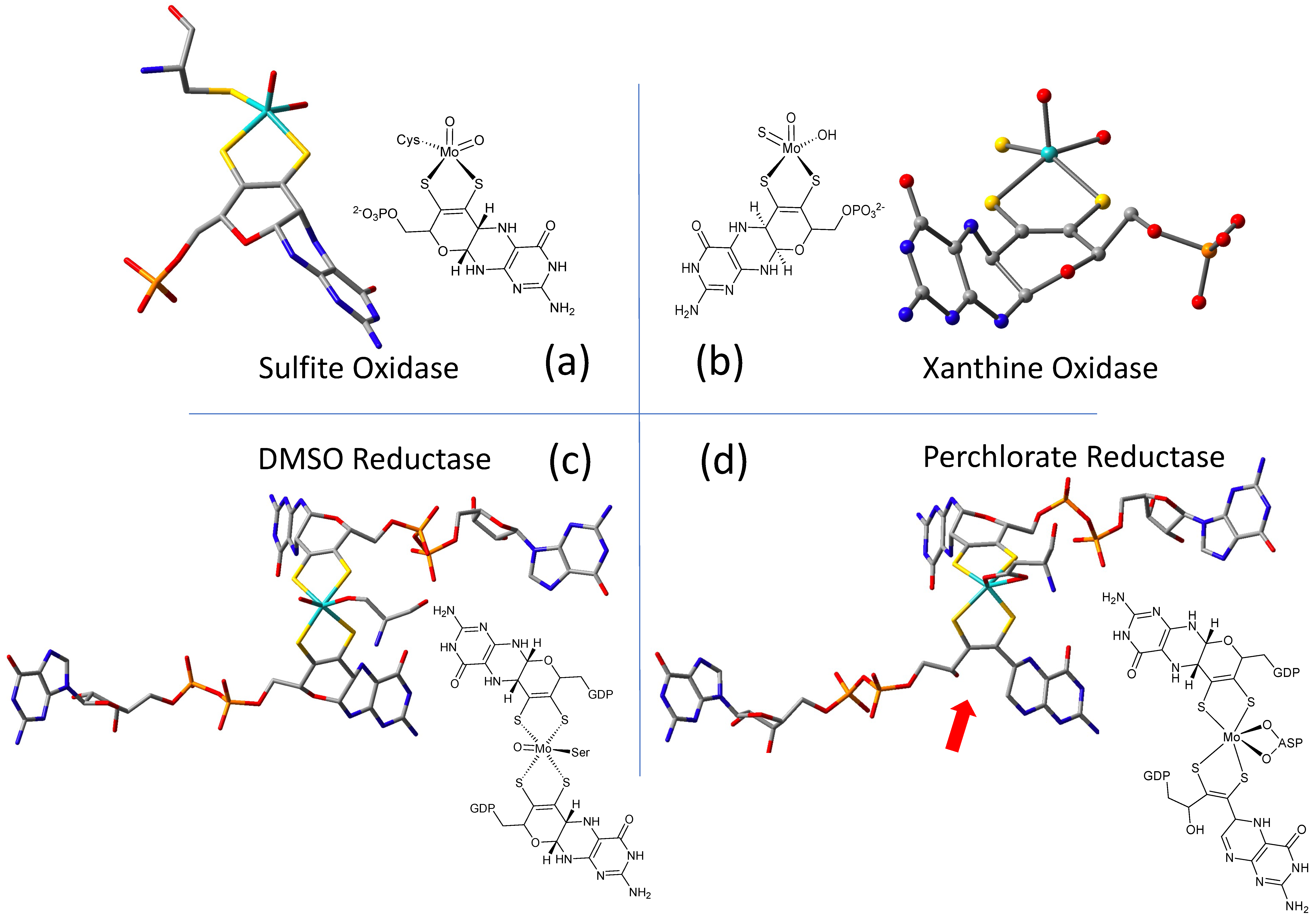
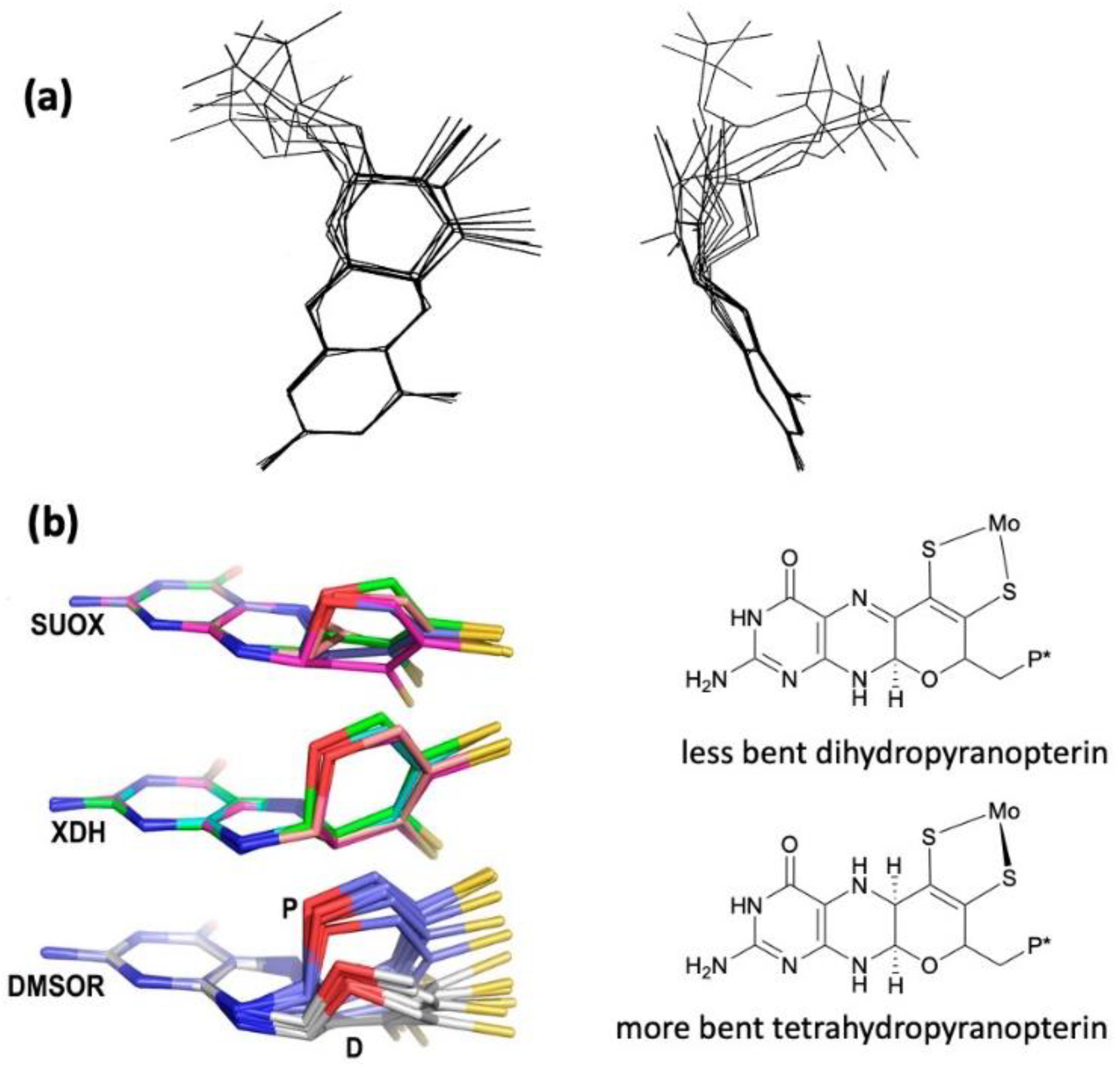
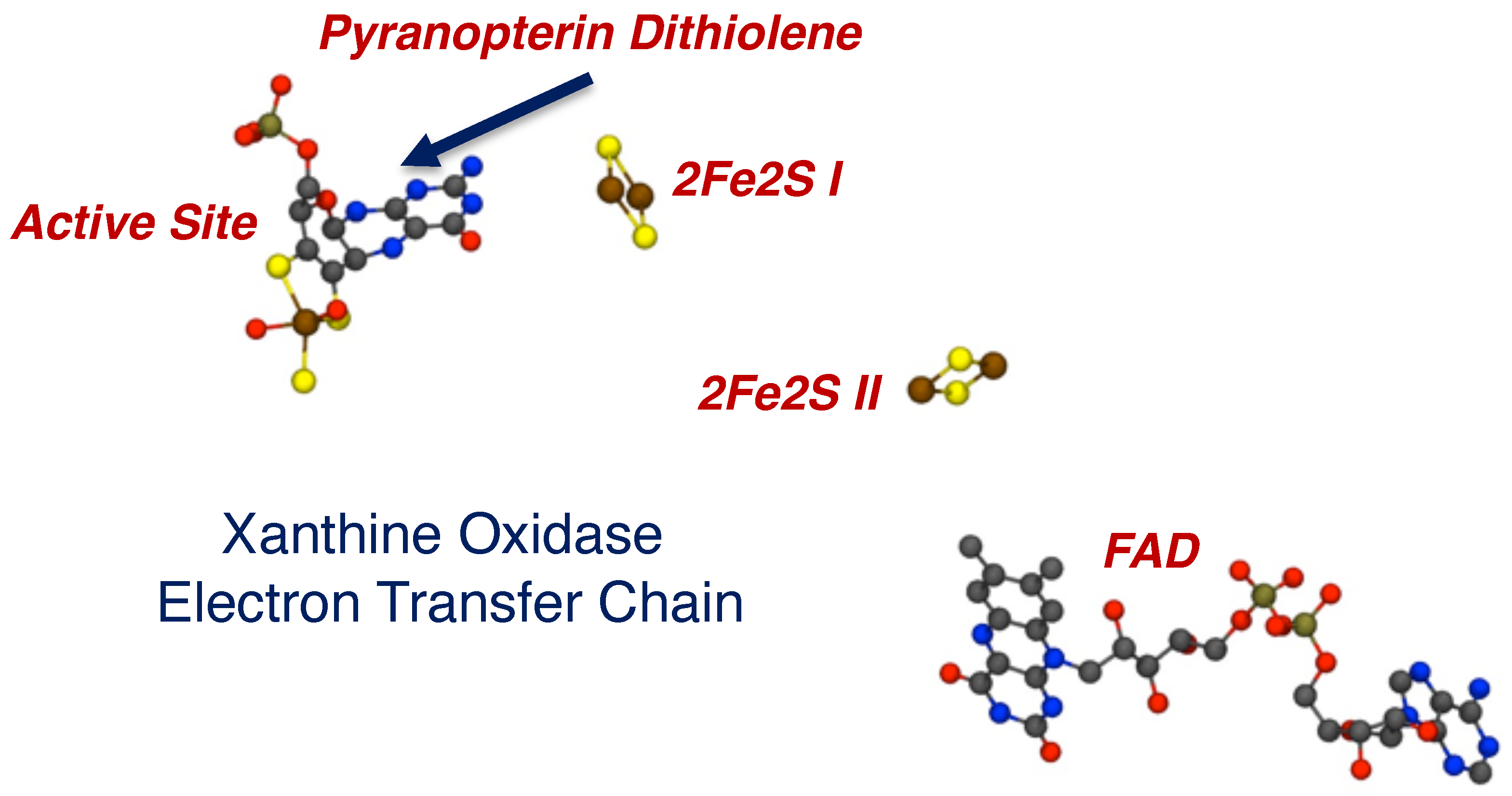
2.1. Protein X-ray Crystallography Gives Atomic Level Views of Moco
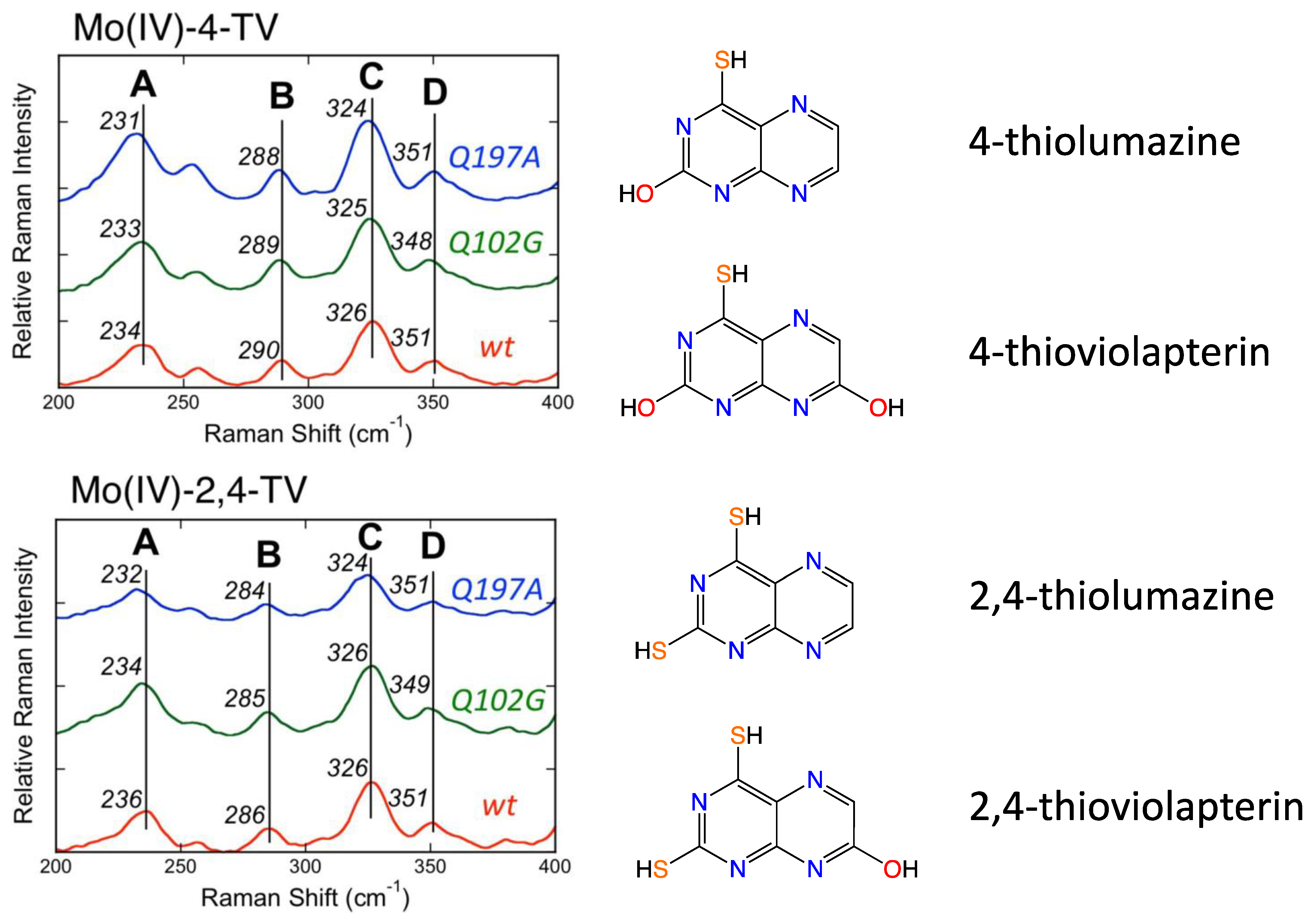
2.2. Information about the PDT of Moco Obtained from Spectroscopy
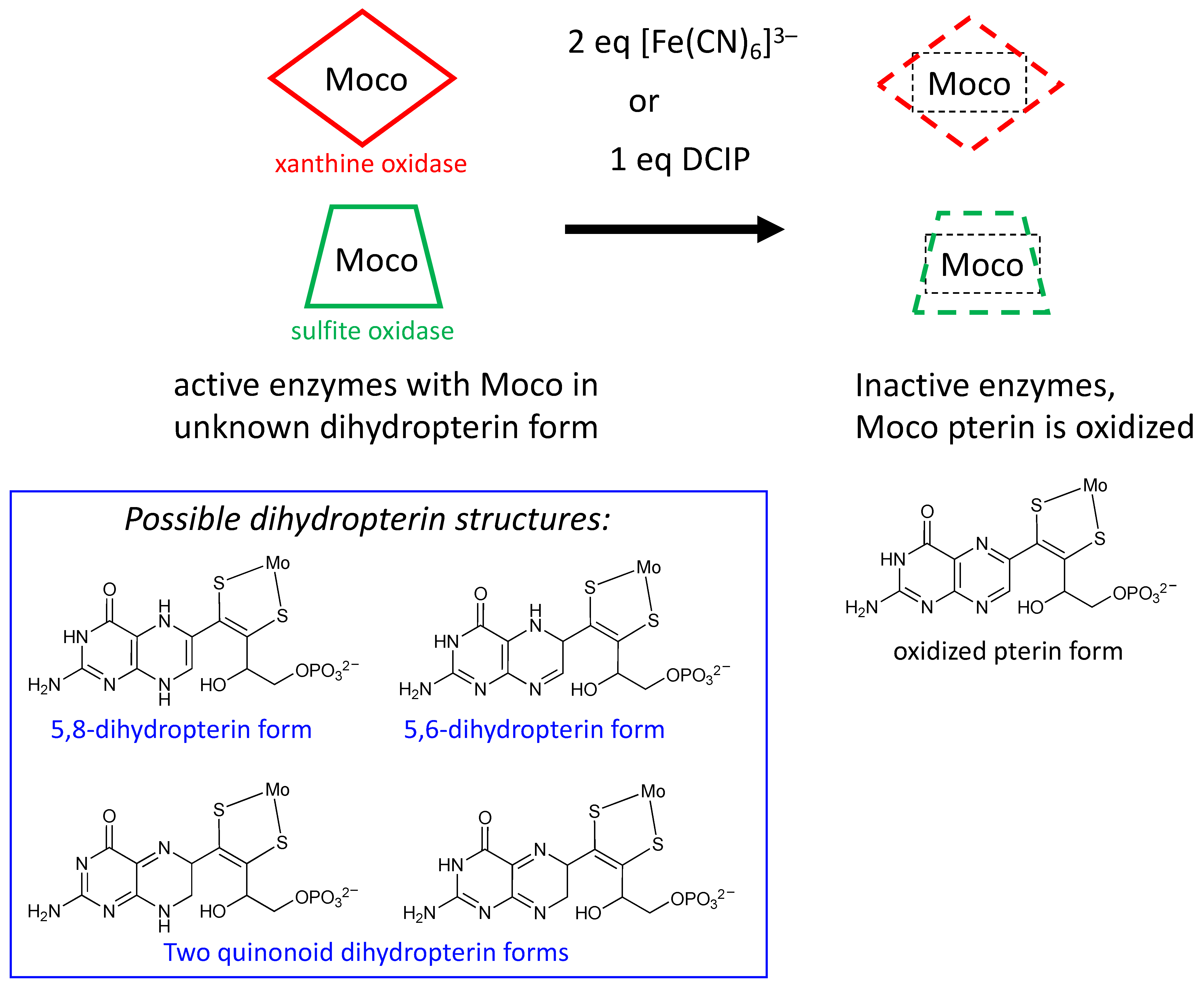
2.3. What Is Known about Pterin Oxidation State and Pterin Redox Reactivity in Moco
2.3.1. Earliest Redox Studies on PDT in Molybdenum Enzymes
2.3.2. Redox Studies on Pyranopterin
2.3.3. Role of PDT Oxidation State in Reductive Activation of DMSO Family Enzymes
2.3.4. Pterin Protein Environment in DMSOR Family Enzymes Correlates with Mo Reduction Potential
3. What Has Been Learned about Moco from Model Studies Directly Probing PDT-Mo Interactions?
3.1. Studies That Define “Simple” Mo-Ditholene Interactions
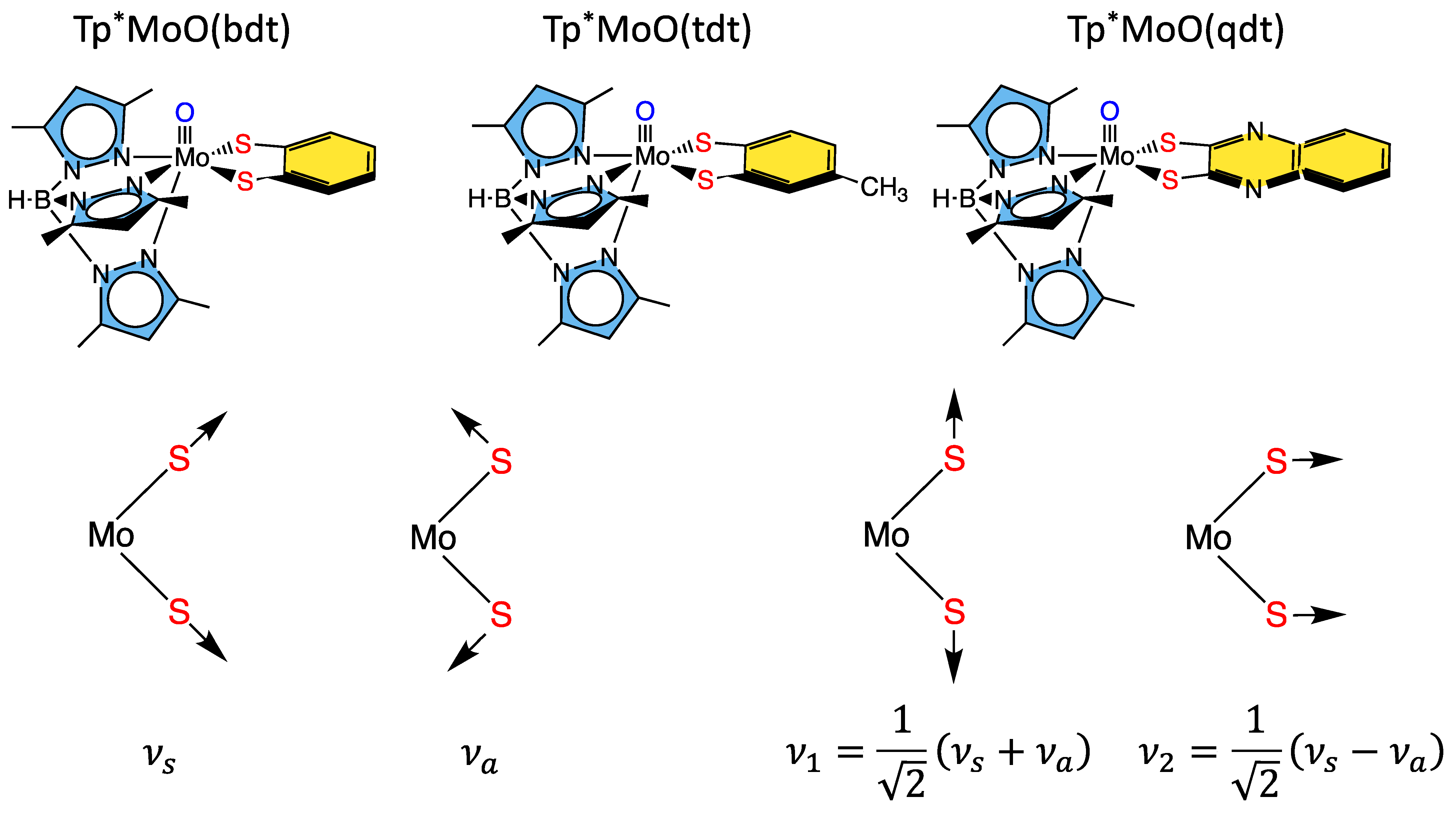
3.1.1. Tp*MoO(bdt)
3.1.2. Remote Charge Effects on Oxygen Atom Transfer Reactivity
3.1.3. Mo-Dithione Interactions Relevant to Molybdoenzymes
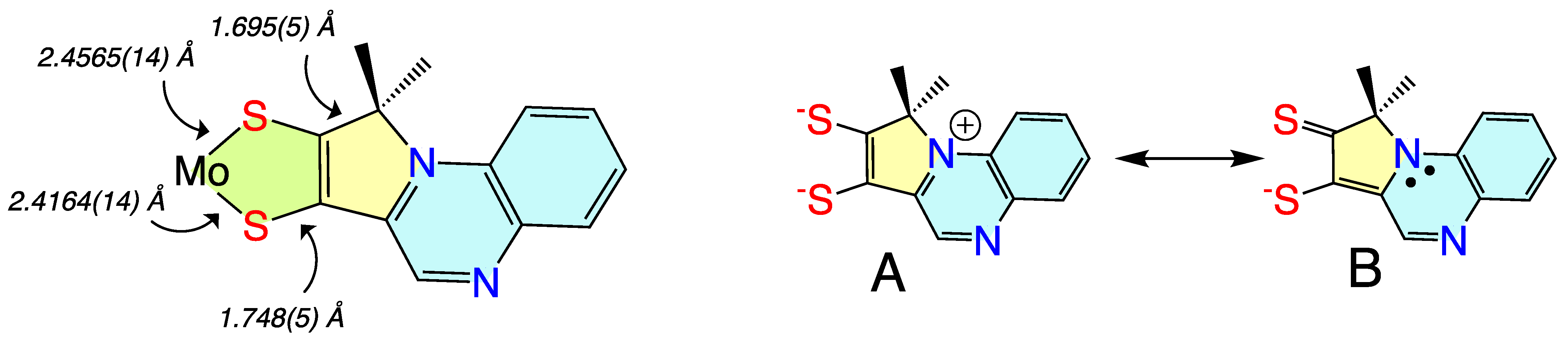
3.1.4. Donor-Acceptor Quinoxaline Dithiolene Ligands
3.2. Model Systems That Incorporate Both Dithiolene and Pyranopterin Structures on Molybdenum
3.2.1. Pyranopterin Impact on Dithiolene
3.2.2. The Electronic Origin of the Thione-Thiol Resonance Form and Implications for Pyranopterin-Mo Enzymes
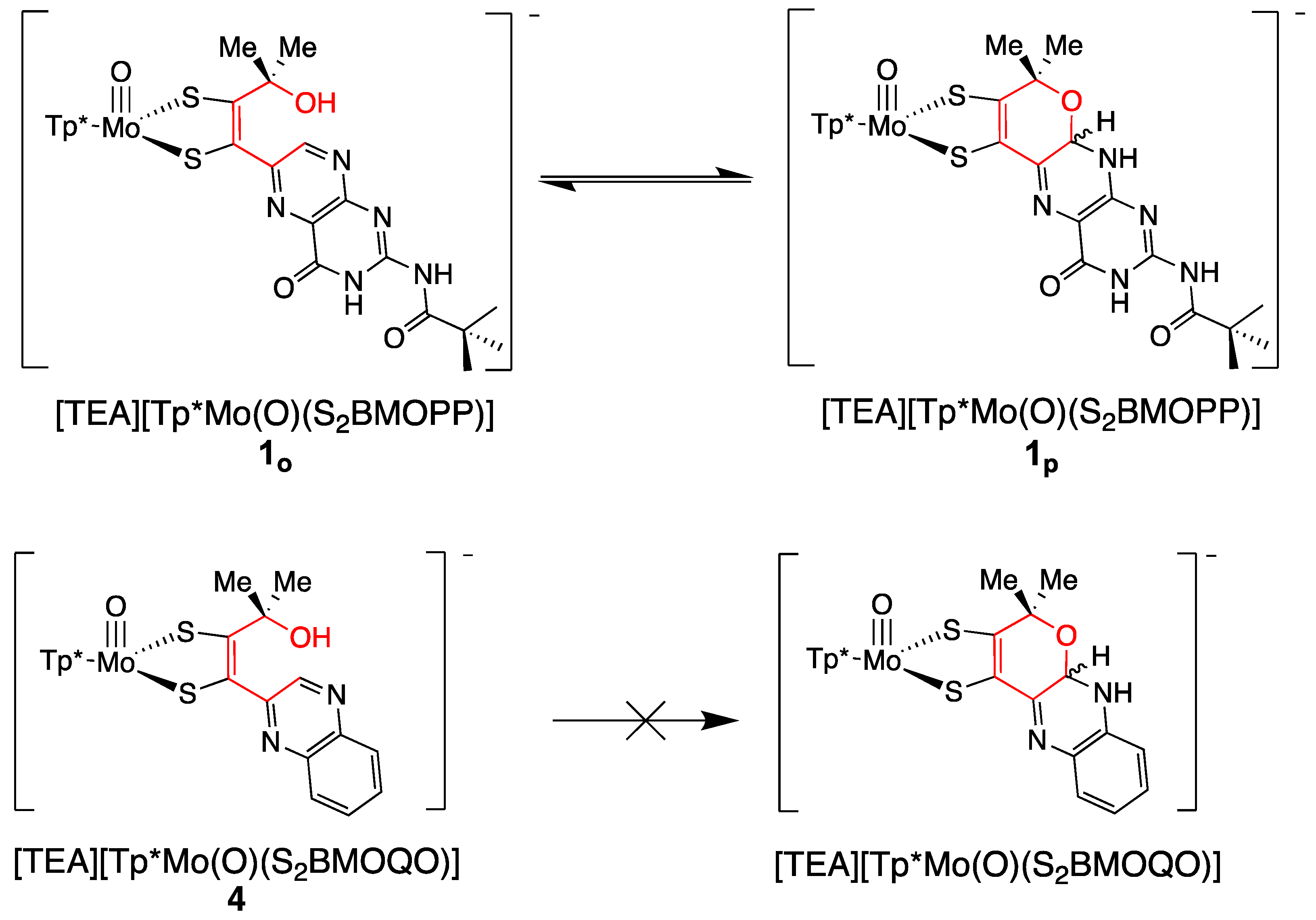
3.2.3. Implications of Pyran Cyclization on PDT Oxidation State and Pterin Conformation in Moco
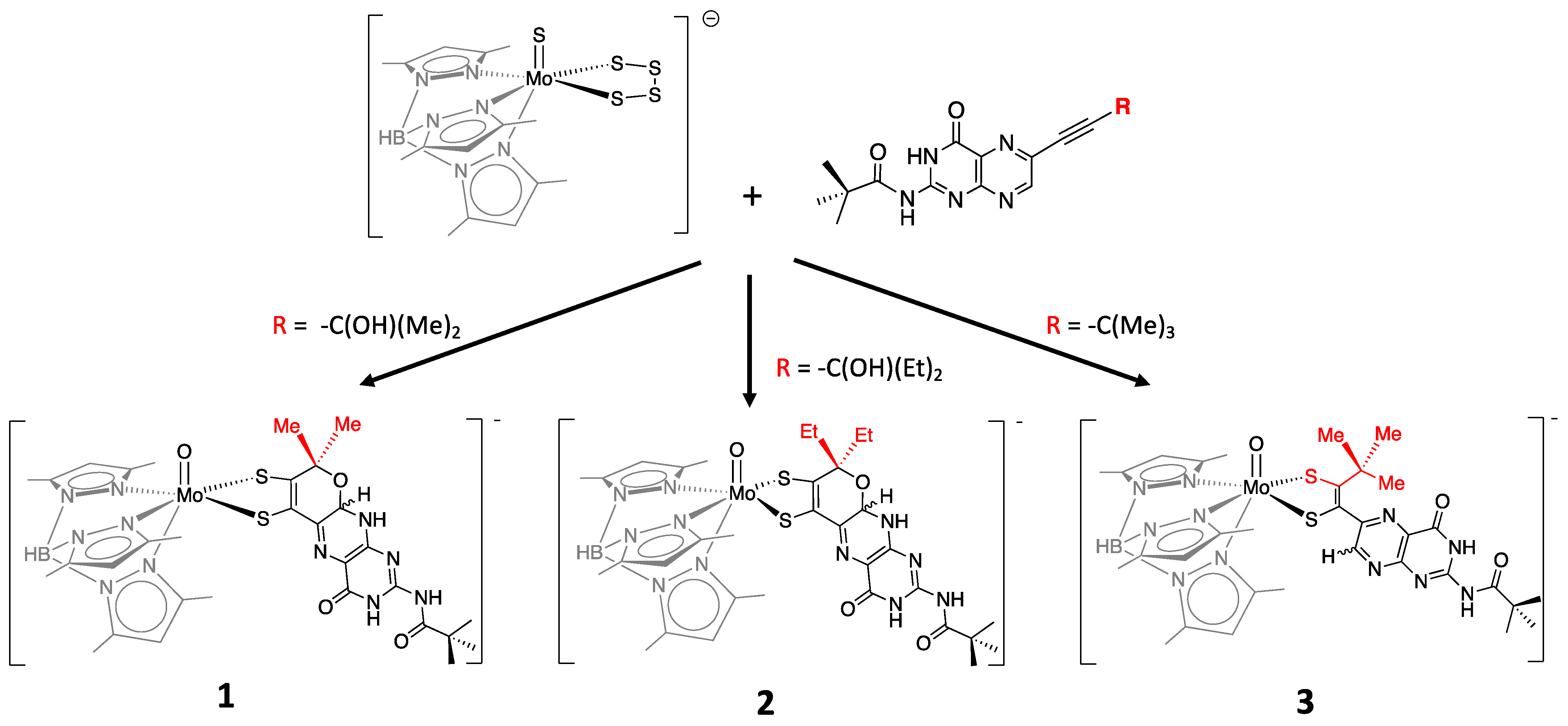
3.3. Modeling Pterin Protonation in PDT Reveals the Indivisible Mo-Pterin-Dithiolene System
3.3.1. Pyranopterin Structure Enhances Pterin Protonation
3.3.2. Pterin Protonation Strongly Affects the Electronic Structure of Mo-Dithiolene
3.3.3. Contrasts between Pyranopterin and Pyranoquinoxaline Model Compounds
4. What We Have Learned from Model Systems That Pertain to Moco in Enzymes? An Update
4.1. Previous Roles of the Pterin Defined
4.2. Recent Results Define New Roles for the PDT in Catalysis
5. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ingersol, L.J.; Kirk, M.L. Structure, Function, and Mechanism of Pyranopterin Molybdenum and Tungsten Enzymes. In Comprehensive Coordination Chemistry III; Constable, E.C., Parkin, G., Que, L., Jr., Eds.; Elsevier: Oxford, UK, 2021; pp. 790–811. [Google Scholar]
- Yang, J.; Enemark, J.H.; Kirk, M.L. Metal-Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity. Inorganics 2020, 8, 19. [Google Scholar] [CrossRef]
- Kirk, M.L.; Kc, K. Molybdenum and Tungsten Cofactors and the Reactions They Catalyze. Transition Metals and Sulfur—A Strong Relationship for Life. In Metal Ions in Life Sciences; Sosa Torres, M., Kroneck, P., Eds.; De Gruyter: Berlin, Germany, 2020; Volume 20, pp. 313–342. [Google Scholar]
- Hille, R.; Schulzke, C.; Kirk, M.L. Molybdenum and Tungsten Enzymes; The Royal Society of Chemistry: Cambridge, UK, 2017. [Google Scholar]
- Kirk, M.L.; Hille, R. Spectroscopic Studies of Mononuclear Molybdenum Enzyme Centers. Molecules 2022, 27, 4802. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Nishino, T.; Bittner, F. Molybdenum enzymes in higher organisms. Coord. Chem. Rev. 2011, 255, 1179–1205. [Google Scholar] [CrossRef] [PubMed]
- Peglow, S.; Toledo, A.H.; Anaya-Prado, R.; Lopez-Neblina, F.; Toledo-Pereyra, L.H. Allopurinol and xanthine oxidase inhibition in liver ischemia reperfusion. J. Hepato-Biliary-Pancreat. Sci. 2011, 18, 137–146. [Google Scholar] [CrossRef]
- Nishino, T. The Conversion of Xanthine Dehydrogenase to Xanthine Oxidase and the Role of the Enzyme in Reperfusion Injury. J. Biochem. 1994, 116, 1–6. [Google Scholar] [CrossRef]
- Kumar, R.; Joshi, G.; Kler, H.; Kalra, S.; Kaur, M.; Arya, R. Toward an Understanding of Structural Insights of Xanthine and Aldehyde Oxidases: An Overview of their Inhibitors and Role in Various Diseases. Med. Res. Rev. 2018, 38, 1073–1125. [Google Scholar] [CrossRef]
- Ahire, D.; Basit, A.; Christopher, L.J.; Iyer, R.; Leeder, J.S.; Prasad, B. Interindividual Variability and Differential Tissue Abundance of Mitochondrial Amidoxime Reducing Component Enzymes in Humans. Drug Metab. Dispos. 2022, 50, 191. [Google Scholar] [CrossRef]
- Mota, C.; Esmaeeli, M.; Coelho, C.; Santos-Silva, T.; Wolff, M.; Foti, A.; Leimkuhler, S.; Romao, M.J. Human aldehyde oxidase (hAOX1): Structure determination of the Moco-free form of the natural variant G1269R and biophysical studies of single nucleotide polymorphisms. FEBS Open Bio 2019, 9, 925–934. [Google Scholar] [CrossRef]
- Mota, C.; Coelho, C.; Leimkuhler, S.; Garattini, E.; Terao, M.; Santos-Silva, T.; Romao, M.J. Critical overview on the structure and metabolism of human aldehyde oxidase and its role in pharmacokinetics. Coord. Chem. Rev. 2018, 368, 35–59. [Google Scholar] [CrossRef]
- Beedham, C.; Miceli, J.J.; Obach, R.S. Ziprasidone metabolism, aldehyde oxidase, and clinical implications. J. Clin. Psychopharmacol. 2003, 23, 229–232. [Google Scholar] [CrossRef]
- Vickers, S.; Polsky, S.L. The biotransformation of nitrogen containing xenobiotics to lactams. Curr. Drug Metab. 2000, 1, 357–389. [Google Scholar] [CrossRef] [PubMed]
- Pritsos, C.A. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem.-Biol. Interact. 2000, 129, 195. [Google Scholar] [CrossRef]
- Maini Rekdal, V.; Bess, E.N.; Bisanz, J.E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 2019, 364, eaau6323. [Google Scholar] [CrossRef]
- Kubitza, C.; Bittner, F.; Ginsel, C.; Havemeyer, A.; Clement, B.; Scheidig, A.J. Crystal structure of human mARC1 reveals its exceptional position among eukaryotic molybdenum enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 11958–11963. [Google Scholar] [CrossRef]
- Ott, G.; Havemeyer, A.; Clement, B. The mammalian molybdenum enzymes of mARC. J. Biol. Inorg. Chem. 2015, 20, 265–275. [Google Scholar] [CrossRef]
- Havemeyer, A.; Lang, J.A.; Clement, B. The fourth mammalian molybdenum enzyme mARC: Current state of research. Drug Metab. Rev. 2011, 43, 524–539. [Google Scholar] [CrossRef]
- Havemeyer, A.; Grünewald, S.; Wahl, B.; Bittner, F.; Mende, R.; Erdélyi, P.; Fischer, J.; Clement, B. Reduction of N-Hydroxy-sulfonamides, Including N-Hydroxy- valdecoxib, by the Molybdenum-Containing Enzyme mARC. Drug Metab. Dispos. 2010, 38, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Struwe, M.; Scheidig, A.; Mengell, J.; Clement, B.; Kirk, M.L. Active Site Structures of the Escherichia coli N-Hydroxylaminopurine Resistance Molybdoenzyme YcbX. Inorg. Chem. 2023, 62, 5315–5319. [Google Scholar] [PubMed]
- Clement, B.; Struwe, M.A. The History of mARC. Molecules 2023, 28, 4713. [Google Scholar] [CrossRef]
- Schwarz, G. Molybdenum cofactor and human disease. Curr. Opin. Chem. Biol. 2016, 31, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Gruenewald, S.; Wahl, B.; Bittner, F.; Hungeling, H.; Kanzow, S.; Kotthaus, J.; Schwering, U.; Mendel, R.R.; Clement, B. The Fourth Molybdenum Containing Enzyme mARC: Cloning and Involvement in the Activation of N-Hydroxylated Prodrugs. J. Med. Chem. 2008, 51, 8173–8177. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Jimenez, M.; Chamizo-Ampudia, A.; Calatrava, V.; Galvan, A.; Fernandez, E.; Llamas, A. From the Eukaryotic Molybdenum Cofactor Biosynthesis to the Moonlighting Enzyme mARC. Molecules 2018, 23, 3287. [Google Scholar] [CrossRef]
- Llamas, A.; Chamizo-Ampudia, A.; Tejada-Jimenez, M.; Galvan, A.; Fernandez, E. The molybdenum cofactor enzyme mARC: Moonlighting or promiscuous enzyme? BioFactors 2017, 43, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.; Moura, J.G. Nitrite reduction by molybdoenzymes: A new class of nitric oxide-forming nitrite reductases. J. Biol. Inorg. Chem. 2015, 20, 403–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Krizowski, S.; Fischer-Schrader, K.; Niks, D.; Tejero, J.; Sparacino-Watkins, C.; Wang, L.; Ragireddy, V.; Frizzell, S.; Kelley, E.E.; et al. Sulfite Oxidase Catalyzes Single-Electron Transfer at Molybdenum Domain to Reduce Nitrite to Nitric Oxide. Antioxid. Redox Signal. 2014, 23, 283–294. [Google Scholar] [CrossRef]
- Sparacino-Watkins, C.E.; Tejero, J.S.; Sun, B.; Gauthier, M.C.; Thomas, J.; Ragireddy, V.; Merchant, B.A.; Wang, J.; Azarov, I.; Basu, P.; et al. Nitrite Reductase and Nitric-oxide Synthase Activity of the Mitochondrial Molybdopterin Enzymes mARC1 and mARC2. J. Biol. Chem. 2014, 289, 10345–10358. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.G. Nitrite reduction by xanthine oxidase family enzymes: A new class of nitrite reductases. J. Biol. Inorg. Chem. 2011, 16, 443–460. [Google Scholar] [CrossRef]
- Yang, J.; Giles, L.J.; Ruppelt, C.; Mendel, R.R.; Bittner, F.; Kirk, M.L. Oxyl and Hydroxyl Radical Transfer in Mitochondrial Amidoxime Reducing Component-Catalyzed Nitrite Reduction. J. Am. Chem. Soc. 2015, 137, 5276–5279. [Google Scholar] [CrossRef]
- Giles, L.J.; Ruppelt, C.; Yang, J.; Mendel, R.R.; Bittner, F.; Kirk, M.L. Molybdenum Site Structure of MOSC Family Proteins. Inorg. Chem. 2014, 53, 9460–9462. [Google Scholar] [CrossRef]
- Kirk, M.L.; Stein, B. The Molybdenum Enzymes. In Comprehensive Inorganic Chemistry II, 2nd ed.; Jan, R., Kenneth, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 263–293. [Google Scholar]
- Hemann, C.; Hood, B.L.; Fulton, M.; Hansch, R.; Schwarz, G.; Mendel, R.R.; Kirk, M.L.; Hille, R. Spectroscopic and kinetic studies of Arabidopsis thaliana sulfite oxidase: Nature of the redox-active orbital and electronic structure contributions to catalysis. J. Am. Chem. Soc. 2005, 127, 16567. [Google Scholar] [CrossRef]
- Enemark, J.H. Consensus structures of the Mo(v) sites of sulfite-oxidizing enzymes derived from variable frequency pulsed EPR spectroscopy, isotopic labelling and DFT calculations. Dalton Trans. 2017, 46, 13202–13210. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R.; Leimkuhler, S. The biosynthesis of the molybdenum cofactors. J. Biol. Inorg. Chem. 2015, 20, 337–347. [Google Scholar] [CrossRef]
- Wollers, S.; Heidenreich, T.; Zarepour, M.; Zachmann, D.; Kraft, C.; Zhao, Y.D.; Mendel, R.R.; Bittner, F. Binding of sulfurated molybdenum cofactor to the C-terminal domain of ABA3 from Arabidopsis thaliana provides insight into the mechanism of molybdenum cofactor sulfuration. J. Biol. Chem. 2008, 283, 9642–9650. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G.; Mendel, R.R. Molybdenum Cofactor Biosynthesis and Molybdoenzymes. Annu. Rev. Plant Biol. 2006, 57, 623–647. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, T.; Wollers, S.; Mendel, R.R.; Bittner, F. Characterization of the NifS-like domain of ABA3 from Arabidopsis thaliana provides insight into the mechanism of molybdenum cofactor sulfuration. J. Biol. Chem. 2005, 280, 4213–4218. [Google Scholar] [CrossRef]
- Wahl, B.; Reichmann, D.; Niks, D.; Krompholz, N.; Havemeyer, A.; Clement, B.; Messerschmidt, T.; Rothkegel, M.; Biester, H.; Hille, R.; et al. Biochemical and spectroscopic characterization of the human mitochondrial amidoxime reducing components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum-enzyme family in eukaryotes. J. Biol. Chem. 2010, 285, 37847–37859. [Google Scholar] [CrossRef]
- Anantharaman, V.; Aravind, L. MOSC domains: Ancient, predicted sulfur-carrier domains, present in diverse metal-sulfur cluster biosynthesis proteins including Molybdenum cofactor sulfurases. FEMS Microbiol. Lett. 2002, 207, 55–61. [Google Scholar] [CrossRef]
- Heider, J.; Ma, K.; Adams, M.W.W. Purification, Characterization, and Metabolic Function of Tungsten-Containing Aldehyde Ferredoxin Oxidoreductase from the Hyperthermophilic and Proteolytic Archaeon Thermococcus Strain ES-1. J. Bacter. 1995, 177, 4757–4764. [Google Scholar] [CrossRef]
- Schut, G.J.; Thorgersen, M.P.; Poole, F.L.; Haja, D.K.; Putumbaka, S.; Adams, M.W.W. Tungsten enzymes play a role in detoxifying food and antimicrobial aldehydes in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2021, 118, e2109008118. [Google Scholar] [CrossRef]
- Rekdal, V.M.; Bernadino, P.N.; Luescher, M.U.; Kiamehr, S.; Le, C.; Bisanz, J.E.; Turnbaugh, P.J.; Bess, E.N.; Balskus, E.P. A widely distributed metalloenzyme class enables gut microbial metabolism of host- and diet-derived catechols. elife 2020, 9, e50845. [Google Scholar] [CrossRef]
- Rekdal, V.M.; Balskus, E.P. Gut Microbiota: Rational Manipulation of Gut Bacterial Metalloenzymes Provides Insights into Dysbiosis and Inflammation. Biochemistry 2018, 57, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Struwe, M.A.; Kalimuthu, P.; Luo, Z.Y.; Zhong, Q.F.; Ellis, D.; Yang, J.; Khadanand, K.C.; Harmer, J.R.; Kirk, M.L.; McEwan, A.G.; et al. Active site architecture reveals coordination sphere flexibility and specificity determinants in a group of closely related molybdoenzymes. J. Biol. Chem. 2021, 296, 100672. [Google Scholar] [CrossRef]
- Ingersol, L.J.; Yang, J.; Khadanand, K.C.; Pokhrel, A.; Astashkin, A.V.; Weiner, J.H.; Johnston, C.A.; Kirk, M.L. Addressing Ligand-Based Redox in Molybdenum-Dependent Methionine Sulfoxide Reductase. J. Am. Chem. Soc. 2021, 142, 2721–2725. [Google Scholar] [CrossRef] [PubMed]
- Dhouib, R.; Othman, D.; Lin, V.; Lai, X.J.; Wijesinghe, H.G.S.; Essilfie, A.T.; Davis, A.; Nasreen, M.; Bernhardt, P.V.; Hansbro, P.M.; et al. A Novel, Molybdenum-Containing Methionine Sulfoxide Reductase Supports Survival of Haemophilus influenzae in an In vivo Model of Infection. Front. Microbiol. 2016, 7, 1743. [Google Scholar] [CrossRef] [PubMed]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Burgmayer, S.J.N.; Kirk, M.L. The Role of the Pyranopterin Dithiolene Component of Moco in Molybdoenzyme Catalysis. In Metallocofactors That Activate Small Molecules: With Focus on Bioinorganic Chemistry; Ribbe, M.W., Ed.; Springer International Publishing: Cham, Switzerland, 2019; Volume 179, pp. 101–151. [Google Scholar]
- Mendel, R.R. The History of the Molybdenum Cofactor-A Personal View. Molecules 2022, 27, 4934. [Google Scholar] [CrossRef]
- Chrysochos, N.; Ahmadi, M.; Trentin, I.; Lokov, M.; Tshepelevitsh, S.; Ullmann, G.M.; Leito, I.; Schulzke, C. Aiding a Better Understanding of Molybdopterin: Syntheses, Structures, and pK(a) Value Determinations of Varied Pterin-Derived Organic Scaffolds Including Oxygen, Sulfur and Phosphorus Bearing Substituents. J. Mol. Struct. 2021, 1230, 129867. [Google Scholar] [CrossRef]
- Leimkühler, S. The biosynthesis of the molybdenum cofactors in Escherichia coli. Environ. Microbiol. 2020, 22, 2007–2026. [Google Scholar] [CrossRef]
- Krausze, J.; Hercher, T.W.; Zwerschke, D.; Kirk, M.L.; Blankenfeldt, W.; Mendel, R.R.; Kruse, T. The functional principle of eukaryotic molybdenum insertases. Biochem. J. 2018, 475, 1739–1753. [Google Scholar] [CrossRef]
- Iobbi-Nivol, C.; Leimkühler, S. Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2013, 1827, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T. Eukaryotic Molybdenum Insertases. Encycl. Inorg. Bioinorg. Chem. 2020, 1–6. [Google Scholar] [CrossRef]
- Hercher, T.W.; Krausze, J.; Hoffmeister, S.; Zwerschke, D.; Lindel, T.; Blankenfeldt, W.; Mendel, R.R.; Kruse, T. Insights into the Cnx1E catalyzed MPT-AMP hydrolysis. Biosci. Rep. 2020, 40, BSR20191806. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R. The Molybdenum Cofactor. J. Biol. Chem. 2013, 288, 13165–13172. [Google Scholar] [CrossRef] [PubMed]
- Probst, C.; Yang, J.; Krausze, J.; Hercher, T.W.; Richers, C.P.; Spatzal, T.; Khadanand, K.C.; Giles, L.J.; Rees, D.C.; Mendel, R.R.; et al. Mechanism of molybdate insertion into pterin-based molybdenum cofactors. Nat. Chem. 2021, 13, 758–765. [Google Scholar] [CrossRef]
- Dobbek, H.; Huber, R. The Molybdenum and Tungsten Cofactors: A Crystallographic View. In Metal Ions in Biological Systems; Sigel, A., Sigel, H., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2002; Volume 39, pp. 227–263. [Google Scholar]
- Johnson, J.L.; Rajagopalan, K.V.; Mukund, S.; Adams, M.W.W. Identification of Molybdopterin As the Organic-Component of the Tungsten Cofactor in 4 Enzymes From Hyperthermophilic Archaea. J. Biol. Chem. 1993, 268, 4848–4852. [Google Scholar] [CrossRef] [PubMed]
- Kaufholdt, D.; Baillie, C.-K.; Meinen, R.; Mendel, R.R.; Hänsch, R. The Molybdenum Cofactor Biosynthesis Network: In vivo Protein-Protein Interactions of an Actin Associated Multi-Protein Complex. Front. Plant Sci. 2017, 8, 1946. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R.; Schwarz, G. Molybdenum cofactor biosynthesis in plants and humans. Coord. Chem. Rev. 2011, 255, 1145–1158. [Google Scholar] [CrossRef]
- Hille, R. The Mononuclear Molybdenum Enzymes. Chem. Rev. 1996, 96, 2757–2816. [Google Scholar] [CrossRef]
- Leimkuhler, S.; Mendel, R. Molybdenum Cofactor Biosynthesis. In Molybdenum and Tungsten Enzymes; Hille, R., Schulzke, C., Kirk, M.L., Eds.; RSC: Cambridge, UK, 2016; Volume 1, pp. 100–111. [Google Scholar]
- Leimkuhler, S.; Wuebbens, M.M.; Rajagopalan, K.V. The history of the discovery of the molybdenum cofactor and novel aspects of its biosynthesis in bacteria. Coord. Chem. Rev. 2011, 255, 1129–1144. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 839–847. [Google Scholar] [CrossRef]
- Kisker, C.; Schindelin, H.; Baas, D.; Retey, J.; Meckenstock, R.U.; Kroneck, P.M.H. A Structural Comparison of Molybdenum Cofactor-Containing Enzymes. FEMS Microbiol. Rev. 1998, 22, 503–521. [Google Scholar] [CrossRef]
- Kirk, M.L. Spectroscopic and Electronic Structure Studies Probing Mechanism: Introduction and Overview. In Molybdenum and Tungsten Enzymes: Spectroscopic and Theoretical Investigations; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–12. [Google Scholar]
- Pushie, M.J.; George, G.N. Spectroscopic studies of molybdenum and tungsten enzymes. Coord. Chem. Rev. 2011, 255, 1055–1084. [Google Scholar] [CrossRef]
- Metz, S.; Thiel, W. Theoretical studies on the reactivity of molybdenum enzymes. Coord. Chem. Rev. 2011, 255, 1085–1103. [Google Scholar] [CrossRef]
- George, G.N. X-Ray Absorption Spectroscopy of Molybdenum and Tungsten Enzymes. In Molybdenum and Tungsten Enzymes: Spectroscopic and Theoretical Investigations; Hille, R., Schulzke, C., Kirk, M.L., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 121–167. [Google Scholar]
- Kirk, M.L.; Peariso, K. Recent applications of MCD spectroscopy to metalloenzymes. Curr. Opin. Chem. Biol. 2003, 7, 220–227. [Google Scholar] [CrossRef]
- Kirk, M.L. Spectroscopic and Electronic Structure Studies of Mo Model Compounds and Enzymes; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 13–67. [Google Scholar]
- Chan, M.K.; Mukund, S.; Kletzin, A.; Adams, M.W.W.; Rees, D.C. Structure of a Hyperthermophilic Tungstopterin Enzyme, Aldehyde Ferredoxin Oxidoreductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef]
- Schindelin, H.; Kisker, C.; Hilton, J.; Rajagopalan, K.V.; Rees, D.C. Crystal structure of DMSO reductase: Redox-linked changes in molybdopterin coordination. Science 1996, 272, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, H.; Kisker, C.; Rees, D.C. The molybdenum-cofactor: A crystallographic perspective. J. Biol. Inorg. Chem. 1997, 2, 773–781. [Google Scholar] [CrossRef]
- Rothery, R.A.; Stein, B.; Solomonson, M.; Kirk, M.L.; Weiner, J.H. Pyranopterin conformation defines the function of molybdenum and tungsten enzymes. Proc. Natl. Acad. Sci. USA 2012, 109, 14773–14778. [Google Scholar] [CrossRef] [PubMed]
- Rothery, R.A.; Weiner, J.H. Shifting the metallocentric molybdoenzyme paradigm: The importance of pyranopterin coordination. J. Biol. Inorg. Chem. 2015, 20, 349–372. [Google Scholar] [CrossRef]
- Bertero, M.G.; Rothery, R.A.; Palak, M.; Hou, C.; Lim, D.; Blasco, F.; Weiner, J.H.; Strynadka, N.C.J. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat. Struct. Biol. 2003, 10, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Kloer, D.P.; Hagel, C.; Heider, J.; Schulz, G.E. Crystal structure of ethylbenzene dehydrogenase from Aromatoleum aromaticum. Structure 2006, 14, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Youngblut, M.D.; Tsai, C.L.; Clark, I.C.; Carlson, H.K.; Maglaqui, A.P.; Gau-Pan, P.S.; Redford, S.A.; Wong, A.; Tainer, J.A.; Coates, J.D. Perchlorate Reductase Is Distinguished by Active Site Aromatic Gate Residues. J. Biol. Chem. 2016, 291, 9190–9202. [Google Scholar] [CrossRef] [PubMed]
- Inscore, F.E.; McNaughton, R.; Westcott, B.L.; Helton, M.E.; Jones, R.; Dhawan, I.K.; Enemark, J.H.; Kirk, M.L. Spectroscopic evidence for a unique bonding interaction in oxo-molybdenum dithiolate complexes: Implications for sigma electron transfer pathways in the pyranopterin dithiolate centers of enzymes. Inorg. Chem. 1999, 38, 1401–1410. [Google Scholar] [CrossRef]
- Kirk, M.L.; McNaughton, R.L.; Helton, M.E. The Electronic Structure and Spectroscopy of Metallo-Dithiolene Complexes. In Progress in Inorganic Chemistry: Synthesis, Properties, and Applications; Stiefel, E.I., Karlin, K.D., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2004; Volume 52, pp. 111–212. [Google Scholar]
- Matz, K.G.; Mtei, R.P.; Leung, B.; Burgmayer, S.J.N.; Kirk, M.L. Noninnocent Dithiolene Ligands: A New Oxomolybdenum Complex Possessing a Donor Acceptor Dithiolene Ligand. J. Am. Chem. Soc. 2010, 132, 7830–7831. [Google Scholar] [CrossRef]
- Mtei, R.P.; Lyashenko, G.; Stein, B.; Rubie, N.; Hille, R.; Kirk, M.L. Spectroscopic and Electronic Structure Studies of a Dimethyl Sulfoxide Reductase Catalytic Intermediate: Implications for Electron- and Atom-Transfer Reactivity. J. Am. Chem. Soc. 2011, 133, 9762–9774. [Google Scholar] [CrossRef]
- Yang, J.; Mogesa, B.; Basu, P.; Kirk, M.L. Large Ligand Folding Distortion in an Oxomolybdenum Donor Acceptor Complex. Inorg. Chem. 2016, 55, 785–793. [Google Scholar] [CrossRef]
- Dong, C.; Yang, J.; Leimkühler, S.; Kirk, M.L. Pyranopterin Dithiolene Distortions Relevant to Electron Transfer in Xanthine Oxidase/Dehydrogenase. Inorg. Chem. 2014, 53, 7077–7079. [Google Scholar] [CrossRef]
- Dong, C.; Yang, J.; Reschke, S.; Leimkühler, S.; Kirk, M.L. Vibrational Probes of Molybdenum Cofactor–Protein Interactions in Xanthine Dehydrogenase. Inorg. Chem. 2017, 56, 6830–6837. [Google Scholar] [CrossRef]
- Stein, B.W.; Yang, J.; Mtei, R.; Wiebelhaus, N.J.; Kersi, D.K.; LePluart, J.; Lichtenberger, D.L.; Enemark, J.H.; Kirk, M.L. Vibrational Control of Covalency Effects Related to the Active Sites of Molybdenum Enzymes. J. Am. Chem. Soc. 2018, 140, 14777–14788. [Google Scholar] [CrossRef]
- Gates, C.; Varnum, H.; Getty, C.; Loui, N.; Chen, J.; Kirk, M.L.; Yang, J.; Nieter Burgmayer, S.J. Protonation and Non-Innocent Ligand Behavior in Pyranopterin Dithiolene Molybdenum Complexes. Inorg. Chem. 2022, 61, 13728–13742. [Google Scholar] [CrossRef] [PubMed]
- Garton, S.D.; Hilton, J.; Oku, H.; Crouse, B.R.; Rajagopalan, K.V.; Johnson, M.K. Active Site Structures and Catalytic Mechanism of Rhodobacter sphaeroides Dimethyl Sulfoxide Reductase as Revealed by Resonance Raman Spectroscopy. J. Am. Chem. Soc. 1997, 119, 12906–12916. [Google Scholar] [CrossRef]
- Johnson, M.K.; Garton, S.D.; Oku, H. Resonance Raman as a Direct Probe for the Catalytic Mechanism of Molybdenum Oxotransferases. J. Biol. Inorg. Chem. 1997, 2, 797–803. [Google Scholar] [CrossRef]
- Bell, A.F.; He, X.; Ridge, J.P.; Hanson, G.R.; McEwan, A.G.; Tonge, P.J. Active site heterogeneity in dimethyl sulfoxide reductase from Rhodobacter capsulatus revealed by Raman spectroscopy. Biochemistry 2001, 40, 440–448. [Google Scholar] [CrossRef]
- Garton, S.D.; Temple, C.A.; Dhawan, I.K.; Barber, M.J.; Rajagopalan, K.V.; Johnson, M.K. Resonance Raman Characterization of Biotin Sulfoxide Reductase: Comparing Oxomolybdenum Enzymes in the Me2SO Reductase Family. J. Biol. Chem. 2000, 275, 6798–6805. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dong, C.; Kirk, M.L. Xanthine oxidase-product complexes probe the importance of substrate/product orientation along the reaction coordinate. Dalton Trans. 2017, 46, 13242–13250. [Google Scholar] [CrossRef]
- Hemann, C.; Ilich, P.; Stockert, A.L.; Choi, E.Y.; Hille, R. Resonance Raman studies of xanthine oxidase: The reduced enzyme—Product complex with violapterin. J. Phys. Chem. B 2005, 109, 3023–3031. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Olson, J.; Palmer, G. The Reaction of Xanthine Oxidase with Lumazine: Characterization of the Reductive Half-reaction. J. Biol. Chem. 1984, 259, 3526–3533. [Google Scholar] [CrossRef]
- Kim, J.H.; Ryan, M.G.; Knaut, H.; Hille, R. The reductive half-reaction of xanthine oxidase: The involvement of prototropic equilibria in the course of the catalytic sequence. J. Biol. Chem. 1996, 271, 6771–6780. [Google Scholar] [CrossRef]
- Hemann, C.; Ilich, P.; Hille, R. Vibrational spectra of lumazine in water at pH 2–13: Ab initio calculation and FTIR/Raman spectra. J. Phys. Chem. B 2003, 107, 2139–2155. [Google Scholar] [CrossRef]
- Pauff, J.M.; Cao, H.; Hille, R. Substrate Orientation and Catalysis at the Molybdenum Site in Xanthine Oxidase Crystal Structures in Complex with Xanthine and Lumazine. J. Biol. Chem. 2009, 284, 8751–8758. [Google Scholar] [CrossRef]
- Basu, P.; Burgmayer, S.J.N. Pterin chemistry and its relationship to the molybdenum cofactor. Coord. Chem. Rev. 2011, 255, 1016–1038. [Google Scholar] [CrossRef]
- Kappock, T.J.; Caradonna, J.P. Pterin-dependent amino acid hydroxylases. Chem. Rev. 1996, 96, 2659–2756. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Johnson, J. The Pterin Molybdenum Cofactors. J. Biol. Chem. 1992, 267, 10199–10202. [Google Scholar] [CrossRef]
- Gardlik, S.; Rajagopalan, K. Oxidation of Molybdopterin in Sulfite Oxidase by Ferricyanide- Effect on Electron Transfer Activities. J. Biol. Chem. 1991, 266, 4889–4895. [Google Scholar] [CrossRef] [PubMed]
- Gardlik, S.; Rajagopalan, K.V. The State of Reduction of Molybdopterin in Xanthine-Oxidase and Sulfite Oxidase. J. Biol. Chem. 1990, 265, 13047–13054. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.; Rajagopalan, K.V.; Hilton, J.; Bastian, N.R.; Stiefel, E.I.; Pilato, R.S.; Spiro, T.G. Resonance Raman Spectroscopic Characterization of the Molybdopterin Active Site of DMSO Reductase. Biochemistry 1995, 34, 3032–3039. [Google Scholar] [CrossRef]
- Helton, M.; Gruhn, N.; McNaughton, R.; Kirk, M. Control of oxo-molybdenum reduction and ionization potentials by dithiolate donors. Inorg. Chem. 2000, 39, 2273–2278. [Google Scholar] [CrossRef]
- Helton, M.E.; Kirk, M.L. A model for ferricyanide-inhibited sulfite oxidase. Inorg. Chem. 1999, 38, 4384–4385. [Google Scholar] [CrossRef] [PubMed]
- Gisewhite, D.R.; Yang, J.; Williams, B.R.; Esmail, A.; Stein, B.; Kirk, M.L.; Burgmayer, S.J.N. Implications of Pyran Cyclization and Pterin Conformation on Oxidized Forms of the Molybdenum Cofactor. J. Am. Chem. Soc. 2018, 140, 12808–12818. [Google Scholar] [CrossRef]
- Williams, B.R.; Gisewhite, D.; Kalinsky, A.; Esmail, A.; Burgmayer, S.J.N. Solvent-Dependent Pyranopterin Cyclization in Molybdenum Cofactor Model Complexes. Inorg. Chem. 2015, 54, 8214–8222. [Google Scholar] [CrossRef]
- Williams, B.R.; Fu, Y.C.; Yap, G.P.A.; Burgmayer, S.J.N. Structure and Reversible Pyran Formation in Molybdenum Pyranopterin Dithiolene Models of the Molybdenum Cofactor. J. Am. Chem. Soc. 2012, 134, 19584–19587. [Google Scholar] [CrossRef][Green Version]
- Wuebbens, M.M.; Rajagopalan, K.V. Structural Characterization of a Molybdopterin Precursor. J. Biol. Chem. 1993, 268, 13493–13498. [Google Scholar] [CrossRef]
- Ceccaldi, P.; Rendon, J.; Leger, C.; Toci, R.; Guigliarelli, B.; Magalon, A.; Grimaldi, S.; Fourmond, V. Reductive activation of E. coli respiratory nitrate reductase. Biochim. Et Biophys. Acta-Bioenerg. 2015, 1847, 1055–1063. [Google Scholar] [CrossRef]
- George, G.N.; Hilton, J.; Temple, C.; Prince, R.C.; Rajagopalan, K.V. Structure of the Molybdenum Site of Dimethyl Sulfoxide Reductase. J. Am. Chem. Soc. 1999, 121, 1256–1266. [Google Scholar] [CrossRef]
- Jacques, J.G.J.; Fourmond, V.; Arnoux, P.; Sabaty, M.; Etienne, E.; Grosse, S.; Biaso, F.; Bertrand, P.; Pignol, D.; Leger, C.; et al. Reductive activation in periplasmic nitrate reductase involves chemical modifications of the Mo-cofactor beyond the first coordination sphere of the metal ion. Biochim. Et Biophys. Acta-Bioenerg. 2014, 1837, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Rothery, R.A.; Weiner, J.H. Pyranopterin Coordination Controls Molybdenum Electrochemistry in Escherichia coli Nitrate Reductase. J. Biol. Chem. 2015, 290, 25164–25173. [Google Scholar] [CrossRef]
- Duval, S.; Santini, J.M.; Lemaire, D.; Chaspoul, F.; Russell, M.J.; Grimaldi, S.; Nitschke, W.; Schoepp-Cothenet, B. The H-bond network surrounding the pyranopterins modulates redox cooperativity in the molybdenum-bisPGD cofactor in arsenite oxidase. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1353. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, B.; Dinsmore, A.; Ajana, W.; Collison, D.; Garner, C.D.; Joule, J.A. Synthesis of the organic ligand of the molybdenum cofactor, in protected form. J. Chem. Soc. Perkin Trans. 1 2001, 3239–3244. [Google Scholar] [CrossRef]
- Pimkov, I.V.; Serli-Mitasev, B.; Peterson, A.A.; Ratvasky, S.C.; Hammann, B.; Basu, P. Designing the Molybdopterin Core through Regioselective Coupling of Building Blocks. Chem. A Eur. J. 2015, 21, 17057–17072. [Google Scholar] [CrossRef]
- Inscore, F.E.; Knottenbelt, S.Z.; Rubie, N.D.; Joshi, H.K.; Kirk, M.L.; Enemark, J.H. Understanding the origin of metal-sulfur vibrations in an oxo-molybdenurn dithiolene complex: Relevance to sulfite oxidase. Inorg. Chem. 2006, 45, 967. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, R.L.; Helton, M.E.; Rubie, N.D.; Kirk, M.L. The oxo-gate hypothesis and DMSO reductase: Implications for a psuedo-sigma bonding interaction involved in enzymatic electron transfer. Inorg. Chem. 2000, 39, 4386. [Google Scholar] [CrossRef]
- Winkler, J.R.; Gray, H.B. Electronic Structures of Oxo-Metal Ions. In Molecular Electronic Structures of Transition Metal Complexes I; Mingos, D.M.P., Day, P., Dahl, J.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 142, pp. 17–28. [Google Scholar]
- Yang, J.; Rothery, R.; Sempombe, J.; Weiner, J.H.; Kirk, M.L. Spectroscopic Characterization of YedY: The Role of Sulfur Coordination in a Mo(V) Sulfite Oxidase Family Enzyme Form. J. Am. Chem. Soc. 2009, 131, 15612–15614. [Google Scholar] [CrossRef] [PubMed]
- Paudel, J.; Pokhrel, A.; Kirk, M.L.; Li, F.F. Remote Charge Effects on the Oxygen-Atom-Transfer Reactivity and Their Relationship to Molybdenum Enzymes. Inorg. Chem. 2019, 58, 2054–2068. [Google Scholar] [CrossRef] [PubMed]
- Kroneck, P.M.H. Acetylene hydratase: A non-redox enzyme with tungsten and iron-sulfur centers at the active site. J. Biol. Inorg. Chem. 2016, 21, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.Z.; Thiel, W. On the Effect of Varying Constraints in the Quantum Mechanics Only Modeling of Enzymatic Reactions: The Case of Acetylene Hydratase. J. Phys. Chem. B 2013, 117, 3954–3961. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.Z.; Yu, J.G.; Himo, F. Mechanism of tungsten-dependent acetylene hydratase from quantum chemical calculations. Proc. Natl. Acad. Sci. USA 2010, 107, 22523. [Google Scholar] [CrossRef]
- Seiffert, G.B.; Ullmann, G.M.; Messerschmidt, A.; Schink, B.; Kroneck, P.M.; Einsle, O. Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc. Natl. Acad. Sci. USA 2007, 104, 3073–3077. [Google Scholar] [CrossRef]
- Mtei, R.P.; Perera, E.; Mogesa, B.; Stein, B.; Basu, P.; Kirk, M.L. A Valence Bond Description of Dizwitterionic Dithiolene Character in an Oxomolybdenum-Bis(dithione) Complex. Eur. J. Inorg. Chem. 2011, 2011, 5467–5470. [Google Scholar] [CrossRef]
- Hsu, J.K.; Bonangelino, C.J.; Kaiwar, S.P.; Boggs, C.M.; Fettinger, J.C.; Pilato, R.S. Direct conversion of alpha-substituted ketones to metallo-1,2-enedithiolates. Inorg. Chem. 1996, 35, 4743–4751. [Google Scholar] [CrossRef]
- Gisewhite, D.R.; Nagelski, A.L.; Cummins, D.C.; Yap, G.P.A.; Burgmayer, S.J.N. Modeling Pyran Formation in the Molybdenum Cofactor: Protonation of Quinoxalyl–Dithiolene Promoting Pyran Cyclization. Inorg. Chem. 2019, 58, 5134–5144. [Google Scholar] [CrossRef]
- Pilato, R.S.; Eriksen, K.A.; Greaney, M.A.; Stiefel, E.I.; Goswami, S.; Kilpatrick, L.; Spiro, T.G.; Taylor, E.C.; Rheingold, A.L. Model Complexes for Molybdopterin-Containing Enzymes: Preparation and Crystallographic Characterization of a Molybdenum-Ene-1-Perthiolate-2-Thiolate (Trithiolate) Complex. J. Am. Chem. Soc. 1991, 113, 9372–9374. [Google Scholar] [CrossRef]
- Pilato, R.S.; Eriksen, K.; Greaney, M.A.; Gea, Y.; Taylor, E.C.; Goswami, S.; Kilpatrick, L.; Spiro, T.G.; Rheingold, A.L.; Stiefel, E.I. Pterins, Quinoxalines, and Metallo-ene-dithiolates—Synthetic Approach to the Molybdenum Cofactor. ACS Symp. Ser. 1993, 535, 83–97. [Google Scholar]
- Dinsmore, A.; Birks, J.H.; Garner, C.D.; Joule, J.A. Synthesis of (eta(5)-cyclopentadienyl)-1-(4-benzyloxycarbonyl-3;4-dihydroquin oxalin-2-yl)ethene-1;2-dithiolatocobalt(III) and (eta(5)-cyclopentadienyl)-1-[2-(N;N-dimethylaminomethyleneamino)-3-methyl-4-oxopteridin-6-yl]ethene-1;2-dithiolatocobalt(III). J. Chem. Soc. -Perkin Trans. 1 1997, 1997, 801–807. [Google Scholar]
- Shultz, D.A.; Kirk, M.L.; Zhang, J.Y.; Stasiw, D.E.; Wang, G.B.; Yang, J.; Habel-Rodriguez, D.; Stein, B.W.; Sommer, R.D. Spectroscopic Signatures of Resonance Inhibition Reveal Differences in Donor-Bridge and Bridge-Acceptor Couplings. J. Am. Chem. Soc. 2020, 142, 4916–4924. [Google Scholar] [CrossRef]
- Guasch, L.; Sitzmann, M.; Nicklaus, M.C. Enumeration of Ring-Chain Tautomers Based on SMIRKS Rules. J. Chem. Inf. Model. 2014, 54, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Matz, K.G.; Mtei, R.P.; Rothstein, R.; Kirk, M.L.; Burgmayer, S.J.N. Study of Molybdenum(4+) Quinoxalyldithiolenes as Models for the Noninnocent Pyranopterin in the Molybdenum Cofactor. Inorg. Chem. 2011, 50, 9804–9815. [Google Scholar] [CrossRef] [PubMed]
- Boyde, S.; Garner, C.D. Electrochemistry of Tris(Quinoxaline-2,3-Dithiolato)Molybdate(IV) in Acidic Solution: Multi-Electron Ligand-Based Redox Activity. J. Chem. Soc. Dalton Trans. 1991, 713–716. [Google Scholar] [CrossRef]
- Dicks, J.P.; Zubair, M.; Davies, E.S.; Garner, C.D.; Schulzke, C.; Wilson, C.; McMaster, J. Synthesis, Structure and Redox Properties of Asymmetric (Cyclopentadienyl)(ene-1,2-dithiolate)cobalt(III) Complexes Containing Phenyl, Pyridyl and Pyrazinyl Units. Eur. J. Inorg. Chem. 2015, 2015, 3550–3561. [Google Scholar] [CrossRef]
- Li, Y.; Gomez-Mingot, M.; Fogeron, T.; Fontecave, M. Carbon Dioxide Reduction: A Bioinspired Catalysis Approach. Acc. Chem. Res. 2021, 54, 4250–4261. [Google Scholar] [CrossRef]
- Porcher, J.P.; Fogeron, T.; Gomez-Mingot, M.; Derat, E.; Chamoreau, L.M.; Li, Y.; Fontecave, M. A Bioinspired Molybdenum Complex as a Catalyst for the Photo- and Electroreduction of Protons. Angew. Chem. Int. Ed. 2015, 54, 14090–14093. [Google Scholar] [CrossRef] [PubMed]
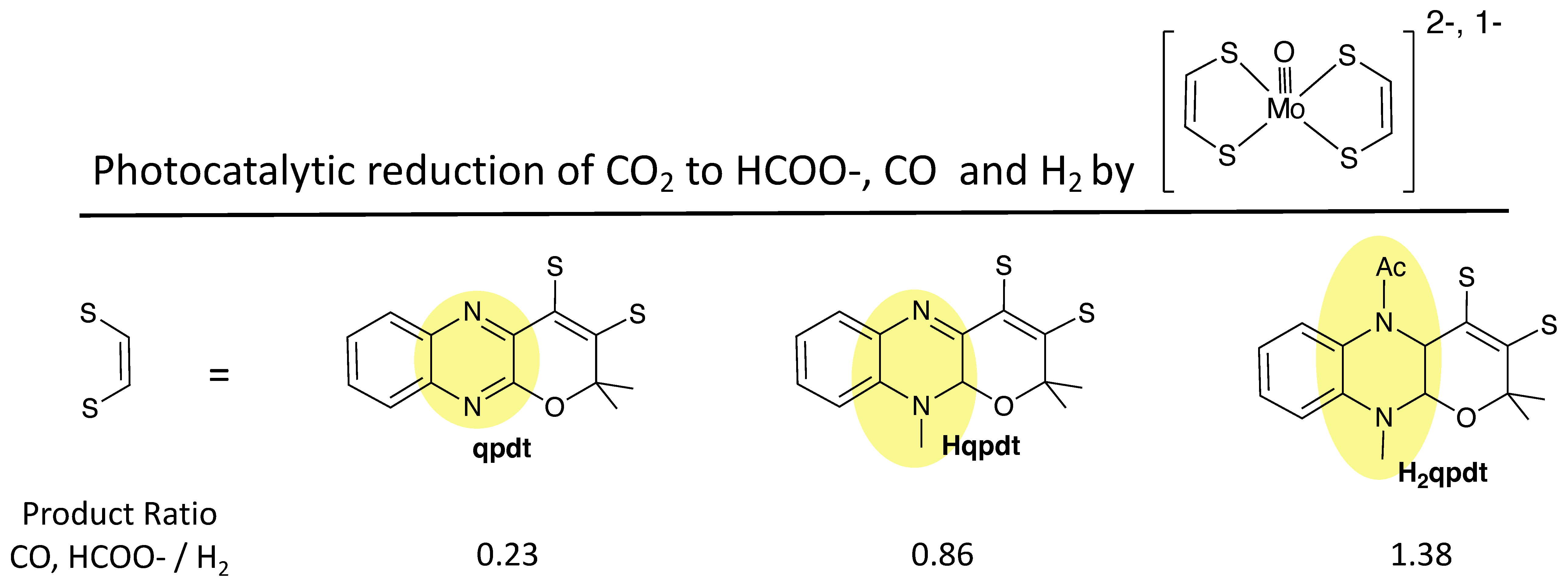
- Fogeron, T.; Retailleau, P.; Chamoreau, L.-M.; Li, Y.; Fontecave, M. Pyranopterin Related Dithiolene Molybdenum Complexes as Homogeneous Catalysts for CO2 Photoreduction. Angew. Chem. Int. Ed. 2018, 57, 17033–17037. [Google Scholar] [CrossRef] [PubMed]
- Porcher, J.-P.; Fogeron, T.; Gomez-Mingot, M.; Chamoreau, L.-M.; Li, Y.; Fontecave, M. Synthesis and Reactivity of a Bio-inspired Dithiolene Ligand and its Mo Oxo Complex. Chem. A Eur. J. 2016, 22, 4447–4453. [Google Scholar] [CrossRef] [PubMed]
- Fogeron, T.; Retailleau, P.; Chamoreau, L.-M.; Fontecave, M.; Li, Y. The unusual ring scission of a quinoxaline-pyran-fused dithiolene system related to molybdopterin. Dalton Trans. 2017, 46, 4161–4164. [Google Scholar] [CrossRef] [PubMed]
- Fogeron, T.; Retailleau, P.; Gomez-Mingot, M.; Li, Y.; Fontecave, M. Nickel Complexes Based on Molybdopterin-like Dithiolenes: Catalysts for CO2 Electroreduction. Organometallics 2019, 38, 1344–1350. [Google Scholar] [CrossRef]
- Hille, R.; Massey, V. Studies on the Oxidative Half-Reaction of Xanthine Oxidase. J. Biol. Chem. 1981, 256, 9090–9095. [Google Scholar] [CrossRef]
- Jones, R.M.; Inscore, F.E.; Hille, R.; Kirk, M.L. Freeze-Quench Magnetic Circular Dichroism Spectroscopic Study of the “Very Rapid” Intermediate in Xanthine Oxidase. Inorg. Chem. 1999, 38, 4963–4970. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Anderson, R.F. Coupled Electron/Proton Transfer in Complex Flavoproteins: Solvent Kinetic Isotope Effect Studies of Electron Transfer in Xanthine Oxidase and Trimethylamine Dehydrogenase. J. Biol. Chem. 2001, 276, 31193–31201. [Google Scholar] [CrossRef]
- Adamson, H.; Simonov, A.N.; Kierzek, M.; Rothery, R.A.; Weiner, J.H.; Bond, A.M.; Parkin, A. Electrochemical evidence that pyranopterin redox chemistry controls the catalysis of YedY, a mononuclear Mo enzyme. Proc. Natl. Acad. Sci. USA 2015, 112, 14506–14511. [Google Scholar] [CrossRef]
























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgmayer, S.J.N.; Kirk, M.L. Advancing Our Understanding of Pyranopterin-Dithiolene Contributions to Moco Enzyme Catalysis. Molecules 2023, 28, 7456. https://doi.org/10.3390/molecules28227456
Burgmayer SJN, Kirk ML. Advancing Our Understanding of Pyranopterin-Dithiolene Contributions to Moco Enzyme Catalysis. Molecules. 2023; 28(22):7456. https://doi.org/10.3390/molecules28227456
Chicago/Turabian StyleBurgmayer, Sharon J. Nieter, and Martin L. Kirk. 2023. "Advancing Our Understanding of Pyranopterin-Dithiolene Contributions to Moco Enzyme Catalysis" Molecules 28, no. 22: 7456. https://doi.org/10.3390/molecules28227456
APA StyleBurgmayer, S. J. N., & Kirk, M. L. (2023). Advancing Our Understanding of Pyranopterin-Dithiolene Contributions to Moco Enzyme Catalysis. Molecules, 28(22), 7456. https://doi.org/10.3390/molecules28227456