
Deep-Learning-Based Mixture Identification for Nuclear Magnetic Resonance Spectroscopy Applied to Plant Flavors

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
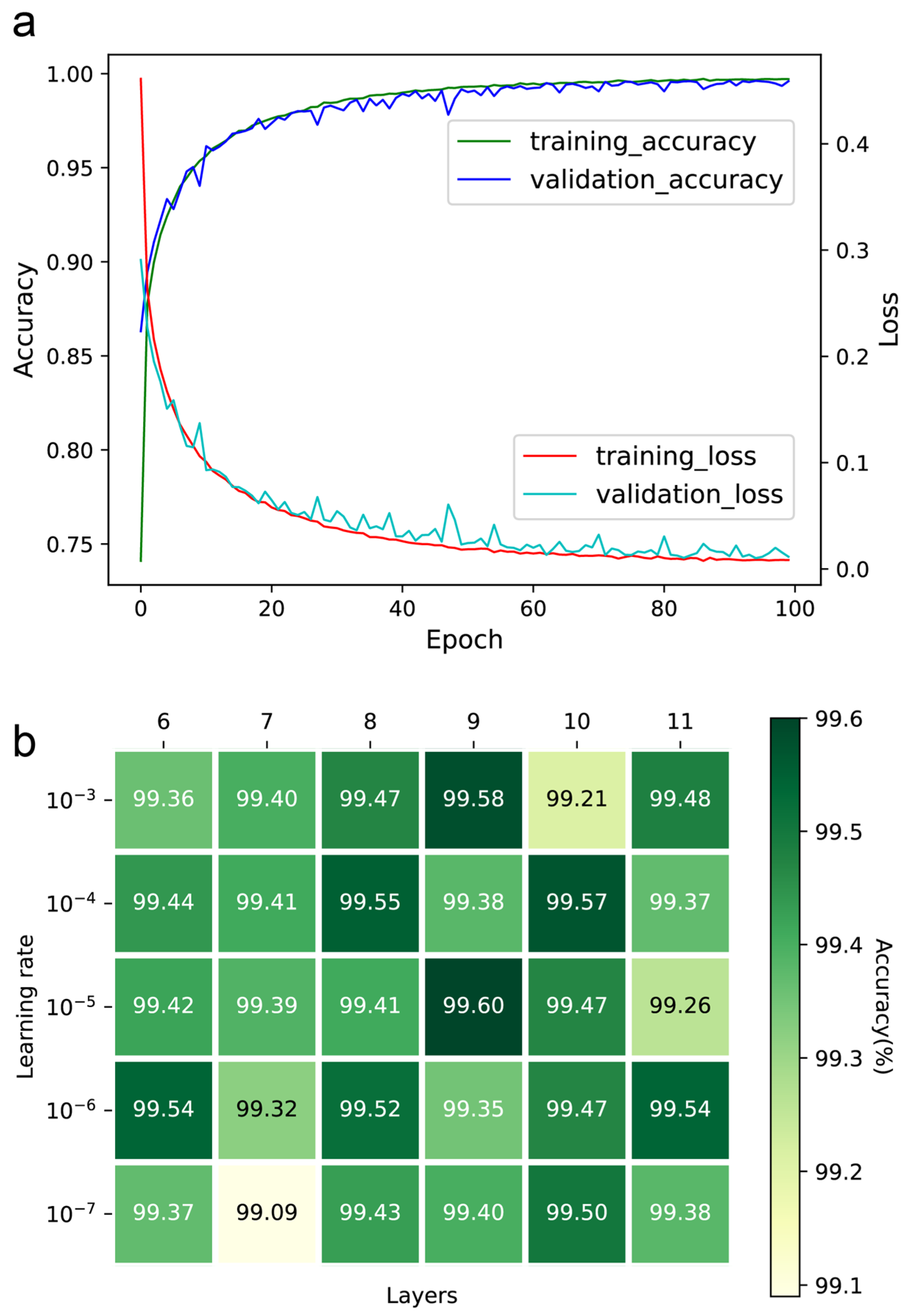
2.1. Implementation, Optimization, and Training of DeepMID
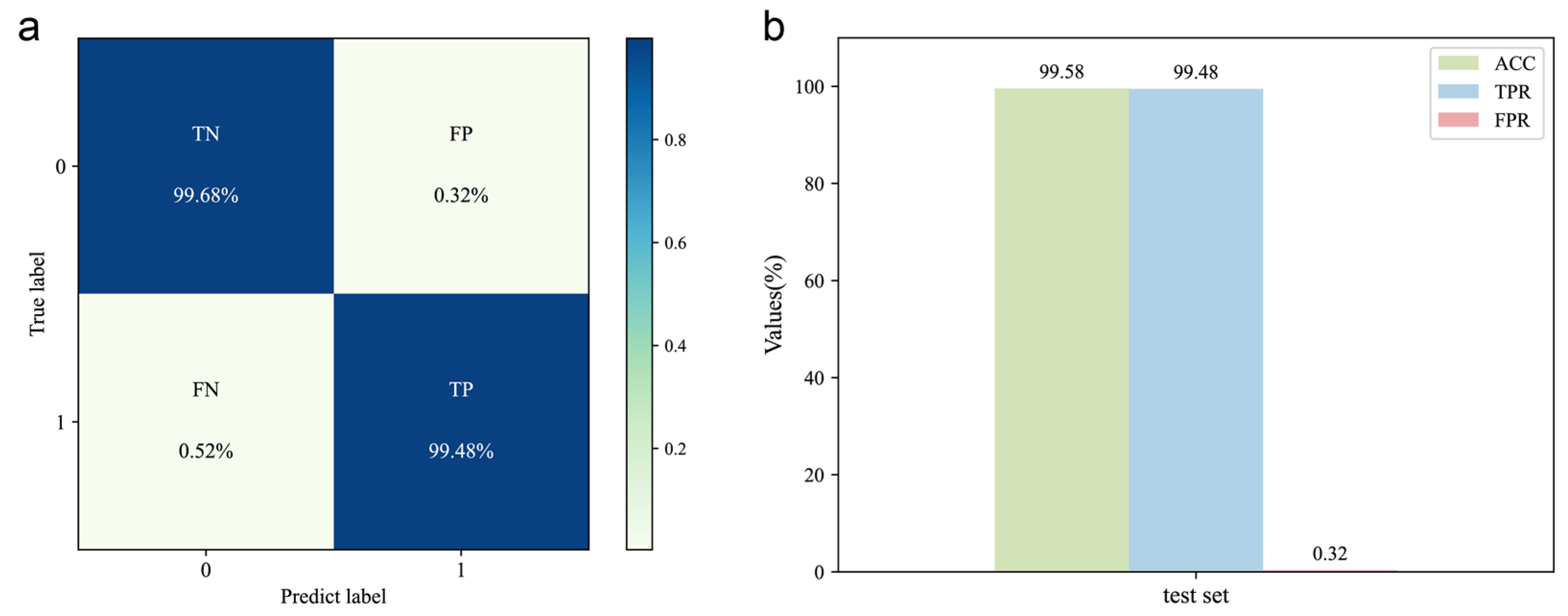
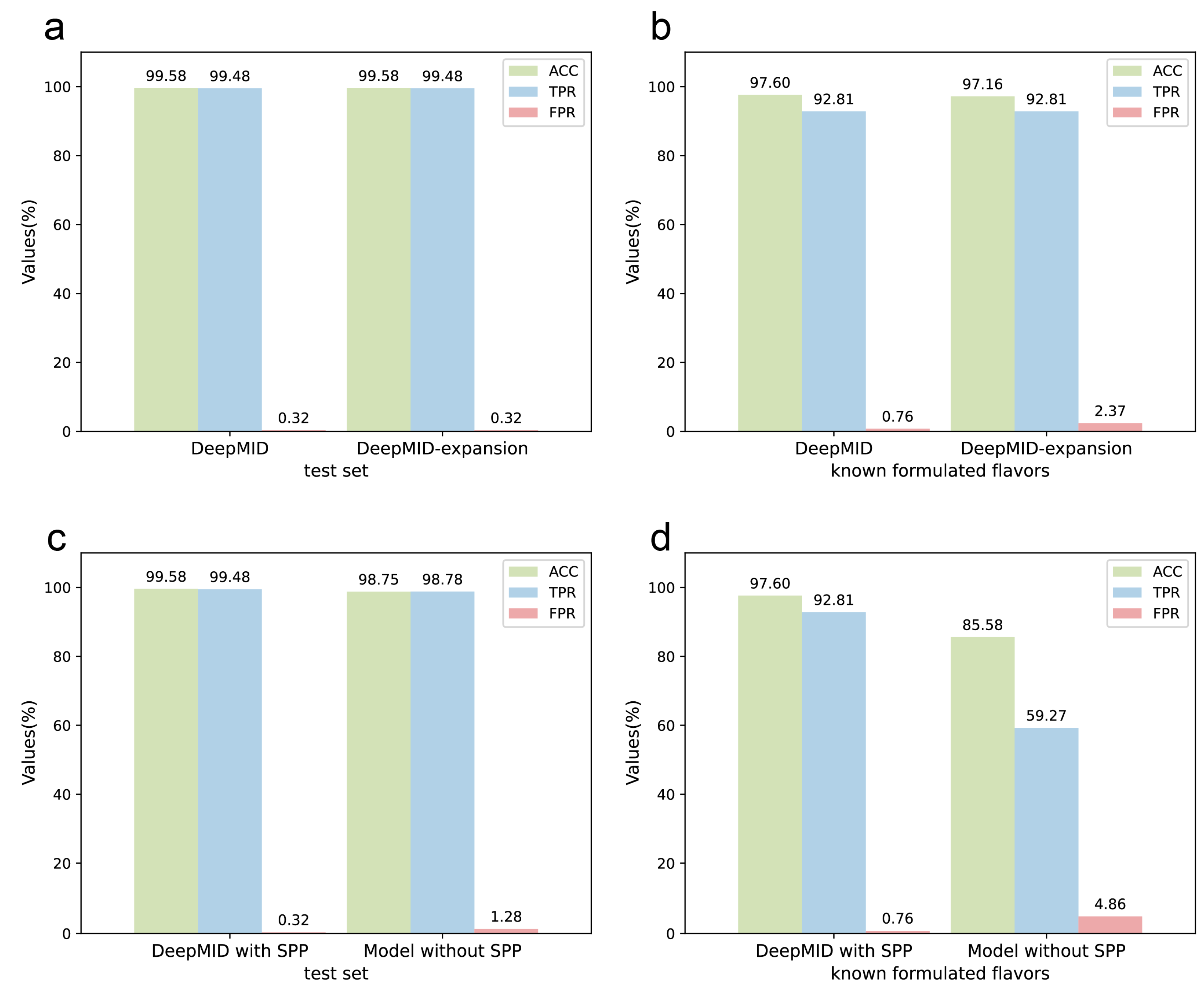
2.2. Evaluation of the DeepMID Model
2.3. Results of Mixture Identification
2.4. Elucidation of Unknown Formulated Flavors
2.5. Stability of the DeepMID Model
2.6. Expansion of the Spectral Database
2.7. Comparison with the Model without the SPP Layer
3. Methods
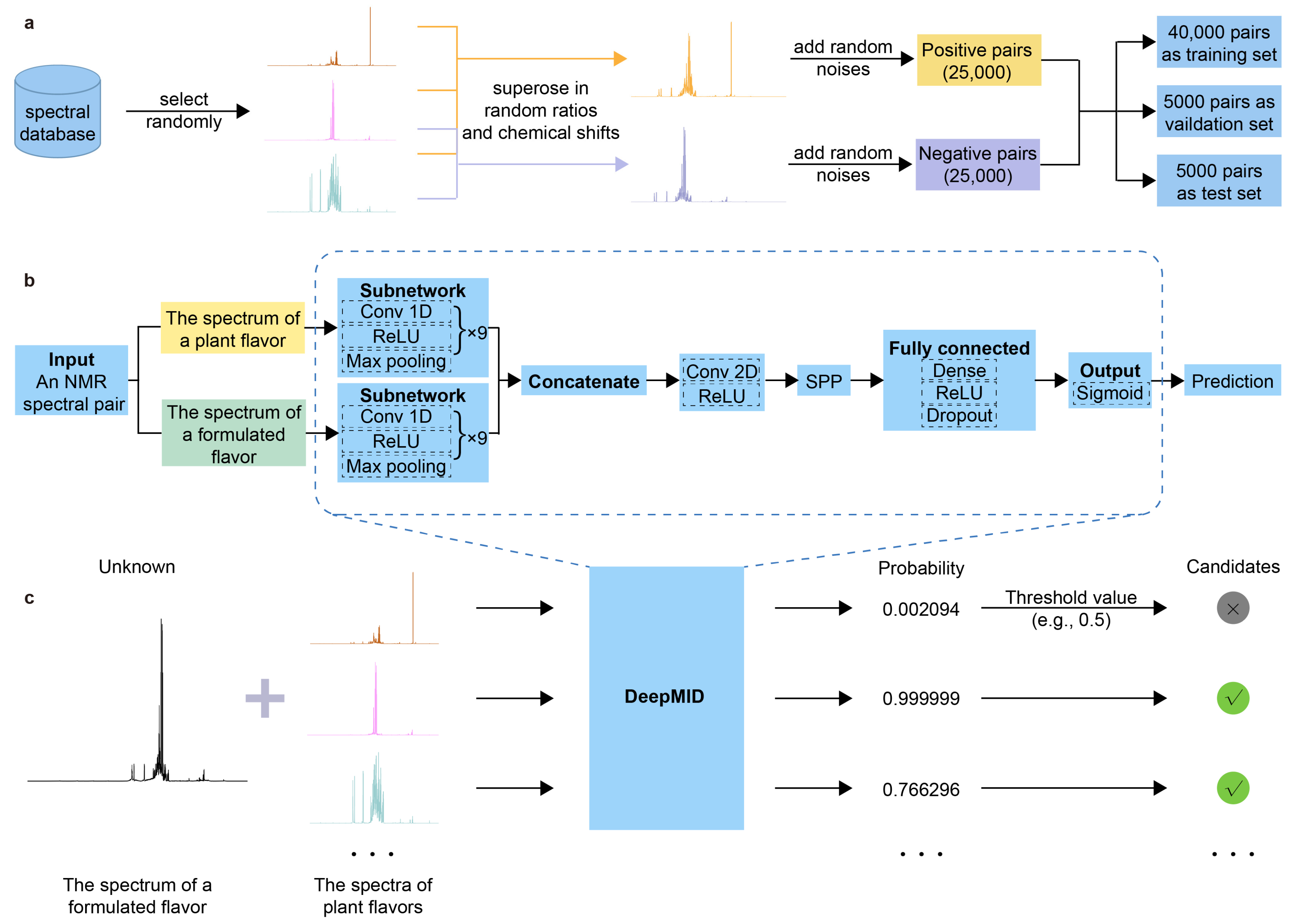
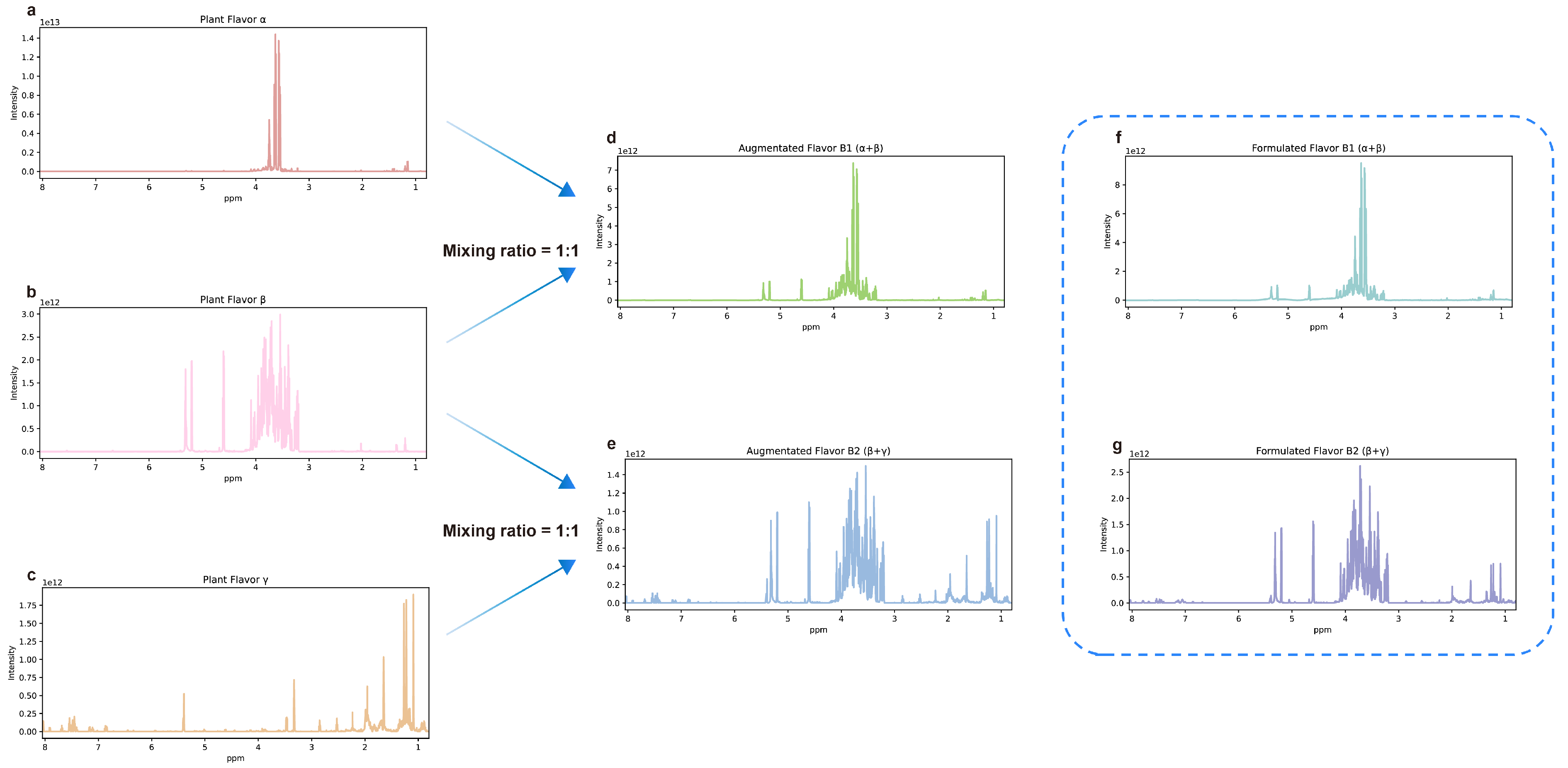
3.1. Data Set Curation
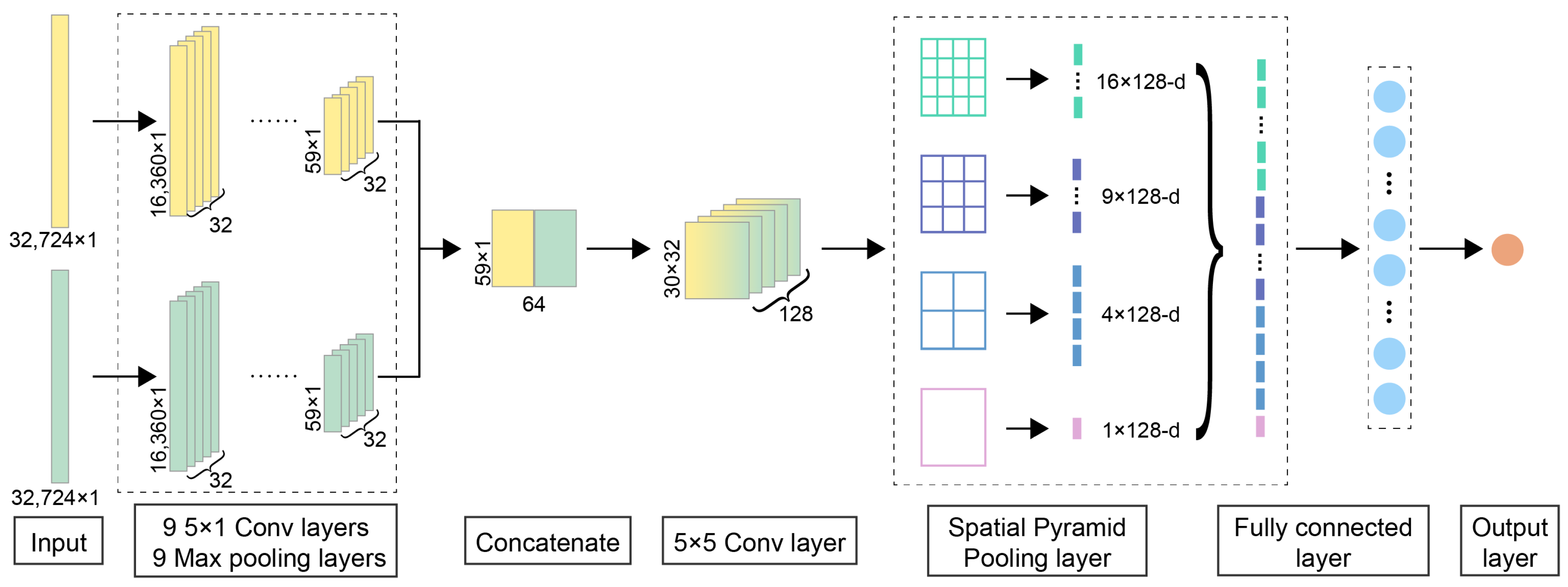
3.2. Pseudo-Siamese Neural Network
3.3. Spatial Pyramid Pooling
3.4. Detailed Network Architecture
3.5. Mixture Identification
3.6. Evaluation Metrics
4. Experiments
4.1. Plant Flavors
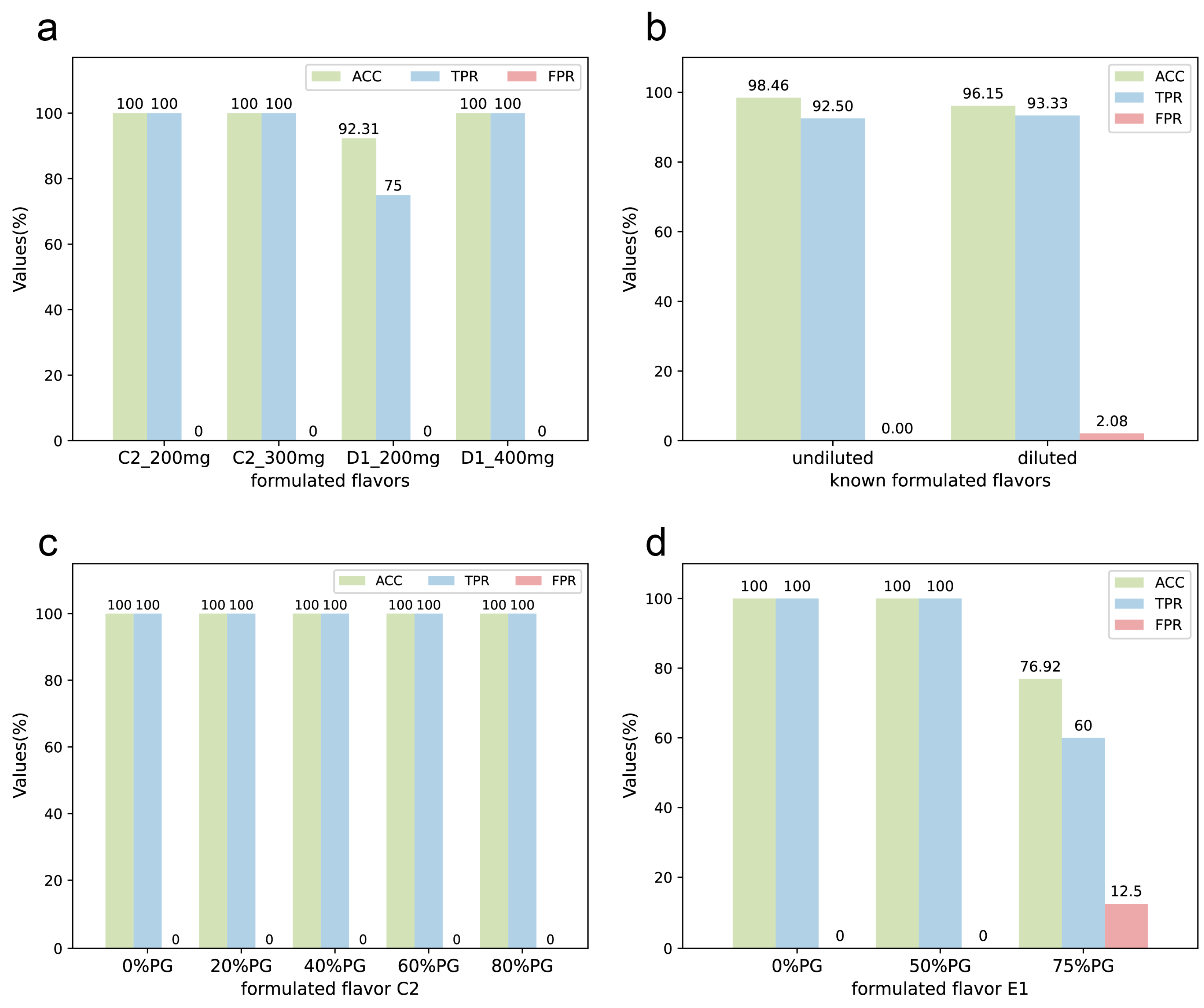
4.2. Known Formulated Flavors
4.3. Unknown Formulated Flavors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug. Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Claridge, T.D.W. Chapter 2—Introducing high-resolution NMR. In High-Resolution NMR Techniques in Organic Chemistry; Claridge, T.D.W., Ed.; Tetrahedron Organic Chemistry Series; Elsevier: Amsterdam, The Netherlands, 2009; Volume 27, pp. 11–34. [Google Scholar]
- Softley, C.A.; Bostock, M.J.; Popowicz, G.M.; Sattler, M. Paramagnetic NMR in drug discovery. J. Biomol. NMR 2020, 74, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Edison, A.S.; Colonna, M.; Gouveia, G.J.; Holderman, N.R.; Judge, M.T.; Shen, X.N.; Zhang, S.C. NMR: Unique Strengths That Enhance Modern Metabolomics Research. Anal. Chem. 2021, 93, 478–499. [Google Scholar] [CrossRef]
- Wishart, D.S. Quantitative metabolomics using NMR. TrAC Trends Anal. Chem. 2008, 27, 228–237. [Google Scholar] [CrossRef]
- Cao, R.G.; Liu, X.R.; Liu, Y.Q.; Zhai, X.Q.; Cao, T.Y.; Wang, A.L.; Qiu, J. Applications of nuclear magnetic resonance spectroscopy to the evaluation of complex food constituents. Food Chem. 2021, 342, 128258. [Google Scholar] [CrossRef]
- Martin, G.J.; Martin, M.L. Thirty Years of Flavor NMR. In Flavor Chemistry: Thirty Years of Progress; Teranishi, R., Wick, E.L., Hornstein, I., Eds.; Springer: Boston, MA, USA, 1999; pp. 19–30. [Google Scholar]
- Tsedilin, A.M.; Fakhrutdinov, A.N.; Eremin, D.B.; Zalesskiy, S.S.; Chizhov, A.O.; Kolotyrkina, N.G.; Ananikov, V.P. How sensitive and accurate are routine NMR and MS measurements? Mendeleev Commun. 2015, 25, 454–456. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K. Essentials of Pharmaceutical Analysis; Springer: Singapore, 2020. [Google Scholar]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Huang, L.; Ho, C.-T.; Wang, Y. Biosynthetic pathways and metabolic engineering of spice flavors. Crit. Rev. Food Sci. Nutr. 2021, 61, 2047–2060. [Google Scholar] [CrossRef]
- Lim, E.-K.; Bowles, D. Plant production systems for bioactive small molecules. Curr. Opin. Biotechnol. 2012, 23, 271–277. [Google Scholar] [CrossRef]
- Sobolev, A.P.; Mannina, L.; Proietti, N.; Carradori, S.; Daglia, M.; Giusti, A.M.; Antiochia, R.; Capitani, D. Untargeted NMR-Based Methodology in the Study of Fruit Metabolites. Molecules 2015, 20, 4088–4108. [Google Scholar] [CrossRef]
- Remaud, G.S.; Akoka, S. A review of flavors authentication by position-specific isotope analysis by nuclear magnetic resonance spectrometry: The example of vanillin. Flavour Fragr. J. 2017, 32, 77–84. [Google Scholar] [CrossRef]
- Galvan, D.; de Aguiar, L.M.; Bona, E.; Marini, F.; Killner, M.H.M. Successful combination of benchtop nuclear magnetic resonance spectroscopy and chemometric tools: A review. Anal. Chim. Acta 2023, 1273, 341495. [Google Scholar] [CrossRef] [PubMed]
- Cobas, C. NMR signal processing, prediction, and structure verification with machine learning techniques. Magn. Reson. Chem. 2020, 58, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, Z.; Guo, D.; Orekhov, V.; Qu, X. Review and Prospect: Deep Learning in Nuclear Magnetic Resonance Spectroscopy. Chem. A Eur. J. 2020, 26, 10391–10401. [Google Scholar] [CrossRef]
- Bengio, Y. Learning deep architectures for AI. Found. Trends Mach. Learn. 2009, 2, 1–127. [Google Scholar] [CrossRef]
- Hinton, G.E.; Salakhutdinov, R. Reducing the Dimensionality of Data with Neural Networks. Science 2006, 313, 504–507. [Google Scholar] [CrossRef]
- Bengio, Y.; Delalleau, O. On the expressive power of deep architectures. In Proceedings of the Algorithmic Learning Theory: 22nd International Conference, ALT 2011, Espoo, Finland, 5–7 October 2011; pp. 18–36. [Google Scholar]
- Bengio, Y.; LeCun, Y. Scaling learning algorithms towards AI. Large-Scale Kernel Mach. 2007, 34, 1–41. [Google Scholar] [CrossRef]
- Teschendorff, A.E. Avoiding common pitfalls in machine learning omic data science. Nat. Mater. 2019, 18, 422–427. [Google Scholar] [CrossRef]
- Ronan, T.; Qi, Z.; Naegle, K.M. Avoiding common pitfalls when clustering biological data. Sci. Signal. 2016, 9, re6. [Google Scholar] [CrossRef]
- Dan, J.; Zhao, X.; Ning, S.; Lu, J.; Loh, K.P.; He, Q.; Loh, N.D.; Pennycook, S.J. Learning motifs and their hierarchies in atomic resolution microscopy. Sci. Adv. 2022, 8, eabk1005. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaría, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of deep learning: Concepts, CNN architectures, challenges, applications, future directions. J. Big Data 2021, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Zhong, G.; Yu, H. A review on the attention mechanism of deep learning. Neurocomputing 2021, 452, 48–62. [Google Scholar] [CrossRef]
- Debus, B.; Parastar, H.; Harrington, P.; Kirsanov, D. Deep learning in analytical chemistry. TrAC Trends Anal. Chem. 2021, 145, 116459. [Google Scholar] [CrossRef]
- Dong, J.-E.; Wang, Y.; Zuo, Z.-T.; Wang, Y.-Z. Deep learning for geographical discrimination of Panax notoginseng with directly near-infrared spectra image. Chemom. Intell. Lab. Syst. 2020, 197, 103913. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Tian, S.; Zhang, H.; Sun, J.; Lu, H.; Zhang, Z. EasyCID: Make component identification easy in Raman spectroscopy. Chemom. Intell. Lab. Syst. 2022, 231, 104657. [Google Scholar] [CrossRef]
- Zeng, H.-T.; Hou, M.-H.; Ni, Y.-P.; Fang, Z.; Fan, X.-Q.; Lu, H.-M.; Zhang, Z.-M. Mixture analysis using non-negative elastic net for Raman spectroscopy. J. Chemom. 2020, 34, e3293. [Google Scholar] [CrossRef]
- Fan, X.; Ming, W.; Zeng, H.; Zhang, Z.; Lu, H. Deep learning-based component identification for the Raman spectra of mixtures. Analyst 2019, 144, 1789–1798. [Google Scholar] [CrossRef]
- Lussier, F.; Thibault, V.; Charron, B.; Wallace, G.Q.; Masson, J.-F. Deep learning and artificial intelligence methods for Raman and surface-enhanced Raman scattering. TrAC Trends Anal. Chem. 2020, 124, 115796. [Google Scholar] [CrossRef]
- Ji, H.; Xu, Y.; Lu, H.; Zhang, Z. Deep MS/MS-Aided Structural-Similarity Scoring for Unknown Metabolite Identification. Anal. Chem. 2019, 91, 5629–5637. [Google Scholar] [CrossRef]
- Ji, H.; Deng, H.; Lu, H.; Zhang, Z. Predicting a Molecular Fingerprint from an Electron Ionization Mass Spectrum with Deep Neural Networks. Anal. Chem. 2020, 92, 8649–8653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, Z.; Fan, X.; Wang, Y.; Yang, Q.; Sun, J.; Wen, M.; Kang, X.; Zhang, Z.; Lu, H. Fusion of Quality Evaluation Metrics and Convolutional Neural Network Representations for ROI Filtering in LC–MS. Anal. Chem. 2023, 95, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.D.; Tsentalovich, Y.P.; Yanshole, V.V. Deep Learning for the Precise Peak Detection in High-Resolution LC–MS Data. Anal. Chem. 2020, 92, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ji, H.; Xu, Z.; Li, Y.; Wang, P.; Sun, J.; Fan, X.; Zhang, H.; Lu, H.; Zhang, Z. Ultra-fast and accurate electron ionization mass spectrum matching for compound identification with million-scale in-silico library. Nat. Commun. 2023, 14, 3722. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Tian, M.; Zhang, H.; Lu, H.; Jiang, Y.; Chen, Y.; Zhang, Z. Highly automatic and universal approach for pure ion chromatogram construction from liquid chromatography-mass spectrometry data using deep learning. J. Chromatogr. A 2023, 1705, 464172. [Google Scholar] [CrossRef]
- Wei, J.N.; Belanger, D.; Adams, R.P.; Sculley, D. Rapid Prediction of Electron–Ionization Mass Spectrometry Using Neural Networks. ACS Cent. Sci. 2019, 5, 700–708. [Google Scholar] [CrossRef]
- Fan, Y.; Yu, C.; Lu, H.; Chen, Y.; Hu, B.; Zhang, X.; Su, J.; Zhang, Z. Deep learning-based method for automatic resolution of gas chromatography-mass spectrometry data from complex samples. J. Chromatogr. A 2023, 1690, 463768. [Google Scholar] [CrossRef]
- Yang, Q.; Ji, H.; Fan, X.; Zhang, Z.; Lu, H. Retention time prediction in hydrophilic interaction liquid chromatography with graph neural network and transfer learning. J. Chromatogr. A 2021, 1656, 462536. [Google Scholar] [CrossRef]
- Fan, X.; Xu, Z.; Zhang, H.; Liu, D.; Yang, Q.; Tao, Q.; Wen, M.; Kang, X.; Zhang, Z.; Lu, H. Fully automatic resolution of untargeted GC-MS data with deep learning assistance. Talanta 2022, 244, 123415. [Google Scholar] [CrossRef]
- Yang, Q.; Ji, H.; Lu, H.; Zhang, Z. Prediction of Liquid Chromatographic Retention Time with Graph Neural Networks to Assist in Small Molecule Identification. Anal. Chem. 2021, 93, 2200–2206. [Google Scholar] [CrossRef]
- Fan, X.; Ma, P.; Hou, M.; Ni, Y.; Fang, Z.; Lu, H.; Zhang, Z. Deep-Learning-Assisted multivariate curve resolution. J. Chromatogr. A 2021, 1635, 461713. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zhang, Y.; Liao, Y.; Yang, Q.; Xie, T.; Fan, X.; Lin, Z.; Chen, Y.; Lu, H.; Zhang, Z. Highly accurate and large-scale collision cross sections prediction with graph neural networks. Commun. Chem. 2023, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Plante, P.-L.; Francovic-Fontaine, É.; May, J.C.; McLean, J.A.; Baker, E.S.; Laviolette, F.; Marchand, M.; Corbeil, J. Predicting Ion Mobility Collision Cross-Sections Using a Deep Neural Network: DeepCCS. Anal. Chem. 2019, 91, 5191–5199. [Google Scholar] [CrossRef]
- Meier, F.; Köhler, N.D.; Brunner, A.-D.; Wanka, J.-M.H.; Voytik, E.; Strauss, M.T.; Theis, F.J.; Mann, M. Deep learning the collisional cross sections of the peptide universe from a million experimental values. Nat. Commun. 2021, 12, 1185. [Google Scholar] [CrossRef] [PubMed]
- Brereton, R.G. Pattern recognition in chemometrics. Chemom. Intell. Lab. Syst. 2015, 149, 90–96. [Google Scholar] [CrossRef]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted Profiling: Quantitative Analysis of 1H NMR Metabolomics Data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. The application of artificial neural networks in metabolomics: A historical perspective. Metabolomics 2019, 15, 142. [Google Scholar] [CrossRef]
- Moritz, B.; Mazin, J.; Anastasiya, K.; Jan, G.K. Deep regression with ensembles enables fast, first-order shimming in low-field NMR. J. Magn. Reson. 2022, 336, 107151. [Google Scholar] [CrossRef]
- Qu, X.; Huang, Y.; Lu, H.; Qiu, T.; Guo, D.; Agback, T.; Orekhov, V.; Chen, Z. Accelerated Nuclear Magnetic Resonance Spectroscopy with Deep Learning. Angew. Chem. Int. Ed. 2020, 59, 10297–10300. [Google Scholar] [CrossRef]
- Li, D.-W.; Hansen, A.L.; Yuan, C.; Bruschweiler-Li, L.; Brüschweiler, R. DEEP picker is a deep neural network for accurate deconvolution of complex two-dimensional NMR spectra. Nat. Commun. 2021, 12, 5229. [Google Scholar] [CrossRef]
- Klukowski, P.; Augoff, M.; Zięba, M.; Drwal, M.; Gonczarek, A.; Walczak, M.J. NMRNet: A deep learning approach to automated peak picking of protein NMR spectra. Bioinformatics 2018, 34, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Luo, J.; Zeng, Q.; Dong, X.; Chen, J.; Zhan, C.; Chen, Z.; Lin, Y. Improvement in Signal-to-Noise Ratio of Liquid-State NMR Spectroscopy via a Deep Neural Network DN-Unet. Anal. Chem. 2021, 93, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, W.; Bratholm, L.A.; Packer, M.J.; Mulholland, A.J.; Glowacki, D.R.; Butts, C.P. IMPRESSION—Prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. Chem. Sci. 2020, 11, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Chongcan, L.; Yong, C.; Weihua, D. Identifying molecular functional groups of organic compounds by deep learning of NMR data. Magn. Reson. Chem. 2022, 60, 1061–1069. [Google Scholar] [CrossRef]
- Piotr, K.; Riek, R.; Güntert, P. Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA. Nat. Commun. 2022, 13, 5785. [Google Scholar] [CrossRef]
- Kavitha, R.; Veera Mohana Rao, K.; Neeraj Praphulla, A.; Vrushali Siddesh, S.; Ramakrishna, V.H.; Satish Chandra, S. Identifying type of sugar adulterants in honey: Combined application of NMR spectroscopy and supervised machine learning classification. Curr. Res. Food Sci. 2022, 5, 272–277. [Google Scholar] [CrossRef]
- Wei, W.; Liao, Y.; Wang, Y.; Wang, S.; Du, W.; Lu, H.; Kong, B.; Yang, H.; Zhang, Z. Deep Learning-Based Method for Compound Identification in NMR Spectra of Mixtures. Molecules 2022, 27, 3653. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Yu, C.; Lv, Y.; Zhang, H.; Yang, Q.; Wen, M.; Lu, H.; Zhang, Z. A Universal and Accurate Method for Easily Identifying Components in Raman Spectroscopy Based on Deep Learning. Anal. Chem. 2023, 95, 4863–4870. [Google Scholar] [CrossRef]
- Helmus, J.J.; Jaroniec, C.P. Nmrglue: An open source Python package for the analysis of multidimensional NMR data. J. Biomol. NMR 2013, 55, 355–367. [Google Scholar] [CrossRef]
- Zhang, Z.-M.; Chen, S.; Liang, Y.-Z. Baseline correction using adaptive iteratively reweighted penalized least squares. Analyst 2010, 135, 1138–1146. [Google Scholar] [CrossRef]
- Bromley, J.; Guyon, I.; LeCun, Y.; Säckinger, E.; Shah, R. Signature verification using a “siamese” time delay neural network. Adv. Neural Inf. Process. Syst. 1993, 6, 737–744. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Spatial pyramid pooling in deep convolutional networks for visual recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2015, 37, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving Deep into Rectifiers: Surpassing Human-Level Performance on ImageNet Classification. In Proceedings of the 2015 IEEE International Conference on Computer Vision (ICCV), Santiago, Chile, 7–13 December 2015; pp. 1026–1034. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2015, arXiv:1412.6980. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wei, W.; Du, W.; Cai, J.; Liao, Y.; Lu, H.; Kong, B.; Zhang, Z. Deep-Learning-Based Mixture Identification for Nuclear Magnetic Resonance Spectroscopy Applied to Plant Flavors. Molecules 2023, 28, 7380. https://doi.org/10.3390/molecules28217380
Wang Y, Wei W, Du W, Cai J, Liao Y, Lu H, Kong B, Zhang Z. Deep-Learning-Based Mixture Identification for Nuclear Magnetic Resonance Spectroscopy Applied to Plant Flavors. Molecules. 2023; 28(21):7380. https://doi.org/10.3390/molecules28217380
Chicago/Turabian StyleWang, Yufei, Weiwei Wei, Wen Du, Jiaxiao Cai, Yuxuan Liao, Hongmei Lu, Bo Kong, and Zhimin Zhang. 2023. "Deep-Learning-Based Mixture Identification for Nuclear Magnetic Resonance Spectroscopy Applied to Plant Flavors" Molecules 28, no. 21: 7380. https://doi.org/10.3390/molecules28217380
APA StyleWang, Y., Wei, W., Du, W., Cai, J., Liao, Y., Lu, H., Kong, B., & Zhang, Z. (2023). Deep-Learning-Based Mixture Identification for Nuclear Magnetic Resonance Spectroscopy Applied to Plant Flavors. Molecules, 28(21), 7380. https://doi.org/10.3390/molecules28217380