
Emerging TACnology: Heterobifunctional Small Molecule Inducers of Targeted Posttranslational Protein Modifications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
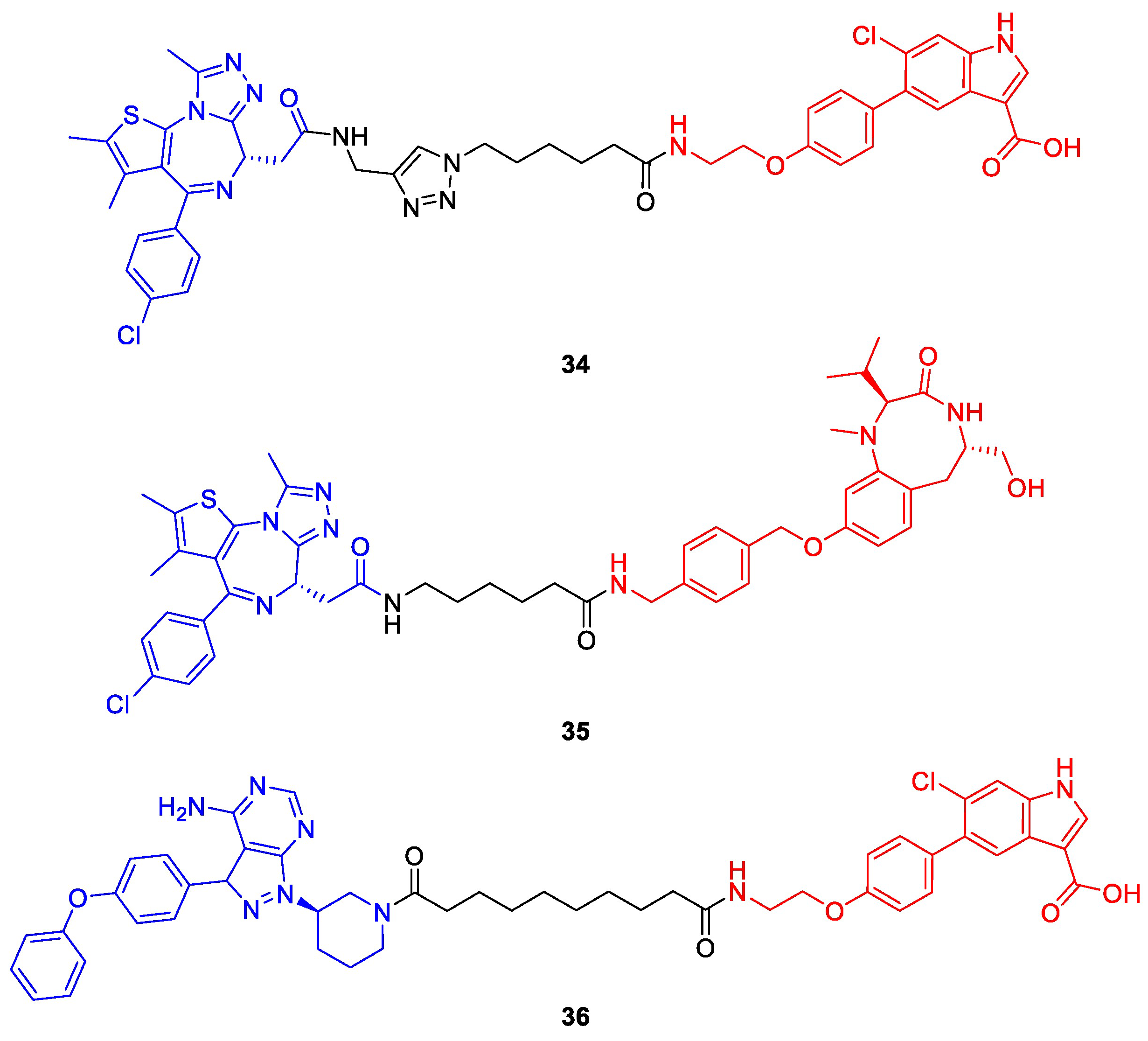{kind=link}
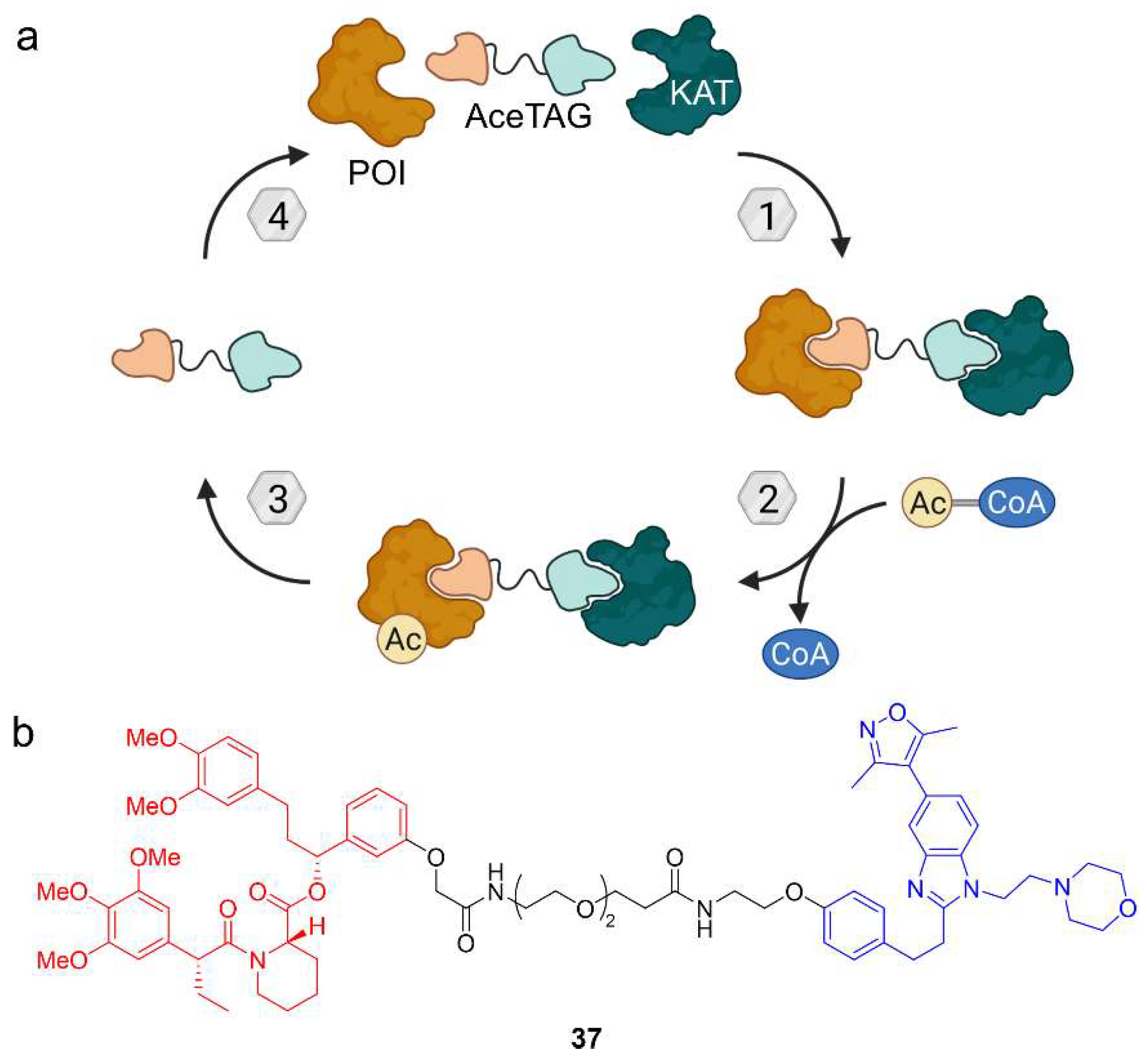{kind=link}
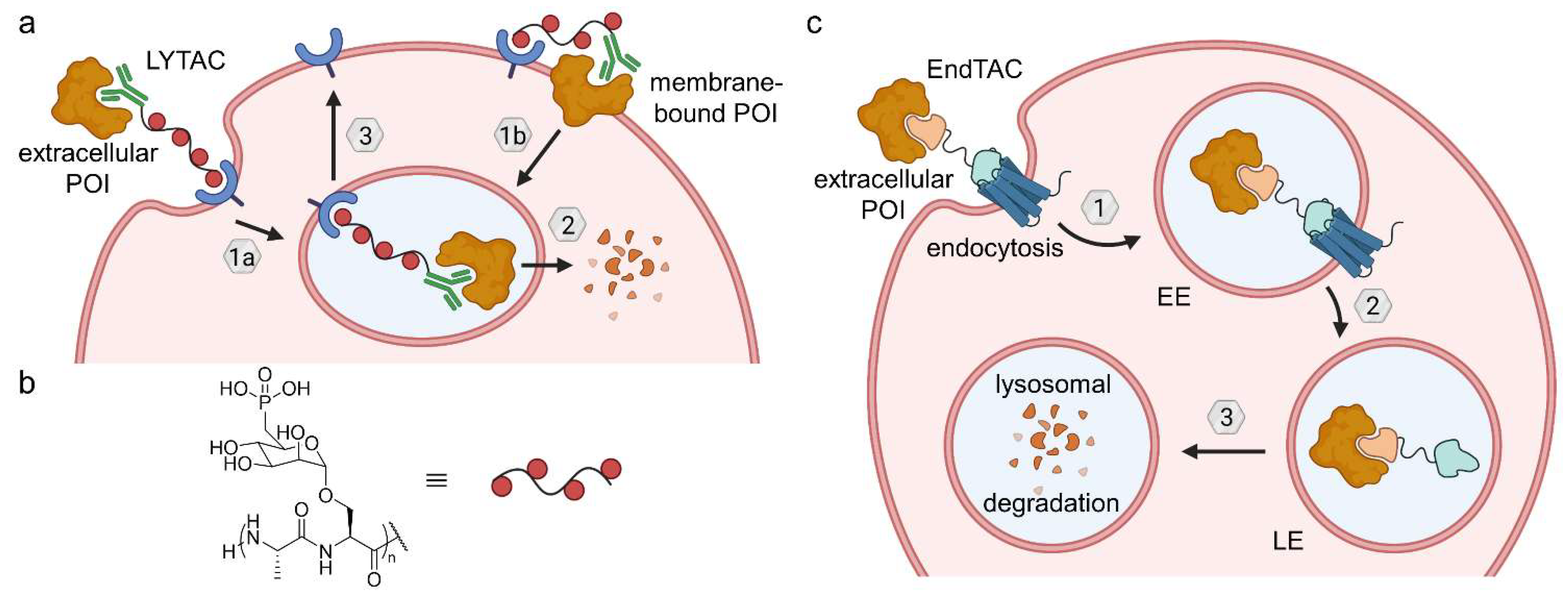{kind=link}
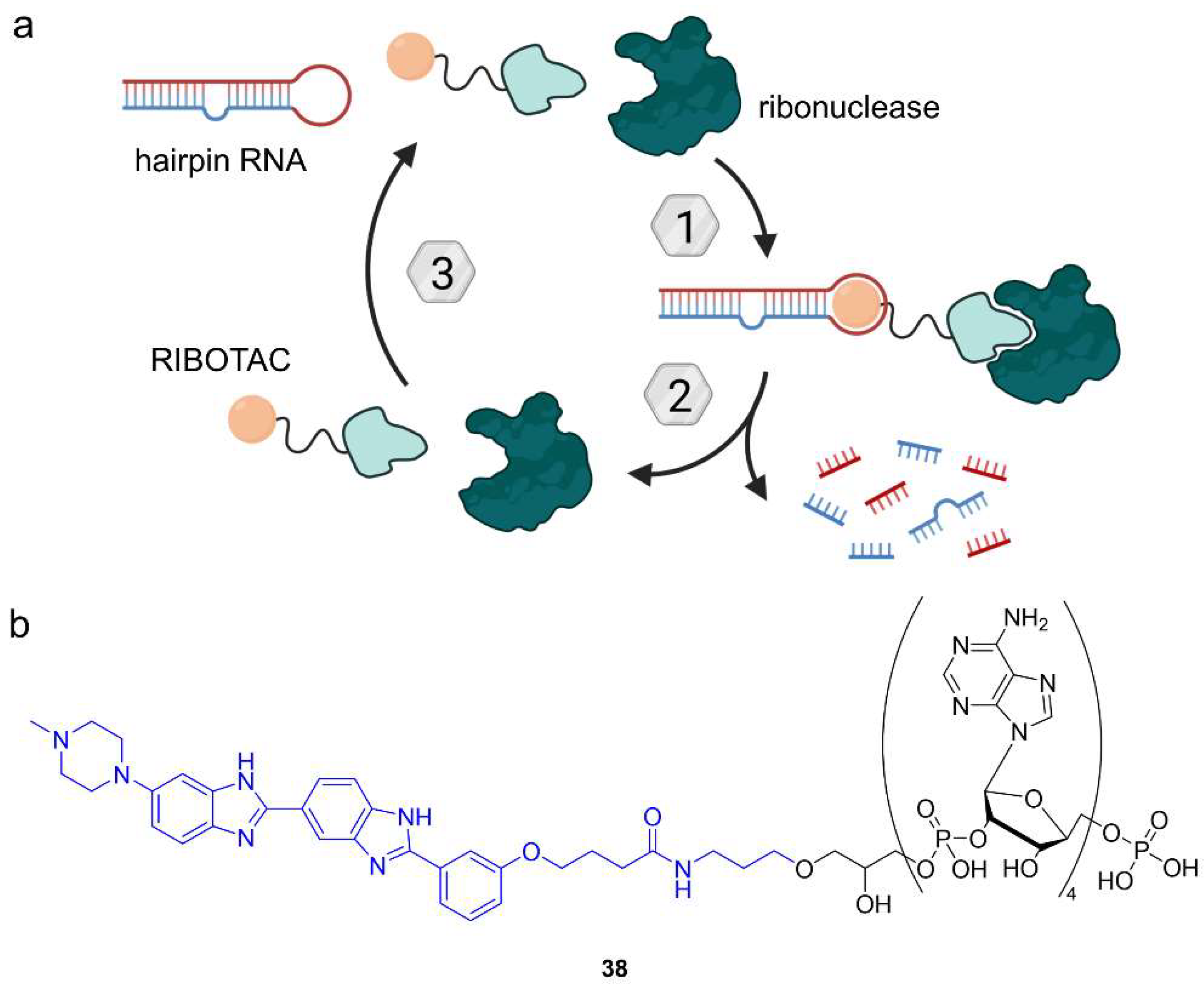{kind=link}
Abstract
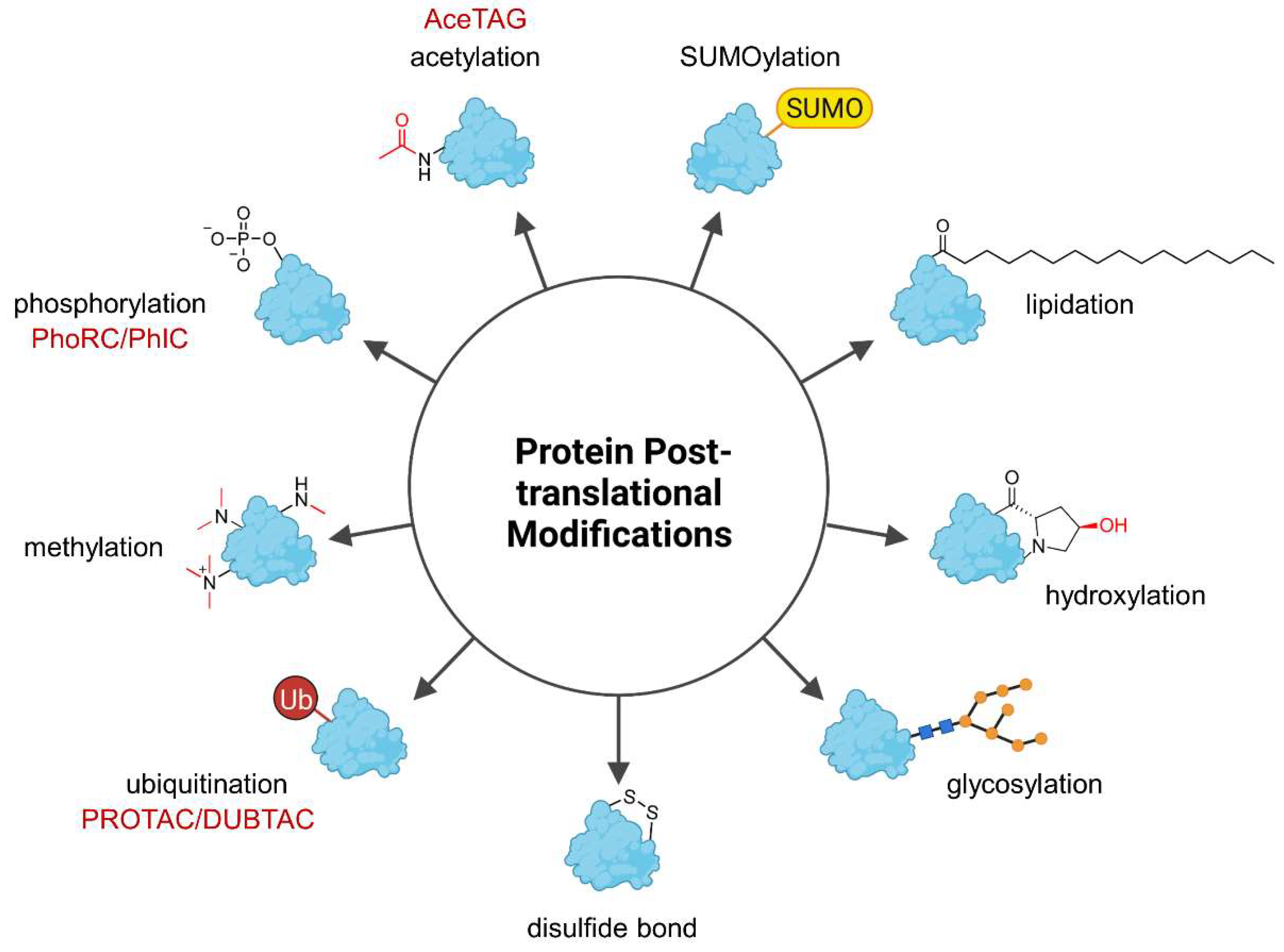
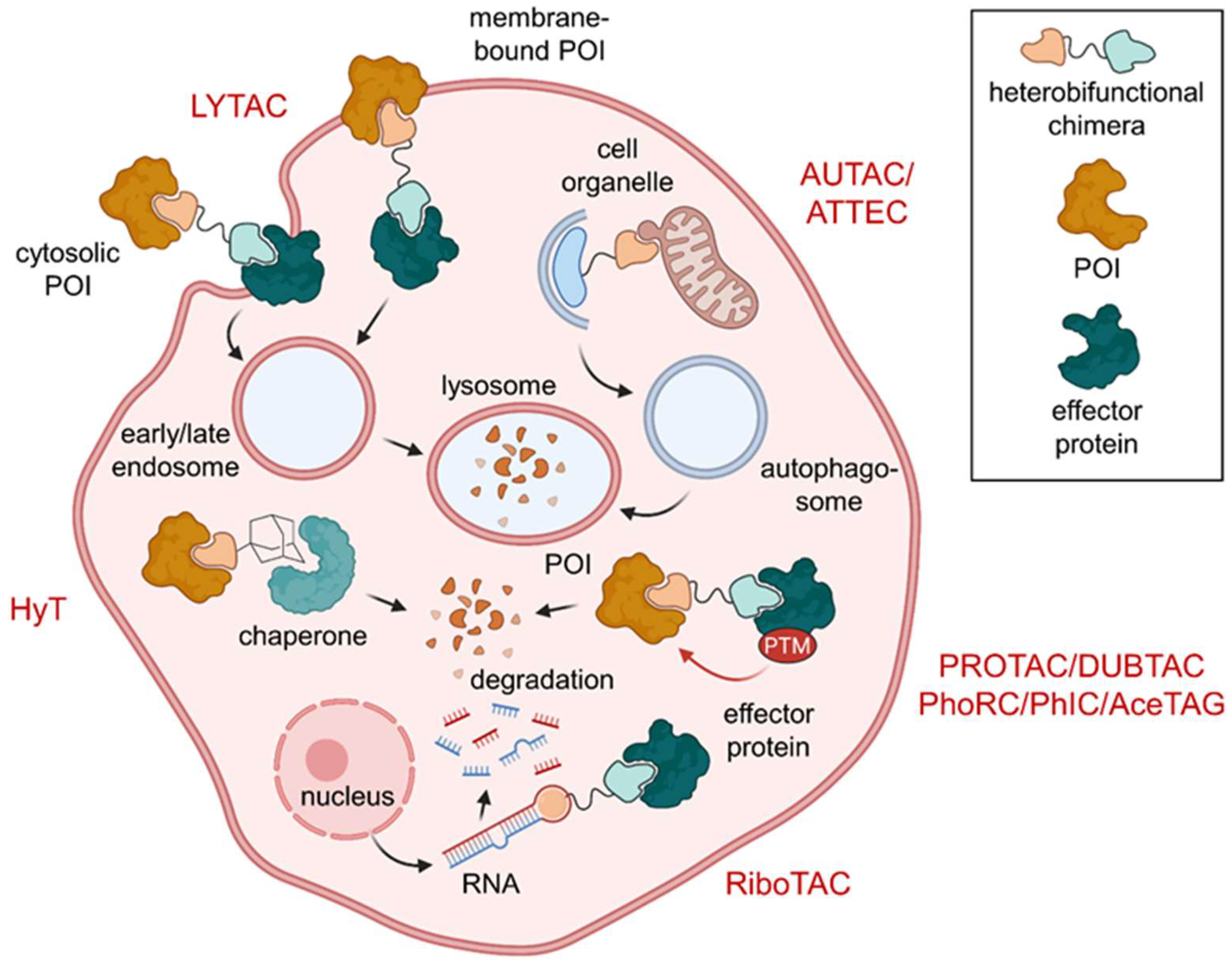
1. Introduction
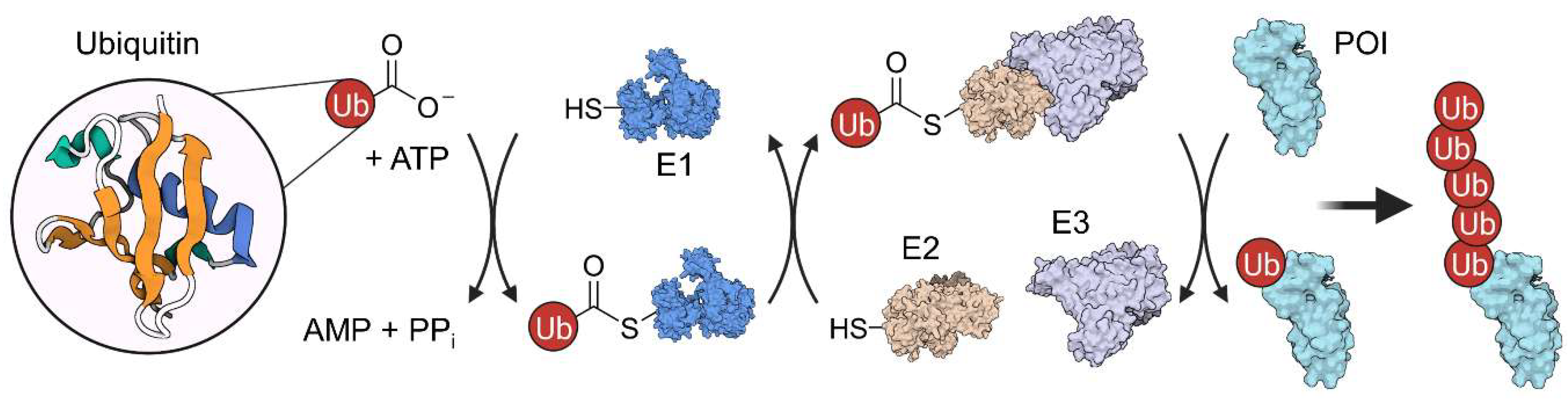
2. Ubiquitination and Autophagy
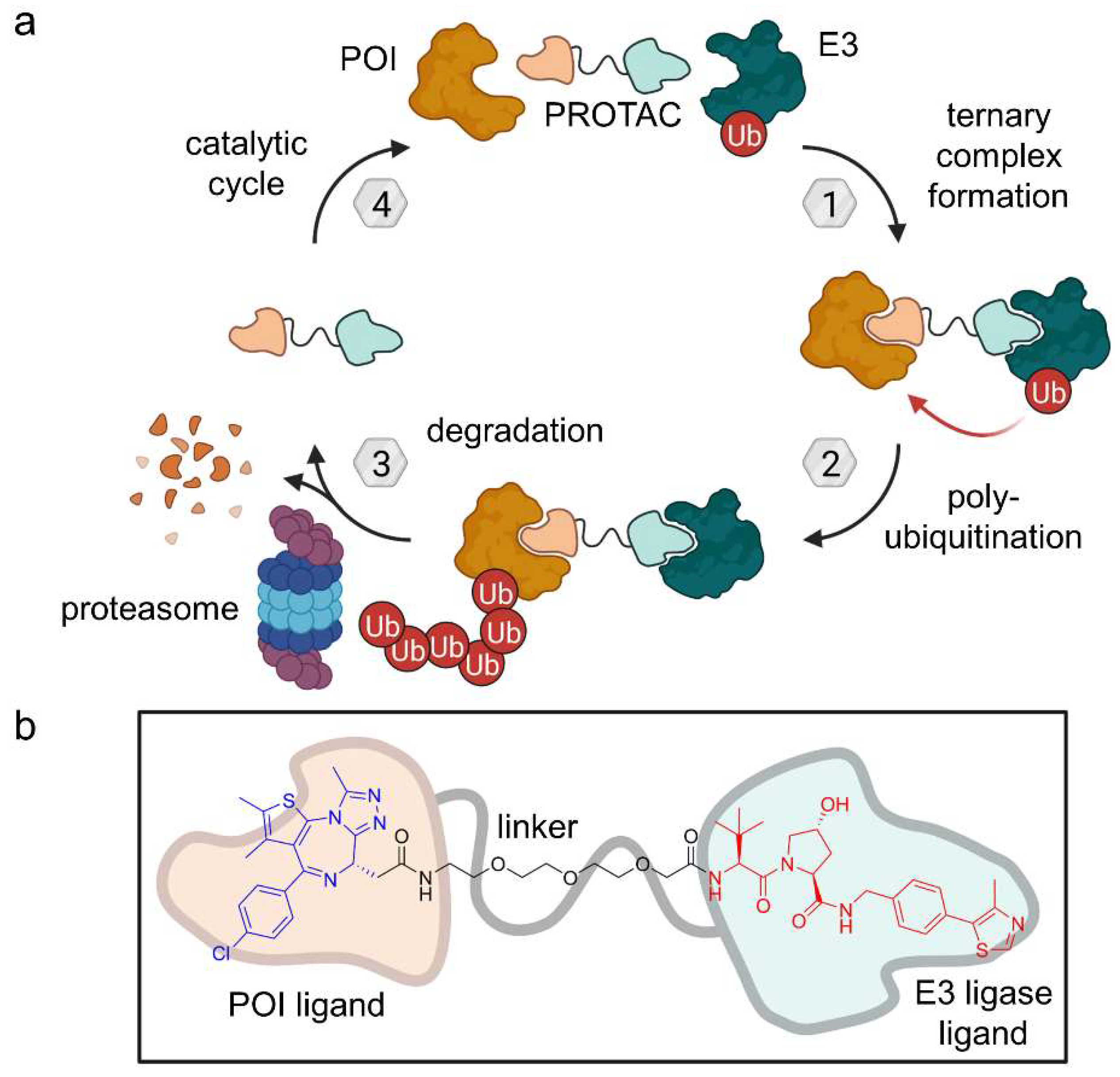
2.1. PROTACs

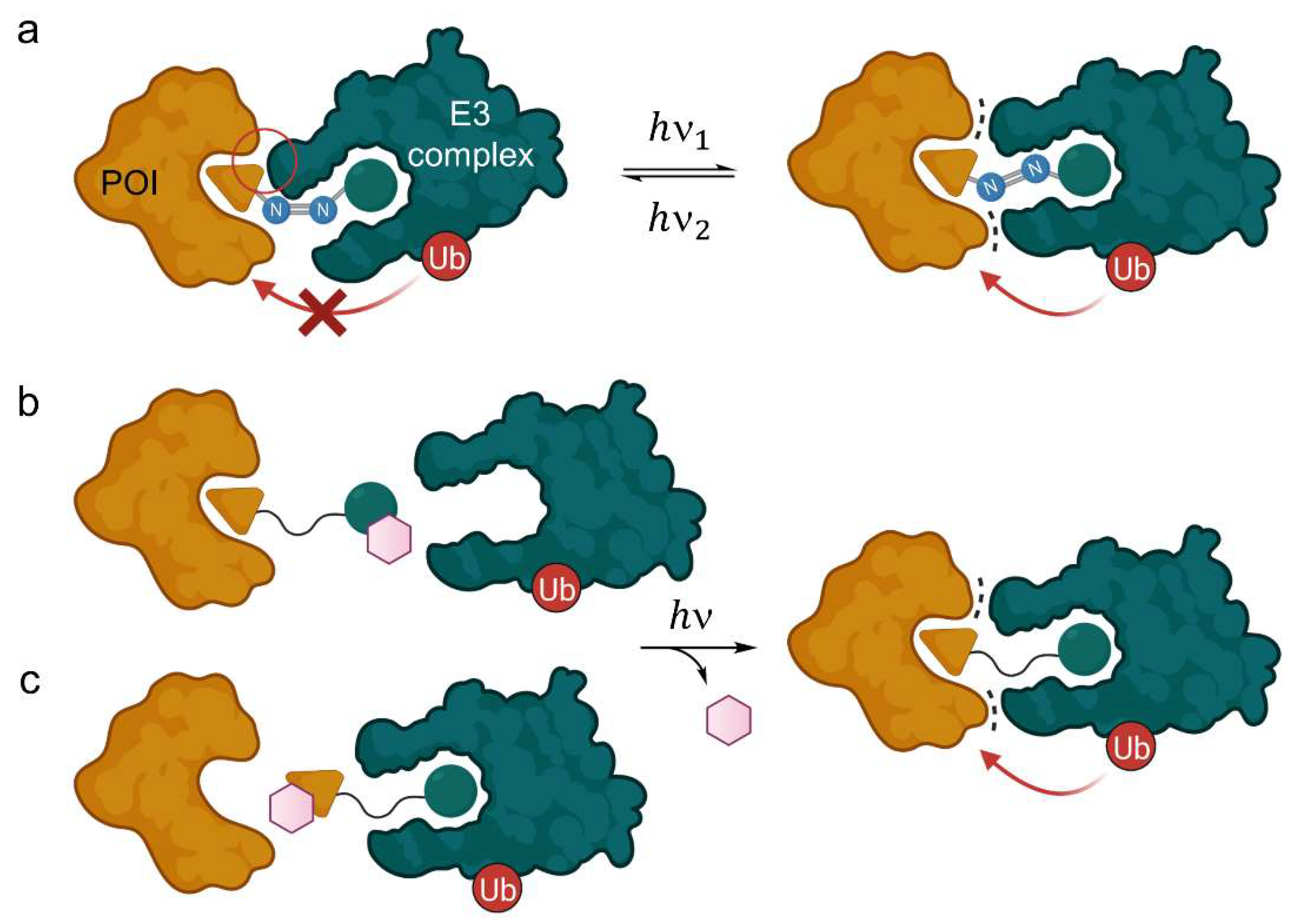
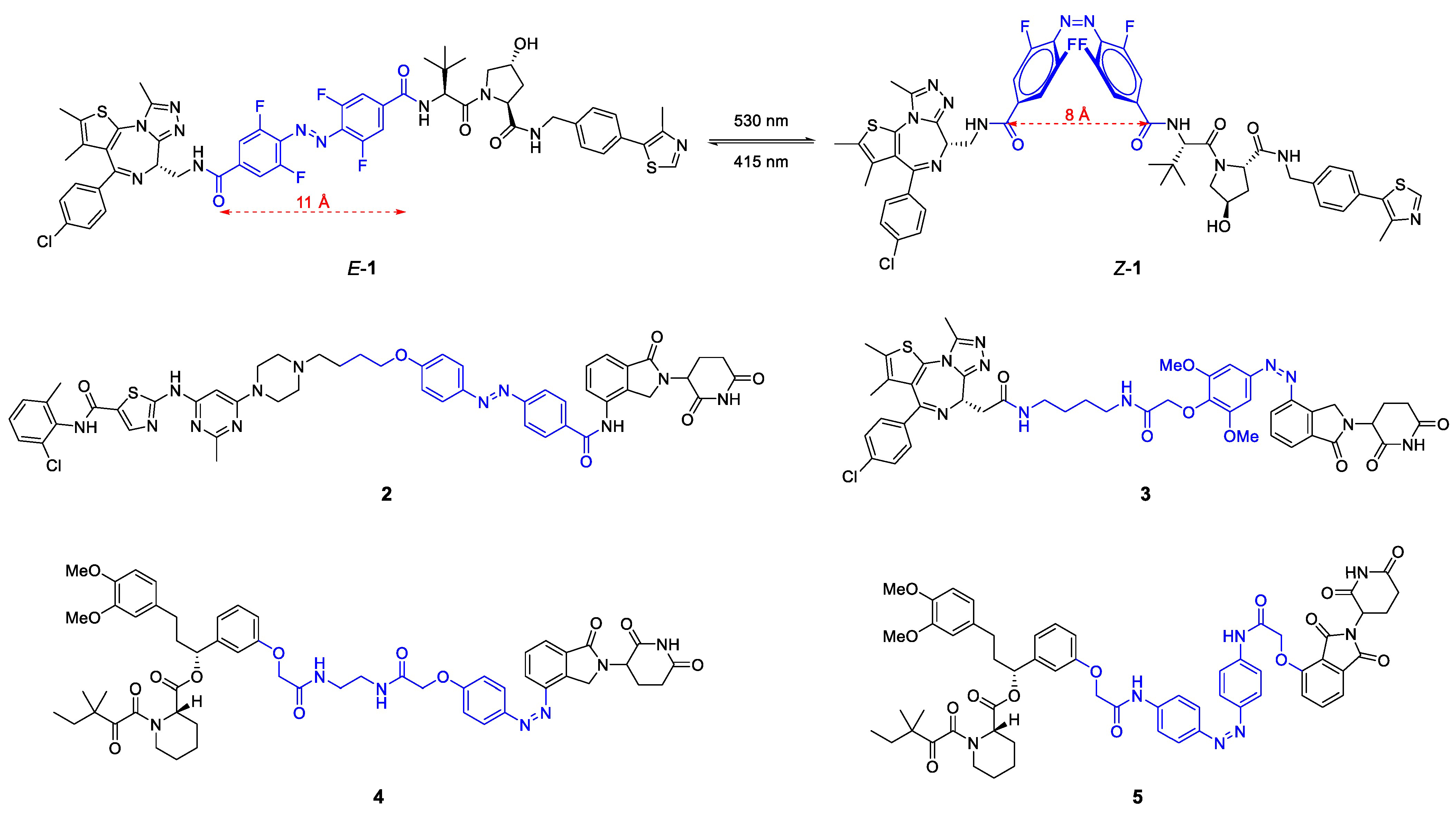
2.1.1. Light-Responsive PROTACs
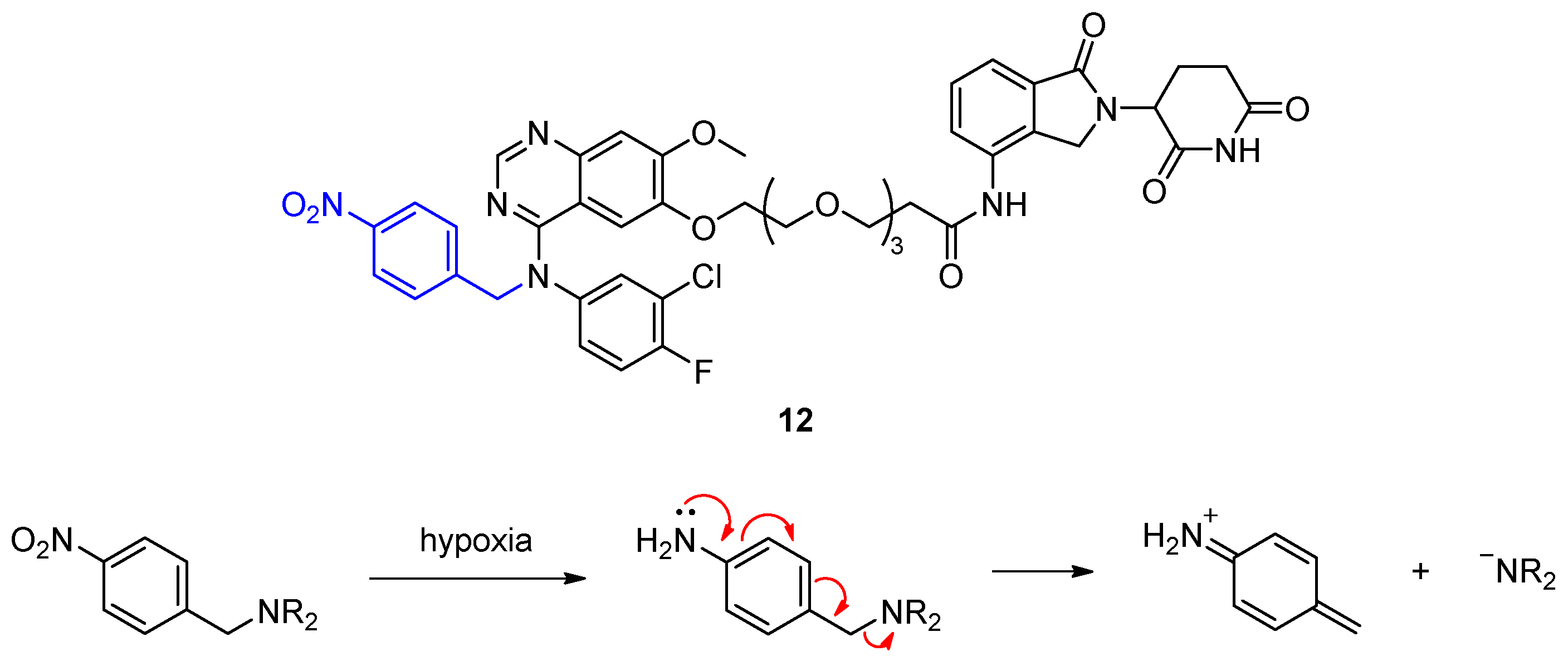
2.1.2. Hypoxia-Activated PROTACs
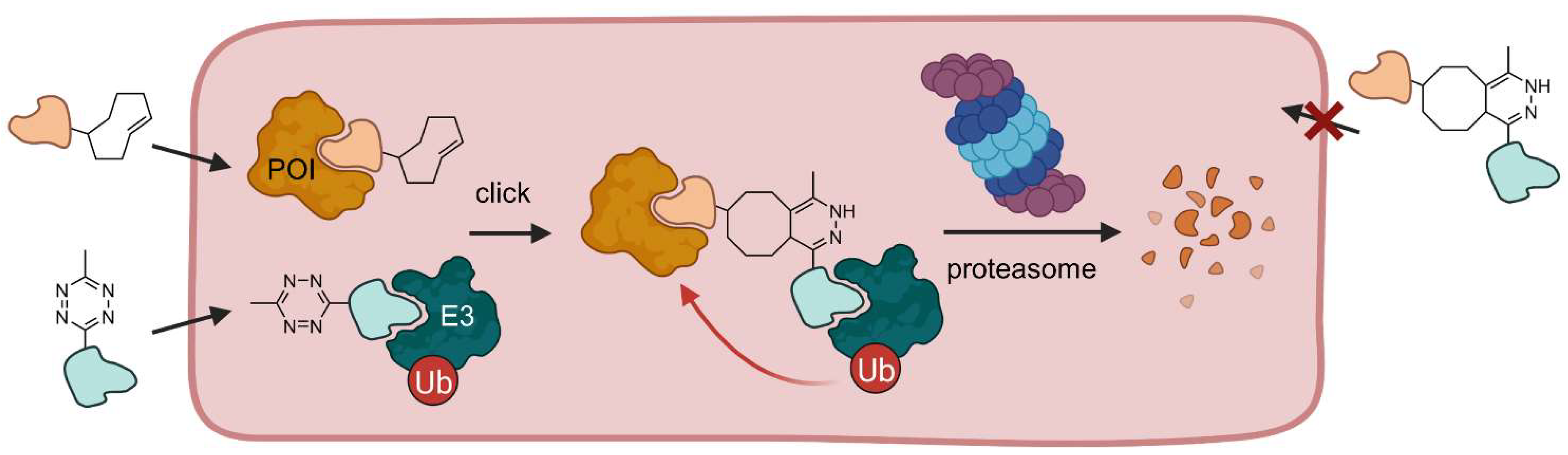
2.1.3. Click-Formed PROTACs (CLIPTACs)
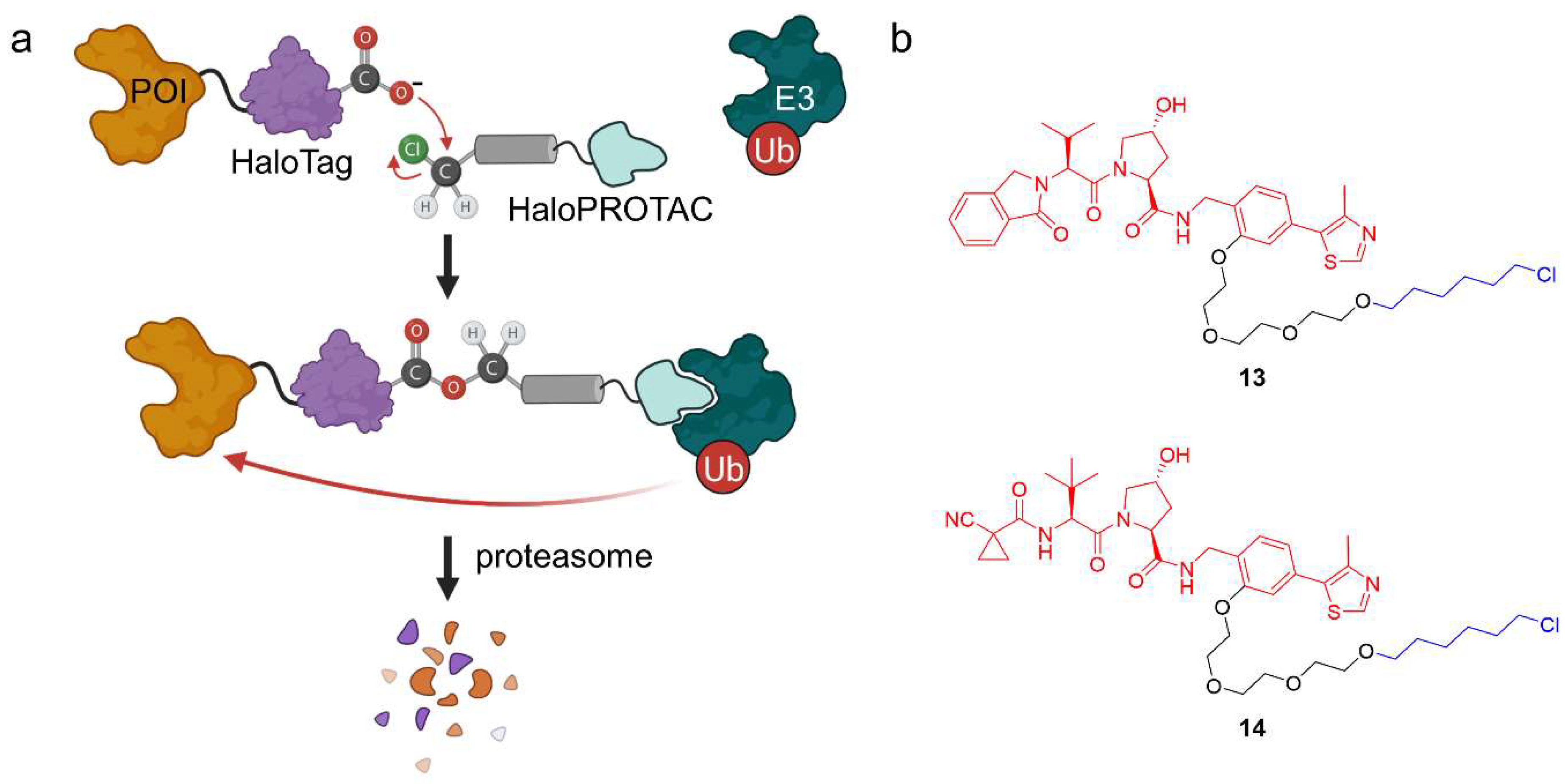
2.1.4. HaloPROTACs
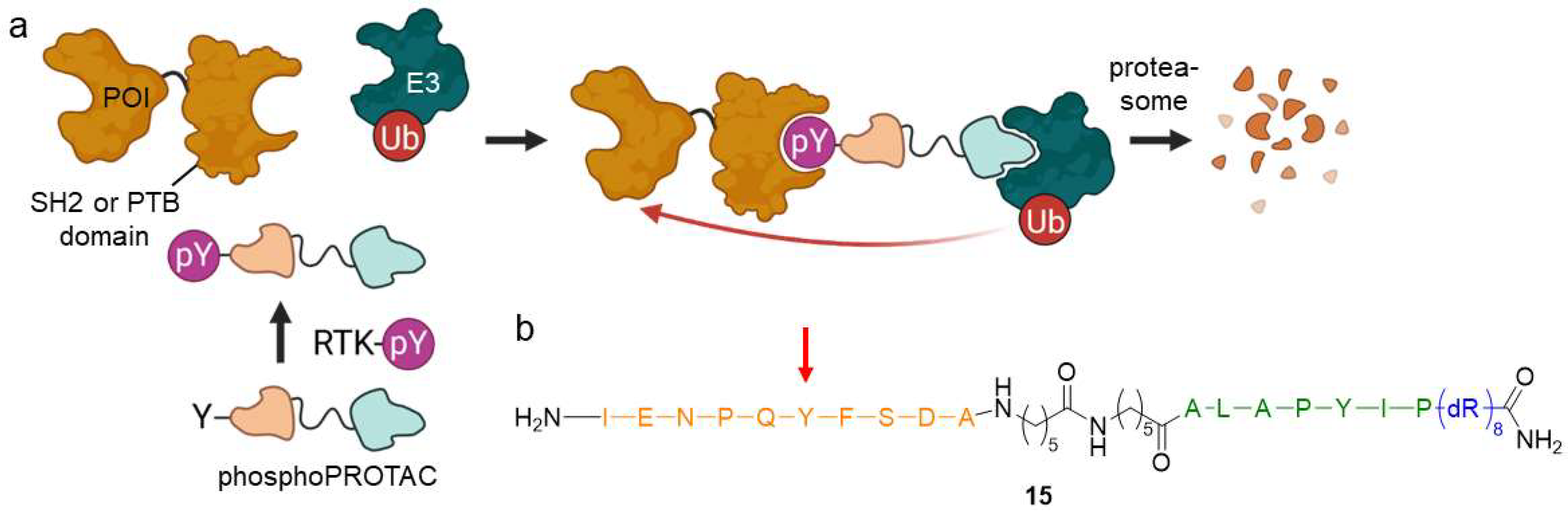
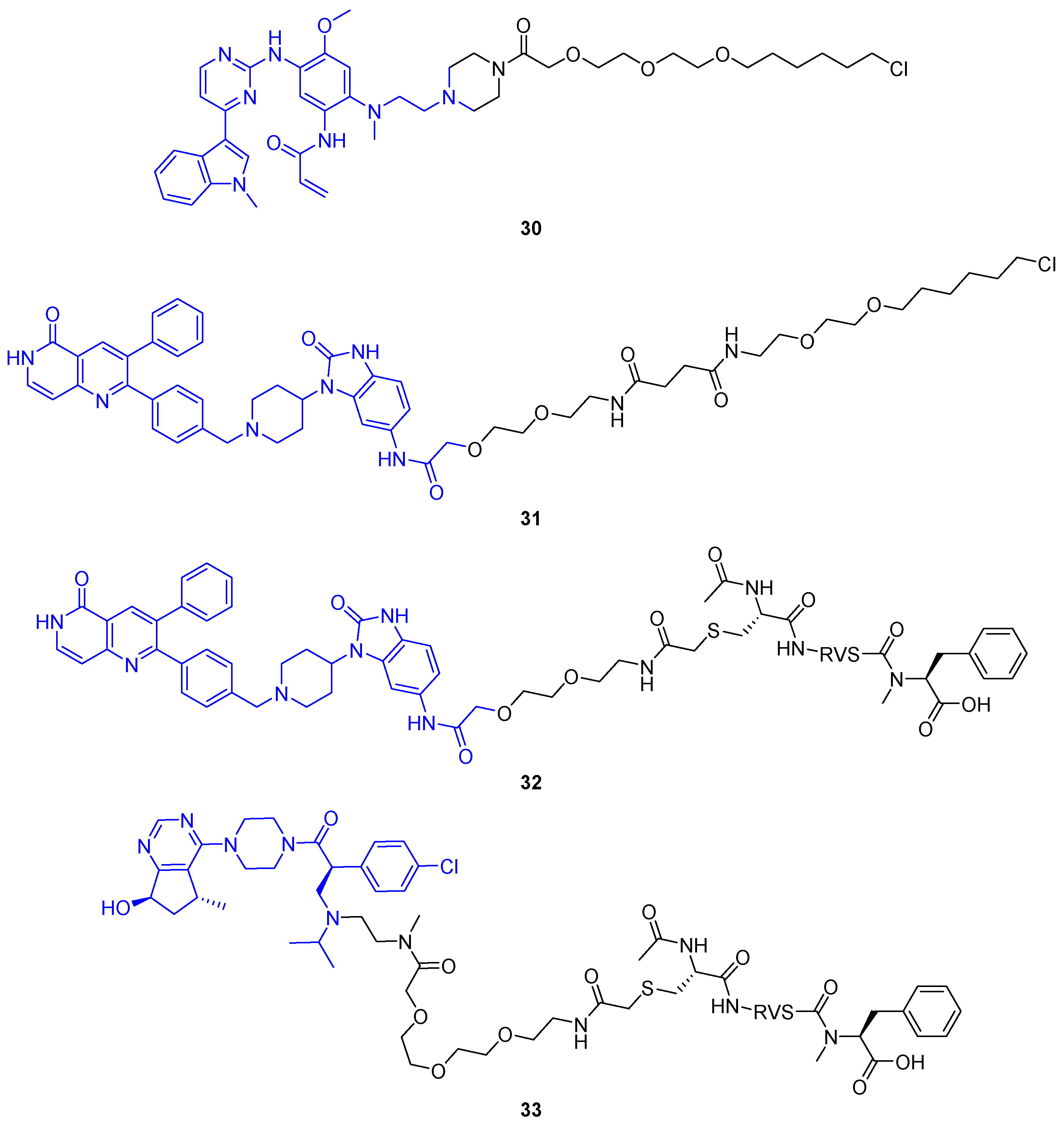
2.1.5. PhosphoPROTACs
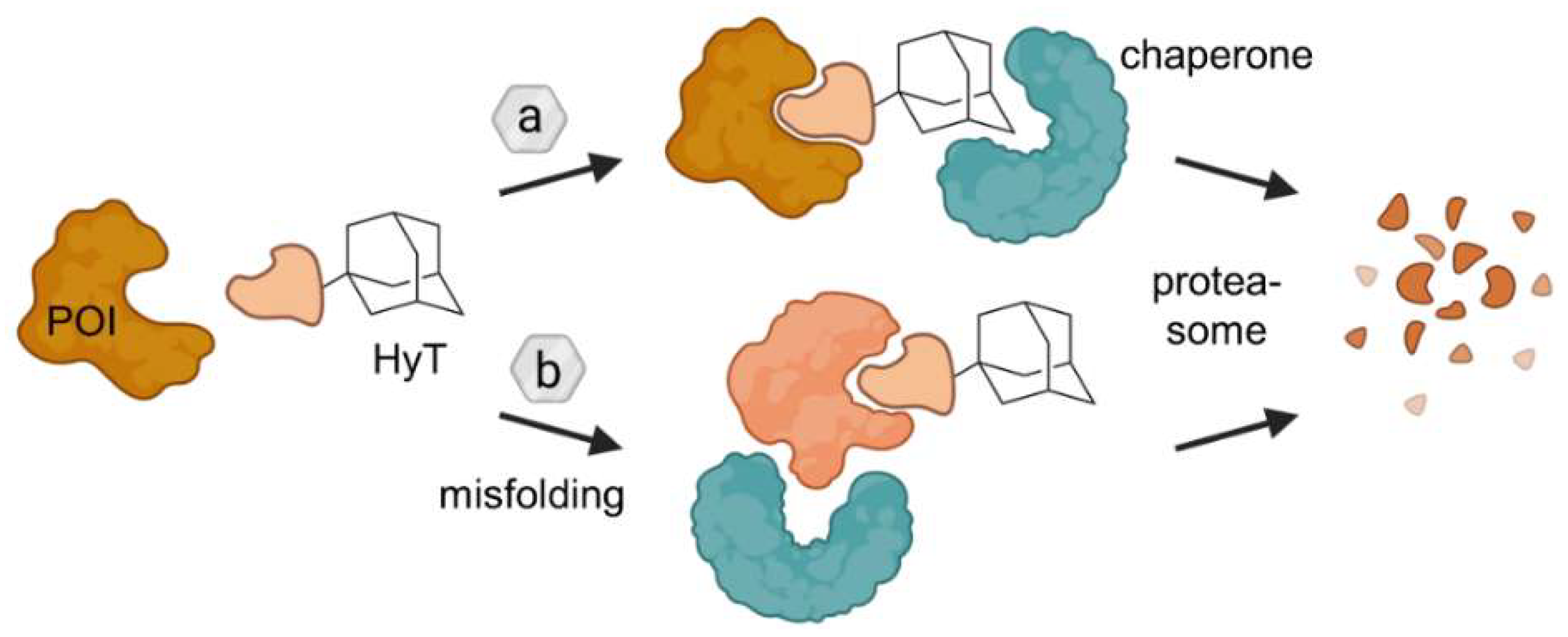
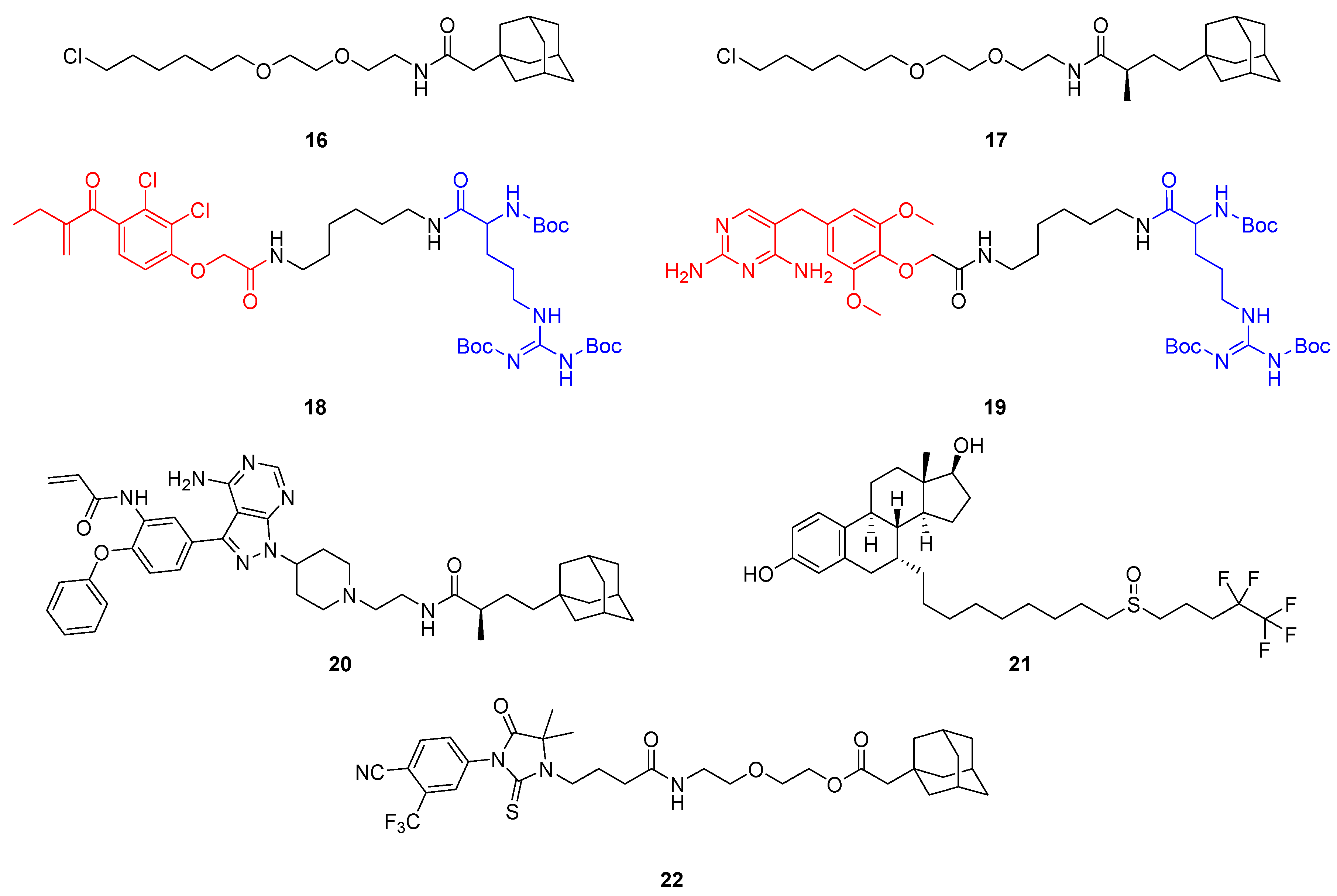
2.2. Hydrophobic Tagging (HyT)

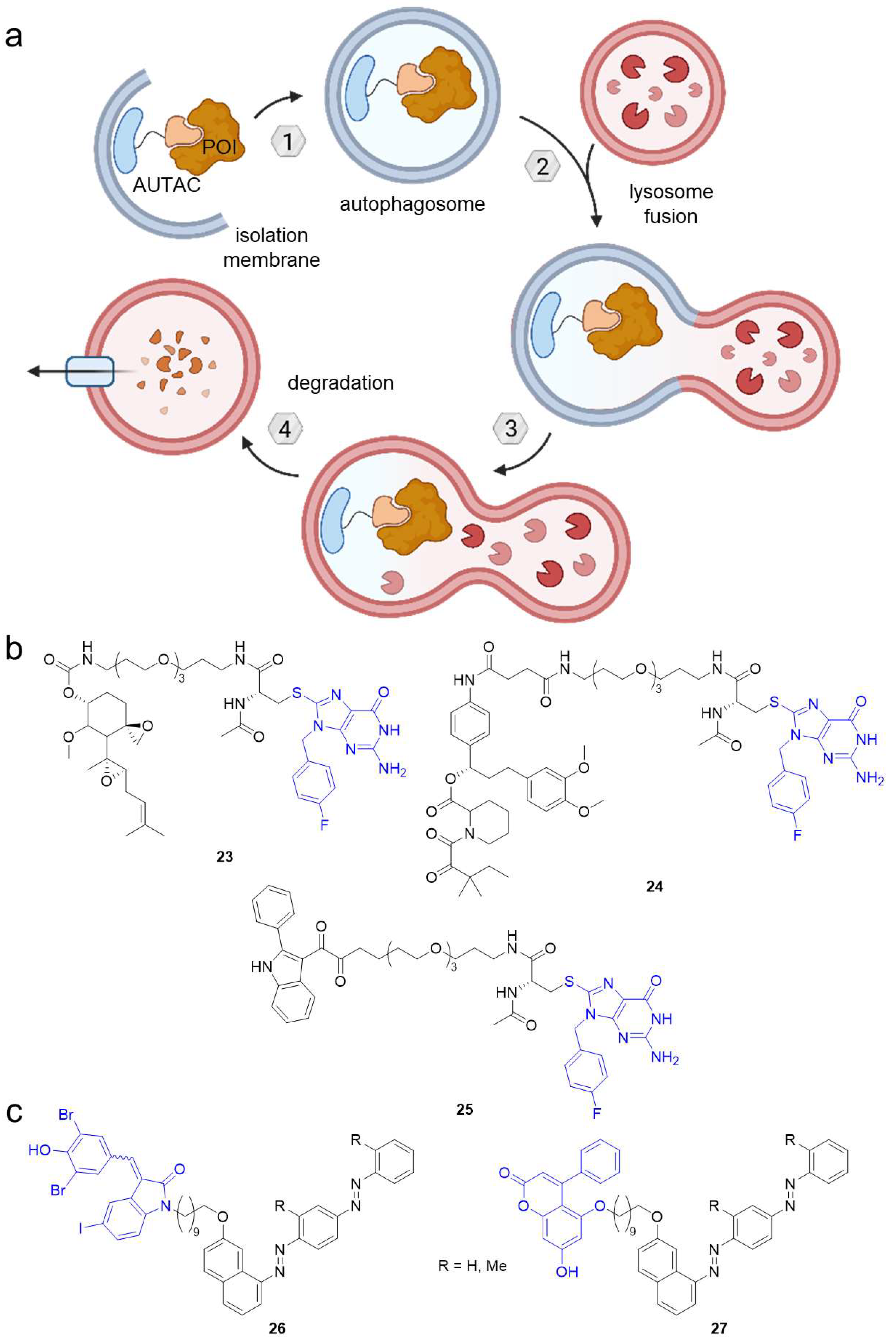
2.3. Autophagy-Targeting Chimeras (AUTACs)
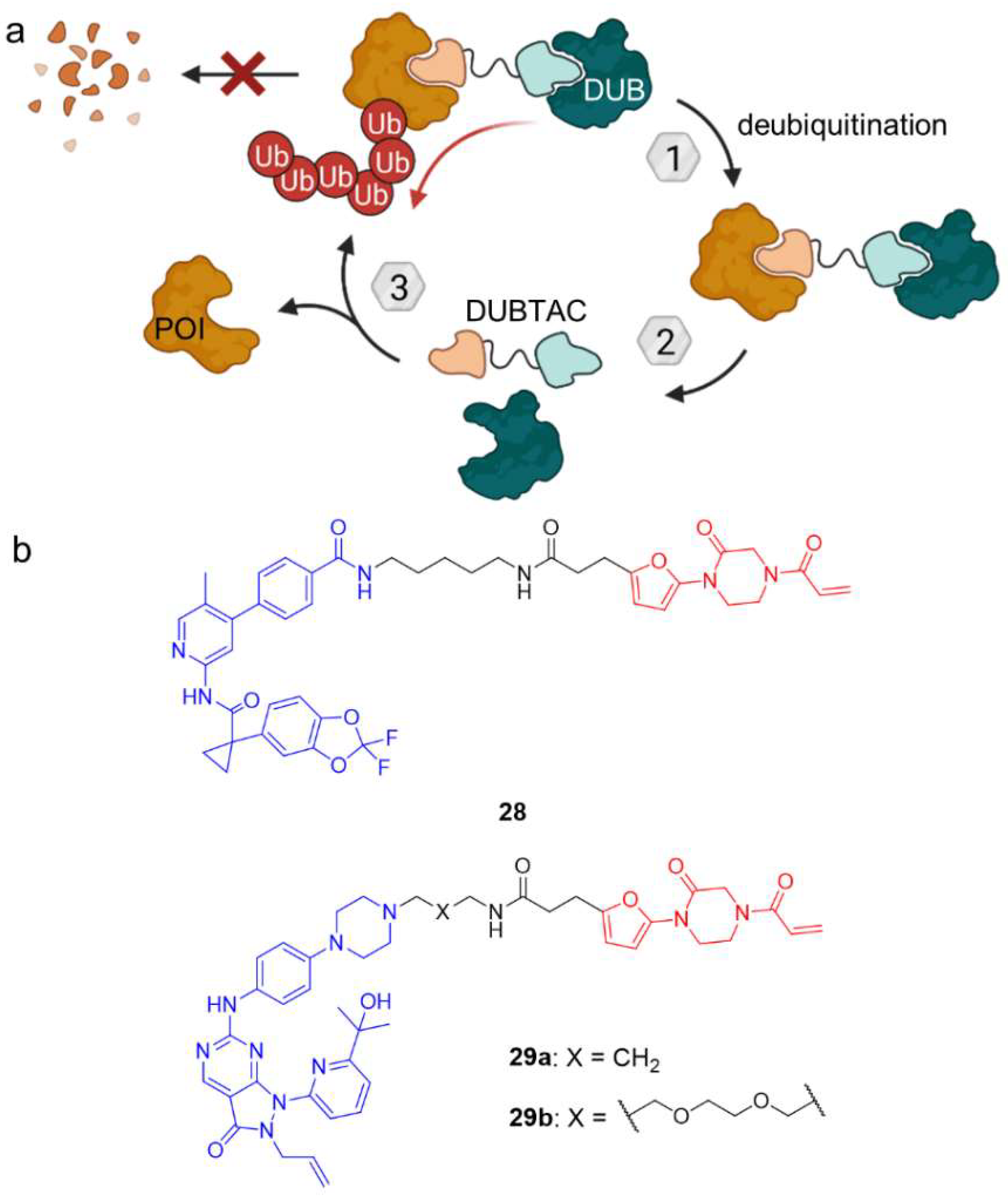
3. Deubiquitination
4. Phosphorylation and Dephosphorylation
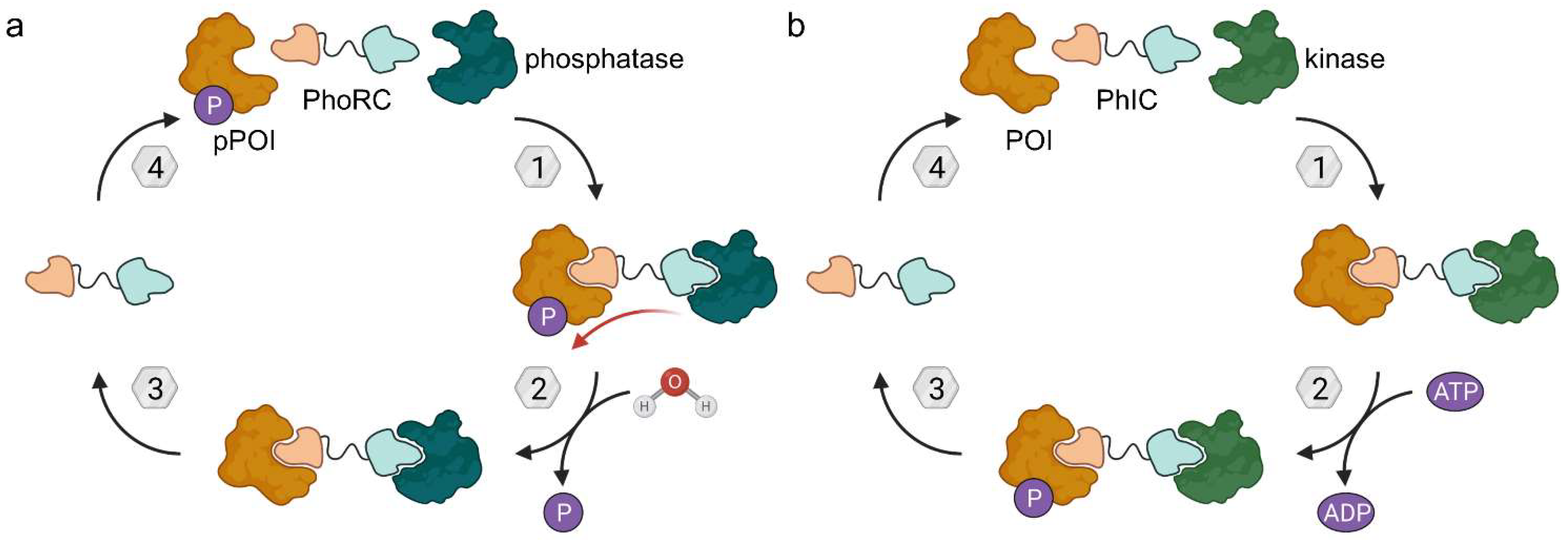
4.1. Phosphatase-Recruiting Chimeras (PhoRCs)
4.2. Phosphorylation-Inducing Chimeras (PhICs)
5. Acetylation
6. Excursus: Induced Degradation of Extracellular and Surface Proteins and RNA
6.1. Lysosome-Targeting Chimeras (LYTACs) and Endosome-Targeting Chimeras (ENDTACs)
6.2. Ribonuclease-Targeting Chimeras (RIBOTACs)
7. Conclusions and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaur, S.; Baldi, B.; Vuong, J.; O’Donoghue, S.I. Visualization and Analysis of Epiproteome Dynamics. J. Mol. Biol. 2019, 431, 1519–1539. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [CrossRef]
- Prabakaran, S.; Lippens, G.; Steen, H.; Gunawardena, J. Post-Translational Modification: Nature’s Escape from Genetic Imprisonment and the Basis for Dynamic Information Encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Minguez, P.; Parca, L.; Diella, F.; Mende, D.R.; Kumar, R.; Helmer-Citterich, M.; Gavin, A.; van Noort, V.; Bork, P. Deciphering a Global Network of Functionally Associated Post-translational Modifications. Mol. Syst. Biol. 2012, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Y.; Lin, S.; Deng, W.; Peng, D.; Cui, Q.; Xue, Y. PTMD: A Database of Human Disease-Associated Post-Translational Modifications. Genom. Proteom. Bioinform. 2018, 16, 244–251. [Google Scholar] [CrossRef]
- Leestemaker, Y.; Ovaa, H. Tools to Investigate the Ubiquitin Proteasome System. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as Attractive Anti-Cancer Targets. Curr. Pharm. Des. 2013, 19, 3215–3225. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted Protein Degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, X.; Feng, F.; Liu, W.; Sun, H. Degradation of Proteins by PROTACs and Other Strategies. Acta Pharm. Sin. B 2020, 10, 207–238. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC Technology in Drug Development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Maniaci, C.; Ciulli, A. Bifunctional Chemical Probes Inducing Protein–Protein Interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156. [Google Scholar] [CrossRef]
- Gu, S.; Cui, D.; Chen, X.; Xiong, X.; Zhao, Y. PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. Bioessays 2018, 40, e1700247. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.-H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Crews, C.M. Targeting the Undruggable Proteome: The Small Molecules of My Dreams. Chem. Biol. 2010, 17, 551–555. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D.F. Optimising Proteolysis-Targeting Chimeras (PROTACs) for Oral Drug Delivery: A Drug Metabolism and Pharmacokinetics Perspective. Drug Discov. Today 2020, 25, 1793–1800. [Google Scholar] [CrossRef]
- Wei, M.; Zhao, R.; Cao, Y.; Wei, Y.; Li, M.; Dong, Z.; Liu, Y.; Ruan, H.; Li, Y.; Cao, S.; et al. First Orally Bioavailable Prodrug of Proteolysis Targeting Chimera (PROTAC) Degrades Cyclin-Dependent Kinases 2/4/6 in Vivo. Eur. J. Med. Chem. 2021, 209, 112903. [Google Scholar] [CrossRef] [PubMed]
- Atilaw, Y.; Poongavanam, V.; Svensson Nilsson, C.; Nguyen, D.; Giese, A.; Meibom, D.; Erdelyi, M.; Kihlberg, J. Solution Conformations Shed Light on PROTAC Cell Permeability. ACS Med. Chem. Lett. 2021, 12, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Aguilar, A.; Liu, Z.; Yang, C.Y.; Wang, S. Simple Structural Modifications Converting a Bona Fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 2019, 62, 9471–9487. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, A.; Pandiella, A. Proteolysis Targeting Chimeras (PROTACs) in Cancer Therapy. J. Exp. Clin. Cancer Res. 2020, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A Selective BCL-XL PROTAC Degrader Achieves Safe and Potent Antitumor Activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Lu, M.-C.; Wang, Y.; Shan, W.-X.; Wang, X.-Y.; You, Q.-D.; Jiang, Z.-Y. Azo-PROTAC: Novel Light-Controlled Small-Molecule Tool for Protein Knockdown. J. Med. Chem. 2020, 63, 4644–4654. [Google Scholar] [CrossRef]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs Enable Optical Control of Protein Degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Naro, Y.; Darrah, K.; Deiters, A. Optical Control of Small Molecule-Induced Protein Degradation. J. Am. Chem. Soc. 2020, 142, 2193–2197. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.Ü.; et al. Light-Induced Control of Protein Destruction by Opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef]
- Yang, C.; Yang, Y.; Li, Y.; Ni, Q.; Li, J. Radiotherapy-Triggered Proteolysis Targeting Chimera Prodrug Activation in Tumors. J. Am. Chem. Soc. 2022. [Google Scholar] [CrossRef]
- Cheng, W.; Li, S.; Wen, X.; Han, S.; Wang, S.; Wei, H.; Song, Z.; Wang, Y.; Tian, X.; Zhang, X. Development of Hypoxia-Activated PROTAC Exerting a More Potent Effect in Tumor Hypoxia than in Normoxia. Chem. Commun. 2021, 57, 12852–12855. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- England, C.G.; Luo, H.; Cai, W. HaloTag Technology: A Versatile Platform for Biomedical Applications. Bioconjug. Chem. 2015, 26, 975–986. [Google Scholar] [CrossRef]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef]
- Ohana, R.F.; Encell, L.P.; Zhao, K.; Simpson, D.; Slater, M.R.; Urh, M.; Wood, K.V. HaloTag7: A Genetically Engineered Tag That Enhances Bacterial Expression of Soluble Proteins and Improves Protein Purification. Protein Expr. Purif. 2009, 68, 110–120. [Google Scholar] [CrossRef]
- Tomoshige, S.; Naito, M.; Hashimoto, Y.; Ishikawa, M. Degradation of HaloTag-Fused Nuclear Proteins Using Bestatin-HaloTag Ligand Hybrid Molecules. Org. Biomol. Chem. 2015, 13, 9746–9750. [Google Scholar] [CrossRef]
- Tomoshige, S.; Hashimoto, Y.; Ishikawa, M. Efficient Protein Knockdown of HaloTag-Fused Proteins Using Hybrid Molecules Consisting of IAP Antagonist and HaloTag Ligand. Bioorg. Med. Chem. 2016, 24, 3144–3148. [Google Scholar] [CrossRef] [PubMed]
- Tovell, H.; Testa, A.; Maniaci, C.; Zhou, H.; Prescott, A.R.; Macartney, T.; Ciulli, A.; Alessi, D.R. Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem. Biol. 2019, 14, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational Protein Knockdown Coupled to Receptor Tyrosine Kinase Activation with PhosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-Molecule Hydrophobic Tagging-Induced Degradation of HaloTag Fusion Proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Tae, H.S.; Sundberg, T.B.; Neklesa, T.K.; Noblin, D.J.; Gustafson, J.L.; Roth, A.G.; Raina, K.; Crews, C.M. Identification of Hydrophobic Tags for the Degradation of Stabilized Proteins. ChemBioChem 2012, 13, 538–541. [Google Scholar] [CrossRef]
- Raina, K.; Crews, C.M. Targeted Protein Knockdown Using Small Molecule Degraders. Curr. Opin. Chem. Biol. 2017, 39, 46–53. [Google Scholar] [CrossRef]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor Mediated Protein Degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef]
- Shi, Y.; Long, M.J.C.; Rosenberg, M.M.; Li, S.; Kobjack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc3Arg-Linked Ligands Induce Degradation by Localizing Target Proteins to the 20S Proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef]
- Xie, T.; Lim, S.M.; Westover, K.D.; Dodge, M.E.; Ercan, D.; Ficarro, S.B.; Udayakumar, D.; Gurbani, D.; Tae, H.S.; Riddle, S.M.; et al. Pharmacological Targeting of the Pseudokinase Her3. Nat. Chem. Biol. 2014, 10, 1006–1012. [Google Scholar] [CrossRef]
- Lim, S.M.; Xie, T.; Westover, K.D.; Ficarro, S.B.; Tae, H.S.; Gurbani, D.; Sim, T.; Marto, J.A.; Jänne, P.A.; Crews, C.M.; et al. Development of Small Molecules Targeting the Pseudokinase Her3. Bioorganic Med. Chem. Lett. 2015, 25, 3382–3389. [Google Scholar] [CrossRef]
- Nietzold, F.; Rubner, S.; Berg, T. The Hydrophobically-Tagged MDM2-P53 Interaction Inhibitor Nutlin-3a-HT Is More Potent against Tumor Cells than Nutlin-3a. Chem. Commun. 2019, 55, 14351–14354. [Google Scholar] [CrossRef]
- Rubner, S.; Scharow, A.; Schubert, S.; Berg, T. Selective Degradation of Polo-like Kinase 1 by a Hydrophobically Tagged Inhibitor of the Polo-Box Domain. Angew. Chem. Int. Ed. 2018, 57, 17043–17047. [Google Scholar] [CrossRef]
- Wittmann, B.M.; Sherk, A.; McDonnell, D.P. Definition of Functionally Important Mechanistic Differences among Selective Estrogen Receptor Down-Regulators. Cancer Res. 2007, 67, 9549–9560. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Wardell, S.E.; Norris, J.D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58, 4883–4887. [Google Scholar] [CrossRef]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo Recognition and Degradation by Selective Autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, N.; Wang, Z.; Luo, S.; Ding, Y.; Lu, B. Degradation of Lipid Droplets by Chimeric Autophagy-Tethering Compounds. Cell Res. 2021, 31, 965–979. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.C.H.; Weissman, A.M. Regulating the P53 System through Ubiquitination. Oncogene 2004, 23, 2096–2106. [Google Scholar] [CrossRef]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef]
- Liu, J.; Yu, X.; Chen, H.; Kaniskan, H.Ü.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-DUBTACs Stabilize Tumor Suppressor Transcription Factors. J. Am. Chem. Soc. 2022, 144, 12934–12941. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, S.; Tom, J.; Fu, Y.; Wu, W.; Zeng, L.; Sun, C.; Liu, Q.; Lin, J.; Lin, K.; Fairbrother, W.J.; et al. Heterobifunctional Molecules Induce Dephosphorylation of Kinases-A Proof of Concept Study. J. Med. Chem. 2020, 63, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, S.U.; Munkanatta Godage, D.N.P.; Shoba, V.M.; Lai, S.; Shi, M.; Wu, P.; Chaudhary, S.K.; Schreiber, S.L.; Choudhary, A. Phosphorylation-Inducing Chimeric Small Molecules. J. Am. Chem. Soc. 2020, 142, 14052–14057. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and Deacetylation of Non-Histone Proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Chen, L.Y.; Wozniak, J.M.; Jadhav, A.M.; Anderson, H.; Malone, T.E.; Parker, C.G. Targeted Protein Acetylation in Cells Using Heterobifunctional Molecules. J. Am. Chem. Soc. 2021, 143, 16700–16708. [Google Scholar] [CrossRef]
- Clackson, T.; Yang, W.; Rozamus, L.W.; Hatada, M.; Amara, J.F.; Rollins, C.T.; Stevenson, L.F.; Magari, S.R.; Wood, S.A.; Courage, N.L.; et al. Redesigning an FKBP-Ligand Interface to Generate Chemical Dimerizers with Novel Specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 10437–10442. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Paiva, S.L.; Rafizadeh, D.N.; Pettersson, M.; Qin, L.; Crews, C.M. Targeted Protein Internalization and Degradation by ENDosome TArgeting Chimeras (ENDTACs). ACS Cent. Sci. 2019, 5, 1079–1084. [Google Scholar]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-Targeting Chimaeras for Degradation of Extracellular Proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Costales, M.G.; Suresh, B.; Vishnu, K.; Disney, M.D. Targeted Degradation of a Hypoxia-Associated Non-Coding RNA Enhances the Selectivity of a Small Molecule Interacting with RNA. Cell Chem. Biol. 2019, 26, 1180–1186. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging Principles for the Therapeutic Exploitation of Glycosylation. Science 2014, 343, 1235681. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S.C. Non-Histone Protein Methylation as a Regulator of Cellular Signalling and Function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heitel, P. Emerging TACnology: Heterobifunctional Small Molecule Inducers of Targeted Posttranslational Protein Modifications. Molecules 2023, 28, 690. https://doi.org/10.3390/molecules28020690
Heitel P. Emerging TACnology: Heterobifunctional Small Molecule Inducers of Targeted Posttranslational Protein Modifications. Molecules. 2023; 28(2):690. https://doi.org/10.3390/molecules28020690
Chicago/Turabian StyleHeitel, Pascal. 2023. "Emerging TACnology: Heterobifunctional Small Molecule Inducers of Targeted Posttranslational Protein Modifications" Molecules 28, no. 2: 690. https://doi.org/10.3390/molecules28020690
APA StyleHeitel, P. (2023). Emerging TACnology: Heterobifunctional Small Molecule Inducers of Targeted Posttranslational Protein Modifications. Molecules, 28(2), 690. https://doi.org/10.3390/molecules28020690