
Drug Metabolism of Hepatocyte-like Organoids and Their Applicability in In Vitro Toxicity Testing

 , , ,
, , ,  , , , and
, , , and 
Abstract
1. Introduction
2. Results
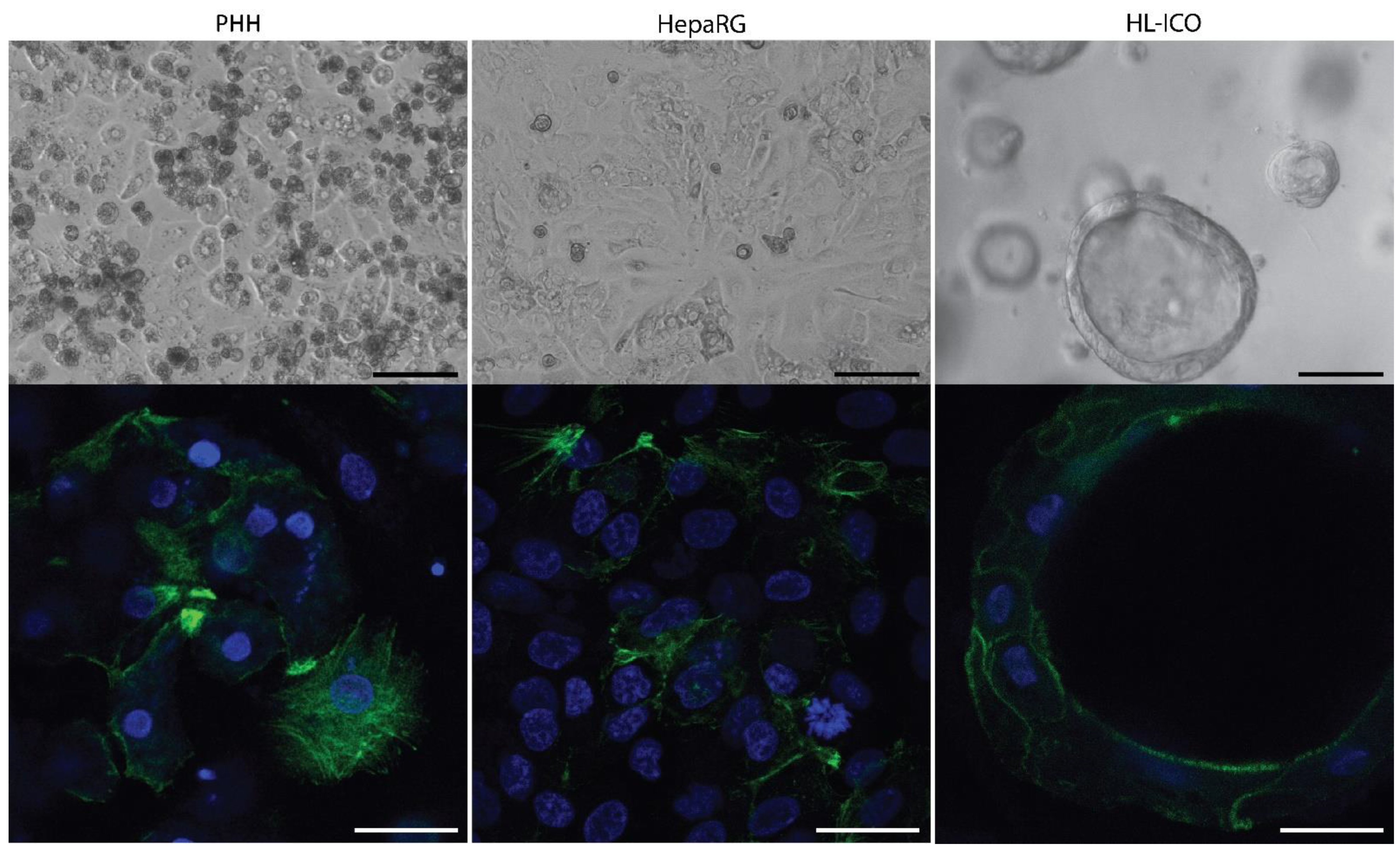
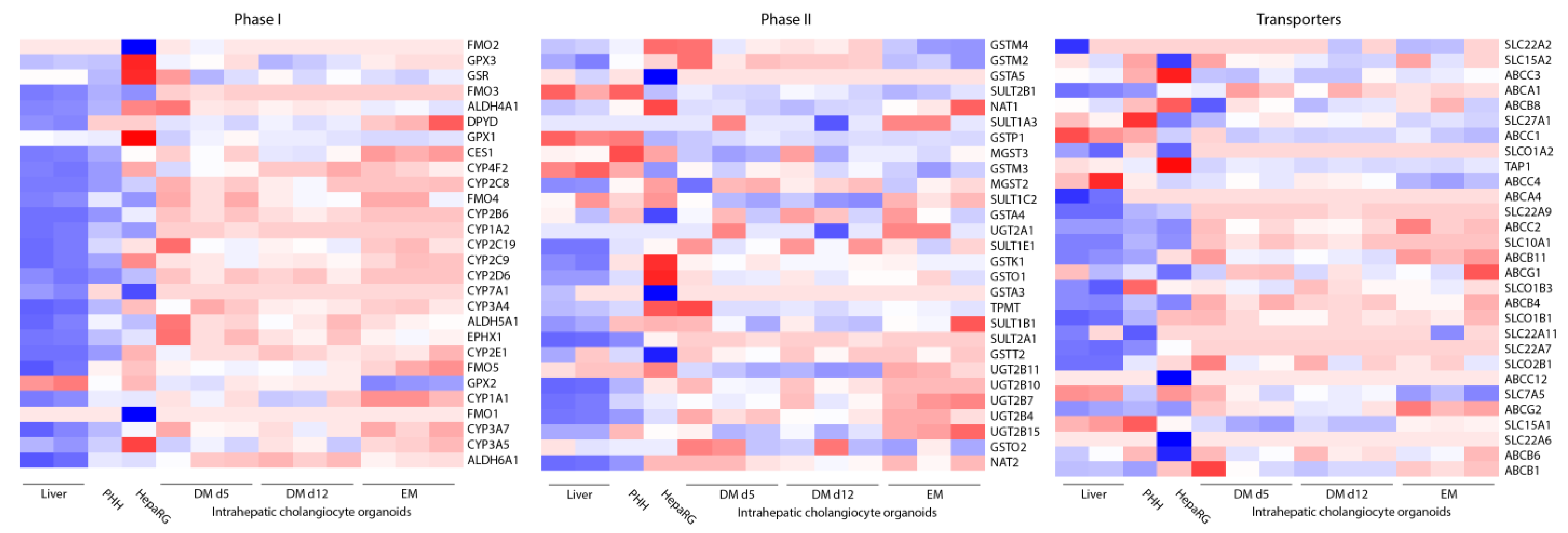
2.1. Expression of Phase I and II Enzymes, and Hepatic Transporters
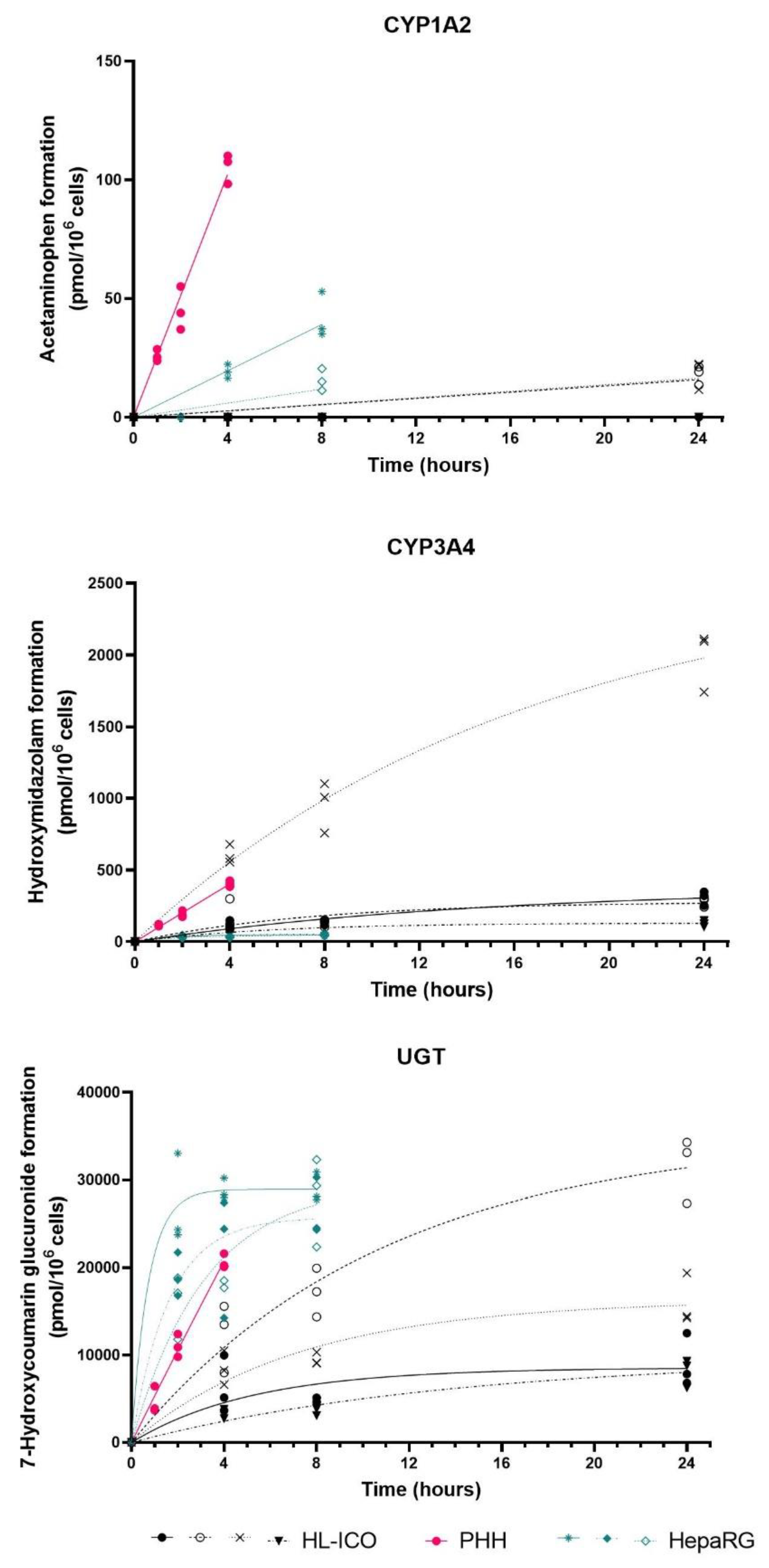
2.2. Phase I and II Enzyme Activity
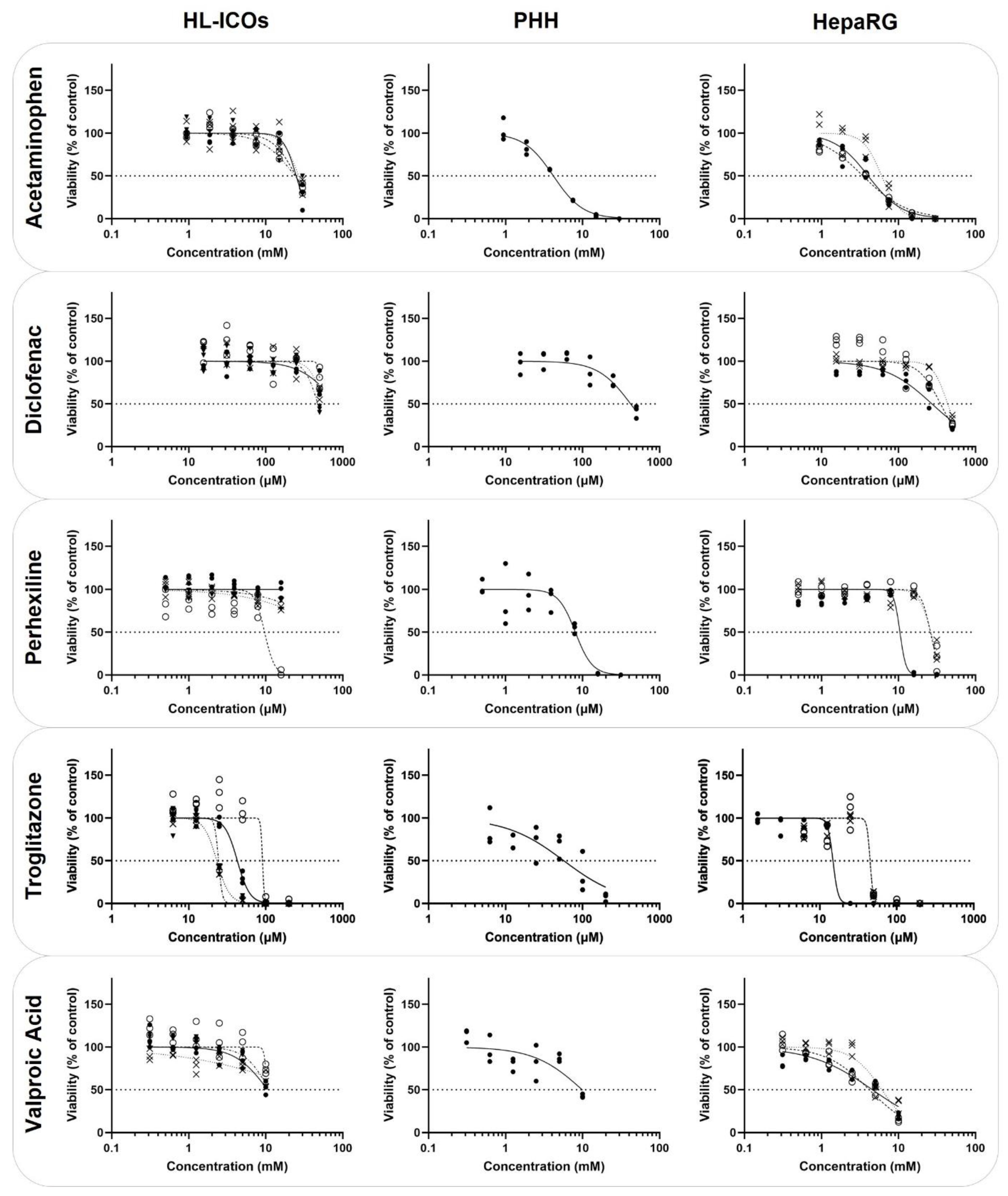
2.3. Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Whole Genome RNA Sequencing
4.3. Cytochrome P450 Activity
4.4. Cytotoxicity
4.5. Cell Viability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Issa, N.T.; Wathieu, H.; Ojo, A.; Byers, S.W.; Dakshanamurthy, S. Drug Metabolism in Preclinical Drug Development: A Survey of the Discovery Process, Toxicology, and Computational Tools. Curr. Drug Metab. 2017, 18, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- García-Cortés, M.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Spanish Group for the Study of Drug-Induced Liver Disease (Grupo de Estudio para las Hepatopatías Asociadas a Medicamentos GEHAM) l Drug-induced liver injury: A safety review. Expert Opin. Drug Saf. 2018, 17, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Craveiro, N.S.; Lopes, B.S.; Tomás, L.; Almeida, S.F. Drug Withdrawal Due to Safety: A Review of the Data Supporting Withdrawal Decision. Curr. Drug Saf. 2020, 15, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Utkarsh, D.; Loretz, C.; Li, A.P. In vitro evaluation of hepatotoxic drugs in human hepatocytes from multiple donors: Identification of P450 activity as a potential risk factor for drug-induced liver injuries. Chem. Biol. Interact. 2016, 255, 12–22. [Google Scholar] [CrossRef]
- Wang, C.W.; Preclaro, I.A.C.; Lin, W.H.; Chung, W.H. An Updated Review of Genetic Associations With Severe Adverse Drug Reactions: Translation and Implementation of Pharmacogenomic Testing in Clinical Practice. Front. Pharmacol. 2022, 13, 886377. [Google Scholar] [CrossRef]
- Zhou, S.F.; Liu, J.P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef]
- Gan, J.; Ma, S.; Zhang, D. Non-cytochrome P450-mediated bioactivation and its toxicological relevance. Drug Metab. Rev. 2016, 48, 473–501. [Google Scholar] [CrossRef]
- Liu, H.; Sahi, J. Role of Hepatic Drug Transporters in Drug Development. J. Clin. Pharmacol. 2016, 56 (Suppl. 7), S11–S22. [Google Scholar] [CrossRef]
- Jetter, A.; Kullak-Ublick, G.A. Drugs and hepatic transporters: A review. Pharmacol. Res. 2020, 154, 104234. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Liang, A.; Liao, G.; To, I.; Shehu, A.; Ma, X. Roles of Cofactors in Drug-Induced Liver Injury: Drug Metabolism and Beyond. Drug Metab. Dispos. 2022, 50, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Ghiciuc, C.; Opritoui, M.; Tursas, L.; Pelkonen, O.; Pasanen, M. Predictive value of animal models for human cytochrome P450 (CYP)-mediated metabolism: A comparative study in vitro. Xenobiotica 2007, 37, 1367–1377. [Google Scholar] [CrossRef]
- Hammer, H.; Schmidt, F.; Marx-Stoelting, P.; Pötz, O.; Braeuning, A. Cross-species analysis of hepatic cytochrome P450 and transport protein expression. Arch. Toxicol. 2021, 95, 117–133. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Acosta, D.; Andersen, M.; Anderson, H.; Bailar, J.C.; Boekelheide, K.; Brent, R.; Charnley, G.; Cheung, V.G.; Green, S.; et al. Toxicity testing in the 21st century: A vision and a strategy. J. Toxicol. Environ. Health Part B 2010, 13, 51–138. [Google Scholar] [CrossRef] [PubMed]
- Zink, D.; Chuah, J.K.C.; Ying, J.Y. Assessing Toxicity with Human Cell-Based In Vitro Methods. Trends Mol. Med. 2020, 26, 570–582. [Google Scholar] [CrossRef]
- Yadav, J.; El Hassani, M.; Sodhi, J.; Lauschke, V.M.; Hartman, J.H.; Russell, L.E. Recent developments in in vitro and in vivo models for improved translation of preclinical pharmacokinetics and pharmacodynamics data. Drug Metab. Rev. 2021, 53, 207–233. [Google Scholar] [CrossRef]
- Serras, A.S.; Rodrigues, J.S.; Cipriano, M.; Rodrigues, A.V.; Oliveira, N.G.; Miranda, J.P. A Critical Perspective on 3D Liver Models for Drug Metabolism and Toxicology Studies. Front. Cell Dev. Biol. 2021, 9, 626805. [Google Scholar] [CrossRef]
- Xu, Q. Human Three-Dimensional Hepatic Models: Cell Type Variety and Corresponding Applications. Front. Bioeng. Biotechnol. 2021, 9, 730008. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, T.; Chen, D.; Wang, Q.; Zhang, L.W. Three-dimensional liver models: State of the art and their application for hepatotoxicity evaluation. Crit. Rev. Toxicol. 2020, 50, 279–309. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S. Three-Dimensional Liver Culture Systems to Maintain Primary Hepatic Properties for Toxicological Analysis In Vitro. Int. J. Mol. Sci. 2021, 22, 10214. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, L.; Liu, W.; Ma, X.; Cen, J.; Sun, Z.; Wang, C.; Feng, S.; Zhang, Z.; Yue, L.; et al. In Vitro Expansion of Primary Human Hepatocytes with Efficient Liver Repopulation Capacity. Cell Stem Cell 2018, 23, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef]
- Kim, Y.; Lasher, C.D.; Milford, L.M.; Murali, T.M.; Rajagopalan, P. A comparative study of genome-wide transcriptional profiles of primary hepatocytes in collagen sandwich and monolayer cultures. Tissue Eng. Part C Methods 2010, 16, 1449–1460. [Google Scholar] [CrossRef]
- Akbari, S.; Arslan, N.; Senturk, S.; Erdal, E. Next-Generation Liver Medicine Using Organoid Models. Front. Cell Dev. Biol. 2019, 7, 345. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver organoids: From basic research to therapeutic applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2020, 3, 100198. [Google Scholar] [CrossRef]
- Lee, J.; Han, H.; Lee, S.; Cho, E.; Lee, H.; Seok, J.; Lim, H.S.; Son, W. Use of 3D Human Liver Organoids to Predict Drug-Induced Phospholipidosis. Int. J. Mol. Sci. 2020, 21, 2982. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Lu, D.; Lin, Z.; Chen, H.; Li, H.; Yang, X.; Yang, M.; Wang, K.; Wei, X.; Zheng, S.; et al. Liver Organoids, Novel and Promising Modalities for Exploring and Repairing Liver Injury. Stem Cell Rev. Rep. 2022. [Google Scholar] [CrossRef]
- Wang, L.; Li, M.; Yu, B.; Shi, S.; Liu, J.; Zhang, R.; Ayada, I.; Verstegen, M.M.A.; van der Laan, L.J.W.; Peppelenbosch, M.P.; et al. Recapitulating lipid accumulation and related metabolic dysregulation in human liver-derived organoids. J. Mol. Med. 2022, 100, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Shiota, J.; Samuelson, L.C.; Razumilava, N. Hepatobiliary Organoids and Their Applications for Studies of Liver Health and Disease: Are We There Yet? Hepatology 2021, 74, 2251–2263. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K.; Sánchez-Romero, N.; Ye, S.; van Steenbeek, F.G.; Oosterhoff, L.A.; Pla Palacin, I.; Chen, C.; van Wolferen, M.E.; van Tienderen, G.; Lieshout, R.; et al. Large-Scale Production of LGR5-Positive Bipotential Human Liver Stem Cells. Hepatology 2020, 72, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Benfenati, E.; Busquet, F.; Castell, J.; Clevert, D.A.; de Kok, T.M.; Dirven, H.; Fritsche, E.; Geris, L.; Gozalbes, R.; et al. Safer chemicals using less animals: Kick-off of the European ONTOX project. Toxicology 2021, 458, 152846. [Google Scholar] [CrossRef]
- Chang, X.; Tan, Y.M.; Allen, D.G.; Bell, S.; Brown, P.C.; Browning, L.; Ceger, P.; Gearhart, J.; Hakkinen, P.J.; Kabadi, S.V.; et al. IVIVE: Facilitating the Use of In Vitro Toxicity Data in Risk Assessment and Decision Making. Toxics 2022, 10, 232. [Google Scholar] [CrossRef]
- Schadt, S.; Simon, S.; Kustermann, S.; Boess, F.; McGinnis, C.; Brink, A.; Lieven, R.; Fowler, S.; Youdim, K.; Ullah, M.; et al. Minimizing DILI risk in drug discovery—A screening tool for drug candidates. Toxicol. In Vitro 2015, 30, 429–437. [Google Scholar] [CrossRef]
- Tolosa, L.; Gómez-Lechón, M.J.; Jiménez, N.; Hervás, D.; Jover, R.; Donato, M.T. Advantageous use of HepaRG cells for the screening and mechanistic study of drug-induced steatosis. Toxicol. Appl. Pharmacol. 2016, 302, 1–9. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of Drug-Induced Hepatotoxicity Using Long-Term Stable Primary Hepatic 3D Spheroid Cultures in Chemically Defined Conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef]
- Arnesdotter, E.; Gijbels, E.; Dos Santos Rodrigues, B.; Vilas-Boas, V.; Vinken, M. Adverse Outcome Pathways as Versatile Tools in Liver Toxicity Testing. Methods Mol. Biol. 2022, 2425, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.Y.; Wen, J.; Stauber, K. In vitro Drug Metabolism Investigation of 7-Ethoxycoumarin in Human, Monkey, Dog and Rat Hepatocytes by High Resolution LC-MS/MS. Drug Metab. Lett. 2018, 12, 33–53. [Google Scholar] [CrossRef]
- Dragovic, S.; Vermeulen, N.P.E.; Gerets, H.H.; Hewitt, P.G.; Ingelman-Sundberg, M.; Park, B.K.; Juhila, S.; Snoeys, J.; Weaver, R.J. Evidence-based selection of training compounds for use in the mechanism-based integrated prediction of drug-induced liver injury in man. Arch. Toxicol. 2016, 90, 2979–3003. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-Induced Hepatotoxicity: A Comprehensive Update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Zhu, Z.; Masse, G.; Duan, S.X.; James, L.P.; Harmatz, J.S.; Greenblatt, D.J. Race, Gender, and Genetic Polymorphism Contribute to Variability in Acetaminophen Pharmacokinetics, Metabolism, and Protein-Adduct Concentrations in Healthy African-American and European-American Volunteers. J. Pharmacol. Exp. Ther. 2017, 362, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, K.; Rasool, M.I.; Alherz, F.A.; El Daibani, A.A.; Bairam, A.F.; Abunnaja, M.S.; Yasuda, S.; Wilson, L.J.; Hui, Y.; Liu, M. SULT genetic polymorphisms: Physiological, pharmacological and clinical implications. Expert Opin. Drug Metab. Toxicol. 2021, 17, 767–784. [Google Scholar] [CrossRef]
- Daly, A.K.; Aithal, G.P.; Leathart, J.B.; Swainsbury, R.A.; Dang, T.S.; Day, C.P. Genetic susceptibility to diclofenac-induced hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 2007, 132, 272–281. [Google Scholar] [CrossRef]
- Ren, Z.; Chen, S.; Pak, S.; Guo, L. A mechanism of perhexiline’s cytotoxicity in hepatic cells involves endoplasmic reticulum stress and p38 signaling pathway. Chem. Biol. Interact. 2021, 334, 109353. [Google Scholar] [CrossRef]
- Sørensen, L.B.; Sørensen, R.N.; Miners, J.O.; Somogyi, A.A.; Grgurinovich, N.; Birkett, D.J. Polymorphic hydroxylation of perhexiline in vitro. Br. J. Clin. Pharmacol. 2003, 55, 635–638. [Google Scholar] [CrossRef]
- Barclay, M.L.; Sawyers, S.M.; Begg, E.J.; Zhang, M.; Roberts, R.L.; Kennedy, M.A.; Elliott, J.M. Correlation of CYP2D6 genotype with perhexiline phenotypic metabolizer status. Pharmacogenetics 2003, 13, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Troglitazone hepatotoxicity: Are we getting closer to understanding idiosyncratic liver injury? Toxicol. Sci. 2007, 97, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, N.J.; Lloyd, S.; Hayden, M.; Butler, R.; Sakai, Y.; Springer, R.; Fackett, A.; Li, A.P. Correlation between troglitazone cytotoxicity and drug metabolic enzyme activities in cryopreserved human hepatocytes. Chem. Biol. Interact. 2002, 142, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; New, L.S.; Ho, H.K.; Chui, W.K.; Chan, E.C.Y. Direct toxicity effects of sulfo-conjugated troglitazone on human hepatocytes. Toxicol. Lett. 2010, 195, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Ezhilarasan, D.; Mani, U. Valproic acid induced liver injury: An insight into molecular toxicological mechanism. Environ. Toxicol. Pharmacol. 2022, 95, 103967. [Google Scholar] [CrossRef]
- Dimitrijevic, D.; Fabian, E.; Nicol, B.; Funk-Weyer, D.; Landsiedel, R. Toward Realistic Dosimetry In Vitro: Determining Effective Concentrations of Test Substances in Cell Culture and Their Prediction by an In Silico Mass Balance Model. Chem. Res. Toxicol. 2022, 35, 1962–1973. [Google Scholar] [CrossRef]
- Kang, H.K.; Sarsenova, M.; Kim, D.; Kim, M.S.; Lee, J.Y.; Sung, E.; Kook, M.G.; Kim, N.G.; Choi, S.W.; Ogay, V.; et al. Establishing a 3D In Vitro Hepatic Model Mimicking Physiologically Relevant to In Vivo State. Cells 2021, 10, 1268. [Google Scholar] [CrossRef]
- Correia, C.; Ferreira, A.; Santos, J.; Lapa, R.; Yliperttula, M.; Urtti, A.; Vale, N. New In Vitro-In Silico Approach for the Prediction of In Vivo Performance of Drug Combinations. Molecules 2021, 26, 4257. [Google Scholar] [CrossRef]
- Di, L. The Impact of Carboxylesterases in Drug Metabolism and Pharmacokinetics. Curr. Drug Metab. 2019, 20, 91–102. [Google Scholar] [CrossRef]
- Nie, Y.; Yang, J.; Liu, S.; Sun, R.; Chen, H.; Long, N.; Jiang, R.; Gui, C. Genetic polymorphisms of human hepatic OATPs: Functional consequences and effect on drug pharmacokinetics. Xenobiotica 2020, 50, 297–317. [Google Scholar] [CrossRef]
- Mennecozzi, M.; Landesmann, B.; Palosaari, T.; Harris, G.; Whelan, M. Sex differences in liver toxicity-do female and male human primary hepatocytes react differently to toxicants in vitro? PLoS ONE 2015, 10, e0122786. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Hengstler, J.G. Characterization of hepatocyte-based in vitro systems for reliable toxicity testing. Arch. Toxicol. 2018, 92, 2981–2986. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, V.M.P.; Wang, Z.; Bakker, W.; Zheng, W.; Spee, B.; Bouwmeester, H. Hepatic bile acid synthesis and secretion: Comparison of in vitro methods. Toxicol. Lett. 2022, 365, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Brecklinghaus, T.; Albrecht, W.; Kappenberg, F.; Duda, J.; Vartak, N.; Edlund, K.; Marchan, R.; Ghallab, A.; Cadenas, C.; Günther, G.; et al. The hepatocyte export carrier inhibition assay improves the separation of hepatotoxic from non-hepatotoxic compounds. Chem. Biol. Interact. 2022, 351, 109728. [Google Scholar] [CrossRef] [PubMed]
- Kasteel, E.E.J.; Darney, K.; Kramer, N.I.; Dorne, J.L.C.M.; Lautz, L.S. Human variability in isoform-specific UDP-glucuronosyltransferases: Markers of acute and chronic exposure, polymorphisms and uncertainty factors. Arch. Toxicol. 2020, 94, 2637–2661. [Google Scholar] [CrossRef]
- den Braver-Sewradj, S.P.; den Braver, M.W.; Baze, A.; Decorde, J.; Fonsi, M.; Bachellier, P.; Vermeulen, N.P.E.; Commandeur, J.N.M.; Richert, L.; Vos, J.C. Direct comparison of UDP-glucuronosyltransferase and cytochrome P450 activities in human liver microsomes, plated and suspended primary human hepatocytes from five liver donors. Eur. J. Pharm. Sci. 2017, 109, 96–110. [Google Scholar] [CrossRef]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schäfer, R.; et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J. Biomol. Screen. 2016, 21, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Faria, J.; van der Laan, L.J.W.; Penning, L.C.; Masereeuw, R.; Spee, B. Human Cholangiocytes Form a Polarized and Functional Bile Duct on Hollow Fiber Membranes. Front. Bioeng. Biotechnol. 2022, 10, 868857. [Google Scholar] [CrossRef]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell-Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef]
- Leite, S.B.; Wilk-Zasadna, I.; Zaldivar, J.M.; Airola, E.; Reis-Fernandes, M.A.; Mennecozzi, M.; Guguen-Guillouzo, C.; Chesne, C.; Guillou, C.; Alves, P.M.; et al. Three-dimensional HepaRG model as an attractive tool for toxicity testing. Toxicol. Sci. 2012, 130, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HL-ICOs | PHH | HepaRG | ||||
|---|---|---|---|---|---|---|
| CYP1A2 | nd | 0.01249 | 0.01293 | nd | 0.4269 | 0.03237 (nd–0.08552) |
| CYP2B6 | nd | nd | nd | nd | 50.13 | 1.228 (0.9913–1.358) |
| CYP2C9 | nd | nd | nd | nd | 15.33 | 2.161 (1.365–2.521) |
| CYP2D6 | nd | nd | nd | nd | 9.601 | 0.1656 (0.1567–0.2866) |
| CYP2E1 | nd | nd | nd | nd | nd | nd |
| CYP3A4 | 0.3294 | 0.3603 | 2.098 | 0.2278 | 1.680 | 0.3136 (0.2645–0.3296) |
| UGT | 13.18 | 38.93 | 22.89 | 8.778 | 86.79 | 158.9 (132.6–225.4) |
| HL-ICOs | PHH | HepaRG | ||||
|---|---|---|---|---|---|---|
| Acetaminophen | 24,870 | 24,630 | 26,840 | 24,010 | 4186 | 4036 (3465–6045) |
| Diclofenac | >500 | >500 | >500 | 475.5 | 421.2 | 351.7 (272.4–434.9) |
| Perhexiline | >31.5 | 9.675 | >31.5 | >31.5 | 8.072 | 25.97 (10.45–26.37) |
| Troglitazone | 42.80 | 90.83 | 23.13 | 24.40 | 57.09 | 45.15 (14.89–45.17) |
| Valproic Acid | >10,000 | >10,000 | >10,000 | >10,000 | 9885 | 4582 (4168–6066) |
| Enzyme | Parent Compound | CAS Number | Dosed Concentration (µM) | |
|---|---|---|---|---|
| Cocktail A | CYP1A2 | Phenacetin | 62-44-2 | 15 |
| Acetaminophen | 103-90-2 | |||
| CYP3A4 | Midazolam | 59467-70-8 | 5 | |
| Midazolam-OH | 59468-90-5 | |||
| CYP2D6 | Dextromethorphan | 125-71-3 | 15 | |
| Dextrorphan | 143-98-6 | |||
| CYP2C9 | Tolbutamide | 64-77-7 | 20 | |
| 4OH-Tolbutamide | 5719-85-7 | |||
| Cocktail B | UGT | 7-OH Coumarin | 93-35-6 | 12 |
| 7-OH Coumarin Glucuronide | 66695-14-5 | |||
| CYP2E1 | Chlorzoxazone | 95-25-0 | 25 | |
| 6OH-Chlorzoxazone | 1750-45-4 | |||
| CYP2B6 | Bupropion | 31677-93-7 | 20 | |
| OH-Bupropion | 92264-81-8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouwmeester, M.C.; Tao, Y.; Proença, S.; van Steenbeek, F.G.; Samsom, R.-A.; Nijmeijer, S.M.; Sinnige, T.; van der Laan, L.J.W.; Legler, J.; Schneeberger, K.; et al. Drug Metabolism of Hepatocyte-like Organoids and Their Applicability in In Vitro Toxicity Testing. Molecules 2023, 28, 621. https://doi.org/10.3390/molecules28020621
Bouwmeester MC, Tao Y, Proença S, van Steenbeek FG, Samsom R-A, Nijmeijer SM, Sinnige T, van der Laan LJW, Legler J, Schneeberger K, et al. Drug Metabolism of Hepatocyte-like Organoids and Their Applicability in In Vitro Toxicity Testing. Molecules. 2023; 28(2):621. https://doi.org/10.3390/molecules28020621
Chicago/Turabian StyleBouwmeester, Manon C., Yu Tao, Susana Proença, Frank G. van Steenbeek, Roos-Anne Samsom, Sandra M. Nijmeijer, Theo Sinnige, Luc J. W. van der Laan, Juliette Legler, Kerstin Schneeberger, and et al. 2023. "Drug Metabolism of Hepatocyte-like Organoids and Their Applicability in In Vitro Toxicity Testing" Molecules 28, no. 2: 621. https://doi.org/10.3390/molecules28020621
APA StyleBouwmeester, M. C., Tao, Y., Proença, S., van Steenbeek, F. G., Samsom, R.-A., Nijmeijer, S. M., Sinnige, T., van der Laan, L. J. W., Legler, J., Schneeberger, K., Kramer, N. I., & Spee, B. (2023). Drug Metabolism of Hepatocyte-like Organoids and Their Applicability in In Vitro Toxicity Testing. Molecules, 28(2), 621. https://doi.org/10.3390/molecules28020621