
Influence of Phosphorus Structures and Their Oxidation States on Flame-Retardant Properties of Polyhydroxyurethanes


Abstract
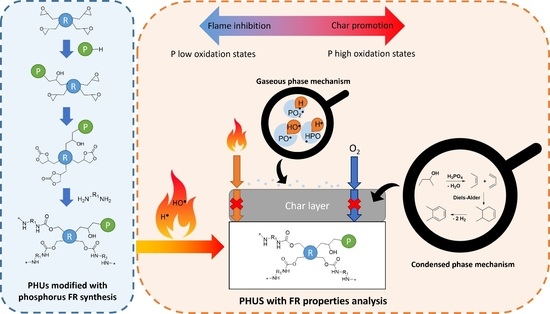
1. Introduction
2. Results and Discussion
2.1. Synthesis of Monomers and Materials
2.1.1. Synthesis of Phosphorylated Cyclic Carbonate MBDAC
2.1.2. Formulations of Phosphorylated-PHU Materials
- Structural analysis
- 2.
- Thermal and flame-retardancy analyses
- Thermogravimetric analyses
- Pyrolysis combustion flow calorimeter analysis
- Cone calorimeter test
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Nuclear Magnetic Resonance
3.2.2. Titration of the Epoxy Equivalent Weight by 1H NMR Spectroscopy
3.2.3. Titration of the Amine Equivalent Weight of Jeffamine EDR-148 by 1H NMR
3.2.4. Titration of the Carbonate Equivalent Weight by 1H NMR
3.2.5. Fourier Transform Infrared Spectroscopy
3.2.6. Differential Scanning Calorimetry
3.2.7. Gel Content and Swelling Rate
3.2.8. Thermogravimetric Analysis
3.2.9. Pyrolysis Combustion Flow Calorimeter Analysis
3.2.10. Cone Calorimeter Test
3.3. Syntheses
3.3.1. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (Thiourea) [49]
3.3.2. Synthesis of 4,4′-(3,6,9,12,15,18,21-heptamethyl-2,5,8,11,14,17,20,23-octaoxatetracosane-1,24-diyl)bis(1,3-dioxolan-2-one) (PPO DC) [59]
3.3.3. Synthesis of 4,4′,4″,4‴-(((methylenebis(4,1-phenylene))bis(azanetriyl))tetrakis (methylene))tetrakis(1,3-dioxolan-2-one) (MBDAC) [45]
3.3.4. Synthesis of 6-(3-((4-(4-(bis(oxiran-2-ylmethyl)amino)benzyl)phenyl)(oxiran-2-ylmethyl) amino)-2-hydroxypropyl)dibenzo[c,e][1,2]oxaphosphinine 6-oxide (MBDA-DOPO) [45]
3.3.5. Synthesis of 4,4′-(((4-(4-((2-hydroxy-3-(6-oxidodibenzo[c,e][1,2]oxaphosphinin-6-yl)propyl) ((2-oxo-1,3-dioxolan-4-yl)methyl)amino)benzyl)phenyl)azanediyl)bis(methylene))bis(1,3-dioxolan-2-one) (MBDAC-DOPO) [45]
3.3.6. Synthesis of Diethyl (3-((4-(4-(bis(oxiran-2-ylmethyl)amino)benzyl)phenyl)(oxiran-2-ylmethyl)amin-o)-2-hydroxypropyl)phosphonate (MBDA-DEP)
3.3.7. Synthesis of Diethyl (3-((4-(4-(bis((2-oxo-1,3-dioxolan-4-yl)methyl)amino)benzyl)phenyl)((2-oxo-1,3-dioxolan-4-yl)methyl)amino)-2-hydroxypropyl)phosphonate (MBDAC-DEP)
3.3.8. Synthesis of Diphenyl (3-((4-(4-(bis(oxiran-2-ylmethyl)amino)benzyl)phenyl)(oxiran-2-ylmethyl)amino)-2-hydroxypropyl)phosphonate (MBDA-DPP)
3.3.9. Synthesis of Diphenyl (3-((4-(4-(bis((2-oxo-1,3-dioxolan-4-yl)methyl)amino)benzyl)phenyl)((2-oxo-1,3-dioxolan-4-yl)methyl)amino)-2-hydroxypropyl)phosphonate (MBDAC-DPP)
3.3.10. Synthesis of Dibenzo[d,f][1,3,2]dioxaphosphepine 6-oxide (BPPO) [61]
3.3.11. Synthesis of 6-(1-((4-(4-(bis(oxiran-2-ylmethyl)amino)benzyl)phenyl)(oxiran-2-ylmethyl)amino)-3-hydroxypropan-2-yl)dibenzo[d,f][1,3,2]dioxaphosphepine 6-oxide (MBDA-BPPO)
3.3.12. Synthesis of 4,4′-(((4-(4-((3-hydroxy-2-(6-oxidodibenzo[d,f][1,3,2]dioxaphosphepin-6-yl)propyl)((2-oxo-1,3-dioxolan-4-yl)methyl)amino)benzyl)phenyl)azanediyl) bis(methylene))bis(1,3-dioxolan-2-one) (MBDAC-BPPO)
3.4. Formulations of the PHU Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications—A review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Polyurethane Market Size, Share & Trends Analysis Report by Product (Flexible Foam, Rigid Foam), by End Use (Construction, Electronics & Appliances), by Region (APAC, North America), and Segment Forecasts, 2021–2028. Available online: https://www.fortunebusinessinsights.com/industry-reports/polyurethane-pu-market-101801 (accessed on 15 May 2022).
- Jeong, J.O.; Park, J.S.; Lim, Y.M. Development of Styrene-Grafted Polyurethane by Radiation-Based Techniques. Materials 2016, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Coste, G.; Negrell, C.; Caillol, S. Cascade (Dithio)Carbonate Ring Opening Reactions for Self-Blowing Polyhydroxythiourethane Foams. Macromol. Rapid Commun. 2022, 43, 2100833. [Google Scholar] [CrossRef]
- Zafar, F.; Ghosal, A.; Sharmin, E.; Chaturvedi, R.; Nishat, N. A Review on Cleaner Production of Polymeric and Nanocomposite Coatings Based on Waterborne Polyurethane Dispersions from Seed Oils. Prog. Org. Coat. 2019, 131, 259–275. [Google Scholar] [CrossRef]
- Pössel, B.; Mülhaupt, R. Lysine-Functionalized Gibbsite Nanoplatelet Dispersions for Nonisocyanate Polyhydroxyurethane Nanocomposites and Translucent Coatings. Macromol. Mater. Eng. 2020, 305, 2000217. [Google Scholar] [CrossRef]
- Quienne, B.; Poli, R.; Pinaud, J.; Caillol, S. Enhanced Aminolysis of Cyclic Carbonates by β-Hydroxylamines for the Production of Fully Biobased Polyhydroxyurethanes. Green Chem. 2021, 23, 1678–1690. [Google Scholar] [CrossRef]
- Carré, C.; Ecochard, Y.; Caillol, S.; Avérous, L. From the Synthesis of Biobased Cyclic Carbonate to Polyhydroxyurethanes: A Promising Route towards Renewable Non-Isocyanate Polyurethanes. ChemSusChem 2019, 12, 3410–3430. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Feng, Y.; Zhang, P.; Hong, W.; Chen, Y.; Fan, H.; Yu, D.; Chen, X. Preparation of Flame-Retardant Polyurethane and Its Applications in the Leather Industry. Polymers 2021, 13, 1730. [Google Scholar] [CrossRef]
- Tirri, T.; Aubert, M.; Wilén, C.E.; Pfaendner, R.; Hoppe, H. Novel Tetrapotassium Azo Diphosphonate (INAZO) as Flame Retardant for Polyurethane Adhesives. Polym. Degrad. Stab. 2012, 97, 375–382. [Google Scholar] [CrossRef]
- Song, L.; Hu, Y.; Tang, Y.; Zhang, R.; Chen, Z.; Fan, W. Study on the Properties of Flame Retardant Polyurethane/Organoclay Nanocomposite. Polym. Degrad. Stab. 2005, 88, 111–116. [Google Scholar] [CrossRef]
- Benin, V.; Gardelle, B.; Morgan, A.B. Heat Release of Polyurethanes Containing Potential Flame Retardants Based on Boron and Phosphorus Chemistries. Polym. Degrad. Stab. 2014, 106, 108–121. [Google Scholar] [CrossRef]
- El-Wahab, H.A.; El-Fattah, M.A.; El-Khalik, N.A.; Sharaby, C.M. Synthesis and performance of flame retardant additives based on cyclodiphosph (V) azane of sulfaguanidine, 1, 3-di-[N/-2-pyrimidinylsulfanilamide]-2, 2, 2.4, 4, 4-hexachlorocyclodiphosph (V) azane and 1, 3-di-[N/-2-pyrimidinylsulfanilamide]-2, 4-di [aminoacetic acid]-2, 4-dichlorocyclodiphosph (V) azane incorporated into polyurethane varnish. Prog. Org. Coat. 2012, 74, 615–621. [Google Scholar]
- Chung, Y.J.; Kim, Y.; Kim, S. Flame Retardant Properties of Polyurethane Produced by the Addition of Phosphorous Containing Polyurethane Oligomers (II). J. Ind. Eng. Chem. 2009, 15, 888–893. [Google Scholar] [CrossRef]
- Bourbigot, S.; Turf, T.; Bellayer, S.; Duquesne, S. Polyhedral Oligomeric Silsesquioxane as Flame Retardant for Thermoplastic Polyurethane. Polym. Degrad. Stab. 2009, 94, 1230–1237. [Google Scholar] [CrossRef]
- Huang, G.; Gao, J.; Li, Y.; Han, L.; Wang, X. Functionalizing Nano-Montmorillonites by Modified with Intumescent Flame Retardant: Preparation and Application in Polyurethane. Polym. Degrad. Stab. 2010, 95, 245–253. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Y.; Jiao, C. Smoke Suppression Properties of Ferrite Yellow on Flame Retardant Thermoplastic Polyurethane Based on Ammonium Polyphosphate. J. Hazard. Mater. 2014, 266, 114–121. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, X.; Tian, Y.; Ding, N.; Wang, Z. Controllable Synthesis of Three Kinds of Zinc Borates and Flame Retardant Properties in Polyurethane Foam. Colloids Surf. A Physicochem. Eng. Asp. 2012, 414, 274–280. [Google Scholar] [CrossRef]
- Singh, H.; Jain, A.K. Ignition, Combustion, Toxicity, and Fire Retardancy of Polyurethane Foams: A Comprehensive Review. J. Appl. Polym. Sci. 2009, 111, 1115–1143. [Google Scholar] [CrossRef]
- Frigione, M.; Maffezzoli, A.; Finocchiaro, P.; Failla, S. Cure Kinetics and Properties of Epoxy Resins Containing a Phosphorous-Based Flame Retardant. Adv. Polym. Technol. 2003, 22, 329–342. [Google Scholar] [CrossRef]
- Van Der Veen, I.; De Boer, J. Phosphorus Flame Retardants: Properties, Production, Environmental Occurrence, Toxicity and Analysis. Chemosphere 2012, 88, 1119–1153. [Google Scholar] [CrossRef]
- Wazarkar, K.; Kathalewar, M.; Sabnis, A. Improvement in Flame Retardancy of Polyurethane Dispersions by Newer Reactive Flame Retardant. Prog. Org. Coat. 2015, 87, 75–82. [Google Scholar] [CrossRef]
- Patel, M.; Mestry, S.; Khuntia, S.P.; Mhaske, S. Gallic Acid-Derived Phosphorus-Based Flame-Retardant Multifunctional Crosslinking Agent for PU Coating. J. Coat. Technol. Res. 2020, 17, 293–303. [Google Scholar] [CrossRef]
- Mestry, S.; Kakatkar, R.; Mhaske, S.T. Cardanol Derived P and Si Based Precursors to Develop Flame Retardant PU Coating. Prog. Org. Coat. 2019, 129, 59–68. [Google Scholar] [CrossRef]
- Shao, C.H.; Wang, T.Z.; Chen, G.N.; Chen, K.J.; Yeh, J.T.; Chen, K.N. Aqueous-Based Polyurethane with Dual-Functional Curing Agent. J. Polym. Res. 2000, 7, 41–49. [Google Scholar] [CrossRef]
- Ding, H.; Xia, C.; Wang, J.; Wang, C.; Chu, F. Inherently Flame-Retardant Flexible Bio-Based Polyurethane Sealant with Phosphorus and Nitrogen-Containing Polyurethane Prepolymer. J. Mater. Sci. 2016, 51, 5008–5018. [Google Scholar] [CrossRef]
- Ding, H.; Huang, K.; Li, S.; Xu, L.; Xia, J.; Li, M. Synthesis of a Novel Phosphorus and Nitrogen-Containing Bio-Based Polyol and Its Application in Flame Retardant Polyurethane Foam. J. Anal. Appl. Pyrolysis 2017, 128, 102–113. [Google Scholar] [CrossRef]
- Bhoyate, S.; Ionescu, M.; Kahol, P.K.; Gupta, R.K. Sustainable Flame-Retardant Polyurethanes Using Renewable Resources. Ind. Crops Prod. 2018, 123, 480–488. [Google Scholar] [CrossRef]
- Liu, X.; Salmeia, K.A.; Rentsch, D.; Hao, J.; Gaan, S. Thermal Decomposition and Flammability of Rigid PU Foams Containing Some DOPO Derivatives and Other Phosphorus Compounds. J. Anal. Appl. Pyrolysis 2017, 124, 219–229. [Google Scholar] [CrossRef]
- Xu, J.; Wu, Y.; Zhang, B.; Zhang, G. Synthesis and Synergistic Flame-Retardant Effects of Rigid Polyurethane Foams Used Reactive DOPO-Based Polyols Combination with Expandable Graphite. J. Appl. Polym. Sci. 2020, 138, 50223. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, C.; Fan, R.; Yuan, Y.; Xing, Y.; Ma, X. Halogen-Free Flame-Retardant Rigid Polyurethane Foam with a Nitrogen-Phosphorus Flame Retardant. J. Fire Sci. 2017, 35, 99–117. [Google Scholar] [CrossRef]
- Qian, X.; Liu, Q.; Zhang, L.; Li, H.; Liu, J.; Yan, S. Synthesis of Reactive DOPO-Based Flame Retardant and Its Application in Rigid Polyisocyanurate-Polyurethane Foam. Polym. Degrad. Stab. 2022, 197, 109852. [Google Scholar] [CrossRef]
- Xi, X.; Pizzi, A.; Gerardin, C.; Lei, H.; Chen, X.; Amirou, S. Preparation and Evaluation of Glucose Based Non-Isocyanate Polyurethane Self-Blowing Rigid Foams. Polymers 2019, 11, 1802. [Google Scholar] [CrossRef] [PubMed]
- 1907/2006/Ec. Règlement (CE) No1907/2006 Du Parlement Européen et Du Conseil Du 18 Décembre 2006. 2006. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:136:0003:0280:fr:PDF (accessed on 5 July 2022).
- Clark, J.H.; Farmer, T.J.; Ingram, I.D.V.; Lie, Y.; North, M. Renewable Self-Blowing Non-Isocyanate Polyurethane Foams from Lysine and Sorbitol. Eur. J. Org. Chem. 2018, 2018, 4265–4271. [Google Scholar] [CrossRef]
- Grignard, B.; Thomassin, J.M.; Gennen, S.; Poussard, L.; Bonnaud, L.; Raquez, J.M.; Dubois, P.; Tran, M.P.; Park, C.B.; Jerome, C.; et al. CO2-Blown Microcellular Non-Isocyanate Polyurethane (NIPU) Foams: From Bio- and CO2-Sourced Monomers to Potentially Thermal Insulating Materials. Green Chem. 2015, 18, 2206–2215. [Google Scholar] [CrossRef]
- Blattmann, H.; Lauth, M.; Mülhaupt, R. Flexible and Bio-Based Nonisocyanate Polyurethane (NIPU) Foams. Macromol. Mater. Eng. 2016, 301, 944–952. [Google Scholar] [CrossRef]
- Steblyanko, A.; Choi, W.; Sanda, F.; Endo, T. Addition of Five-Membered Cyclic Carbonate with Amine and Its Application to Polymer Synthesis. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2375–2380. [Google Scholar] [CrossRef]
- Tomita, H.; Sanda, F.; Endo, T.J. Reactivity Comparison of Five- and Six-Membered Cyclic Carbonates with Amines: Basic Evaluation for Synthesis of Poly(Hydroxyurethane). Polym. Sci. Part A Polym. Chem. 2001, 39, 162–168. [Google Scholar] [CrossRef]
- Kihara, N.; Endo, T. Synthesis and Properties of Poly(Hydroxyurethane)S. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2765–2773. [Google Scholar] [CrossRef]
- Kihara, N.; Kushida, Y.; Endo, T. Optically Active Poly(Hydroxyurethane)s Derived from Cyclic Carbonate and L-Lysine Derivatives. J. Polym. Sci. Part A Polym. Chem. 1996, 34, 2173–2179. [Google Scholar] [CrossRef]
- Benyahya, S.; Boutevin, B.; Caillol, S.; Lapinte, V.; Habas, J.P. Optimization of the Synthesis of Polyhydroxyurethanes Using Dynamic Rheometry. Polym. Int. 2012, 61, 918–925. [Google Scholar] [CrossRef]
- Błażek, K.; Datta, J. Renewable Natural Resources as Green Alternative Substrates to Obtain Bio-Based Non-Isocyanate Polyurethanes-Review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 173–211. [Google Scholar] [CrossRef]
- Yadav, N.; Seidi, F.; Crespy, D.; D’Elia, V. Polymers Based on Cyclic Carbonates as Trait d’Union Between Polymer Chemistry and Sustainable CO2 Utilization. ChemSusChem 2019, 12, 724–754. [Google Scholar] [CrossRef] [PubMed]
- Coste, G.; Denis, M.; Sonnier, R.; Negrell, C.; Caillol, S. Synthesis of Reactive Phosphorus-Based Carbonate for Flame Retardant Polyhydroxyurethane Foams. Polym. Degrad. Stab. 2022, 202, 110031. [Google Scholar] [CrossRef]
- Cornille, A.; Dworakowska, S.; Bogdal, D.; Boutevin, B.; Caillol, S. A New Way of Creating Cellular Polyurethane Materials: NIPU Foams. Eur. Polym. J. 2015, 66, 129–138. [Google Scholar] [CrossRef]
- Mora, A.-S.; Tayouo, R.; Boutevin, B.; David, G.; Caillol, S. A Perspective Approach of the Influence of H Bonds in Epoxy Networks, from Synthesis to Final Properties. Eur. Polym. J. 2019, 123, 109460. [Google Scholar] [CrossRef]
- Wilk, I.J. The Effect of Hydrogen Bonding on Steric Conformation. J. Mol. Struct. 1968, 2, 420–422. [Google Scholar] [CrossRef]
- Schartel, B. Phosphorus-Based Flame Retardancy Mechanisms-Old Hat or a Starting Point for Future Development? Materials 2010, 3, 4710–4745. [Google Scholar] [CrossRef]
- Braun, U.; Balabanovich, A.I.; Schartel, B.; Knoll, U.; Artner, J.; Ciesielski, M.; Döring, M.; Perez, R.; Sandler, J.K.W.; Altstädt, V.; et al. Influence of the Oxidation State of Phosphorus on the Decomposition and Fire Behaviour of Flame-Retarded Epoxy Resin Composites. Polymer 2006, 47, 8495–8508. [Google Scholar] [CrossRef]
- Velencoso, M.M.; Battig, A.; Markwart, J.C.; Schartel, B.; Wurm, F.R. Molecular Firefighting—How Modern Phosphorus Chemistry Can Help Solve the Flame Retardancy. Angew. Chemie Int. Ed. 2018, 57, 10450–10467. [Google Scholar] [CrossRef]
- Röchow, E.T.; Häußler, L.; Korwitz, A.; Pospiech, D. Thermal Decomposition of Phosphonate-Containing Methacrylate Based Copolymers. Polym. Degrad. Stab. 2018, 152, 235–243. [Google Scholar] [CrossRef]
- Liang, S.; Neisius, M.; Mispreuve, H.; Naescher, R.; Gaan, S. Flame Retardancy and Thermal Decomposition of Flexible Polyurethane Foams: Structural Influence of Organophosphorus Compounds. Polym. Degrad. Stab. 2012, 97, 2428–2440. [Google Scholar] [CrossRef]
- Qi, J.; Wen, Q.; Zhu, W. Research Progress on Flame-Retarded Silicone Rubber. IOP Conf. Ser. Mater. Sci. Eng. 2018, 392, 032007. [Google Scholar] [CrossRef]
- Hamdani, S.; Longuet, C.; Perrin, D.; Lopez-cuesta, J.M.; Ganachaud, F. Flame Retardancy of Silicone-Based Materials. Polym. Degrad. Stab. 2009, 94, 465–495. [Google Scholar] [CrossRef]
- Fang, F.; Huo, S.; Shen, H.; Ran, S.; Wang, H.; Song, P.; Fang, Z. A Bio-Based Ionic Complex with Different Oxidation States of Phosphorus for Reducing Flammability and Smoke Release of Epoxy Resins. Compos. Commun. 2020, 17, 104–108. [Google Scholar] [CrossRef]
- Laoutid, F.; Bonnaud, L.; Alexandre, M.; Lopez-Cuesta, J.M.; Dubois, P. New Prospects in Flame Retardant Polymer Materials: From Fundamentals to Nanocomposites. Mater. Sci. Eng. R Rep. 2009, 63, 100–125. [Google Scholar] [CrossRef]
- Huggett, C. Estimation of Rate of Heat Release by Means of Oxygen Consumption Measurements. Fire Mater. 1980, 4, 61–65. [Google Scholar] [CrossRef]
- Cornille, A.; Guillet, C.; Benyahya, S.; Negrell, C.; Boutevin, B.; Caillol, S. Room Temperature Flexible Isocyanate-Free Polyurethane Foams. Eur. Polym. J. 2016, 84, 873–888. [Google Scholar] [CrossRef]
- Ghoteimi, R.; Braka, A.; Rodriguez, C.; Cros-Perrial, E.; Tai Nguyen, V.; Uttaro, J.P.; Mathé, C.; Chaloin, L.; Ménétrier-Caux, C.; Jordheim, L.P.; et al. 4-Substituted-1,2,3-Triazolo Nucleotide Analogues as CD73 Inhibitors, Their Synthesis, in Vitro Screening, Kinetic and in Silico Studies. Bioorg. Chem. 2021, 107, 10457. [Google Scholar] [CrossRef]
- Lenz, J.; Pospiech, D.; Komber, H.; Paven, M.; Albach, R.; Mentizi, S.; Langstein, G.; Voit, B. Synthesis of the H-Phosphonate Dibenzo[d,f][1,3,2]Dioxaphosphepine 6-Oxide and the Phospha-Michael Addition to Unsaturated Compounds. Tetrahedron 2019, 75, 1306–1310. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Tg (°C) * | Swelling Rates (%) | Gel Content (%) |
|---|---|---|---|
| MBDAC | 2 | 190 | 86 |
| MBDAC-DOPO 1% P | 21 | 300 | 92 |
| MBDAC-DOPO 2% P | 4 | 340 | 88 |
| MBDAC-BPPO 1% P | 33 | 220 | 84 |
| MBDAC-BPPO 2% P | 23 | 240 | 74 |
| MBDAC DPP 1% P | 24 and 63 | 200 | 90 |
| MBDAC-DPP 2% P | −3 and 63 | 210 | 87 |
| MBDAC-DEP 1% P | 22 | 220 | 92 |
| MBDAC-DEP 2% P | 26 | 230 | 90 |
| Sample 1 | Td,5wt% (°C) | Td,50wt% (°C) | Residue at 750 °C (%) |
|---|---|---|---|
| MBDAC | 199 | 349 | 6 |
| MBDAC-DOPO 1% P | 200 | 349 | 11 |
| MBDAC-DOPO 2% P | 197 | 354 | 12 |
| MBDAC-BPPO 1% P | 198 | 348 | 15 |
| MBDAC-BPPO 2% P | 210 | 366 | 22 |
| MBDAC DPP 1% P | 189 | 340 | 18 |
| MBDAC-DPP 2% P | 196 | 340 | 23 |
| MBDAC-DEP 1% P | 188 | 340 | 11 |
| MBDAC-DEP 2% P | 201 | 340 | 20 |
| pHRR (W/g) | TpHRR (°C) | THR (kJ/g) | Residue Fraction | ΔH (kJ/g) | |
|---|---|---|---|---|---|
| MBDAC | 157 | 403 | 19.3 | 0.01 | 19.6 |
| MBDAC-DOPO 1% | 169 | 379 | 18.9 | 0.06 | 20.1 |
| MBDAC-DOPO 2% | 162 | 366 | 18.9 | 0.09 | 20.8 |
| MBDAC-BPPO 1% | 138 | 352 | 17.7 | 0.09 | 19.3 |
| MBDAC-BPPO 2% | 159 | 334 | 19 | 0.15 | 22.3 |
| MBDAC-DPP 1% | 129 | 356 | 17.0 | 0.09 | 18.6 |
| MBDAC-DPP 2% | 142 | 340 | 18.4 | 0.10 | 20.4 |
| MBDAC-DEP 1% | 168 | 344 | 17.2 | 0.10 | 19.1 |
| MBDAC-DEP 2% | 171 | 337 | 16.2 | 0.13 | 18.7 |
| TTI (s)° | pHRR1 (kW·m−2) | TpHRR1 (s) | pHRR2 (kW·m−2) | TpHRR2 (s) | |
|---|---|---|---|---|---|
| MBDAC | 36 | 1644 | 90 | / | / |
| MBDAC-DOPO 1% | 32 | 1362 | 85 | / | / |
| MBDAC-DOPO 2% | 41 | 1134 | 95 | / | / |
| MBDAC-BPPO 1% | 37 | 1270 | 58 | 957 | 118 |
| MBDAC-BPPO 2% | 23 | 626 | 40 | 789 | 143 |
| MBDAC-DPP 1% | 32 | 629 | 55 | 1066 | 125 |
| MBDAC-DPP 2% | 48 | 509 | 68 | 1205 | 123 |
| MBDAC-DEP 1% | 28 | 427 | 38 | 922 | 155 |
| MBDAC-DEP 2% | 36 | 390 | 45 | 1065 | 165 |
| Residue Fraction | EHC (kJ·g−1) | X | THR (kJ·g−1) | TSR (m2·m−2) | |
|---|---|---|---|---|---|
| MBDAC | 0.02 | 20.1 | 1.03 | 19.8 | 820 |
| MBDAC-DOPO 1% | 0.09 | 20.1 | 1.00 | 18.4 | 1472 |
| MBDAC-DOPO 2% | 0.11 | 18.6 | 0.9 | 16.6 | 2320 |
| MBDAC-BPPO 1% | 0.09 | 19.9 | 1.03 | 18.1 | 1440 |
| MBDAC-BPPO 2% | 0.11 | 19.7 | 0.89 | 17.5 | 1976 |
| MBDAC-DPP 1% | 0.07 | 20 | 1.08 | 18.7 | 1475 |
| MBDAC-DPP 2% | 0.09 | 20.9 | 0.99 | 18.7 | 1585 |
| MBDAC-DEP 1% | 0.1 | 17.2 | 0.90 | 15.6 | 1180 |
| MBDAC-DEP 2% | 0.15 | 18.1 | 0.97 | 15.4 | 1163 |
| PPO DC | MBDAC | MBDAC-DOPO | MBDAC-BPPO | MBDAC-DEP | MBDAC-DPP | |
|---|---|---|---|---|---|---|
| Functionality | 2.0 | 4.0 | 3.0 | 3.0 | 3.0 | 3.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denis, M.; Coste, G.; Sonnier, R.; Caillol, S.; Negrell, C. Influence of Phosphorus Structures and Their Oxidation States on Flame-Retardant Properties of Polyhydroxyurethanes. Molecules 2023, 28, 611. https://doi.org/10.3390/molecules28020611
Denis M, Coste G, Sonnier R, Caillol S, Negrell C. Influence of Phosphorus Structures and Their Oxidation States on Flame-Retardant Properties of Polyhydroxyurethanes. Molecules. 2023; 28(2):611. https://doi.org/10.3390/molecules28020611
Chicago/Turabian StyleDenis, Maxinne, Guilhem Coste, Rodolphe Sonnier, Sylvain Caillol, and Claire Negrell. 2023. "Influence of Phosphorus Structures and Their Oxidation States on Flame-Retardant Properties of Polyhydroxyurethanes" Molecules 28, no. 2: 611. https://doi.org/10.3390/molecules28020611
APA StyleDenis, M., Coste, G., Sonnier, R., Caillol, S., & Negrell, C. (2023). Influence of Phosphorus Structures and Their Oxidation States on Flame-Retardant Properties of Polyhydroxyurethanes. Molecules, 28(2), 611. https://doi.org/10.3390/molecules28020611