
Molecular Dynamics Study of the Curvature-Driven Interactions between Carbon-Based Nanoparticles and Amino Acids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
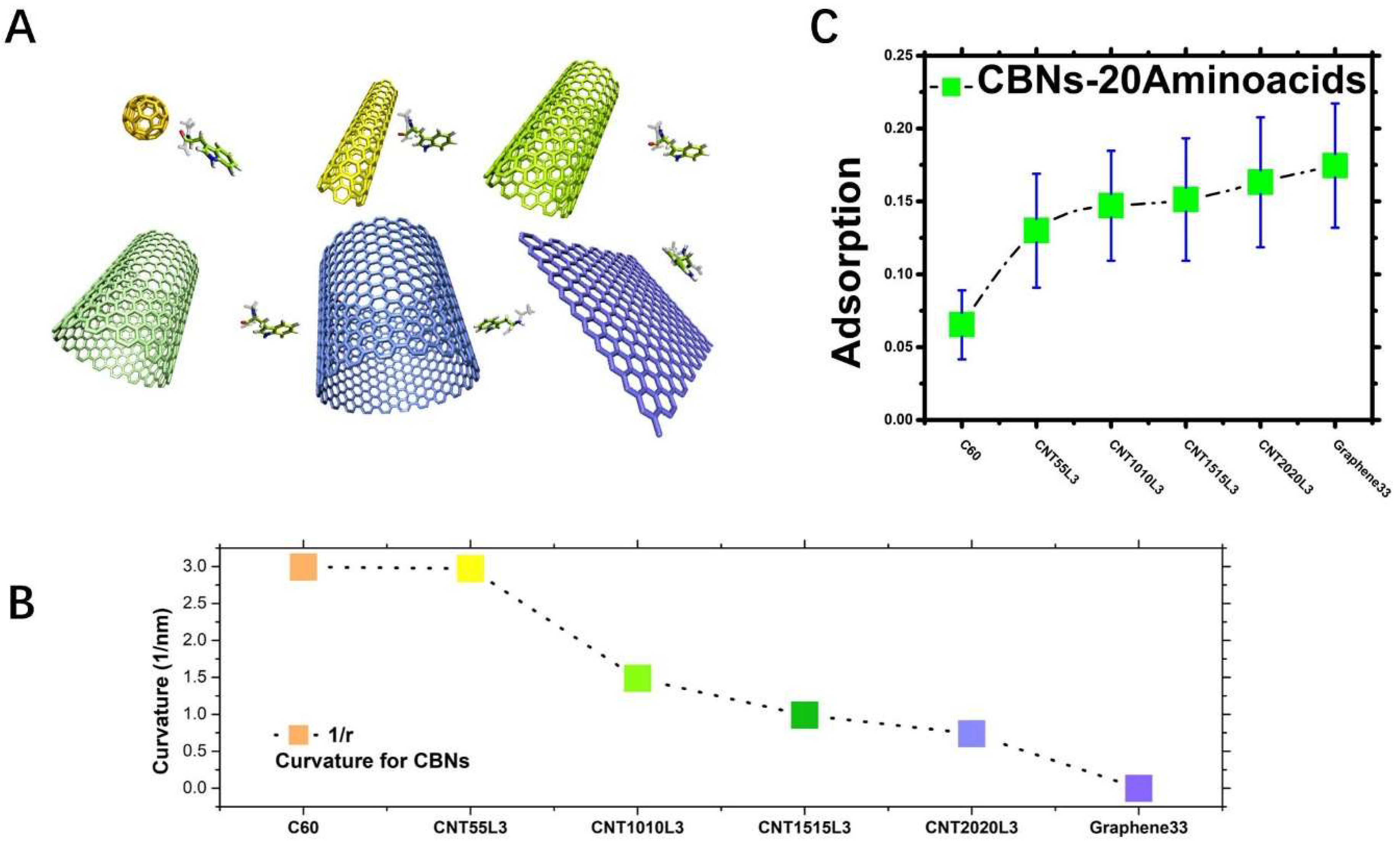
2.1. CBNs Adsorption Trend Analysis
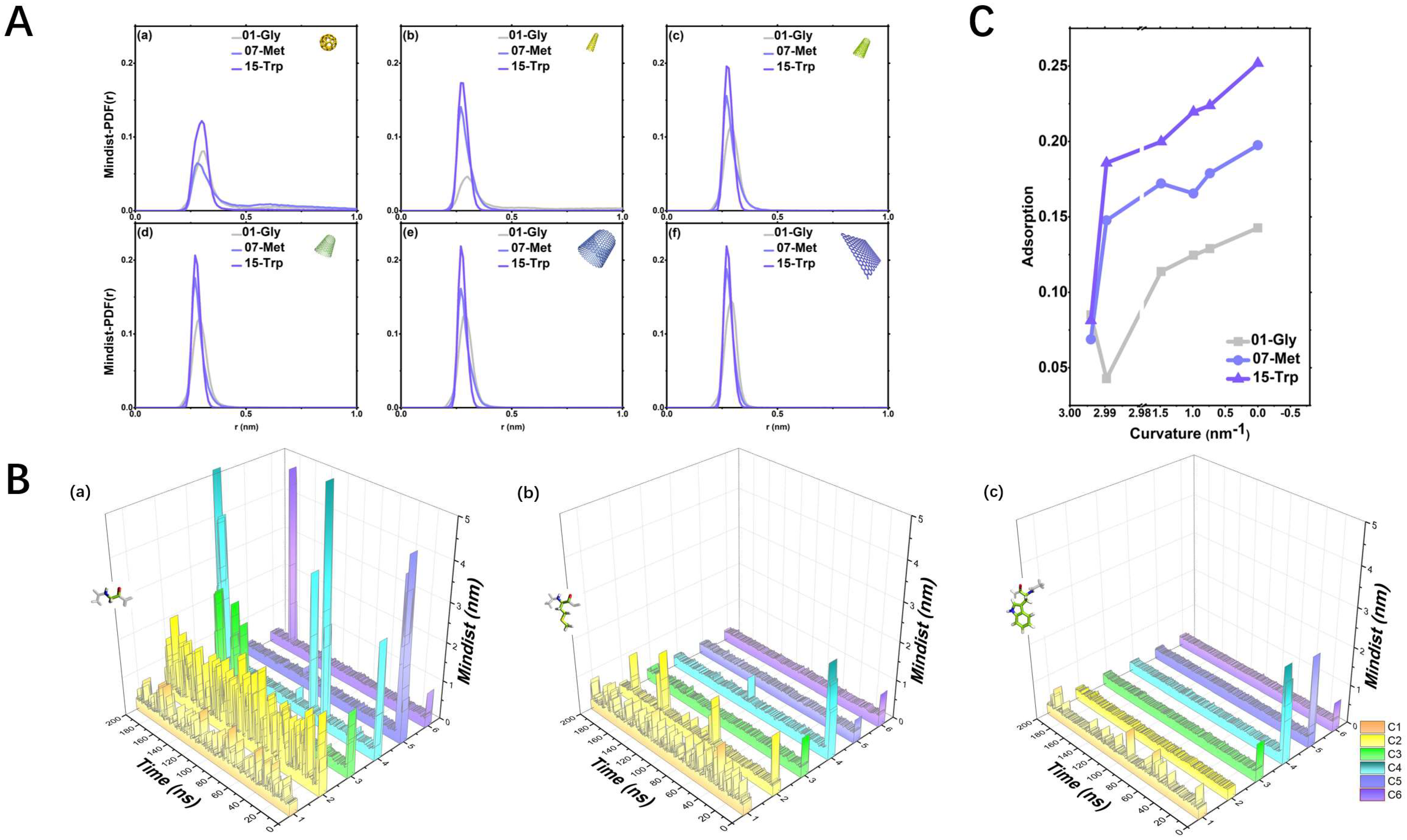
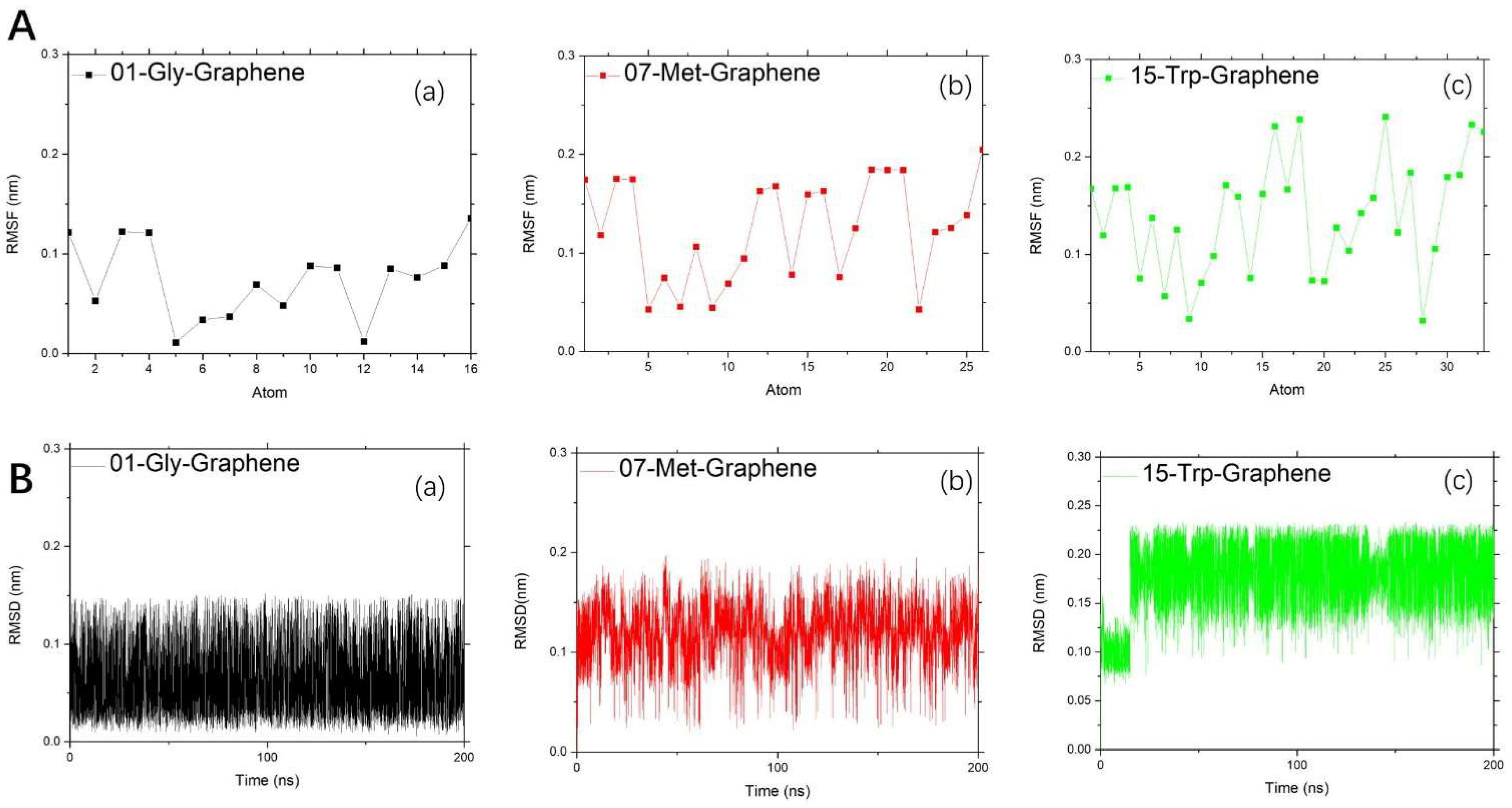
2.2. Kinetics and Statistical Analysis
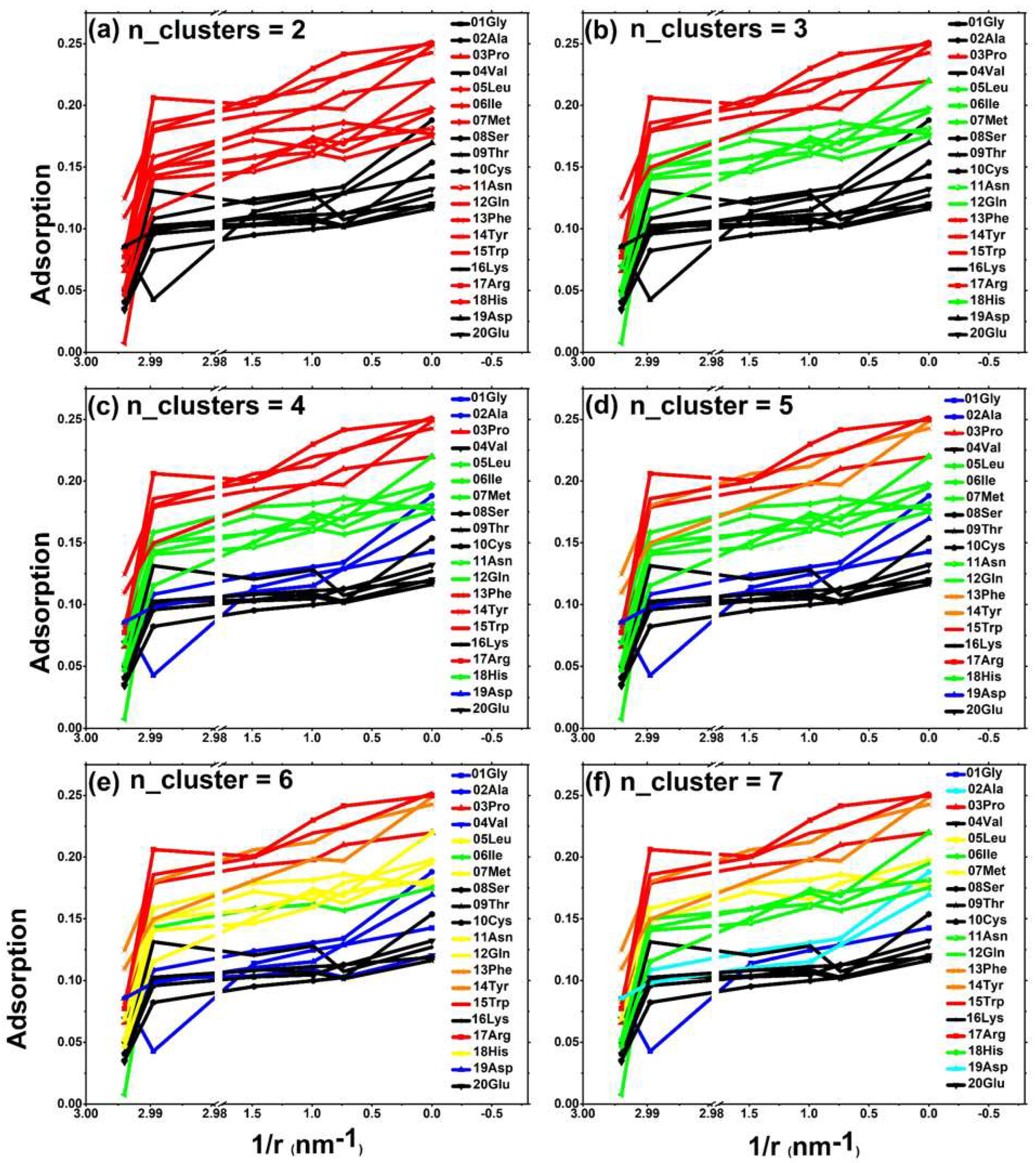
2.3. Multi-Dimensional Cluster Analysis
3. Discussion
4. Materials and Methods
- (1)
- Simulate system MD parameters
- (2)
- Analysis method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. In silico design and validation of high-affinity RNA aptamers targeting epithelial cellular adhesion molecule dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Meng, X.-Y.; Perez-Aguilar, J.M.; Zhou, R. An in silico study of TiO2 nanoparticles interaction with twenty standard amino acids in aqueous solution. Sci. Rep. 2016, 6, 37761. [Google Scholar] [CrossRef]
- Alimohammadi, E.; Khedri, M.; Jahromi, A.M.; Maleki, R.; Rezaian, M. Graphene-Based Nanoparticles as Potential Treatment Options for Parkinson’s Disease A Molecular Dynamics Study. Int. J. Nanomed. 2020, 15, 6887–6903. [Google Scholar] [CrossRef] [PubMed]
- Alimohammadi, E.; Nikzad, A.; Khedri, M.; Rezaian, M.; Jahromi, A.M.; Rezaei, N.; Maleki, R. Potential treatment of Parkinson’s disease using new-generation carbon nanotubes: A biomolecular in silico study. Nanomedicine 2021, 16, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Maleki, R.; Khedri, M.; Rezvantalab, S.; Afsharchi, F.; Musaie, K.; Shafiee, S.; Shahbazi, M.A. β-Amyloid Targeting with Two-Dimensionl Covalent Organic Frameworks Multi-Scale In-Silico Dissection of Nano-Biointerface. ChemBioChem 2021, 22, 2306–2318. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, D.; Ji, B.; Shi, X.; Gao, H. Structure-based design of carbon nanotubes as HIV-1 protease inhibitors: Atomistic and coarse-grained simulations. J. Mol. Graph. Model. 2010, 29, 171–177. [Google Scholar] [CrossRef]
- Rajesh, C.; Majumder, C.; Mizuseki, H.; Kawazoe, Y. A theoretical study on the interaction of aromatic amino acids with graphene and single walled carbon nanotube. J. Chem. Phys. 2009, 130, 124911. [Google Scholar] [CrossRef]
- Zuo, G.; Kang, S.-G.; Xiu, P.; Zhao, Y.; Zhou, R. Interactions Between Proteins and Carbon-Based Nanoparticles: Exploring the Origin of Nanotoxicity at the Molecular Level. Small 2012, 9, 1546–1556. [Google Scholar] [CrossRef]
- An, D.; Su, J.; Weber, J.K.; Gao, X.; Zhou, R.; Li, J. A Peptide-Coated Gold Nanocluster Exhibits Unique Behavior in Protein Activity Inhibition. J. Am. Chem. Soc. 2015, 137, 8412–8418. [Google Scholar] [CrossRef]
- He, Z.; Zhou, R. Exploring an In-Plane Graphene and Hexagonal Boron Nitride Array for Separation of Single Nucleotides. ACS Nano 2021, 15, 11704–11710. [Google Scholar] [CrossRef]
- Georgakilas, V.; Tagmatarchis, N.; Pantarotto, D.; Bianco, A.; Briand, J.-P.; Prato, M. Amino acid functionalisation of water soluble carbon nanotubes. Chem. Commun. 2002, 3050–3051. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Liu, Q.; Li, Y. Interaction of Amino Acids and Single-Wall Carbon Nanotubes. J. Phys. Chem. C 2012, 116, 1724–1731. [Google Scholar] [CrossRef]
- Roman, T.; Diño, W.A.; Nakanishi, H.; Kasai, H. Amino acid adsorption on single-walled carbon nanotubes. Eur. Phys. J. D 2006, 38, 117–120. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Z.; Tian, X.; Xiu, P.; Zhou, R. Amino acid analogues bind to carbon nanotube via pi-pi interactions: Comparison of molecular mechanical and quantum mechanical calculations. J. Chem. Phys. 2012, 136, 025103. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Mui, K.; Daub, E.; Leung, T.; Honek, J. Single-Walled Carbon Nanotube Binding Peptides: Probing Tryptophan’s Importance by Unnatural Amino Acid Substitution. J. Phys. Chem. B 2007, 111, 14411–14417. [Google Scholar] [CrossRef]
- Yu, D.; Dai, L. Self-Assembled Graphene/Carbon Nanotube Hybrid Films for Supercapacitors. J. Phys. Chem. Lett. 2010, 1, 467–470. [Google Scholar] [CrossRef]
- He, Z.; Zhou, J. Probing carbon nanotube–amino acid interactions in aqueous solution with molecular dynamics simulations. Carbon 2014, 78, 500–509. [Google Scholar] [CrossRef]
- Nechaeva, L.S.; Butyrskaya, E.V.; Zapryagaev, S.A. Computer simulation of amino acid sorption on carbon nanotubes. J. Struct. Chem. 2017, 58, 217–225. [Google Scholar] [CrossRef]
- Lovat, V.; Pantarotto, D.; Lagostena, L.; Cacciari, B.; Grandolfo, M.; Righi, M.; Spalluto, G.; Prato, M.; Ballerini, L. Carbon Nanotube Substrates Boost Neuronal Electrical Signaling. Nano Lett. 2005, 5, 1107–1110. [Google Scholar] [CrossRef]
- Wang, J. Carbon-Nanotube Based Electrochemical Biosensors: A Review. Electroanalysis 2005, 17, 7–14. [Google Scholar] [CrossRef]
- Moore, R.R.; Banks, C.E.; Compton, R.G. Basal Plane Pyrolytic Graphite Modified Electrodes: Comparison of Carbon Nanotubes and Graphite Powder as Electrocatalysts. Anal. Chem. 2004, 76, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, J.; Zhang, J.; Takada, S.; Wang, W. Overcoming the Bottleneck of the Enzymatic Cycle by Steric Frustration. Phys. Rev. Lett. 2019, 122, 238102. [Google Scholar] [CrossRef] [PubMed]
- Zorgati, H.; Larsson, M.; Ren, W.; Sim, A.Y.L.; Gettemans, J.; Grimes, J.M.; Li, W.; Robinson, R.C. The role of gelsolin domain 3 in familial amyloidosis (Finnish type). Proc. Natl. Acad. Sci. USA 2019, 116, 13958–13963. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Li, J.; Lu, J.; Li, W.; Wang, W. Role of substrate-product frustration on enzyme functional dynamics. Phys. Rev. E 2019, 100, 052409. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.G.; Marzinek, J.K.; Holdbrook, D.A.; Bond, P.J. Multiscale molecular dynamics simulation approaches to the structure and dynamics of viruses. Prog. Biophys. Mol. Biol. 2017, 128, 121–132. [Google Scholar] [CrossRef]
- Deo, R.P.; Lawrence, N.S.; Wang, J. Electrochemical detection of amino acids at carbon nanotube and nickel–carbon nanotube modified electrodes. Anal. 2004, 129, 1076–1081. [Google Scholar] [CrossRef]
- Bianco, A.; Kostarelos, K.; Prato, M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 2005, 9, 674–679. [Google Scholar] [CrossRef]
- Lins, R.D.; Hünenberger, P.H. A new GROMOS force field for hexopyranose-based carbohydrates. J. Comput. Chem. 2005, 26, 1400–1412. [Google Scholar] [CrossRef]
- Scott, W.R.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Chandler, D.; Percus, J.K. Introduction to Modern Statistical Mechanics. Phys. Today 1988, 41, 114–118. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Verkuil, R.; Liu, J.; Lin, Z.; Hie, B.; Sercu, T.; Lerer, A.; Rives, A. Learning inverse folding from millions of predicted structures. bioRxiv 2022. [Google Scholar] [CrossRef]
- Shields, B.J.; Stevens, J.; Li, J.; Parasram, M.; Damani, F.; Alvarado, J.I.M.; Janey, J.M.; Adams, R.P.; Doyle, A.G. Bayesian reaction optimization as a tool for chemical synthesis. Nature 2021, 590, 89–96. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.; Wang, Z.; Luo, J. Molecular Dynamics Study of the Curvature-Driven Interactions between Carbon-Based Nanoparticles and Amino Acids. Molecules 2023, 28, 482. https://doi.org/10.3390/molecules28020482
Huang W, Wang Z, Luo J. Molecular Dynamics Study of the Curvature-Driven Interactions between Carbon-Based Nanoparticles and Amino Acids. Molecules. 2023; 28(2):482. https://doi.org/10.3390/molecules28020482
Chicago/Turabian StyleHuang, Wanying, Zhenyu Wang, and Junyan Luo. 2023. "Molecular Dynamics Study of the Curvature-Driven Interactions between Carbon-Based Nanoparticles and Amino Acids" Molecules 28, no. 2: 482. https://doi.org/10.3390/molecules28020482
APA StyleHuang, W., Wang, Z., & Luo, J. (2023). Molecular Dynamics Study of the Curvature-Driven Interactions between Carbon-Based Nanoparticles and Amino Acids. Molecules, 28(2), 482. https://doi.org/10.3390/molecules28020482