
In Silico Studies of Four Compounds of Cecropia obtusifolia against Malaria Parasite


Abstract
:
1. Introduction
2. Results
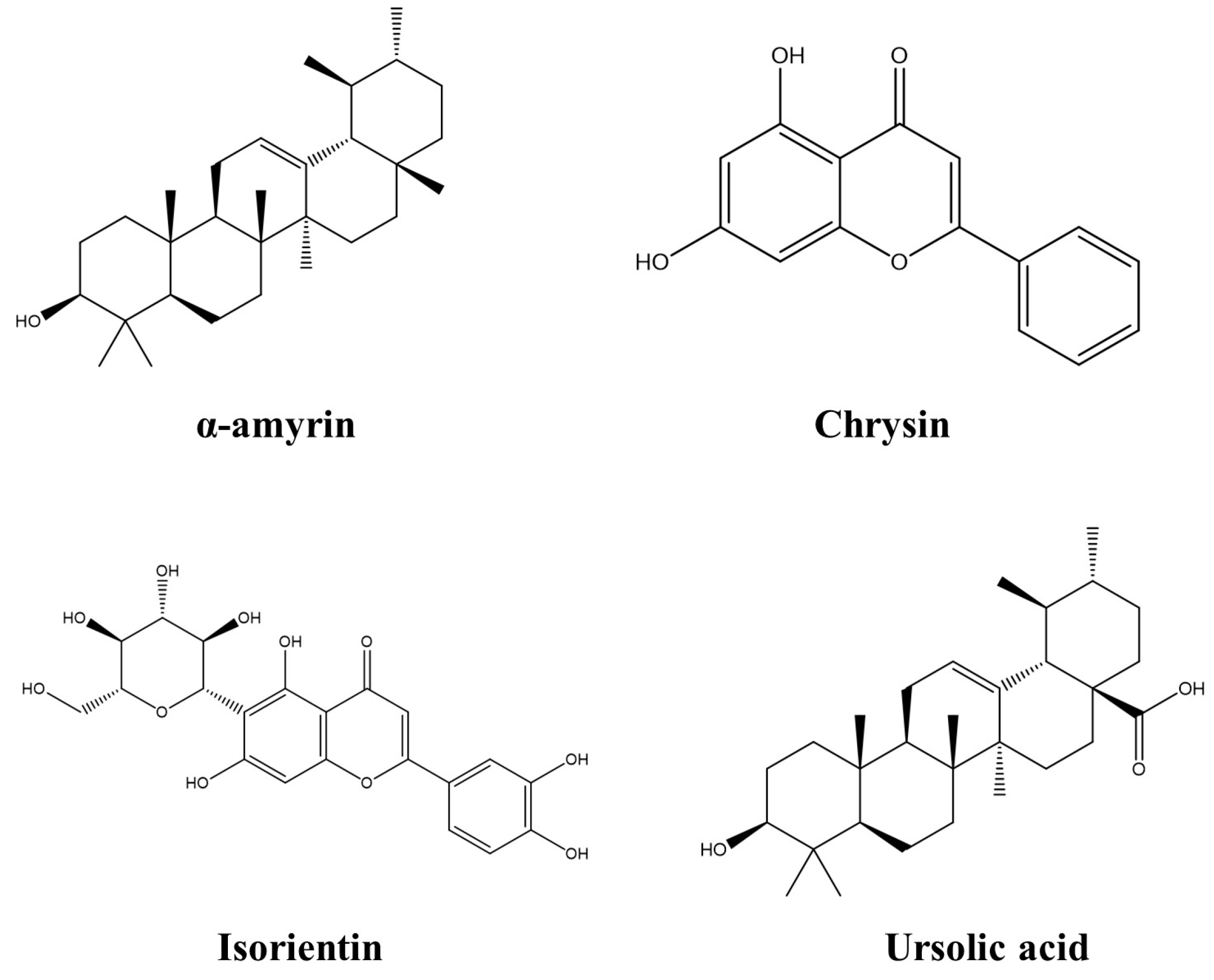
2.1. Compounds of Cecropia obtusifolia
2.2. Molecular Docking
2.3. Molecular Dynamics
2.4. Pharmacokinetic Properties
3. Discussion
4. Materials and Methods
4.1. Proteins Preparation
4.2. Compound Retrieval and Preparation
4.3. Molecular Docking Analysis
4.4. Molecular Dynamics Simulation
4.5. Pharmacokinetic Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Informe Mundial Sobre el Paludismo 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Organización Mundial de la Salud. Malaria. 2023. Available online: https://www.who.int/es/news-room/fact-sheets/detail/malaria (accessed on 29 March 2023).
- Pereira, Á.; Ríos, M.P. Epidemiología y Tratamiento del Paludismo. OFFARM 2002, 21, 110–116. [Google Scholar]
- Rodríguez, M.H.; Betanzos-Reyes, Á.F. Plan de mejoramiento del control de la malaria hacia su eliminación en Mesoamérica. Salud Pública México 2011, 53, 333–348. [Google Scholar]
- Organización Mundial de la Salud. Informe Mundial Sobre la Malaria 2021 OMS. 2021. Available online: https://cdn.who.int/media/docs/default-source/malaria/world-malaria-reports/world-malaria-report-2021-regional-briefing-kit-spa.pdf?sfvrsn=338167b6_25&download=true (accessed on 29 March 2023).
- Boletin Epiodemiológico de la Secretaria de Salud. Situación Epidemiológica de Malaria en México, Hasta la Semana 27 del 2020. Available online: http://www.anhp.org.mx/archivoseventos/03-Panorama-Epidemiol%C3%B3gico-Paludismo_16-07-2020.pdf (accessed on 21 August 2021).
- Alonso-Castro, A.J.; Miranda-Torres, A.C.; González-Chávez, M.M.; Salazar-Olivo, L.A. Cecropia obtusifolia Bertol and its active compound, chlorogenic acid, stimulate 2-NBDglucose uptake in both insulin-sensitive and insulin-resistant 3T3 adipocytes. J. Ethnopharmacol. 2008, 120, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Vidal Durango, J.; Marrugo Negrete, J.; Jaramillo Colorado, B.; Pérez Castro, L. Remediation of soils contaminated with mercury using guarumo (Cecropia peltata). Eng. Dev. Univ. North 2010, 27, 113–129. [Google Scholar]
- Medrano-Sánchez, E.J.; Hernández-Bolio, G.I.; Lobato-García, C.E.; González-Cortazar, M.; Antunez-Mojica, M.; Gallegos-García, A.J.; Barredo-Hernández, C.O.; López-Rodríguez, R.; Aguilar-Sánchez, N.C.; Gómez-Rivera, A. Intra- and Interspecies Differences of Two Cecropia Species from Tabasco, Mexico, Determined through the Metabolic Analysis and 1H-NMR-Based Fingerprinting of Hydroalcoholic Extracts. Plants 2023, 12, 2440. [Google Scholar] [CrossRef]
- Trejo, G.M. Phytochemical Study of Guarumbo (Cecropia obtusifolia) as a Hypoglycemic Agent. Bachelor’s Thesis, National Polytechnic Institute (IPN), National School of Biological Sciences (ENCB), La Ciudad de México, Mexico, 1983; pp. 1–55. [Google Scholar]
- Sanchez, M.B.; Miranda-Pérez, E.; Verjan, J.C.G.; de Los Angeles Fortis Barrera, M.; Pérez-Ramos, J.; Alarcón-Aguilar, F.J. Potential of the chlorogenic acid as multitarget agent: Insulin-secretagogue and PPAR α/γ dual agonist. Biomed. Pharmacother. 2017, 94, 169–175. [Google Scholar] [CrossRef]
- Cadena-Zamudio, J.D.; Nicasio-Torres, P.; Monribot-Villanueva, J.L.; Guerrero-Analco, J.A.; Ibarra-Laclette, E. Integrated Analysis of the Transcriptome and Metabolome of Cecropia obtusifolia: A Plant with High Chlorogenic Acid Content Traditionally Used to Treat Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 7572. [Google Scholar] [CrossRef]
- Costa, G.M.; Schenkel, E.P.; Reginatto, F.H. Chemical and pharmacological aspects of the genus Cecropia. Nat. Prod. Common. 2011, 6, 913–920. [Google Scholar] [CrossRef]
- Rivera-Mondragón, A.; Tuenter, E.; Bijttebier, S.; Cos, P.; Apers, S.; Caballero-George, C.; Foubert, K.; Pieters, L. Two new antiplasmodial flavonolignans from the leaves of Cecropia obtusifolia. Phytochem. Lett. 2019, 31, 118–120. [Google Scholar] [CrossRef]
- Bilsland, E.; Van Vliet, L.; Williams, K.; Feltham, J.; Carrasco, M.P.; Fotoran, W.L.; Cubillos, E.F.G.; Wunderlich, G.; Grøtli, M.; Hollfelder, F.; et al. Plasmodium dihydrofolate reductase is a second enzyme target for the antimalarial action of triclosan. Sci. Rep. 2018, 8, 1038. [Google Scholar] [CrossRef]
- Sharma, D.; Lather, M.; Mallick, P.K.; Adak, T.; Dang, A.S.; Valecha, N.; Sing, O.P. Polymorphism in drug resistance genes dihydrofolate reductase and dihydropteroate synthase in Plasmodium falciparum in some states of India. Parasit. Vectors 2015, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Kongsaeree, P.; Khongsuk, P.; Leartsakulpanich, U.; Chitnumsub, P.; Tarnchompoo, B.; Walkinshaw, M.D.; Yuthavong, Y. Crystal Structure of Dihydrofolate Reductase from Plasmodium Vivax: Pyrimethamine Displacement Linked with Mutation-Induced Resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 13046. [Google Scholar] [CrossRef] [PubMed]
- Hodder, A.N.; Sleebs, B.E.; Czabotar, P.E.; Gazdik, M.; Xu, Y.; O’Neill, M.T.; Lopaticki, S.; Nebl, T.; Triglia, T.; Smith, B.J.; et al. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat. Struct. Mol. Biol. 2015, 22, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bhardwaj, V.; Purohit, R. Identification of a novel binding mechanism of Quinoline based molecules with lactate dehydrogenase of Plasmodium falciparum. J. Biomol. Struct. 2019, 39, 348–356. [Google Scholar] [CrossRef]
- Liu, P. Plasmepsin: Function, characterization and targeted antimalarial drug development. In Natural Remedies in the Fight Against Parasites; Books on Demand: Norderstedt, Germany, 2017; pp. 183–218. [Google Scholar]
- Read, J.A.; Wilkinson, K.W.; Tranter, R.; Sessions, R.B.; Brady, R.L. Chloroquine binds in the cofactor binding site of Plasmodium falciparum lactate dehydrogenas. J. Biol. Chem. 1999, 274, 10213–10218. [Google Scholar] [CrossRef]
- Plucinski, M.M.; McElroy, P.D.; Dimbu, P.R.; Fortes, F.; Nace, D.; Halsey, E.S.; Rogier, E. Clearance dynamics of lactate dehydrogenase and aldolase following antimalarial treatment for Plasmodium falciparum infection. Parasites Vectors 2019, 12, 293. [Google Scholar] [CrossRef]
- Arora, G.; Chuang, Y.; Sinnis, P.; Dimopoulos, G.; Fikrig, E. Malaria: Influence of Anopheles mosquito saliva on Plasmodium infection. Trends Immunol. 2023, 44, 256–265. [Google Scholar] [CrossRef]
- WHO. Report of the WHO Strategic Advisory Group on Malaria Eradication; A Report of the Strategic Advisory Group on Malaria Eradication; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Van der Plujim, R.W.; Amaratunga, C.; Dhorda, M.; Dondorp, A.M. Triple Artemisinin-Based Combination Therapies for Malaria—A New Paradigm? Trends Parasitol. 2021, 37, 15–24. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, Y.; Wang, M. Bioactive Substances of Plant Origin. In Handbook of Food Chemistry; Cheung, P.C.K., Mehta, B.M., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2020; pp. 967–1008. [Google Scholar]
- Qasim, M.; Abideen, Z.; Adnan, M.; Gulzar, S.; Gul, B.; Rasheed, M.; Khan, M. Antioxidant properties, phenolic composition, bioactive compounds and nutritive value of medicinal halophytes commonly used as herbal teas. S. Afr. J. Bot. 2017, 110, 240–250. [Google Scholar] [CrossRef]
- Chandran, H.; Meena, M.; Barupal, T.; Sharma, K. Plant tissue culture as a perpetual source for production of industrially important bioactive compounds. Biotechnol. Rep. 2020, 26, e00450. [Google Scholar] [CrossRef]
- Jug, G.; Anderluh, M.; Tomašič, T. Comparative evaluation of several docking tools for docking small molecule ligands to DC-SIGN. J. Mol. Model. 2015, 21, 164. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Rosero, Y.J.; Barrios-Correa, L.; Anaya-Gil, J. La cromatografía en capafina: Una alternativa vigente en la industria farmacéutica. Rev. Química PUCP 2021, 35, 19–25. [Google Scholar]
- Yu, W.; Mackerell, A. Computer-aided drug design methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [PubMed]
- Prieto-Martínez, F.; Medina-Franco, J. Computer-aided drug design: When informatics, chemistry and art meets. Tip. Rev. Espec. Cienc. 2018, 21, 124–134. [Google Scholar]
- Anwar, T.; Kumar, P.; Khan, A. Modern Tools and Techniques in Computer-Aided Drug Design. Curr. Comput. Aided Drug Des. 2021, 1–30. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Saikia, S.; Bordoloi, M. Molecular Docking: Challenges, Advances and its Use in Drug Discovery Perspective. Curr. Drug Targets 2019, 20, 501–521. [Google Scholar] [CrossRef]
- Stanzione, F.; Giangreco, I.; Cole, J. Use of molecular docking computational tools in drug discovery. Prog. Med. Chem. 2021, 60, 273–343. [Google Scholar]
- Chawla, P.; Teli, G.; Gill, R.K.; Narang, R.K. An Insight into Synthetic Strategies and Recent Developments of Dihydrofolate Reductase Inhibitors. ChemistrySelect 2021, 6, 12101–12145. [Google Scholar] [CrossRef]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; Matthews, D. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. USA 2012, 109, 16823–16828. [Google Scholar] [CrossRef]
- Polino, A.J.; Nasamu, A.S.; Niles, J.C.; Goldberg, D.E. Assessment of biological role and insight into druggability of the Plasmodium falciparum protease plasmepsin V. ACS Infect. Dis. 2020, 6, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.; Coronado, L.; Caballero, Z.; Faral-Tello, P.; Robello, C.; Spadafora, C. Extracellular vesicles carrying lactate dehydrogenase induce suicide in increased population density of Plasmodium falciparum in vitro. Sci. Rep. 2019, 9, 5042. [Google Scholar] [CrossRef] [PubMed]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 2006, 71, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Polino, A.J.; Miller, J.J.; Bhakat, S.; Mukherjee, S.; Bobba, S.; Bowman, G.R.; Goldberg, D.E. The nepenthesin insert in the Plasmodium falciparum aspartic protease plasmepsin V is necessary for enzyme function. J. Biol. Chem. 2022, 298, 102355. [Google Scholar] [CrossRef]
- Shadrack, D.M.; Nyandoro, S.S.; Munissi, J.J.; Mubofu, E.B. In silico evaluation of anti-malarial agents from Hoslundia opposita as inhibitors of Plasmodium falciparum lactate dehydrogenase (PfLDH) enzyme. Comput. Mol. Biosci. 2016, 6, 23–32. [Google Scholar] [CrossRef]
- Sivaramakrishnan, M.; Kandaswamy, K.; Natesan, S.; Devarajan, R.; Ramakrishnan, S.; Kothandan, R. Molecular docking and dynamics studies on plasmepsin V of malarial parasite Plasmodium vivax. Inform. Med. Unlocked 2020, 19, 100331. [Google Scholar] [CrossRef]
- Chaniad, P.; Mungthin, M.; Payaka, A.; Viriyavejakul, P.; Punsawad, C. Antimalarial properties and molecular docking analysis of compounds from Dioscorea bulbifera L. as new antimalarial agent candidates. BMC Complement. Med. Ther. 2021, 21, 144. [Google Scholar] [CrossRef]
- Stompor-gorący, M.; Bajek-bil, A.; Machaczka, M. Chrysin: Perspectives on Contemporary Status and Future Possibilities as Pro-Health Agent. Nutrients 2021, 13, 2038. [Google Scholar] [CrossRef]
- Jin, H.; Xu, Z.; Cui, K.; Zhang, T.; Lu, W.; Huang, J. Dietary flavonoids fisetin and myricetin: Dual inhibitors of Plasmodium falciparum falcipain-2 and plasmepsin II. Fitoterapia 2014, 94, 55–61. [Google Scholar] [CrossRef]
- Lehane, A.M.; Saliba, K.J. Common dietary flavonoids inhibit the growth of the intraerythrocytic malaria parasite. BMC Res. Notes 2008, 18, 26. [Google Scholar] [CrossRef]
- Ziqubu, K.; Dludla, P.; Joubert, E.; Muller, C.; Louw, J.; Tiano, L.; Nkambule, B.; Kappo, A.; Mazibuko-Mbeje, S. Isoorientin: A dietary flavone with the potential to ameliorate diverse metabolic complications. Pharmacol. Res. 2020, 158, 104867. [Google Scholar] [CrossRef] [PubMed]
- Ziqubu, K.; Muller, C.; Dludla, P.; Mthembu, S.; Obonye, N.; Louw, J.; Kappo, A.; Silvestri, S.; Orlando, P.; Tiano, L.; et al. Impact of Isoorientin on Metabolic Activity and Lipid Accumulation in Differentiated Adipocytes. Molecules 2020, 25, 1773. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, K.; Reddy, G.; Azad, R.; Yarla, N.; Dharmapuri, G.; Srivastava, A.; Kamal, M.; Pallu, R. Evaluation of Anti-Inflammatory Properties of Isoorientin Isolated from Tubers of Pueraria tuberosa. Oxid. Med. Cell Longev. 2017, 2017, 5498054. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wei, J.; Chen, Y.; He, P.; Lin, J.; Tan, S.; Nie, J.; Lu, S.; He, M.; Lu, Z.; et al. Isoorientin from Gypsophila elegans induces apoptosis in liver cancer cells via mitochondrial-mediated pathway. J. Ethnopharmacol. 2016, 187, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Y.; Wang, S.; Wang, J.; Luo, Y.; Jin, C. Research progress on anti-tumor pharmacological effects and mechanism of isoorientin. Anim. Feed. Sci. 2019, 40, 86–88. [Google Scholar]
- Atay, I.; Kirmizibekmez, H.; Kaiser, M.; Akaydin, G.; Yesilada, E.; Tasdemir, D. Evaluation of in vitro antiprotozoal activity of Ajuga laxmannii and its secondary metabolites. Pharm. Biol. 2016, 54, 1808–1814. [Google Scholar] [CrossRef]
- Salo-Ahen, O.; Alanko, I.; Bhadane, R.; Alexandre, A.; Honorato, R.; Hossain, S.; Juffer, A.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Kalimuthu, A.; Panneerselvam, T.; Pavadai, P.; Pandian, S.; Sundar, K.; Murugesan, S.; Ammunje, D.; Kumar, S.; Arunachalam, S.; Kunjiappan, S. Pharmacoinformatics-based investigation of bioactive compounds of Rasam (South Indian recipe) against human cancer. Sci. Rep. 2021, 11, 21488. [Google Scholar] [CrossRef]
- Van den Anker, J.; Reed, M.; Allegaert, K.; Kearns, G. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25. [Google Scholar] [CrossRef]
- Parikh, P.; Savjani, J.; Gajjar, A.; Chhabria, M. Bioinformatics and Cheminformatics Tools in Early Drug Discovery. In Bioinformatics Tools for Pharmaceutical Drug Product Development; Chavda, V., Anand, K., Apostolopoulos, V., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2023. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Alqahtani, M.; Kazi, M.; Alsenaidy, M.; Ahmad, M. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Rénia, L.; Howland, S.; Claser, C.; Gruner, A.; Suwanarusk, R.; Teo, T.; Russell, B.; Lisa, N. Cerebral malaria: Mysteries at the blood-brain barrier. Virulence 2012, 3, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Sproston, J.; Barton, P.; Riley, R. Estimating human ADME properties, pharmacokinetic parameters and likely clinical dose in drug discovery. Expert. Opin. Drug Discov. 2019, 14, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K.; et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef]
- Basile, A.; Yahi, A.; Tatonetti, N. Artificial Intelligence for Drug Toxicity and Safety. Trends Pharmacol. 2019, 40, 624–635. [Google Scholar] [CrossRef]
- Garrido, A.; Lepailleur, A.; Mignani, S.; Dallemagne, P.; Rochais, C. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef]
- Hanwell, M.; Curtis, D.; Lonie, D.; Vandermeerschd, T.; Zurek, E.; Hutchison, G. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool, CCP4 Newsletter. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef]
- Van Aalten, D.M. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996, 10, 255–262. [Google Scholar] [CrossRef]
- Turner, P.J. XMGRACE, Version 5.1.19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Ligand | Binding Affinities (Kcal/mol) | Amino Acid Interactions |
|---|---|---|---|
| 2BL9 | α-amyrin Chrysin Isoorientin Ursolic acid Pyrimithamine | −7.9 −8.7 −8.5 −7.8 −7.6 | K55, Y56, V60, Y63, K169, Y192, S220, Y223, F232, C14, A15, D53, F57, S117, S120, I121, I173, G175, Y179 L39, L45, M54, F57, S116, S120, I121, Y125, I173, G175 K55, Y56, V60, Y63, Y192, S220, E221, F232 I13, C14, A15, L45, D53, M54, Y56, F57, I173, Y179 |
| 4ZL4 | α-amyrin Chrysin Isoorientin Ursolic acid Wehi | −8.2 −9.6 −8.3 −7.8 −8.2 | I24, D25, T285, F286, H288, K390, E399, L400, V402 Y27, D48, Y107, E109, L147, F148, V156 A28, G50, S51, C108, E109, D281, G283, T285, H288 D25, I24, E109, T285, F286, V354, K390 Y29, D48, S51, E109, Q151, G283, S284, T285, F286, H288, V354, V357 |
| 1CET | α-amyrin Chrysin Isoorientin Ursolic acid Chloroquine | −7.9 −7.8 −9.1 −7.7 −6.3 | G11, G13, M14, P34, D35, I36, V37, A80, G81, F82, I105 G11, G13, D35, I36, A80, Y67, F82, G81, I105 S12, C13, I36, G11, N35, Y67, A80, G81, T83, E108 F34, N35, I36, V37, Y67, A80, G81, I105, I109 G11, F34, D35, A80, G81, F82, I36, Y67, I105 |
| Parameter | α-Amyrin | Chrysin | Isoorientin | Ursolic Acid |
|---|---|---|---|---|
| Formula | C30H50O | C15H10O4 | C21H20O11 | C30H48O3 |
| MW (g/mol−1) | 426.39 | 254.06 | 448.1 | 456.36 |
| Lipinski | Accepted | Accepted | Rejected | Accepted |
| LogP (log mol/L) | 7.603 | 3.58 | 0.708 | 6.453 |
| HIA | 0.003 | 0.012 | 0.909 | 0.004 |
| Caco-2 | −5.131 | −4.874 | −6.251 | −5.396 |
| PPB (%) | 97.63 | 98.03 | 90.58 | 97.44 |
| VD (L/kg) | 1.251 | 0.493 | 0.834 | 0.672 |
| BBB | 0.056 | 0.02 | 0.011 | 0.265 |
| CL (mL/min/kg) | 5.609 | 5.131 | 4.067 | 3.538 |
| T1/2 | 0.075 | 0.787 | 0.823 | 0.07 |
| hERG | 0.042 | 0.037 | 0.214 | 0.007 |
| H-HT | 0.156 | 0.079 | 0.133 | 0.435 |
| Carcinogencity | 0.012 | 0.317 | 0.037 | 0.085 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobato-Tapia, C.A.; Moreno-Hernández, Y.; Olivo-Vidal, Z.E. In Silico Studies of Four Compounds of Cecropia obtusifolia against Malaria Parasite. Molecules 2023, 28, 6912. https://doi.org/10.3390/molecules28196912
Lobato-Tapia CA, Moreno-Hernández Y, Olivo-Vidal ZE. In Silico Studies of Four Compounds of Cecropia obtusifolia against Malaria Parasite. Molecules. 2023; 28(19):6912. https://doi.org/10.3390/molecules28196912
Chicago/Turabian StyleLobato-Tapia, Carlos Alberto, Yolotl Moreno-Hernández, and Zendy Evelyn Olivo-Vidal. 2023. "In Silico Studies of Four Compounds of Cecropia obtusifolia against Malaria Parasite" Molecules 28, no. 19: 6912. https://doi.org/10.3390/molecules28196912
APA StyleLobato-Tapia, C. A., Moreno-Hernández, Y., & Olivo-Vidal, Z. E. (2023). In Silico Studies of Four Compounds of Cecropia obtusifolia against Malaria Parasite. Molecules, 28(19), 6912. https://doi.org/10.3390/molecules28196912