Effects of Me–Solvent Interactions on the Structure and Infrared Spectra of MeTFSI (Me = Li, Na) Solutions in Carbonate Solvents—A Test of the GFN2-xTB Approach in Molecular Dynamics Simulations


Abstract
:
1. Introduction
2. Results
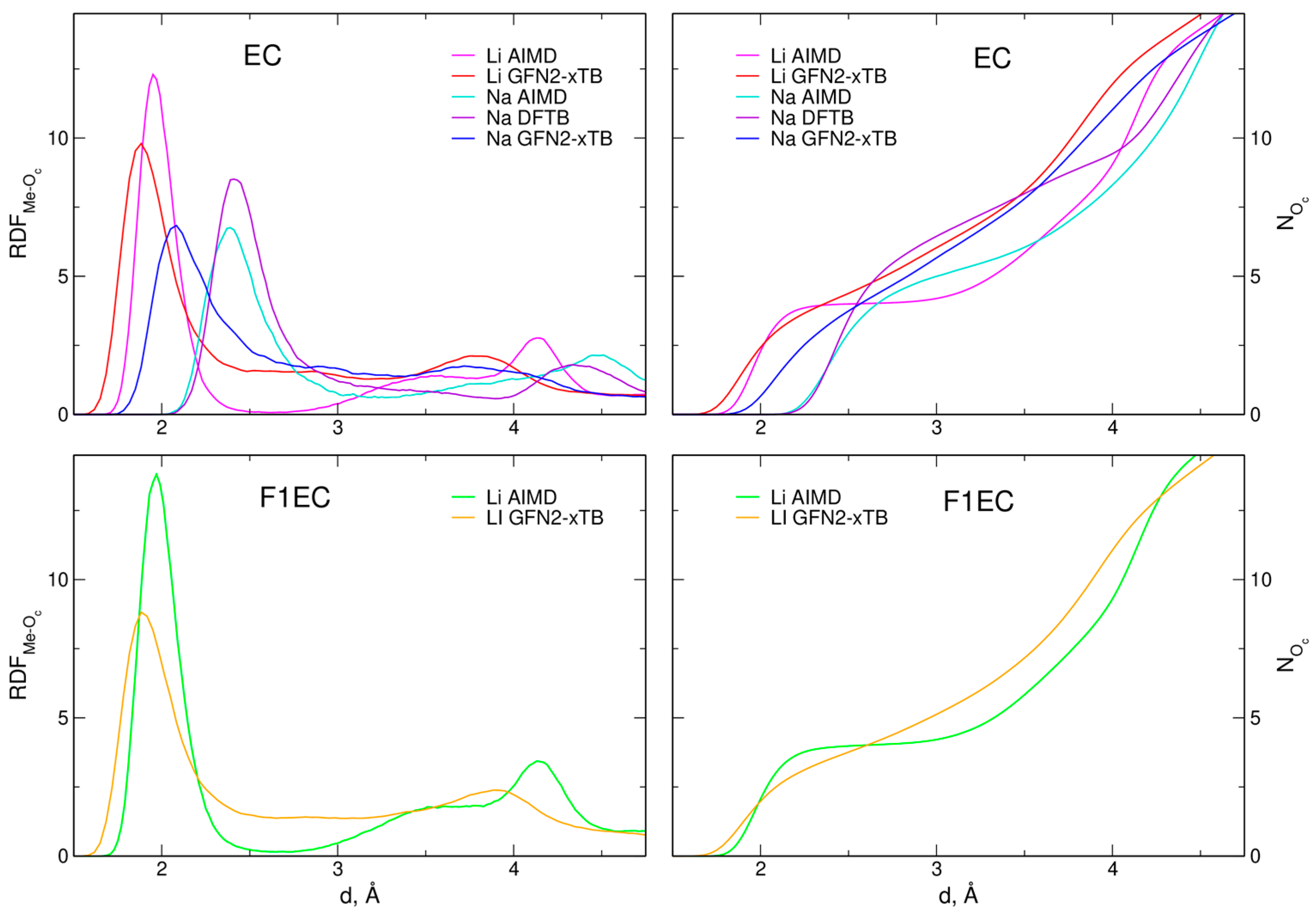
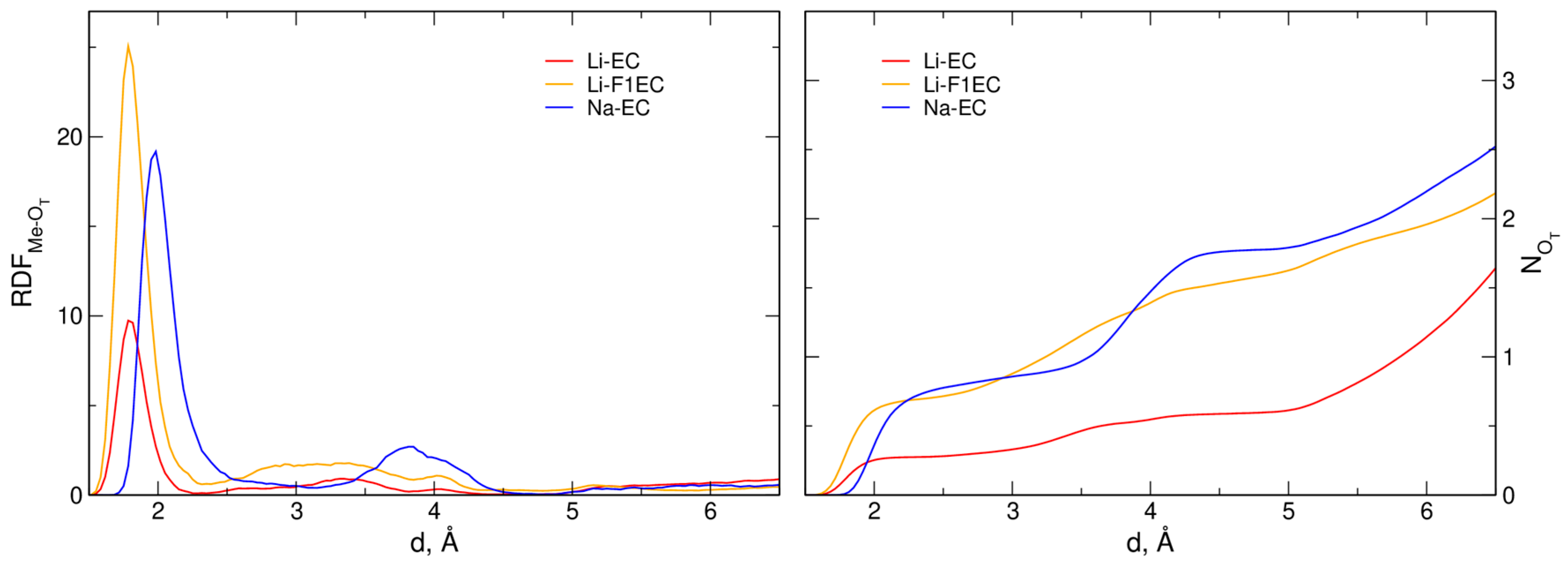
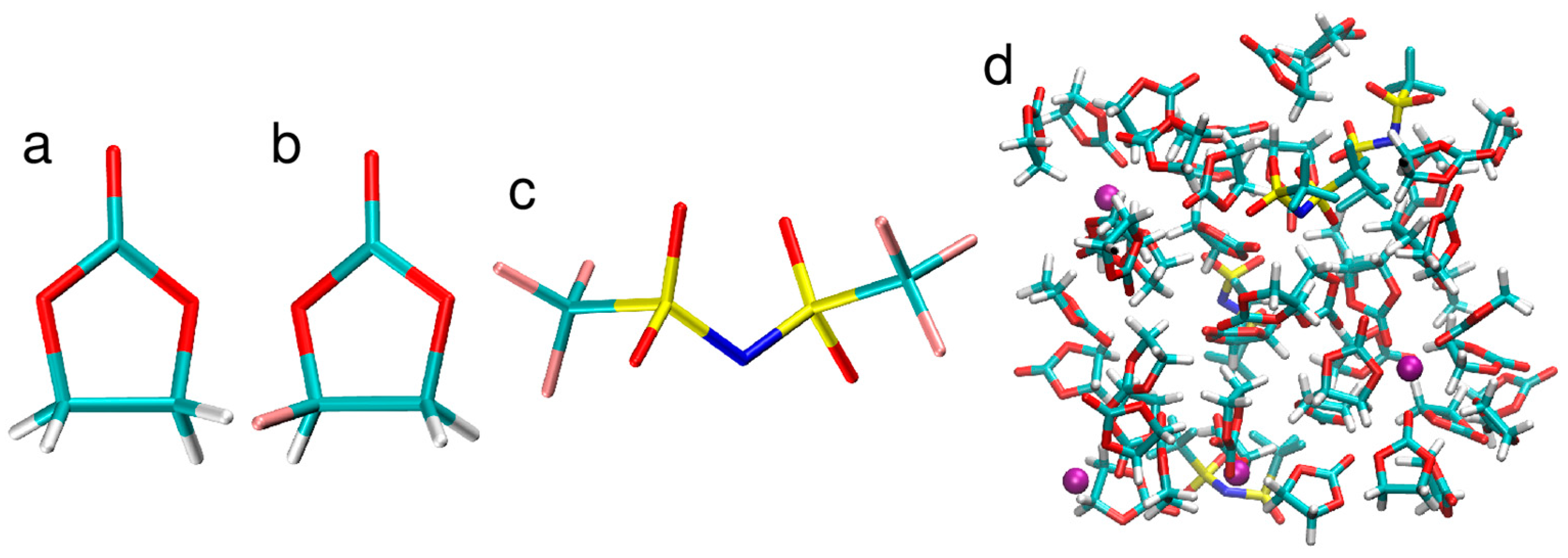
2.1. Structure of the Electrolytes
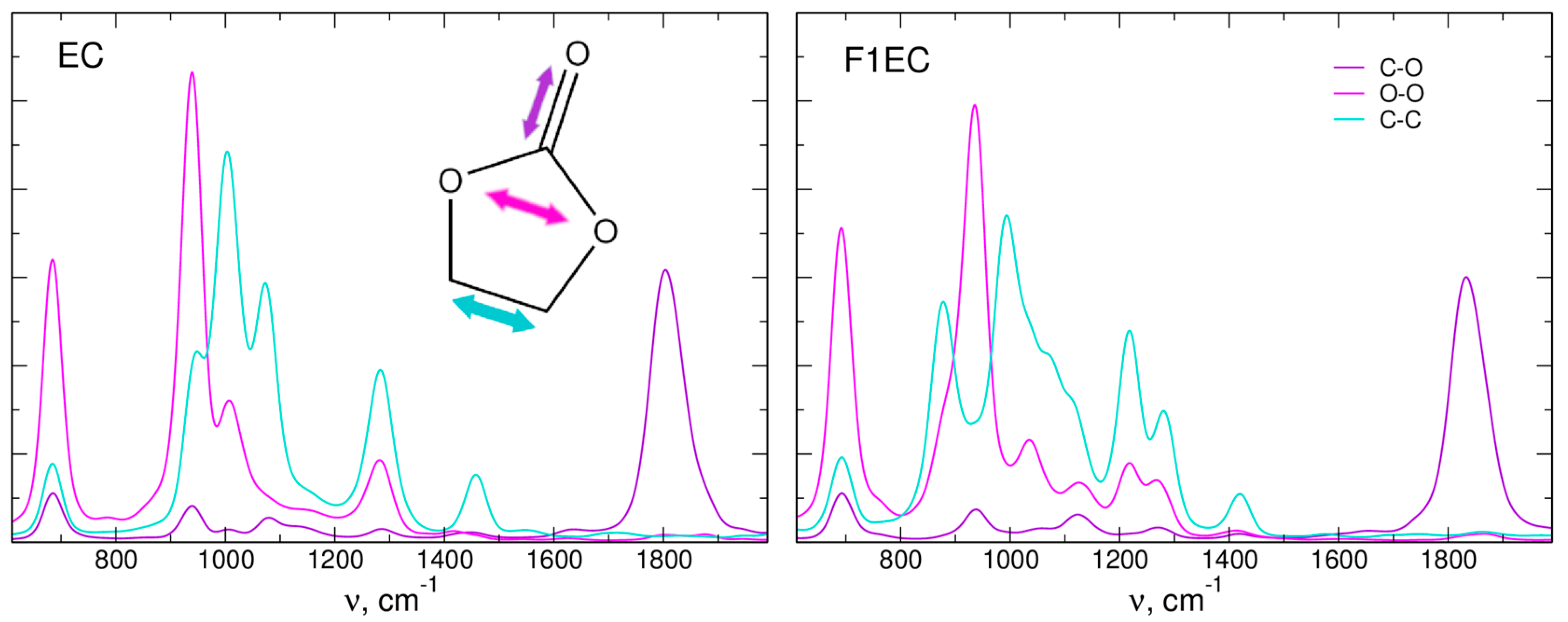
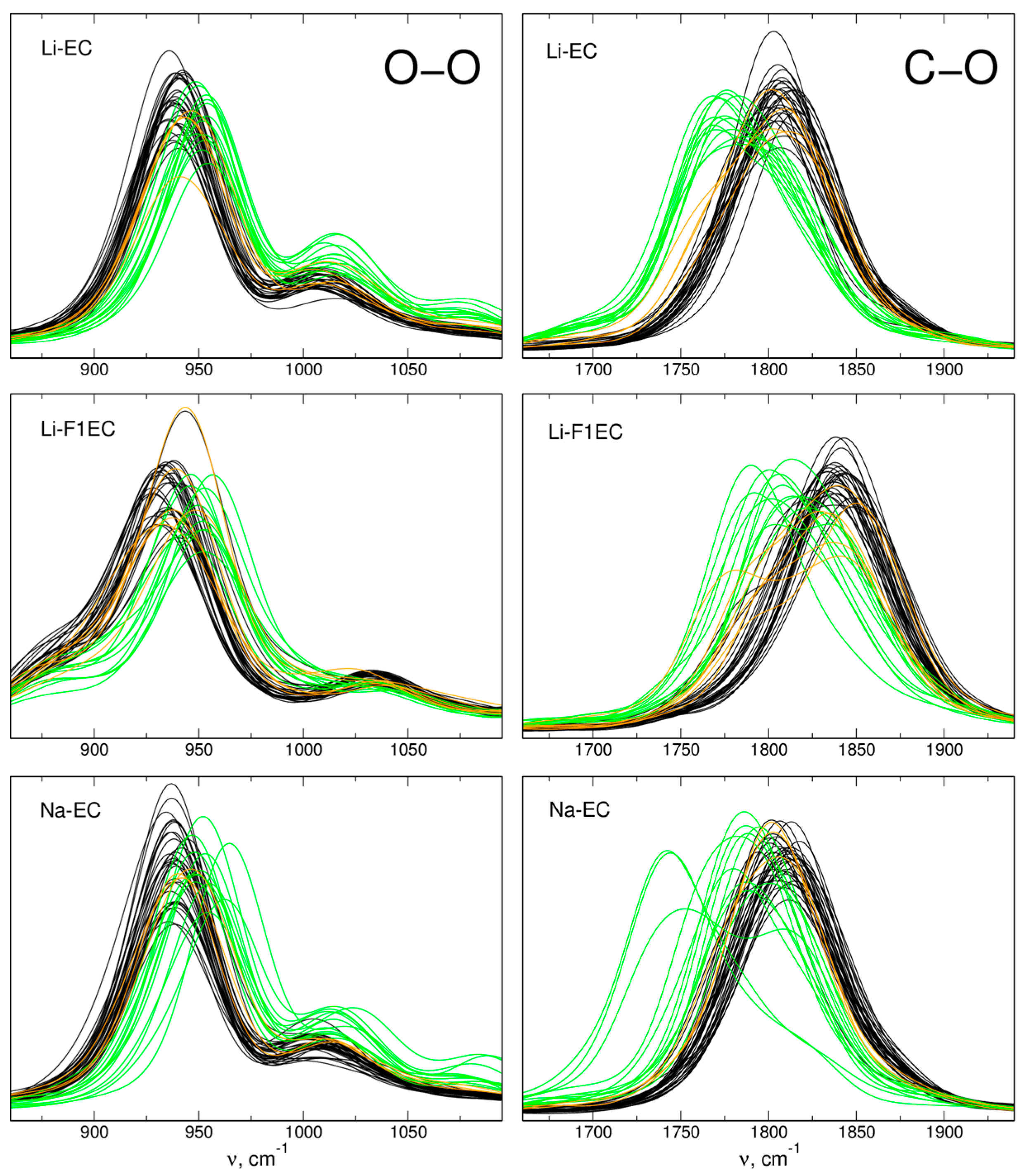
2.2. IR Spectra
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Li, M.; Lu, J.; Chen, Z.; Amine, K. 30 Years of Lithium-Ion Batteries. Adv. Mater. 2018, 30, 1800561. [Google Scholar] [CrossRef]
- Zubi, G.; Dufo-López, R.; Carvalho, M.; Pasaoglu, G. The Lithium-Ion Battery: State of the Art and Future Perspectives. Renew. Sustain. Energy Rev. 2018, 89, 292–308. [Google Scholar] [CrossRef]
- Grey, C.P.; Hall, D.S. Prospects for Lithium-Ion Batteries and Beyond—A 2030 Vision. Nat. Commun. 2020, 11, 6279. [Google Scholar] [CrossRef] [PubMed]
- Palomares, V.; Serras, P.; Villalunega, L.; Hueso, K.B.; Carretero-González, J.; Rojo, T. Na-Ion Batteries, Recent Advances and Present Challenges to Become Low Cost Energy Storage Systems. Energy Environ. Sci. 2012, 5, 5884–5901. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef]
- Ponrouch, A.; Monti, D.; Boschin, A.; Steen, B.; Johansson, P.; Palacín, M.R. Non-Aqueous Electrolytes for Sodium-Ion Batteries. J. Mater. Chem. A 2015, 3, 22–42. [Google Scholar] [CrossRef]
- Nayak, P.K.; Yang, L.; Brehm, W.; Adelhelm, P. From Lithium-Ion to Sodium-Ion Batteries: Advantages, Challenges, and Surprises. Angew. Chem. Int. Ed. 2018, 57, 102–120. [Google Scholar] [CrossRef]
- Yoo, H.D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg Rechargeable Batteries: An On-Going Challenge. Energy Environ. Sci. 2013, 6, 2265–2279. [Google Scholar] [CrossRef]
- Shah, R.; Mittal, V.; Matsil, E.; Rosenkranz, A. Magnesium-Ion Batteries for Electric Vehicles: Current Trends and Future Perspectives. Adv. Mech. Eng. 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Guan, S.; Huo, H.; Ma, Y.; Gao, Y.; Zuo, P.; Yin, G. A Review on Magnesium Aluminum Chloride Complex Electrolytes for Mg Batteries. Adv. Funct. Mater. 2021, 31, 2100650. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hu, L.; Wu, H.; Weng, W.; Koh, M.; Redfern, P.C.; Curtiss, L.A.; Amine, K. Fluorinated Electrolytes for 5 V Lithium-Ion Battery Chemistry. Energy Environ. Sci. 2013, 6, 1806–1810. [Google Scholar] [CrossRef]
- Xu, C.; Lindgren, F.; Philippe, B.; Gorgoi, M.; Björefors, F.; Edström, K.; Gustafsson, T. Improved Performance of the Silicon Anode for Li-ion Batteries: Understanding the Surface Modification Mechanism of Fluoroethylene Carbonate as an Effective Electrolyte Additive. Chem. Mater. 2015, 27, 2591–2599. [Google Scholar] [CrossRef]
- von Aspern, N.; Röschenthaler, G.-V.; Winter, M.; Cekic-Laskovic, I. Fluorine and Lithium: Ideal Partners for High-Performance Rechargeable Battery Electrolytes. Angew. Chem. Int. Ed. 2019, 58, 15978–16000. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Wang, G.; Qu, L.; Luo, D.; Yang, L.; Horano, S.-I. A Novel Mixture of Diethylene Glycol Diethylether and Non-flammable Methyl-nonafluorobutyl Ether as a Safe Electrolyte for Lithium Ion Batteries. J. Mater. Chem. A 2015, 3, 21159–21166. [Google Scholar] [CrossRef]
- Bolloli, M.; Kalhoff, J.; Alloin, F.; Bresser, D.; Le, M.L.P.; Langlois, B.; Passerini, S.; Sanchez, J.-Y. Fluorinated Carbamates as Suitable Solvents for LiTFSI-Based Lithium-Ion Electrolytes: Physicochemical Properties and Electrochemical Characterization. J. Phys. Chem. C 2015, 119, 22404–22414. [Google Scholar] [CrossRef]
- Bolloli, M.; Alloin, F.; Kalhoff, J.; Bresser, D.; Passerini, S.; Judeinstein, P.; Leprêtre, J.-C.; Sanchez, J.-Y. Effect of Carbonates Fluorination on the Properties of LiTFSI-Based Electrolytes for Li-Ion Batteries. Electrochim. Acta 2015, 161, 159–170. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, Z.; Li, L.; Yang, J.; Xiong, S.; Qian, Y. Ether-Based Nonflammable Electrolyte for Room Temperature Sodium Battery. J. Power Sources 2015, 284, 222–226. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.; Kim, H.; Kang, K.; Choi, N.-S. Highly Stable Linear Carbonate-Containing Electrolytes with Fluoroethylene Carbonate for High-Performance Cathodes in Sodium-Ion Batteries. J. Power Sources 2016, 320, 49–58. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, B.; Xue, R.; Chen, L.; Huang, X. Ion Association and Solvation Studies of LiClO4/Ethylene Carbonate Electrolyte by Raman and Infrared Spectroscopy. J. Electrochem. Soc. 1998, 145, 3346–3350. [Google Scholar] [CrossRef]
- Aroca, R.; Nazri, M.; Nazri, G.A.; Camargo, A.J.; Trsic, M. Vibrational Spectra and Ion-Pair Properties of Lithium Hexafluorophosphate in Ethylene Carbonate Based Mixed-Solvent Systems for Lithium Batteries. J. Solution Chem. 2000, 29, 1047–1060. [Google Scholar] [CrossRef]
- Allen, J.L.; Borodin, O.; Seo, D.M.; Henderson, W.A. Combined Quantum Chemical/Raman Spectroscopic Analyses of Li+ Cation Solvation: Cyclic Carbonate Solvents-Ethylene Carbonate and Propylene Carbonate. J. Power Sources 2014, 267, 821–830. [Google Scholar] [CrossRef]
- Kameda, Y.; Saito, S.; Umebayashi, Y.; Fujji, K.; Arno, Y.; Usuki, T. Local Structure of Li+ in Concentrated LIPF6-Dimethyl Carbonate Solutions. J. Mol. Liquids 2016, 217, 17–22. [Google Scholar] [CrossRef]
- Cresce, A.V.; Russell, S.M.; Borodin, O.; Allen, J.A.; Schroeder, M.A.; Dai, M.; Peng, J.; Gobet, M.P.; Greenbaum, S.G.; Rogers, R.E.; et al. Solvation Behavior of Carbonate-Based Electrolytes in Sodium Ion Batteries. Phys. Chem. Chem. Phys. 2017, 19, 574–586. [Google Scholar] [CrossRef]
- Maeda, S.; Kameda, Y.; Amo, Y.; Usuki, T.; Ikeda, K.; Otomo, T.; Yanagisawa, M.; Seki, S.; Arai, N.; Watanabe, H.; et al. Local Structure of Li+ in Concentrated Ethylene Carbonate Solutions Studied by Low-Frequency Raman Scattering and Neutron Diffraction with 6Li/7Li Isotopic Substitution Methods. J. Phys. Chem. B 2017, 121, 10979–10987. [Google Scholar] [CrossRef] [PubMed]
- Masia, M.; Probst, M.; Rey, R. Ethylene Carbonate-Li+: A Theoretical Study of Structural and Vibrational Properties in Gas and Liquid Phases. J. Phys. Chem. B 2004, 108, 2016–2027. [Google Scholar] [CrossRef]
- Arslanargin, A.; Powers, A.; Beck, T.L.; Rick, S.W. Models of Ion Solvation Thermodynamics in Ethylene Carbonate and Propylene Carbonate. J. Phys. Chem. B 2016, 120, 1497–1508. [Google Scholar] [CrossRef]
- Ding, W.; Lei, X.; Ouyang, C. Coordination of Lithium Ion with Ethylene Carbonate Electrolyte Solvent: A Computational Study. Int. J. Quantum Chem. 2016, 116, 97–102. [Google Scholar] [CrossRef]
- Borodin, O.; Olguin, M.; Ganesh, P.; Kent, P.R.C.; Allen, J.L.; Henderson, W.A. Competitive Lithium Solvation of Linear and Cyclic Carbonates from Quantum Chemistry. Phys. Chem. Chem. Phys. 2016, 18, 164–175. [Google Scholar] [CrossRef]
- Eilmes, A. Li+ Binding to Fluorinated Carbonate Solvents Studied by Ab Initio Quantum Chemical Calculations. J. Mol. Liquids 2016, 224, 452–459. [Google Scholar] [CrossRef]
- Okoshi, M.; Yamada, Y.; Komaba, S.; Yamada, A.; Nakai, H. Theoretical Analysis of Interactions between Potassium Ions and Organic Electrolyte Solvents: A Comparison with Lithium, Sodium, and Magnesium Ions. J. Electrochem. Soc. 2017, 164, A54–A60. [Google Scholar] [CrossRef]
- Eilmes, A.; Kubisiak, P.; Wróbel, P. Explicit and Hybrid Solvent Models for Estimates of Parameters Relevant to the Reduction Potential of Ethylene Carbonate. Int. J. Mol. Sci. 2022, 23, 15590. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D. LiTFSI Structure and Transport in Ethylene Carbonate from Molecular Dynamics Simulations. J. Phys. Chem. B 2006, 110, 4971–4977. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O.; Smith, G.D. Development of Many-Body Polarizable Force Fields for Li-Battery Applications: 2 LiTFSI-Doped Oligoether, Polyether, and Carbonate-Based Electrolytes. J. Phys. Chem. B 2006, 110, 6293–6299. [Google Scholar] [CrossRef]
- Takenaka, N.; Sakai, H.; Suzuki, Y.; Uppula, P.; Nagaoka, M. A Computational Chemical Insight into Microscopic Additive Effect on Solid Electrolyte Interphase Film Formation in Sodium-Ion Batteries: Suppresion of Unstable Film Growth by Intact Fluoroethylene Carbonate. J. Phys. Chem. C 2015, 119, 18046–18055. [Google Scholar] [CrossRef]
- Chang, T.-M.; Dang, L.X. Li+ Solvation and Kinetics of Li+-BF4−/PF6− Ion Pairs in Ethylene Carbonate Molecular Dynamics Study with Classical Rate Theories. J. Chem. Phys. 2017, 147, 161709. [Google Scholar] [CrossRef] [PubMed]
- Karatrantos, A.V.; Ohba, T.; Cai, Q. Diffusion of Ions and Solvent in Propylene Carbonate Solutions for Lithium-Ion Battery Applications. J. Mol. Liquids 2020, 320, 114351. [Google Scholar] [CrossRef]
- Wróbel, P.; Kubisiak, P.; Eilmes, A. MeTFSI (Me = Li, Na) Solvation in Ethylene Carbonate and Fluorinated Ethylene Carbonate: A Molecular Dynamics Study. J. Phys. Chem. B 2021, 125, 1248–1258. [Google Scholar] [CrossRef]
- Thomas, M.; Brehm, M.; Fligg, R.; Vöhringer, P.; Kirchner, B. Computing Vibrational Spectra from Ab Initio Molecular Dynamics. Phys. Chem. Chem. Phys. 2013, 15, 6608–6622. [Google Scholar] [CrossRef]
- Fischer, S.A.; Ueltschi, T.W.; El-Khoury, P.Z.; Miffin, A.L.; Hess, W.P.; Wang, H.-F.; Cramer, C.J.; Govind, N. Infrared and Raman Spectroscopy from Ab Initio Molecular Dynamics and Static Normal Mode Analysis: The C-H Region of DMSO as a Case Study. J. Phys. Chem. B 2016, 120, 1429–1436. [Google Scholar] [CrossRef]
- Eilmes, A.; Alves, W.A. Combining Experimental and Theoretical Vibrational Spectroscopy to Study Magnesium Aluminum Chloride Complex Electrolytes. J. Mol. Liquids 2021, 333, 116053. [Google Scholar] [CrossRef]
- Yan, F.; Mukherjee, K.; Maroncelli, M.; Kim, H.J. Infrared Spectroscopy of Li+ Solvation in EmimBF4 and in Propylene Carbonate: Ab Initio Molecular Dynamics and Experiment. J. Phys. Chem. B 2022, 126, 9643–9662. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-Consistent-Charge Density Functional Tight-Binding Method for Simulations of Complex Material Properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. [Google Scholar] [CrossRef]
- Zentel, T.; Kühn, O. Properties of Hydrogen Bonds in the Protic Ionic Liquid Ethylammonium Nitrate. Theor. Chem. Acc. 2017, 136, 87. [Google Scholar] [CrossRef]
- Sakti, A.W.; Wahyudi, S.T.; Ahmad, F.; Darmawan, N.; Hardhienata, H.; Alatas, H. Effects of Salt Concentration on the Water and Ion Self-Diffusion Coefficients of a Model Aqueous Sodium-Ion Battery Electrolyte. J. Phys. Chem. B 2022, 126, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, P.; Kubisiak, P.; Eilmes, A. Hydrogen Bonding and Infrared Spectra of Ethyl-3-methylimidazolium Bis(trifluorosulfonyl)imide/Water Mixtures: A View from Molecular Dynamics Simulations. J. Phys. Chem. B 2022, 126, 10922–10932. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1994, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, A Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | rm(Me–Oc) | CN(Me–Oc) | ||
|---|---|---|---|---|
| AIMD 1 | GFN2-xTB | AIMD | GFN2-xTB | |
| Li-EC | 1.95 | 1.90 | 4.00 | 4.40 |
| Li-F1EC | 1.97 | 1.90 | 3.98 | 3.75 |
| Na-EC | 2.39 | 2.09 | 5.00 | 5.65 |
| System | rm(Me–OT) | CN(Me–OT) | ||
|---|---|---|---|---|
| AIMD 1 | GFN2-xTB | AIMD | GFN2-xTB | |
| Li-EC | - | 1.78 | 0.0 | 0.28 |
| Li-F1EC | - | 1.78 | 0.0 | 0.71 |
| Na-EC | 2.4 | 1.99 | 0.2 | 0.85 |
| Vibration/System | ΔIR | ΔFT | Δexp | ||
|---|---|---|---|---|---|
| AIMD 1 | GFN2-xTB | AIMD 1 | GFN2-xTB | ||
| C=O/Li-EC | −33 | −27 | −35 | −32 | −30 2 |
| C=O/Li-F1EC | −25 | −28 | −33 | −32 | −30 2, −36 3 |
| C=O/Na-EC | −16 | −15 | −21 | −21 | −20 3 |
| O-O/Li-EC | - | - | 9 | 14 | 12 4 |
| O-O/Li-F1EC | 17 | - | 15 | 15 | 17 4 |
| O-O/Na-EC | - | - | 4 | 14 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróbel, P.; Eilmes, A. Effects of Me–Solvent Interactions on the Structure and Infrared Spectra of MeTFSI (Me = Li, Na) Solutions in Carbonate Solvents—A Test of the GFN2-xTB Approach in Molecular Dynamics Simulations. Molecules 2023, 28, 6736. https://doi.org/10.3390/molecules28186736
Wróbel P, Eilmes A. Effects of Me–Solvent Interactions on the Structure and Infrared Spectra of MeTFSI (Me = Li, Na) Solutions in Carbonate Solvents—A Test of the GFN2-xTB Approach in Molecular Dynamics Simulations. Molecules. 2023; 28(18):6736. https://doi.org/10.3390/molecules28186736
Chicago/Turabian StyleWróbel, Piotr, and Andrzej Eilmes. 2023. "Effects of Me–Solvent Interactions on the Structure and Infrared Spectra of MeTFSI (Me = Li, Na) Solutions in Carbonate Solvents—A Test of the GFN2-xTB Approach in Molecular Dynamics Simulations" Molecules 28, no. 18: 6736. https://doi.org/10.3390/molecules28186736
APA StyleWróbel, P., & Eilmes, A. (2023). Effects of Me–Solvent Interactions on the Structure and Infrared Spectra of MeTFSI (Me = Li, Na) Solutions in Carbonate Solvents—A Test of the GFN2-xTB Approach in Molecular Dynamics Simulations. Molecules, 28(18), 6736. https://doi.org/10.3390/molecules28186736