
Lipase-Catalyzed Strategies for the Preparation of Enantiomeric THIQ and THβC Derivatives: Green Aspects


Abstract
:
1. Introduction
2. Enzymatic Strategies for the Synthesis of THIQ and THβC Enantiomers
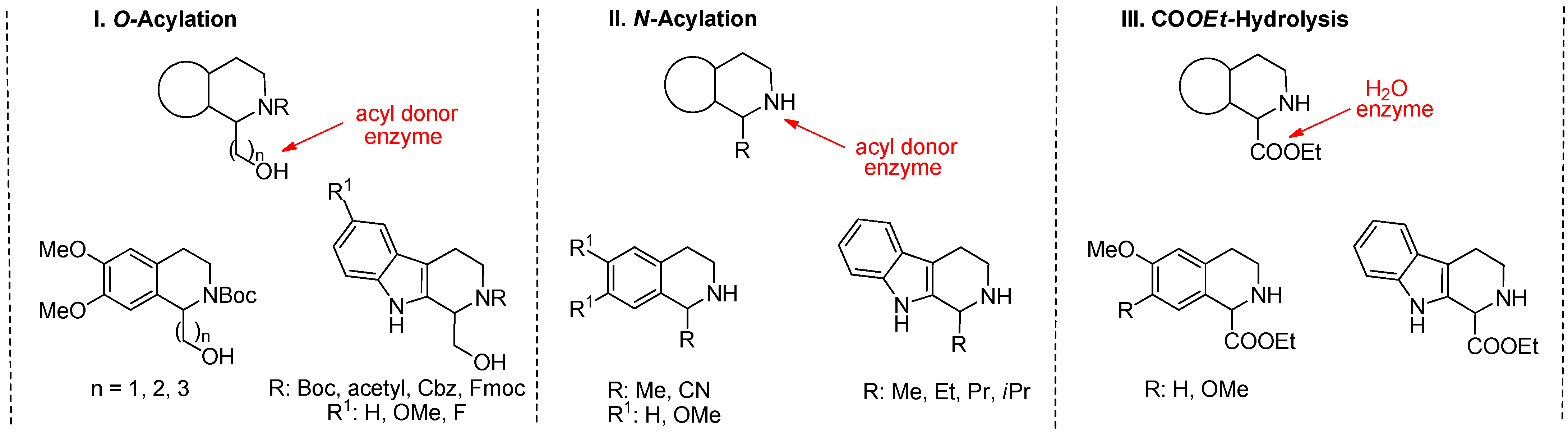
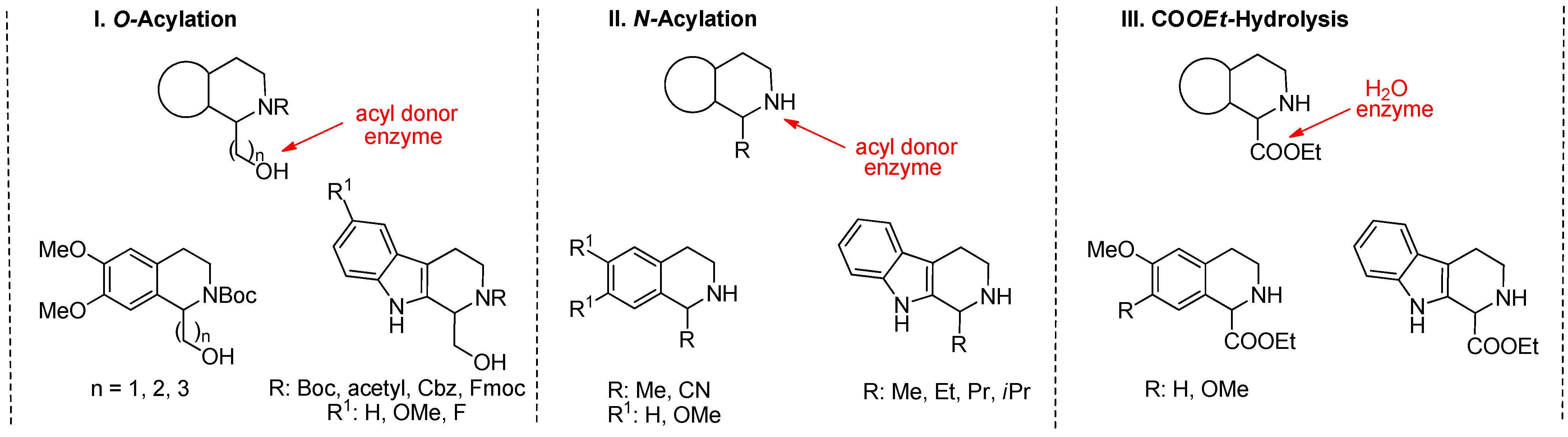
2.1. KR through O-Acylation
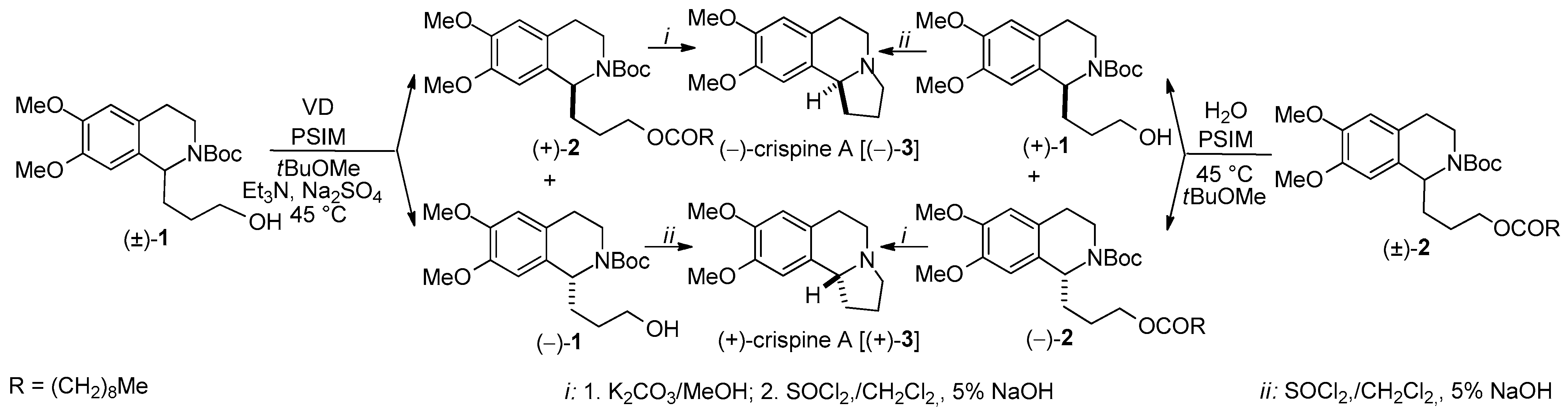
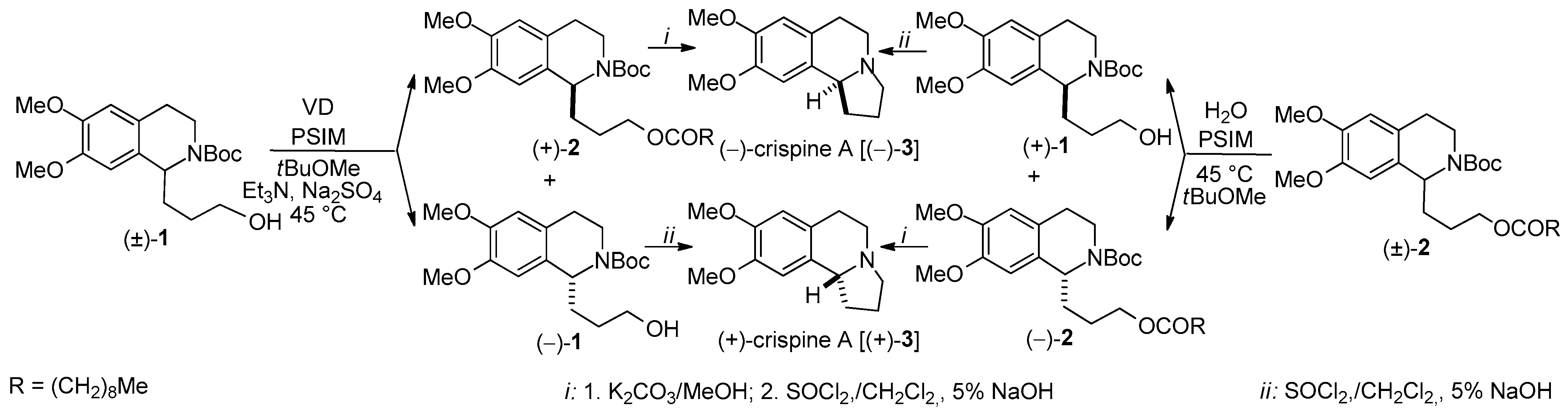
- A new total synthesis of crispine A enantiomers via Burkholderia cepacia lipase-catalyzed acylation of the primary hydroxy group of N-Boc-protected 1-(3-hydroxypropyl)-6,7-bis(methyloxy)-1,2,3,4-THIQ [(±)-1] and enantioselective hydrolysis of the corresponding O-decanoate [(±)-2], with a remote, four-atom-distant stereogenic center was reported [47].
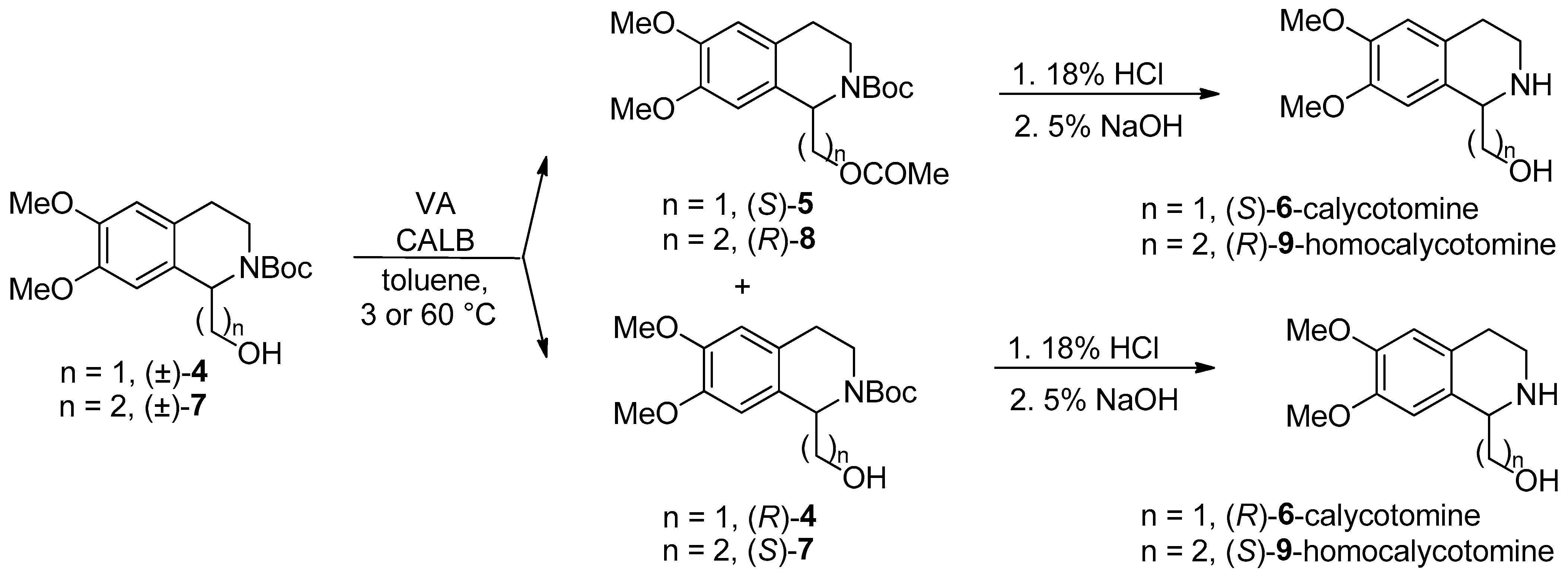
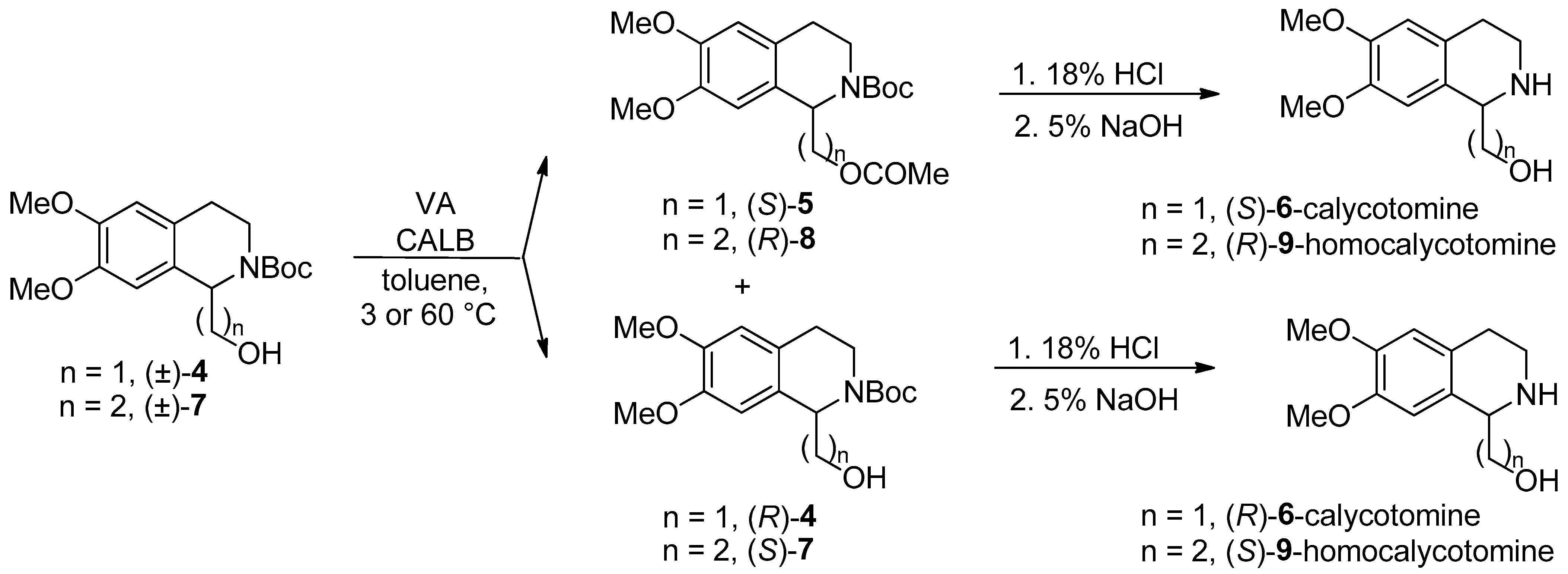
- Both enantiomers of calycotomine [49] and homocalycotomine [50] (one of them is the intermediate of emetine) through Candida antarctica lipase B-catalyzed asymmetric O-acylation of N-Boc-protected (6,7-dimethoxy-1,2,3,4-THIQ-1-yl)methanol ((±)-4)), with a remote, two-atom-distant stereogenic center and (6,7-dimethoxy-1,2,3,4-THIQ-1-yl)ethanol ((±)-7)), with a remote, three-atom-distant stereogenic center were described.
- A systematic study on the Candida antarctica lipase B-catalyzed O-acylation of THIQ amino alcohol homologues ((±)-1, (±)-4, and (±)-7) containing a remote stereogenic center was reported [50].
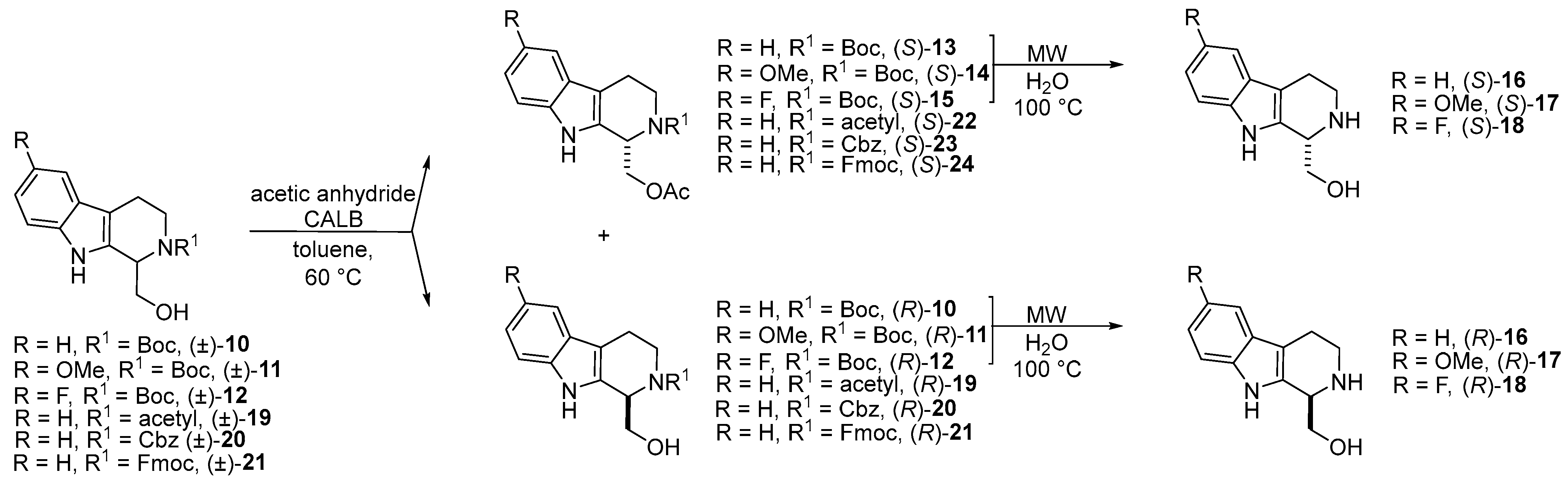
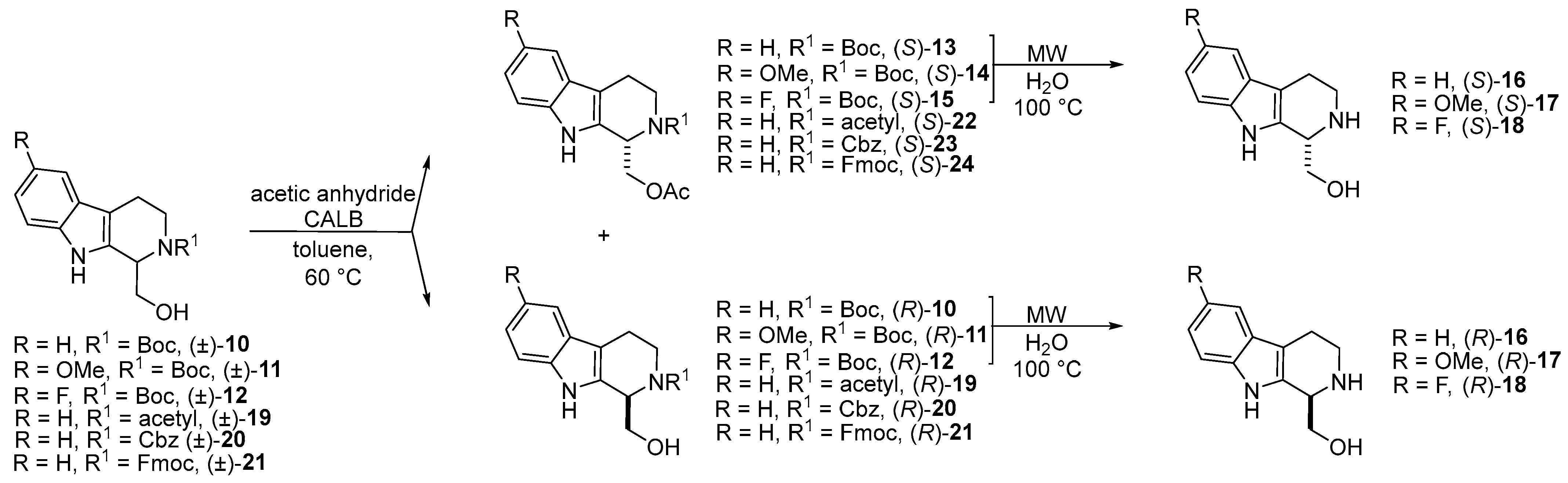
- The synthesis of new enantiomers of 1,2,3,4-THβC derivatives through Candida antarctica lipase B-catalyzed O-acylation of N-Boc-protected 1-hydroxymethyl-1,2,3,4-THβC ((±)-10), 1-hydroxymethyl-6-methoxy-1,2,3,4-THβC ((±)-11), and 1-hydroxymethyl-6-fluoro-1,2,3,4-THβC ((±)-12) was described [52].
- In the frame of substrate engineering, the steric effect of different N-protecting groups (N-Boc, N-acetyl, N-Cbz, N-Fmoc) on the enantioselectivity and reaction rate of Candida antarctica lipase B-catalyzed O-acylation of 1-hydroxymethyl-THβCs [(±)-10, (±)-19, (±)-20, and (±)-21] was investigated [53].
2.1.1. KR through N-Acylation
2.1.2. DKR through N-Acylation
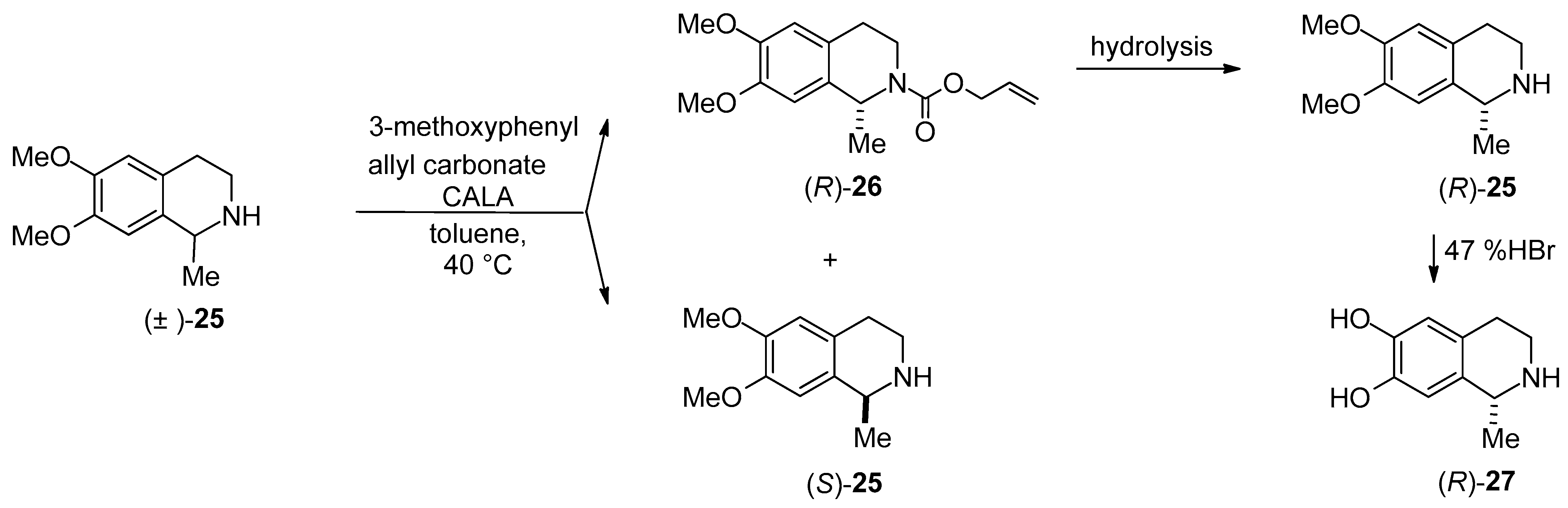
- Preparation of (R)-6,7-dihydroxy-1-methyl-1,2,3,4-THIQ (salsolinol) through Candida antarctica lipase A-catalyzed N-acylation of 1-methyl-6,7-dimethoxy-1,2,3,4-THIQ [(±)-25] was reported [54].
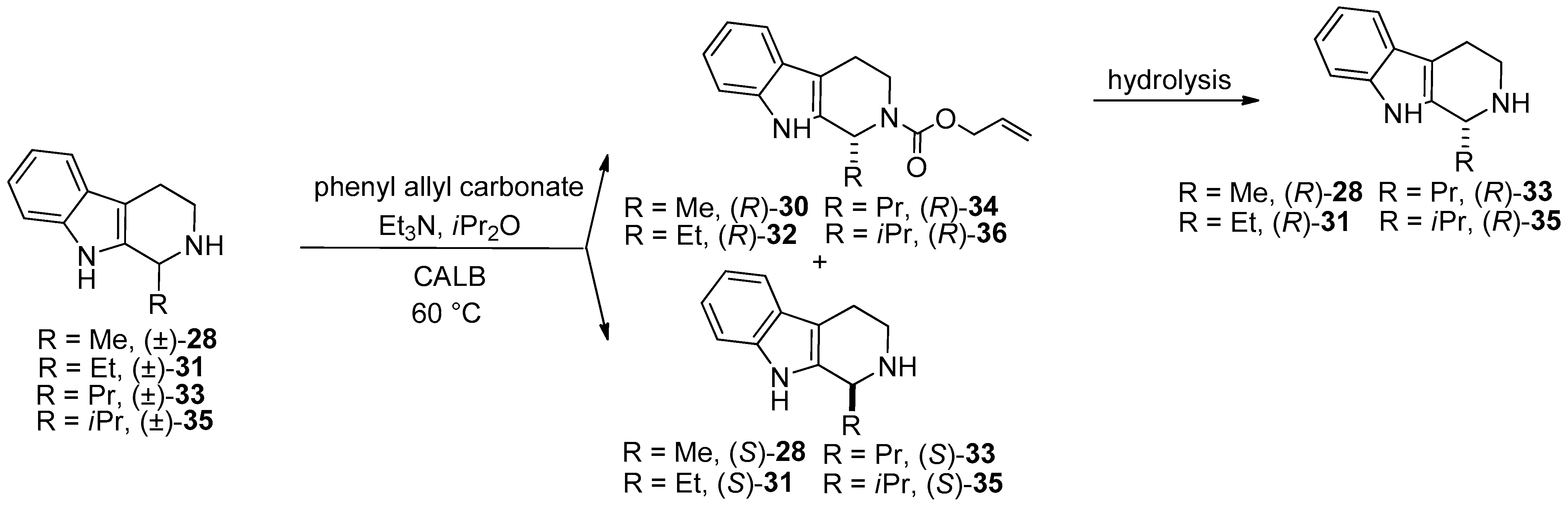
- The synthesis of both enantiomers of salsolidine (the (R)-enantiomer has a monoamine oxidase A (MAO A) inhibiting effect) and its analogue 1-methyl-1,2,3,4-THβC carried out through Candida rugosa lipase- and Candida antarctica lipase B-catalyzed N-alkoxycarbonylation of racemic 1-methyl-6,7-dimethoxy-1,2,3,4-THIQ ((±)-25) and 1-methyl-1,2,3,4-THβC ((±)-28) was described [55].
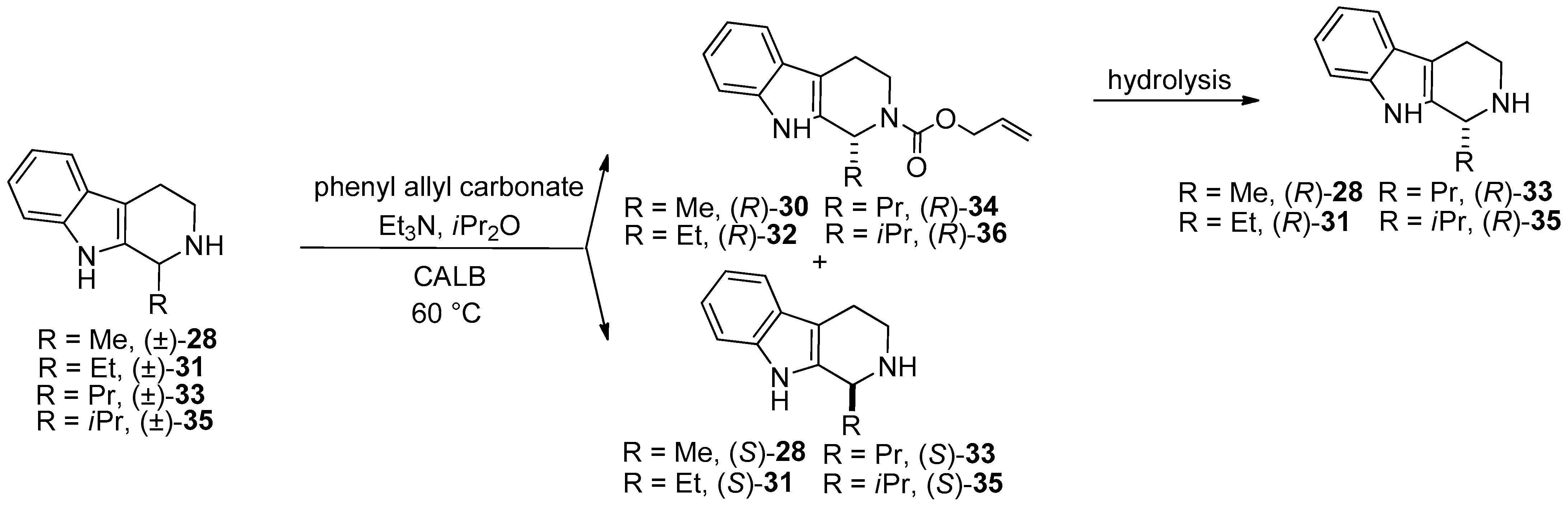
- In the frame of substrate specificity, the influence of different alkyl chain substituents (Me, Et, Pr, iPr) on the enantioselectivity and reaction rate of Candida antarctica lipase B-catalyzed N-alkoxycarbonylation of 1-substituted THβCs (Me ((±)-28), Et ((±)-31), Pr ((±)-33), iPr ((±)-35)) was studied [56].
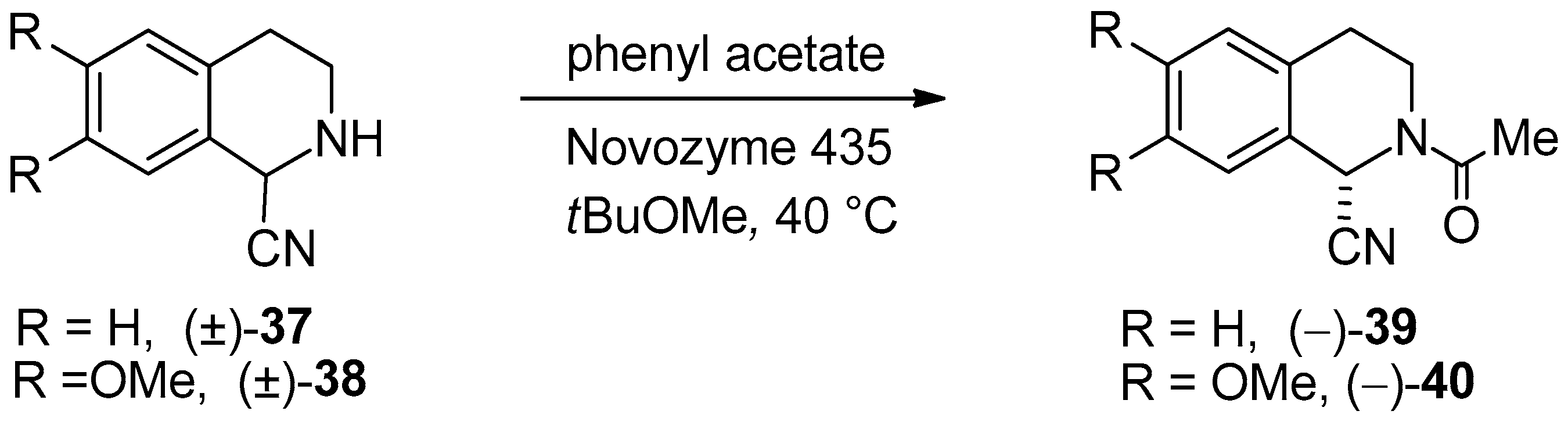
- The synthesis of 1-cyano-1,2,3,4-THIQ enantiomers through Candida lipase-catalyzed DKR (racemization and asymmetric amidation) of racemic 1-cyano-1,2,3,4-THIQ (((±)-39)) and 1-cyano-6,7-dimethoxy-1,2,3,4-THIQ (((±)-40)) was described [57].
2.2. DKR through COOEt Hydrolysis
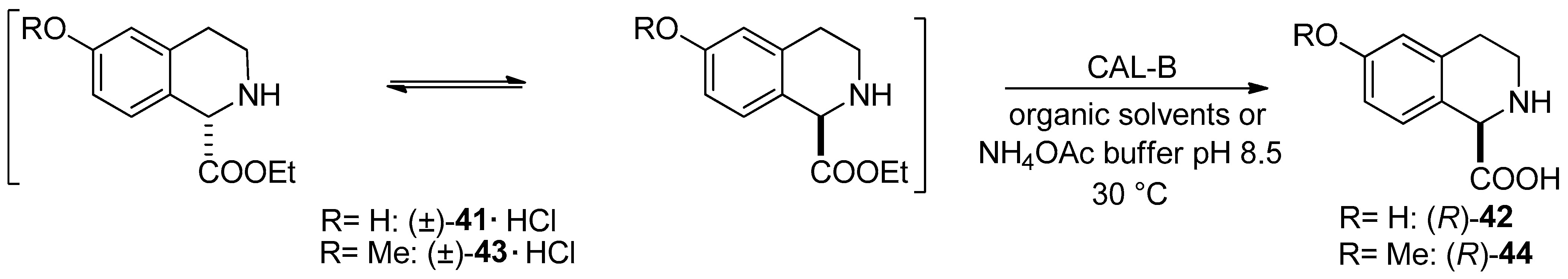
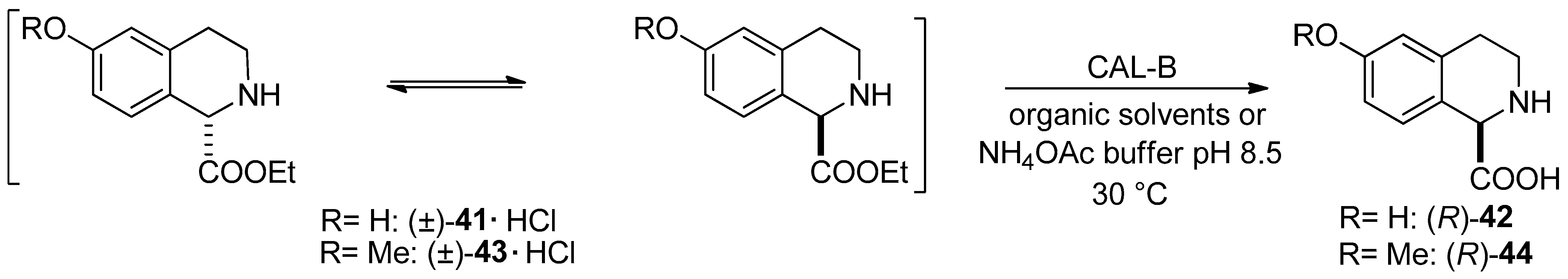
- Preparation of (R)-6-hydroxy-1,2,3,4-THIQ-1-CA through Candida antarctica lipase B-catalyzed DKR of the hydrochloride salt of ethyl 6-hydroxy- and 6-methoxy-1,2,3,4-THIQ-1-carboxylate ((±)-41·HCl and (±)-43·HCl) was reported. Note that the starting material can be transformed into (R)-2-tert-butyl-1-methyl-6-hydroxy-3,4-dihydroisoquinoline-1,2(1H)-dicarboxylate, which is the key intermediate of a Liver X Receptor agonist [58].
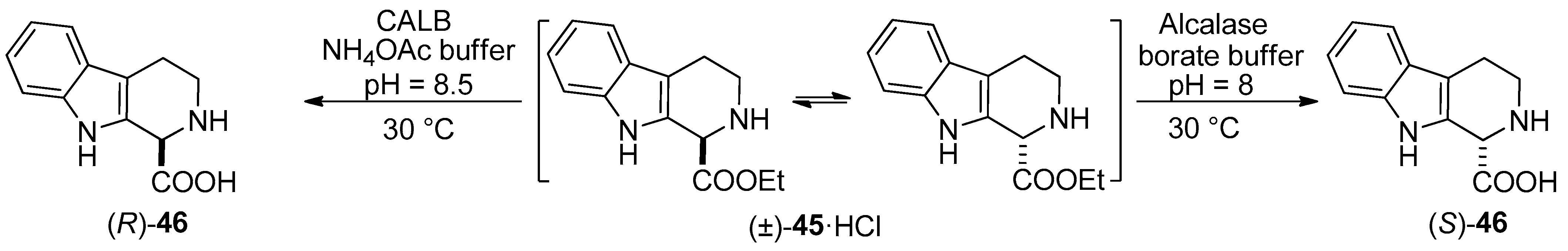
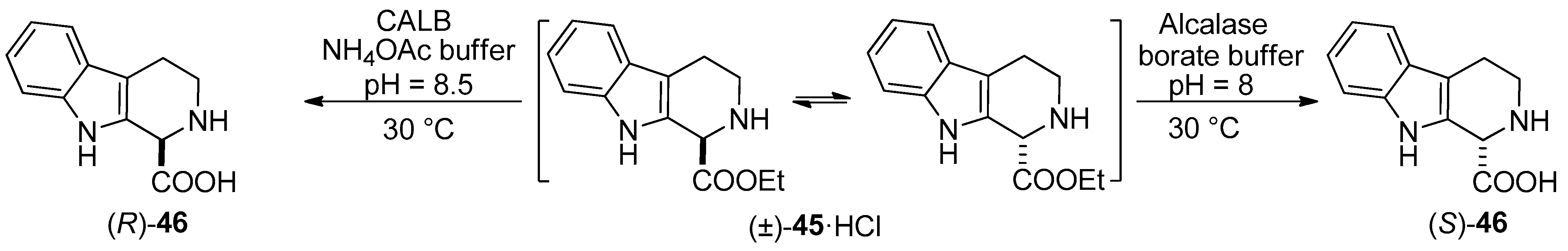
- Both enantiomers of 1,2,3,4-THβC were prepared through Candida antarctica lipase B-catalyzed stereocomplementary new DKR processes. The enzymatic hydrolysis of the hydrochloride salts as starting amino CE racemates furnished the desired enantiomeric AAs quantitatively [61].
3. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muthukrishnan, I.; Sridharan, V.; Menéndez, J.C. Progress in the chemistry of tetrahydroquinoline. Chem. Rev. 2019, 119, 5057–5191. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, F.; Liang, T.; Xie, Z.; Yang, Y.; Cao, C.; Gao, J.; Lu, T.; Chen, X. A review of synthetic bioactive tetrahydro-β-carbolines: A medicinal chemistry perspective. Eur. J. Med. Chem. 2021, 225, 113815. [Google Scholar] [CrossRef] [PubMed]
- Kaufm, T.S. Synthetic pathways to salsolidine. Tetrahedron Asymmetry 2004, 15, 1203–1237. [Google Scholar] [CrossRef]
- Boyd, E.M.; Knight, L.M. The expectorant action of cephaeline, emetine and 2-dehydroemetine. J. Pharm. Pharmacol. 1964, 16, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Al-Yahya, M.A.; Hassan, M.M.A. Noscapine. Anal. Profiles Drug Subst. 1982, 11, 407–461. [Google Scholar] [CrossRef]
- Poveda, A.; Ray-Coquard, I.; Romero, I.; Lopez-Guerrero, J.A.; Colombo, N. Emerging treatment strategies in recurrent platinum-sensitive ovarian cancer: Focus on trabectedin. Cancer Treat. Rev. 2014, 40, 366–375. [Google Scholar] [CrossRef]
- Liu, Z.-Z.; Wang, Y.; Tang, Y.-F.; Chen, S.-Z.; Chen, X.-G.; Li, H.-Y. Synthesis and antitumor activity of simplified ecteinascidin–saframycin analogs. Bioorg. Med. Chem. Lett. 2006, 16, 1282–1285. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Aoshima, A.; Ikeshiro, Y.; Chen, Y.-P.; Furukawa, H.; Itoigawa, M.; Fujioka, T.; Mihashi, K.; Cosentino, L.M.; Morris-Natschke, S.-L.; et al. Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure-activity correlations with related alkaloids. Bioorg. Med. Chem. 2005, 13, 443–448. [Google Scholar] [CrossRef]
- Leal, J.F.M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, E.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; Cuevas, C.; et al. Molecular pharmacology and antitumor activity of Zalypsis® in several human cancer cell lines. Biochem. Pharmacol. 2009, 78, 162–170. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Yang, J.; He, J.; Wu, X.-D.; Shao, L.-D.; Li, Y.; Huang, S.-X.; Li, R.-T.; Zhao, Q.-S. Vincamajorines A and B, monoterpenoid indole alkaloids with new carbon skeletons from Vinca major. Tetrahedron Lett. 2014, 55, 6490–6494. [Google Scholar] [CrossRef]
- Cong, H.-J.; Zhao, Q.; Zhang, S.-W.; Wei, J.-J.; Wang, W.-Q.; Xuan, L.-J. Terpenoid indole alkaloids from Mappianthus iodoides Hand.-Mazz. Phytochemistry 2014, 100, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.-S.; Sim, K.-M. Alkaloids from Kopsia griffithii. Phytochemistry 1998, 47, 145–147. [Google Scholar] [CrossRef]
- Davis, R.A.; Duffy, S.; Avery, V.M.; Camp, D.; Hooper, J.N.A.; Quinn, R.J. (+)-7-Bromotrypargine: An antimalarial β-carboline from the Australian marine sponge Ancorina sp. Tetrahedron Lett. 2010, 51, 583–585. [Google Scholar] [CrossRef]
- Brock, G.B.; McMahon, C.G.; Chen, K.K.; Costigan, T.; Shen, W.; Watkins, V.; Anglin, G.; Whitaker, S. Effect of tadalafil in chronic renal failure rabbits: Relevance to erectile dysfunction. J. Urol. 2002, 168, 1332–1336. [Google Scholar] [CrossRef]
- He, S.; Dobbelaar, P.H.; Guo, L.; Ye, Z.; Liu, J.; Jian, T.; Truong, Q.; Shah, S.K.; Du, W.; Qi, H.; et al. SAR exploration at the C-3 position of tetrahydro-β-carboline sstr3 antagonists. Bioorg. Med. Chem. Lett. 2016, 26, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Brokamp, R.; Bergmann, B.; Müller, I.B.; Bienz, S. Stereoselective preparation of pyridoxal 1,2,3,4-tetrahydro-β-carboline derivatives and the influence of their absolute and relative configuration on the proliferation of the malaria parasite Plasmodium falciparum. Bioorganic Med. Chem. 2014, 22, 1832–1837. [Google Scholar] [CrossRef]
- Spindler, A.; Stefan, K.; Wiese, M. Synthesis and investigation of tetrahydro-β-carboline derivatives as inhibitors of the Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2016, 59, 6121–6135. [Google Scholar] [CrossRef]
- Shankaraiah, N.; da Silva, W.A.; Andrade, C.K.R.; Santos, L.S. Enantioselective total synthesis of (S)-(−)-quinolactacin B. Tetrahedron Lett. 2008, 49, 4289–4291. [Google Scholar] [CrossRef]
- Szabó, T.; Volk, B.; Milen, M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules 2021, 26, 663. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Rozwadowska, M.D. Asymmetric Synthesis of Isoquinoline Alkaloids. Chem. Rev. 2004, 104, 3341–3370. [Google Scholar] [CrossRef]
- Miyazaki, M.; Ando, N.; Sugai, K.; Seito, Y.; Fukuoka, H.; Kanemitsu, T.; Nagata, K.; Odanaka, Y.; Nakamura, K.T.; Itoh, T. Catalytic asymmetric allylation of 3,4-dihydroisoquinolines and its application to the synthesis of isoquinoline alkaloids. J. Org. Chem. 2011, 76, 534–542. [Google Scholar] [CrossRef]
- Dai, J.; Dan, W.; Schneider, U.; Wang, J. β-Carboline alkaloid monomers and dimers: Occurrence, structural diversity, and biological activities. Eur. J. Med. Chem. 2018, 157, 622–656. [Google Scholar] [CrossRef]
- Buaban, K.; Phutdhawong, W.; Taechowisan, T.; Phutdhawong, W.S. Synthesis and investigation of tetrahydro-β-carboline derivatives as inhibitors of plant pathogenic fungi. Molecules 2021, 26, 207. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, G.K.; Ortega-Rojas, M.A.; Chavelas-Hernández, L.; Razo-Hernández, R.S.; Valdéz-Camacho, J.R.; Escalante, J. Solvent-free lipase-catalyzed transesterification of alcohols with methyl esters under vacuum-assisted conditions. ChemistrySelect 2022, 7, e202202643. [Google Scholar] [CrossRef]
- Dolzhenko, A.V.; Dolzhenko, A.V. Green solvents for eco-friendly synthesis of bioactive heterocyclic compounds. In Green Synthetic Approaches for Biologically Relevant Heterocycles, 1st ed.; Brahmachari, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 5; pp. 101–139. ISBN 9780128000700. [Google Scholar]
- Sousa, R.R.; Silva, A.S.; Fernandez-Lafuente, R.; Ferreira-Leitão, V.S. Solvent-free esterifications mediated by immobilized lipases: A review from thermodynamic and kinetic perspectives. Catal. Sci. Technol. 2021, 11, 5696–5711. [Google Scholar] [CrossRef]
- Gustini, L.; Finzel, L.; Lavilla, C.; Noordover, B.A.J.; Hendrix, M.M.R.M.; Gehrels, C.; Koning, C.E. Understanding the limitations of the solvent-free enzymatic synthesis of sorbitol-containing polyesters. ACS Sustain. Chem. Eng. 2016, 4, 2259–2268. [Google Scholar] [CrossRef]
- Serrano-Arnaldos, M.; Máximo-Martín, M.F.; Montiel-Morte, M.C.; Ortega-Requena, S.; Gómez-Gómez, E.; Bastida-Rodríguez, J. Solvent-free enzymatic production of high quality cetyl esters. Bioprocess Biosyst. Eng. 2016, 39, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Brady, D. Green chemistry, biocatalysis, and the chemical industry of the future. ChemSusChem 2022, 15, e202102628. [Google Scholar] [CrossRef] [PubMed]
- Godoy, C.A.; Pardo-Tamayo, J.S.; Barbosa, O. Microbial lipases and their potential in the production of pharmaceutical building blocks. Int. J. Mol. Sci. 2022, 23, 9933. [Google Scholar] [CrossRef]
- Belafriekh, A.; Secundo, F.; Serra, S.; Djeghaba, Z. Enantioselective enzymatic resolution of racemic alcohols by lipases in green organic solvents. Tetrahedron Asymmetry 2017, 28, 473–478. [Google Scholar] [CrossRef]
- Farrán, A.; Cai, C.; Sandoval, M.; Xu, Y.; Liu, J.; Hernáiz, M.J.; Linhardt, R.J. Green solvents in carbohydrate chemistry: From raw materials to fine chemicals. Chem. Rev. 2015, 115, 6811–6853. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Nakashima, K.; Kamiya, N.; Goto, M. Recent advances of enzymatic reactions in ionic liquids. Biochem. Eng. J. 2010, 48, 295–314. [Google Scholar] [CrossRef]
- Hernáiz, M.J.; Alcántara, A.R.; García, J.I.; Sinisterra, J.V. Applied biotransformations in green solvents. Chem. Eur. J. 2010, 16, 9422–9437. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Pan, W. Ionic liquids: Green solvents for nonaqueous biocatalysis. Enzyme Microb. Technol. 2005, 37, 19–28. [Google Scholar] [CrossRef]
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and techniques for solvent selection: Green solvent selection guides. Tools and techniques for solvent selection: Green solvent selection guides. Sustain. Chem. Process. 2016, 4, 7. [Google Scholar] [CrossRef]
- De Santis, P.; Meyer, L.-E.; Kara, S. The rise of continuous flow biocatalysis—Fundamentals, very recent developments and future perspectives. React. Chem. Eng. 2020, 5, 2155–2184. [Google Scholar] [CrossRef]
- Thompson, M.P.; Peñafiel, I.; Cosgrove, S.C.; Turner, N.J. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org. Process Res. Dev. 2019, 23, 9–18. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, M.; Liu, Z.; Shi, J.; Huang, F.; Luo, X. Constructing a continuous flow bioreactor based on a hierarchically porous cellulose monolith for ultrafast and nonstop enzymatic esterification/transesterification. ACS Sustain. Chem. Eng. 2019, 7, 2056–2063. [Google Scholar] [CrossRef]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef]
- Gruber, P.; Carvalho, F.; Marques, M.P.C.; O’Sullivan, B.; Subrizi, F.; Dobrijevic, D.; Ward, J.; Hailes, H.C.; Fernandes, P.; Wohlgemuth, R.; et al. Enzymatic synthesis of chiral amino-alcohols by coupling transketolase and transaminase-catalyzed reactions in a cascading continuous-flow microreactor system. Biotechnol. Bioeng. 2018, 115, 586–596. [Google Scholar] [CrossRef]
- Woodcock, L.L.; Wiles, C.; Greenway, G.M.; Watts, P.; Wells, A.; Eyley, S. Enzymatic synthesis of a series of alkyl esters using Novozyme 435 in a packed-bed, miniaturized, continuous flow reactor. Biocatal. Biotransform. 2008, 26, 466–472. [Google Scholar] [CrossRef]
- Zdun, B.; Kopińska, I.; Dranka, M.; Reiter, T.; Kroutil, W.; Borowiecki, P. Chemoenzymatic synthesis of optically active alcohols possessing 1,2,3,4-tetrahydroquinoline moiety employing lipases or variants of the acyltransferase from Mycobacterium smegmatis. Catalysts 2022, 12, 1610. [Google Scholar] [CrossRef]
- Breen, G.F. Enzymatic resolution of a secondary amine using novel acylating reagents. Tetrahedron Asymmetry 2004, 15, 1427–1430. [Google Scholar] [CrossRef]
- Stirling, M.; Blacker, J.; Page, M.I. Chemoenzymatic dynamic kinetic resolution of secondary amines. Tetrahedron Lett. 2007, 48, 1247–1250. [Google Scholar] [CrossRef]
- Zhang, Q.; Tu, G.; Zhao, Y.; Cheng, T. Total synthesis of the antitumor active pyrrolo[2,1-a]isoquinoline alkaloid (±)-crispine A. Tetrahedron 2002, 58, 6795–6798. [Google Scholar] [CrossRef]
- Forró, E.; Schönstein, L.; Fülöp, F. Total synthesis of crispine A enantiomers through a Burkholderia cepacia lipase-catalysed kinetic resolution. Tetrahedron Asymmetry 2011, 22, 1255–1260. [Google Scholar] [CrossRef]
- Kanemitsu, T.; Yamashita, Y.; Nagata, K.; Itoh, T. Synthesis of (-)-Trolline,(-)-Crispin A and (-)-Crispine E. Heterocycles 2007, 74, 199–203. [Google Scholar] [CrossRef]
- Schönstein, L.; Forró, E.; Fülöp, F. Continuous-flow enzymatic resolution strategy for the acylation of amino alcohols with a remote stereogenic centre: Synthesis of calycotomine enantiomers. Tetrahedron Asymmetry 2013, 24, 202–206. [Google Scholar] [CrossRef]
- Schönstein, L.; Forró, E.; Fülöp, F. Enzymatic reaction for the preparation of homocalycotomine enantiomers. Tetrahedron Asymmetry 2013, 24, 1059–1062. [Google Scholar] [CrossRef]
- Yamada, M.; Azuma, K.; Takizawa, I.; Ejima, Y.; Yamano, M.; Satoh, K.; Doi, T.; Ueda, H.; Tokuyama, H. Efficient and scalable I asymmetric total synthesis of (−)-Emetine with parmaceutical grade quality; First multigram scale synthesis. Org. Process Res. Dev. 2023, 27, 343–357. [Google Scholar] [CrossRef]
- Megyesi, R.; Forró, E.; Fülöp, F. Enzymatic strategy for the resolution of new 1-hydroxymethyl tetrahydro-β-carboline derivatives in batch and continuous-flow systems. ChemistryOpen 2016, 5, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, R.; Forró, E.; Fülöp, F. Substrate engineering: Effects of different N-protecting groups in the CAL-B catalysed asymmetric O-acylation of 1-hydroxymethyl-tetrahydro-β-carbolines. Tetrahedron 2018, 74, 2634–2640. [Google Scholar] [CrossRef]
- Ding, W.; Li, M.; Dai, R.; Deng, Y. Lipase-catalyzed synthesis of the chiral tetrahydroisoquinoline (R)-salsolinol. Tetrahedron Asymmetry 2012, 23, 1376–1379. [Google Scholar] [CrossRef]
- Kovács, B.; Megyesi, R.; Forró, E.; Fülöp, F. Efficient lipase-catalyzed route for the kinetic resolution of salsolidine and its ß-carboline analogue. Tetrahedron Asymmetry 2017, 28, 1829–1833. [Google Scholar] [CrossRef]
- Kovács, B.; Forró, E.; Fülöp, F. Candida antarctica lipase B catalyzed kinetic resolution of 1,2,3,4-tetrahydro-ß-carbolines: Substrate specificity. Tetrahedron 2018, 74, 6873–6877. [Google Scholar] [CrossRef]
- Sakulsombat, M.; Vongvilai, P.; Ramström, O. Efficient asymmetric synthesis of 1-cyano-tetrahydroisoquinolines from lipase dual activity and opposite enantioselectivities in α-aminonitrile resolution. Chem. Eur. J. 2014, 20, 11322–11325. [Google Scholar] [CrossRef]
- Forró, E.; Megyesi, R.; Paál, T.A.; Fülöp, F. Efficient dynamic kinetic resolution method for the synthesis of enantiopure 6-hydroxy- and 6-methoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid. Tetrahedron Asymmetry 2016, 27, 1213–1216. [Google Scholar] [CrossRef]
- Anderson, W.K.; McPherson, H.L., Jr.; New, J.S.; Rick, A.C. Synthesis and murine antineoplastic activity of bis[carbamoyloxymethyl] derivatives of pyrrolo[2,1-a]isoquinoline. J. Med. Chem. 1984, 27, 1321–1325. [Google Scholar] [CrossRef]
- Grill, I.S.; Kick, E.; Richlin-Zack, K.; Yang, W.; Wang, Y.; Patel, R.N. Enzymatic resolution of methyl (1RS)-N-tBoc-6-hydroxy-3,4-dihydro-1H-isoquinoline-1-carboxylate by Seaprose S. Tetrahedron Asymmetry 2007, 18, 2147–2154. [Google Scholar] [CrossRef]
- Megyesi, R.; Mándi, A.; Kurtán, T.; Forró, E.; Fülöp, F. Dynamic kinetic resolution of ethyl 1,2,3,4-tetrahydro-β-carboline-1-carboxylate. Use of different hydrolases for stereocomplementary processes. Eur. J. Org. Chem. 2017, 32, 4713–4718. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Reaction Time (min) | eeamino alcohol (%) b | eeester (%) b | Conv. (%) | E |
|---|---|---|---|---|---|
| (±)-10 * | 15 | 86 | 99 | 47 | >200 |
| (±)-20 * | 60 | 69 | 99 | 41 | >200 |
| (±)-21 * | 60 | 79 | 98 | 45 | >200 |
| Substrate | Reaction Time (day) | eeamine (%) b | eecarbamide (%) b | Conv. (%) | E |
|---|---|---|---|---|---|
| (±)-28 * | 1 | 99 | 97 | 50 | >200 |
| (±)-31 * | 3 | 99 | 99 | 50 | >200 |
| (±)-33 * | 5 | 99 | 99 | 50 | >200 |
| (±)-35 * | 7 | 27 | 99 | 21 | >200 |
| Type of the Enzymatic Reaction | Enzymatic Strategies with Green Aspects * | Schemes | Refs |
|---|---|---|---|
| KR through O-acylation | Total synthesis of the antitumor-active alkaloid crispine A enantiomers, KRs in tBuOMe | Scheme 2 | [47] |
| Synthesis of calycotomine enantiomers, preliminary experiments of KR in a continuous-flow system | Scheme 3 | [49] | |
| Synthesis of homocalycotomine enantiomers. A systematic study on the O-acylation of THIQ amino alcohol homologues containing a remote stereogenic center, in a continuous-flow system | Scheme 3 | [50] | |
| KR of new 1-hydroxymethyl THβC derivatives in batch and continuous-flow systems | Scheme 4 | [52] | |
| KR through N-acylation | KR of salsolidine and its analogue 1-methyl-1,2,3,4-THβC in tBuOMe in a continuous-flow system | Scheme 6 | [55] |
| DKR through N-acylation | DKR of 1-cyano-1,2,3,4-THIQ derivatives in tBuOMe | Scheme 8 | [57] |
| DKR through COOEt hydrolysis | DKR of 6-hydroxy- and 6-methoxy-1,2,34-THIQ-1-CE hydrochlorides in aqueous NH4OAc buffer (pH 8.5) | Scheme 9 | [58] |
| Directed DKR of 1,2,3,4-THβC-1-CE hydrochloride (in aqueous NH4OAc buffer (pH 8.5), borate buffer (pH 8.0)) | Scheme 10 | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsy, G.; Forró, E. Lipase-Catalyzed Strategies for the Preparation of Enantiomeric THIQ and THβC Derivatives: Green Aspects. Molecules 2023, 28, 6362. https://doi.org/10.3390/molecules28176362
Orsy G, Forró E. Lipase-Catalyzed Strategies for the Preparation of Enantiomeric THIQ and THβC Derivatives: Green Aspects. Molecules. 2023; 28(17):6362. https://doi.org/10.3390/molecules28176362
Chicago/Turabian StyleOrsy, György, and Enikő Forró. 2023. "Lipase-Catalyzed Strategies for the Preparation of Enantiomeric THIQ and THβC Derivatives: Green Aspects" Molecules 28, no. 17: 6362. https://doi.org/10.3390/molecules28176362
APA StyleOrsy, G., & Forró, E. (2023). Lipase-Catalyzed Strategies for the Preparation of Enantiomeric THIQ and THβC Derivatives: Green Aspects. Molecules, 28(17), 6362. https://doi.org/10.3390/molecules28176362