
Sampling and Mapping Chemical Space with Extended Similarity Indices

 ,
,  and
and
Abstract
:1. Introduction
2. Theory
2.1. Extended Similarity
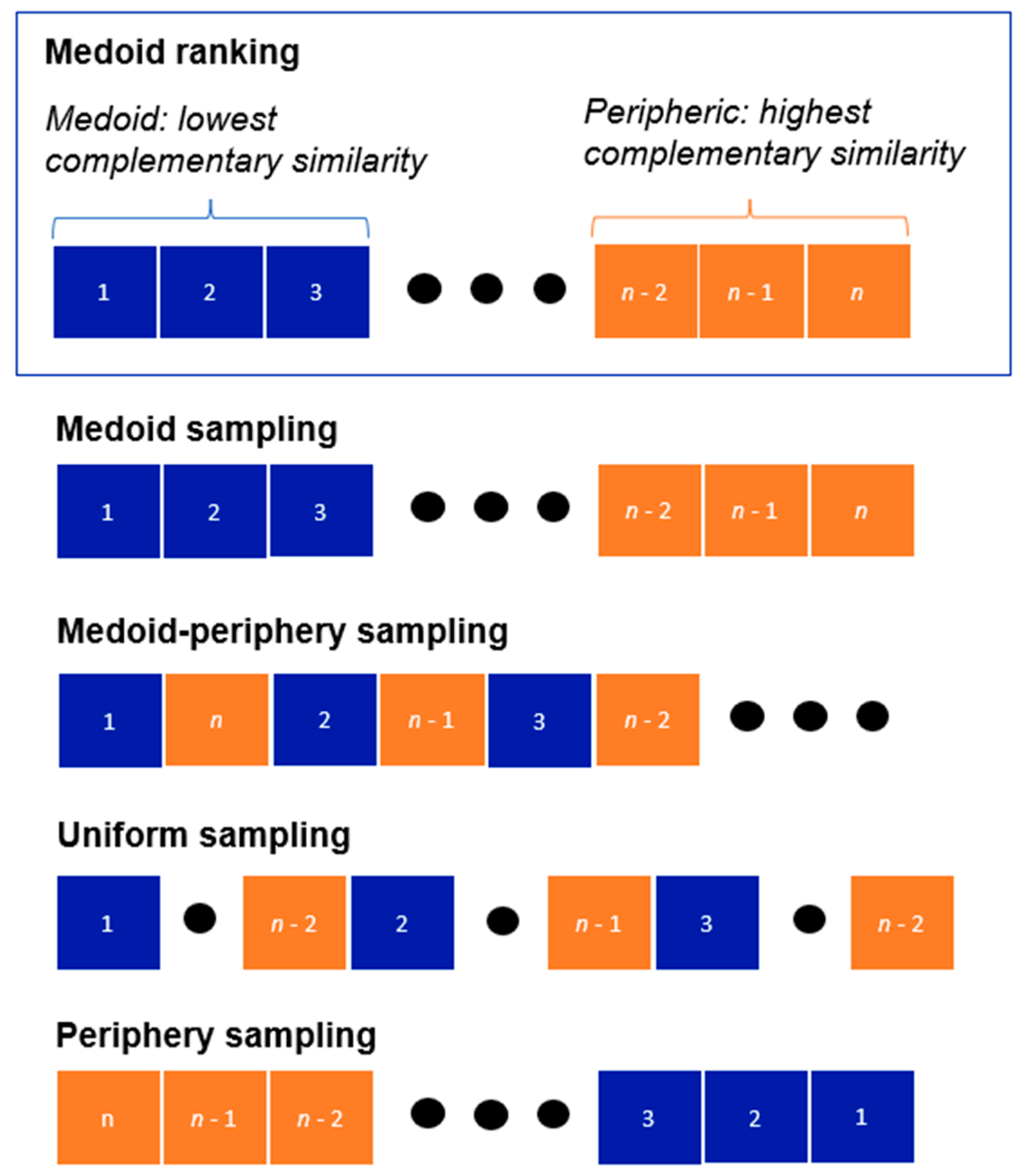
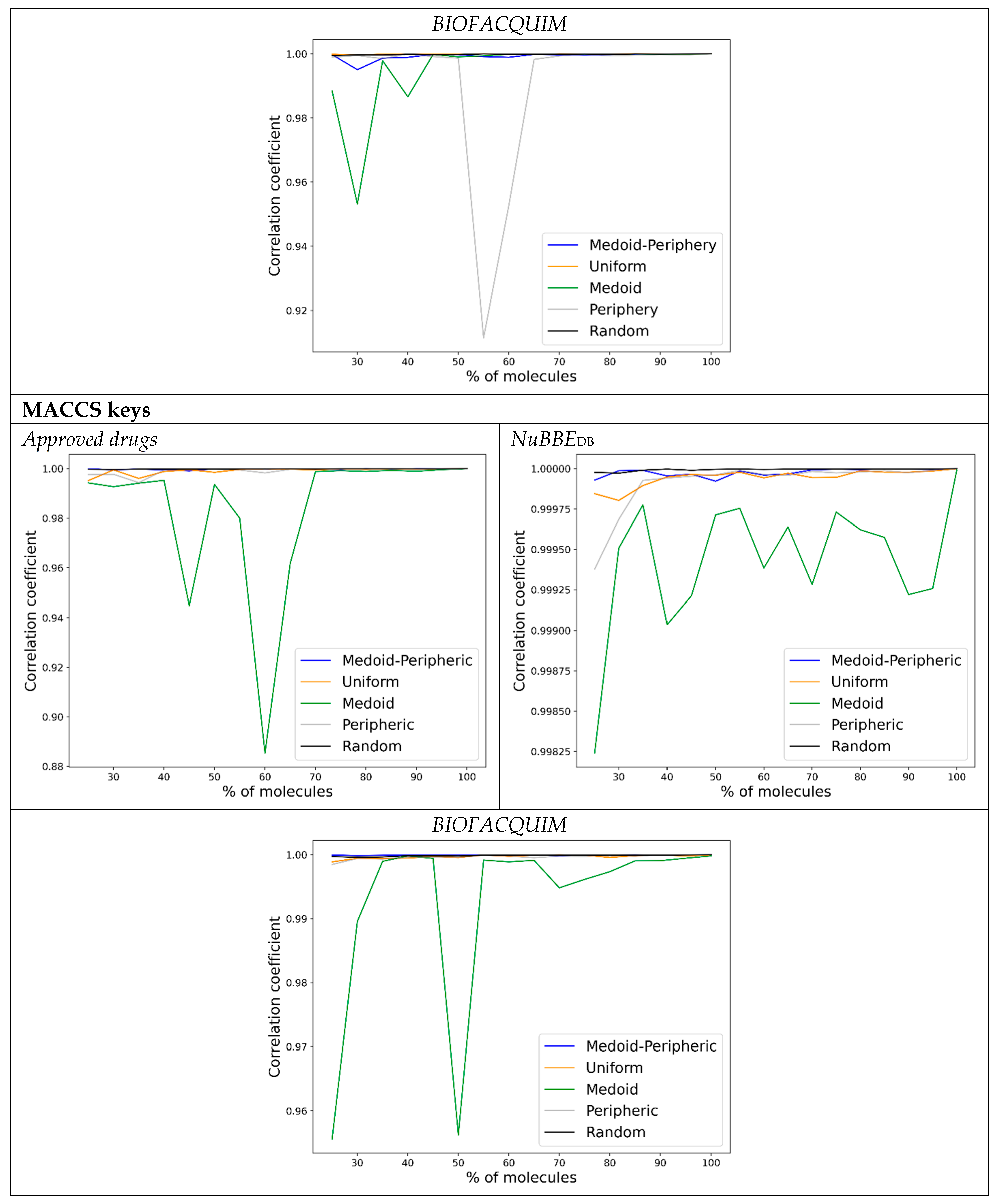
2.2. Sampling Techniques
- Medoid sampling: selecting molecules in increasing order of their complementary similarity values (sampling chemical space from the center-to-the-outside).
- Medoid–periphery sampling: selecting molecules in an alternating pattern, with odd selections (1, 3, 5, …) coming from the medoid region, and even selections (2, 4, 6, …) coming from the outlier region.
- Uniform sampling: the data are separated into five batches, and then we take one molecule from each of them in increasing order of complementary similarity within each batch.
- Periphery sampling: selecting molecules in decreasing order of their complementary similarity values (sampling chemical space from the outside-to-the-center).
3. Results
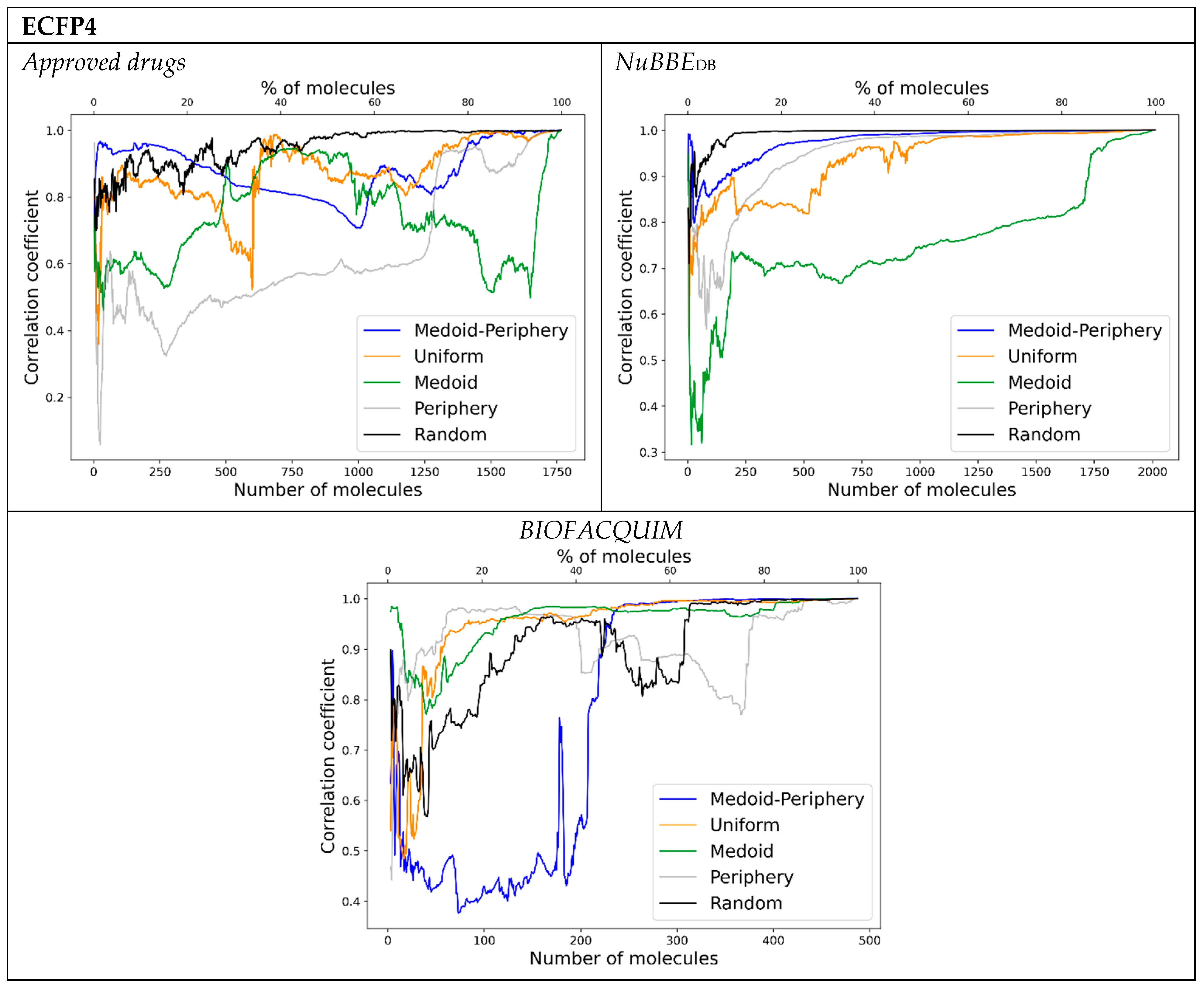
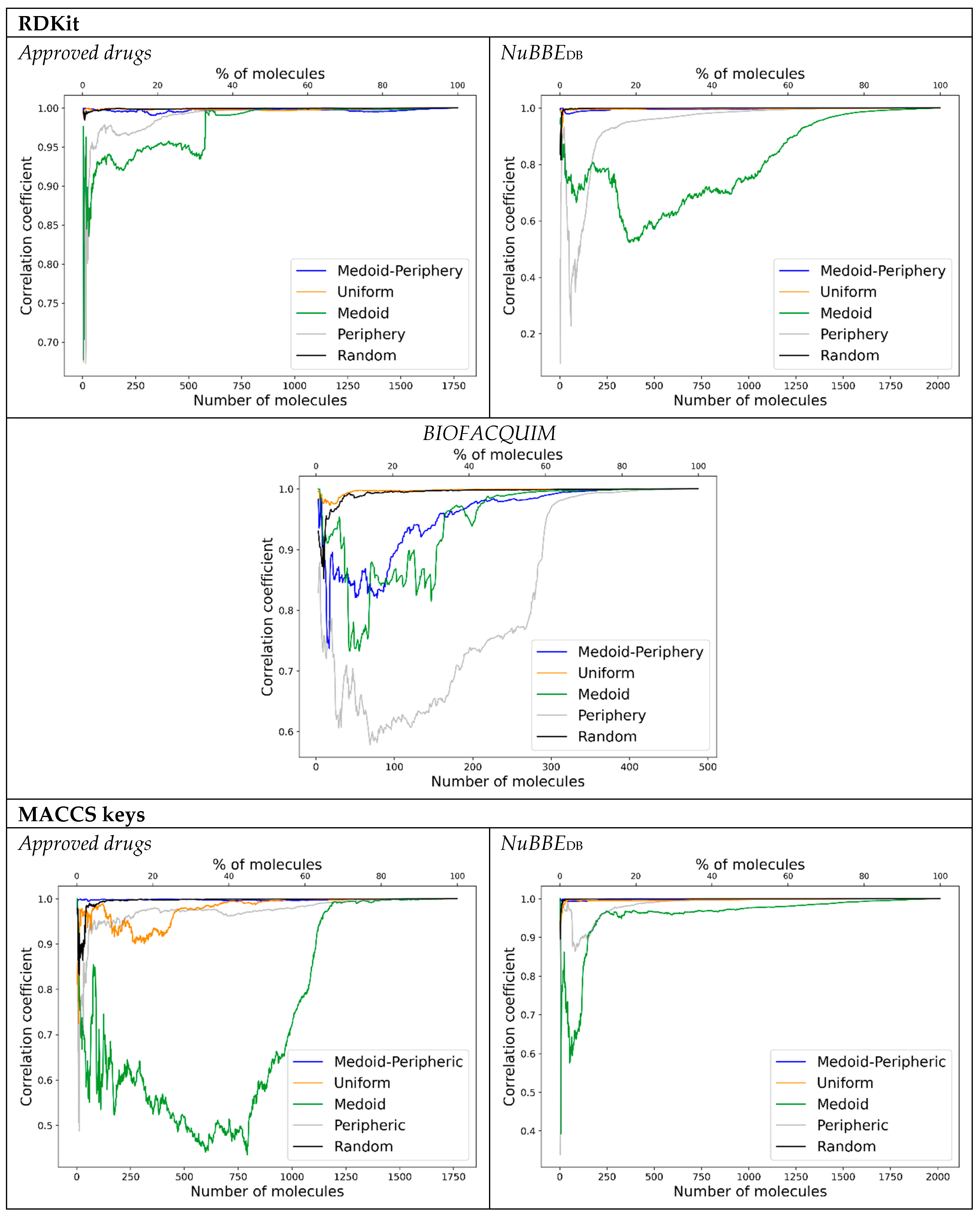
3.1. Overall Diversity
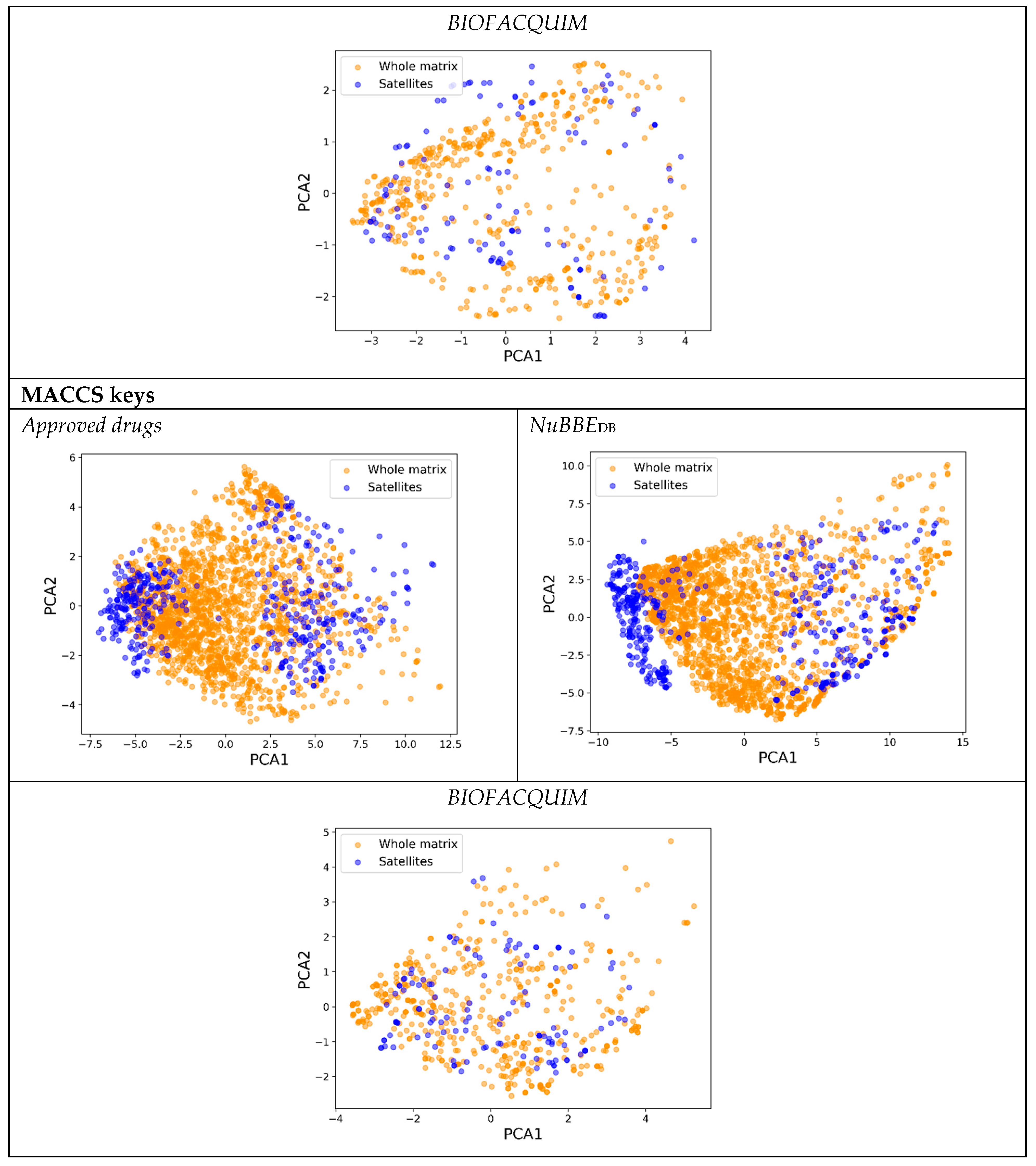
3.2. ChemMaps with Backward Approach
3.3. ChemMaps with Forward Approach
4. Methods
4.1. Molecular Libraries and Computational Conditions
4.2. ChemMaps
- Generate the similarity matrix using the Jaccard-Tanimoto index.
- Perform PCA on the given matrix with two PCs.
- Compute all pairwise Euclidean distances based on PC scorings. These distances will be used as reference values.
- Choose the first three satellites () according to the sampling method chosen. (i.e., for medoid sampling the three compounds with the lowest complementary similarity).
- Perform PCA with the similarity matrix and obtain the pairwise Euclidean distances based on those PC scores.
- Calculate the correlation between distances with the whole matrix (step 3) and the satellite’s matrix (step 5).
- Iterate over steps 4 to 6 adding one satellite at the time, based on the chosen sampling method.
- Establish the proportion of satellites required to preserve a high correlation (of at least 0.90).
- Start taking 25% of the database as satellites () by the sampling method of choice, having then a similarity matrix. This percentage is used as demonstrated in previous work to be the lowest percentage needed to render high correlation coefficients [24].
- Perform PCA with 2 PCs and use the scorings to calculate the Euclidean distances.
- Add the next 5% to the satellites according to the sampling method and perform step 2 with the updated satellite matrix.
- Calculate the correlation between the updated satellite Euclidean distances (of the elements in common) and the distances from the former satellite matrix (i.e., 30–25%).
- Repeat steps 3 and 4 until a high correlation (greater than 0.90) or to 100%.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rarey, M.; Nicklaus, M.C.; Warr, W. Special Issue on Reaction Informatics and Chemical Space. J. Chem. Inf. Model. 2022, 62, 2009–2010. [Google Scholar] [CrossRef]
- Warr, W.A.; Nicklaus, M.C.; Nicolaou, C.A.; Rarey, M. Exploration of Ultralarge Compound Collections for Drug Discovery. J. Chem. Inf. Model. 2022, 62, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Chávez-Hernández, A.L.; López-López, E.; Saldívar-González, F.I. Chemical Multiverse: An Expanded View of Chemical Space. Mol. Inform. 2022, 41, 2200116. [Google Scholar] [CrossRef]
- Lipinski, C.; Hopkins, A. Navigating Chemical Space for Biology and Medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef]
- Virshup, A.M.; Contreras-García, J.; Wipf, P.; Yang, W.; Beratan, D.N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. [Google Scholar] [CrossRef]
- Osolodkin, D.I.; Radchenko, E.V.; Orlov, A.A.; Voronkov, A.E.; Palyulin, V.A.; Zefirov, N.S. Progress in Visual Representations of Chemical Space. Expert. Opin. Drug Discov. 2015, 10, 959–973. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Naveja, J.J.; López-López, E. Reaching for the Bright StARs in Chemical Space. Drug Discov. Today 2019, 24, 2162–2169. [Google Scholar] [CrossRef] [PubMed]
- Bro, R.; Smilde, A.K. Principal Component Analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef]
- Gaytán-Hernández, D.; Chávez-Hernández, A.L.; López-López, E.; Miranda-Salas, J.; Saldívar-González, F.I.; Medina-Franco, J.L. Art Driven by Visual Representations of Chemical Space. ChemRxiv Chem. Educ. 2023. [Google Scholar] [CrossRef]
- Viarengo-Baker, L.A.; Brown, L.E.; Rzepiela, A.A.; Whitty, A. Defining and Navigating Macrocycle Chemical Space. Chem. Sci. 2021, 12, 4309–4328. [Google Scholar] [CrossRef]
- Cihan Sorkun, M.; Mullaj, D.; Koelman, J.M.V.A.; Er, S. ChemPlot, a Python Library for Chemical Space Visualization**. Chem. Methods 2022, 2, e202200005. [Google Scholar] [CrossRef]
- Bonachera, F.; Marcou, G.; Kireeva, N.; Varnek, A.; Horvath, D. Using Self-Organizing Maps to Accelerate Similarity Search. Bioorg. Med. Chem. 2012, 20, 5396–5409. [Google Scholar] [CrossRef] [PubMed]
- Takács, G.; Sándor, M.; Szalai, Z.; Kiss, R.; Balogh, G.T. Analysis of the Uncharted, Druglike Property Space by Self-Organizing Maps. Mol. Divers. 2022, 26, 2427–2441. [Google Scholar] [CrossRef]
- Achenbach, J.; Klingler, F.-M.; Blöcher, R.; Moser, D.; Häfner, A.-K.; Rödl, C.B.; Kretschmer, S.; Krüger, B.; Löhr, F.; Stark, H.; et al. Exploring the Chemical Space of Multitarget Ligands Using Aligned Self-Organizing Maps. ACS Med. Chem. Lett. 2013, 4, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Andronov, M.; Fedorov, M.V.; Sosnin, S. Exploring Chemical Reaction Space with Reaction Difference Fingerprints and Parametric T-SNE. ACS Omega 2021, 6, 30743–30751. [Google Scholar] [CrossRef]
- Gaspar, H.A.; Baskin, I.I.; Marcou, G.; Horvath, D.; Varnek, A. Chemical Data Visualization and Analysis with Incremental Generative Topographic Mapping: Big Data Challenge. J. Chem. Inf. Model. 2015, 55, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.; Marcou, G.; Varnek, A. Generative Topographic Mapping in Drug Design. Drug Discov. Today Technol. 2019, 32–33, 99–107. [Google Scholar] [CrossRef]
- Kireeva, N.; Baskin, I.I.; Gaspar, H.A.; Horvath, D.; Marcou, G.; Varnek, A. Generative Topographic Mapping (GTM): Universal Tool for Data Visualization, Structure-Activity Modeling and Dataset Comparison. Mol. Inform. 2012, 31, 301–312. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Sánchez-Cruz, N.; López-López, E.; Díaz-Eufracio, B.I. Progress on Open Chemoinformatic Tools for Expanding and Exploring the Chemical Space. J. Comput. Aided Mol. Des. 2022, 36, 341–354. [Google Scholar] [CrossRef]
- Dunn, T.B.; Seabra, G.M.; Kim, T.D.; Juárez-Mercado, K.E.; Li, C.; Medina-Franco, J.L.; Miranda-Quintana, R.A. Diversity and Chemical Library Networks of Large Data Sets. J. Chem. Inf. Model. 2022, 62, 2186–2201. [Google Scholar] [CrossRef]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. ChemGPS-NP: Tuned for Navigation in Biologically Relevant Chemical Space. J. Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Gottfries, J. Chemography: The Art of Navigating in Chemical Space. J. Comb. Chem. 2001, 3, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Naveja, J.J.; Medina-Franco, J.L. ChemMaps: Towards an Approach for Visualizing the Chemical Space Based on Adaptive Satellite Compounds. F1000Research 2017, 6, 1134. [Google Scholar] [CrossRef]
- Awale, M.; Reymond, J.-L. Similarity Mapplet: Interactive Visualization of the Directory of Useful Decoys and ChEMBL in High Dimensional Chemical Spaces. J. Chem. Inf. Model. 2015, 55, 1509–1516. [Google Scholar] [CrossRef]
- Pikalyova, R.; Zabolotna, Y.; Horvath, D.; Marcou, G.; Varnek, A. The Chemical Library Space and Its Application to DNA-Encoded Libraries. ChemRxiv Theor. Comput. Chem. 2023. [Google Scholar] [CrossRef]
- Pikalyova, R.; Zabolotna, Y.; Horvath, D.; Marcou, G.; Varnek, A. Chemical Library Space: Definition and DNA-Encoded Library Comparison Study Case. J. Chem. Inf. Model. 2023, 63, 4042–4055. [Google Scholar] [CrossRef]
- Borrel, A.; Conway, M.; Nolte, S.Z.; Unnikrishnan, A.; Schmitt, C.P.; Kleinstreuer, N.C. ChemMaps.Com v2.0: Exploring the Environmental Chemical Universe. Nucleic Acids Res. 2023, 51, W78–W82. [Google Scholar] [CrossRef]
- Borrel, A.; Kleinstreuer, N.C.; Fourches, D. Exploring Drug Space with ChemMaps.Com. Bioinformatics 2018, 34, 3773–3775. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Quintana, R.A.; Rácz, A.; Bajusz, D.; Héberger, K. Extended Similarity Indices: The Benefits of Comparing More than Two Objects Simultaneously. Part 2: Speed, Consistency, Diversity Selection. J. Cheminform. 2021, 13, 33. [Google Scholar] [CrossRef]
- Miranda-Quintana, R.A.; Bajusz, D.; Rácz, A.; Héberger, K. Extended Similarity Indices: The Benefits of Comparing More than Two Objects Simultaneously. Part 1: Theory and Characteristics†. J. Cheminform. 2021, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Flores-Padilla, E.A.; Juárez-Mercado, K.E.; Naveja, J.J.; Kim, T.D.; Alain Miranda-Quintana, R.; Medina-Franco, J.L. Chemoinformatic Characterization of Synthetic Screening Libraries Focused on Epigenetic Targets. Mol. Inform. 2022, 41, 2100285. [Google Scholar] [CrossRef]
- Bajusz, D.; Miranda-Quintana, R.A.; Rácz, A.; Héberger, K. Extended Many-Item Similarity Indices for Sets of Nucleotide and Protein Sequences. Comput. Struct. Biotechnol. J. 2021, 19, 3628–3639. [Google Scholar] [CrossRef]
- Rácz, A.; Mihalovits, L.M.; Bajusz, D.; Héberger, K.; Miranda-Quintana, R.A. Molecular Dynamics Simulations and Diversity Selection by Extended Continuous Similarity Indices. J. Chem. Inf. Model. 2022, 62, 3415–3425. [Google Scholar] [CrossRef]
- Chang, L.; Perez, A.; Miranda-Quintana, R.A. Improving the Analysis of Biological Ensembles through Extended Similarity Measures. Phys. Chem. Chem. Phys. 2022, 24, 444–451. [Google Scholar] [CrossRef]
- Rácz, A.; Dunn, T.B.; Bajusz, D.; Kim, T.D.; Miranda-Quintana, R.A.; Héberger, K. Extended Continuous Similarity Indices: Theory and Application for QSAR Descriptor Selection. J. Comput. Aided Mol. Des. 2022, 36, 157–173. [Google Scholar] [CrossRef]
- Pilón-Jiménez, B.; Saldívar-González, F.; Díaz-Eufracio, B.; Medina-Franco, J. BIOFACQUIM: A Mexican Compound Database of Natural Products. Biomolecules 2019, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Valli, M.; dos Santos, R.N.; Figueira, L.D.; Nakajima, C.H.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. Development of a Natural Products Database from the Biodiversity of Brazil. J. Nat. Prod. 2013, 76, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Pilon, A.C.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An Updated Database to Uncover Chemical and Biological Information from Brazilian Biodiversity. Sci. Rep. 2017, 7, 7215. [Google Scholar] [CrossRef]
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES. 2. Algorithm for Generation of Unique SMILES Notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Landrum, G.; Penzotti, J. RDKit. 2018. Available online: http://www.rdkit.org/ (accessed on 17 January 2022).
- Dunn, T.B.; López-López, E.; Kim, T.D.; Medina-Franco, J.L.; Miranda-Quintana, R.A. Exploring Activity Landscapes with Extended Similarity: Is Tanimoto Enough? Mol. Inform. 2023, 42, 2300056. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Pairwise Similarity | Average PCA Distance | ||||||
|---|---|---|---|---|---|---|---|
| Dataset | N | ECFP4 | RDKit | MACCS Keys | ECFP4 | RDKit | MACCS Keys |
| Approved drugs | 1768 | 0.09 | 0.21 | 0.32 | 1.39 | 4.48 | 4.67 |
| NuBBEDB | 2013 | 0.12 | 0.24 | 0.42 | 2.34 | 5.93 | 7.42 |
| BIOFACQUIM | 488 | 0.12 | 0.25 | 0.46 | 1.21 | 2.88 | 3.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Pérez, K.; López-López, E.; Medina-Franco, J.L.; Miranda-Quintana, R.A. Sampling and Mapping Chemical Space with Extended Similarity Indices. Molecules 2023, 28, 6333. https://doi.org/10.3390/molecules28176333
López-Pérez K, López-López E, Medina-Franco JL, Miranda-Quintana RA. Sampling and Mapping Chemical Space with Extended Similarity Indices. Molecules. 2023; 28(17):6333. https://doi.org/10.3390/molecules28176333
Chicago/Turabian StyleLópez-Pérez, Kenneth, Edgar López-López, José L. Medina-Franco, and Ramón Alain Miranda-Quintana. 2023. "Sampling and Mapping Chemical Space with Extended Similarity Indices" Molecules 28, no. 17: 6333. https://doi.org/10.3390/molecules28176333
APA StyleLópez-Pérez, K., López-López, E., Medina-Franco, J. L., & Miranda-Quintana, R. A. (2023). Sampling and Mapping Chemical Space with Extended Similarity Indices. Molecules, 28(17), 6333. https://doi.org/10.3390/molecules28176333