

Drug Reprofiling to Identify Potential HIV-1 Protease Inhibitors

 ,
, 
Abstract
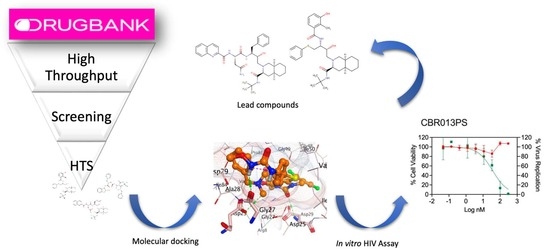
1. Introduction
2. Results
2.1. Molecular Docking Study
2.2. In Vitro Anti-HIV-1 Assay
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. In Silico Anti-HIV Activity Prediction
4.3. In Vitro Anti-HIV Assays to Measure Selective Activity
4.3.1. Cells and Virus
4.3.2. Cytotoxicity
4.3.3. Anti-HIV-1 Activity
- CEM.SS cells were seeded in clear 96-well U-bottom plates (Thermofisher Scientific) following the procedure described for the cytotoxicity assay. Triplicate wells received 50 μL of each compound dilution at 4X. Virus and cell control triplicates received 50 μL and 100 μL of complete medium (RPMI 1640 medium supplemented with 10% FBS and with 50 U/mL of penicillin and 50 μg/mL of streptomycin), respectively. The same nine concentrations of each compound tested in the cytotoxicity assay were applied in the antiviral assay, with all dilutions performed in complete medium. Briefly 50 μL of HIV-1MN was added to all the wells except for cell controls at a concentration of 200 infectious particles per well. The plates were incubated at 37 °C, 5% CO2, and 98% humidity for 72 h.
- Briefly, 24 h prior to completing the incubation of step 1, white opaque 96-well flat-bottom plates were seeded with TZM-bl cells at a concentration of 1 × 105 cells/mL (100 μL per well) in complete medium. The plates were incubated at 37 °C, 5% CO2, and 98% humidity overnight. On the following day, 100 μL of the supernatant from each well was carefully transferred to the TZM-bl plates. The cells were spinoculated by spinning down the plates at 1740 g, for 1 h and 40 min, at 23 °C in a Sigma 4-16K centrifuge using a plate rotor at 2 × 96 (Qiagen, Hilden, Germany). After spinoculation, 100 μL of complete fresh medium was added to all wells. The plates were incubated at 37 °C, 5% CO2, and 98% humidity for 72 h and then stained by performing the multinuclear-activated galactosidase indicator (MAGI) assay as previously described [34]. The number of infected cells per well was estimated using C.T.L. ImmunoSpot (Cellular Technology Ltd., Shaker Heights, OH, USA).
4.4. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. The Top 10 Causes of Death [Fact Sheet]. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 17 March 2023).
- World Health Organization. HIV/AIDS. Available online: https://www.who.int/health-topics/hiv-aids/ (accessed on 16 December 2022).
- Bartlett, J.A.; DeMasi, R.; Quinn, J.; Moxham, C.; Rousseau, F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS 2001, 15, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Meibohm, A.; Condra, J.H.; Valentine, F.T.; McMahon, D.; Gonzalez, C.; Jonas, L.; et al. 3-year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann. Intern. Med. 2000, 133, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to imProve R&D Productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar]
- Reddy, G.S.; Ali, A.; NAlam, M.N.; Anjum, S.G.; Cao, H.; Nathans, R.S.; Schiffer, C.A.; Rana, T.M. Design and synthesis of HIV-1 protease inhibitors incorporating oxazolidinones as P2/P2′ ligands in pseudosymmetric dipeptide isosteres. J. Med. Chem. 2007, 50, 4316–4328. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Duarte, R.R.R.; Copertino, D.C., Jr.; Iñiguez, L.P.; Marston, J.L.; Nixon, D.F.; Powell, T.R. Repurposing FDA-ApProved Drugs for COVID-19 Using a Data-Driven ApProach. Mol. Med. 2021, 27, 105. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges, and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Koch, D.C.; Bisson, W.H.; Jang, H.S.; Kolluri, S.K. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. 2014, 5, e1038. [Google Scholar] [CrossRef]
- Cihlar, T.; Ray, A.S. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antivir. Res. 2010, 85, 39–58. [Google Scholar] [CrossRef]
- Kalra, S.; Kalra, B.; Agrawal, N.; Unnikrishnan, A. Understanding diabetes in patients with HIV/AIDS. Diabetol. Metab. Syndr. 2011, 3, 2. [Google Scholar] [CrossRef]
- Richmond, S.R.; Carper, M.J.; Lei, X.; Zhang, S.; Yarasheski, K.E.; Ramanadham, S. HIV-protease inhibitors suppress skeletal muscle fatty acid oxidation by reducing CD36 and CPT1 fatty acid transporters. Biochim. Biophys. Acta 2010, 1801, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Hruz, P.W. HIV protease inhibitors and insulin resistance: Lessons from in-vitro, rodent and healthy human volunteer models. Curr. Opin. HIV AIDS 2008, 3, 660–665. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, S.; Schow, S.R.; Manchem, V.P.; Spevak, W.R.; Cristobal, C.P.; Shi, S.; Macsata, R.W.; Lum, R.T.; Goldfine, I.D.; et al. In vitro and in vivo prevention of HIV protease inhibitor-induced insulin resistance by a novel small molecule insulin receptor activator. J. Cell Biochem. 2004, 92, 1234–1245. [Google Scholar] [CrossRef]
- Nolte, L.A.; Yarasheski, K.E.; Kawanaka, K.; Fisher, J.; Le, N.; Holloszy, J.O. The HIV protease inhibitor indinavir decreases insulin- and contraction-stimulated glucose transport in skeletal muscle. Diabetes 2001, 50, 1397–1401. [Google Scholar] [CrossRef]
- Noor, M.A.; Seneviratne, T.; Aweeka, F.T.; Lo, J.C.; Schwarz, J.M.; Mulligan, K.; Schambelan, M.; Grunfeld, C. Indinavir acutely inhibits insulin-stimulated glucose disposal in humans: A randomized, placebo-controlled study. AIDS 2002, 16, F1–F8. [Google Scholar] [CrossRef][Green Version]
- Cade, W.T.; Reeds, D.N.; Lassa-Claxton, S.; Davila-Roman, V.G.; Waggoner, A.D.; Powderly, W.G.; Yarasheski, K.E. Post-exercise heart rate recovery in HIV-positive individuals on highly active antiretroviral therapy. Early indicator of cardiovascular disease? HIV Med. 2008, 9, 96–100. [Google Scholar] [CrossRef]
- Luzi, L.; Perseghin, G.; Tambussi, G.; Meneghini, E.; Scifo, P.; Pagliato, E.; Del Maschio, A.; Testolin, G.; Lazzarin, A. Intramyocellular lipid accumulation and reduced whole body lipid oxidation in HIV lipodystrophy. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E274–E280. [Google Scholar] [CrossRef]
- Hruz, P.W. Molecular Mechanisms for Altered Glucose Homeostasis in HIV Infection. Am. J. Infect. Dis. 2006, 2, 187–192. [Google Scholar] [CrossRef]
- Kim, R.J.; Wilson, C.G.; Wabitsch, M.; Lazar, M.A.; Steppan, C.M. HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism. Obesity 2006, 14, 994–1002. [Google Scholar] [CrossRef]
- Condra, J.H.; Schleif, W.A.; Blahy, O.M.; Gabryelski, L.J.; Graham, D.J.; Quintero, J.C.; Rhodes, A.; Robbins, H.L.; Roth, E.; Shivaprakash, M.; et al. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 1995, 374, 569–571. [Google Scholar] [CrossRef]
- Clavel, F.; Hance, A.J. HIV drug resistance. N. Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Muzammil, S.; Ohtaka, H.; Schon, A.; Vega, S.; Freire, E. Structural and thermodynamic basis of resistance to HIV-1 protease inhibition: Implications for inhibitor design. Curr. Drug Targets Infect. Disord. 2003, 3, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Okafor, S.N.; Angsantikul, P.; Ahmed, H. Discovery of Novel HIV protease Inhibitors Using Modern Computational Techniques. Int. J. Mol. Sci. 2022, 23, 12149. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Ceftaroline Fosamil: A Review of its Use in the Treatment of Complicated Skin and Soft Tissue Infections and Community-Acquired Pneumonia. Drugs 2013, 73, 1067–1094. [Google Scholar] [CrossRef] [PubMed]
- Szkudlinski, M.W. Challenges and opportunities of trapping ligands. Mol. Pharmacol. 2007, 72, 231–234. [Google Scholar] [CrossRef]
- Nolan, D. Metabolic complications associated with HIV protease inhibitor therapy. Drugs 2003, 63, 2555–2574. [Google Scholar] [CrossRef]
- Song, M.; Wu, H.; Wu, S.; Ge, T.; Wang, G.; Zhou, Y.; Sheng, S.; Jiang, J. Antibiotic drug levofloxacin inhibits Proliferation and induces apoptosis of lung cancer cells Through inducing mitochondrial dysfunction and oxidative damage. Biomed. Pharmacother. 2016, 84, 1137–1143. [Google Scholar] [CrossRef]
- AlsAlahat, I.; Al-Majdoub, Z.M.; Taha, M.O.; Barber, J.; Aojula, H.; Hodson, N.; Freeman, S. Inhibition of aggregation of amyloid-beta through coValent modification with benzylpenicillin; potential relevance to Alzheimer’s disease. Biochem. Biophys. Rep. 2021, 26, 100943. [Google Scholar]
- Khan, A.N.; Qureshi, I.A.; Khan, U.K.; Uversky, V.N.; Khan, R.H. Inhibition and disruption of amyloid formation by the antibiotic levofloxacin: A new direction for antibiotics in an era of multi-drug resistance. Arch. Biochem. Biophys. 2021, 714, 109077. [Google Scholar] [CrossRef]
- Qureshi, A.; Rajput, A.; Kaur, G.; Kumar, M. HIVProtI: An integrated web-based platform for prediction and design of HIV Proteins inhibitors. J. Cheminform. 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Begay, O.; Jean-Pierre, N.; Abraham, C.J.; Chudolij, A.; Seidor, S.; Rodriguez, A.; Ford, B.E.; Henderson, M.; Katz, D.; Zydowsky, T.; et al. Identification of personal lubricants that can cause rectal epithelial cell damage and enhance HIV type 1 replication in vitro. AIDS Res. Hum. Retroviruses 2011, 27, 1019–1024. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Drug | Current Indication | Docking Scores (kcal/mol) | Anti-HIV Protease Activity c | ||
|---|---|---|---|---|---|---|
| GBVI/WSA dG b | London dG | IC50 d (µM) | % Inhibition | |||
| 1 | Saquinavir | HIV-1 protease inhibitor | −10.15 | −7.81 | 4.58 | 58.4 |
| 2 | CBR001PS | Thrombin inhibitor | −16.75 | −7.70 | 20.68 | 62.43 |
| 3 | CBR002PS a | Antiviral—Hepatitis C Virus | −17.35 | −6.31 | −4.19 e | 52.52 |
| 4 | CBR003PS | Antibiotic | −19.04 | −8.73 | 0.42 | 58.74 |
| 5 | CBR004PS | Antibiotic | −21.57 | −9.20 | −9.05 e | 59.05 |
| 6 | CBR005PS | Vitamin | −17.61 | −9.17 | 23.61 | 53.96 |
| 7 | CBR006PS a | Treatment of atherosclerosis | −15.05 | −8.96 | 0.03 | 52.04 |
| 8 | CBR007PS | Anti-hypertension | −16.12 | −8.37 | 0.04 | 51.89 |
| 9 | CBR008PS | Treatment of neurogenic bladder dysfunction or myasthenia gravis | −14.47 | −8.72 | 0.01 | 61.89 |
| 10 | CBR009PS a | Anti-cancer | −19.32 | −8.84 | 23.38 | 60.97 |
| 11 | CBR010PS | Diabetes mellitus, Type 2 | −16.89 | −9.88 | −81.75 e | 54.08 |
| 12 | CBR011PS | Antihypertensive drug (ACEI) | −15.22 | −8.78 | 5.43 | 56.75 |
| 13 | CBR012PS | ACEI | −17.28 | −8.08 | 0.04 | 52.48 |
| 14 | CBR013PS a | Treatment in endometriosis and uterine fibroids | −16.61 | −10.51 | −10.75 e | 60.55 |
| Drug | Ligand | Receptor a | Interaction | Distance (Å) | Energy (kcal/mol) |
|---|---|---|---|---|---|
| CBR001PS | N 8 | O Gly48 (B) | H-donor | 3.04 | −1.5 |
| O 3 | O HOH 226 (A) | H-acceptor | 2.75 | −3.6 | |
| O 4 | CB Pro81 (A) | H-acceptor | 3.39 | −0.3 | |
| N 12 | OD2 Asp25 (A) | Ionic | 3.38 | −0.8 | |
| N 14 | OD1 Asp25 (A) | Ionic | 3.93 | −0.6 | |
| N 14 | OD1 Asp25 (A) | Ionic | 3.07 | −4.0 | |
| 6-ring | CA Ala28 (B) | pi-H | 4.25 | −0.2 | |
| 6-ring | N Asp29 (B) | pi-H | 3.65 | −0.9 | |
| 6-ring | N Asp30 (B) | pi-H | 4.39 | −0.6 | |
| CBR003PS | C 53 | OD2 Asp30 (A) | H-donor | 3.12 | −0.8 |
| O 8 | CA Ala28 (A) | H-acceptor | 3.11 | −0.5 | |
| O 13 | N Asp29 (B) | H-acceptor | 3.06 | −1.6 | |
| O 13 | N Asp30 (B) | H-acceptor | 3.51 | −2.0 | |
| CBR009PS | N 9 | OD2 Asp29 (A) | H-donor | 3.01 | −0.7 |
| N 14 | OD2 Asp29 (B) | H-donor | 3.18 | −1.4 | |
| N 16 | OD2 Asp30 (B) | H-donor | 3.17 | −5.0 | |
| S 2 | N Asp30 (B) | H-acceptor | 3.71 | −0.8 | |
| S 2 | CB Asp30 (B) | H-acceptor | 3.52 | −0.5 | |
| N 9 | OD2 Asp29 (A) | Ionic | 3.01 | −4.4 | |
| 6-ring | CA Ala28 (B) | pi-H | 4.15 | −0.9 | |
| 6-ring | N Asp29 (B) | pi-H | 4.43 | −0.5 | |
| CBR013PS | C 19 | OD1 Asp25 (B) | H-donor | 3.30 | −1.3 |
| O 6 | N Asp30 (A) | H-acceptor | 3.32 | −0.8 | |
| 6-ring | N Asp29 (B) | pi-H | 3.75 | −0.6 | |
| 6-ring | N Asp30 (B) | pi-H | 4.12 | −0.5 | |
| Saquinavir | N 11 | O GLY27 (A) | H-donor | 3.02 | −2.3 |
| C 16 | OD2 ASP29 (A) | H-donor | 3.27 | −1.1 | |
| C 33 | O GLY27 (A) | H-donor | 3.41 | −0.7 | |
| O 1 | N GLY48 (A) | H-acceptor | 3.01 | −2.5 | |
| 6-ring | CA ALA28 (B) | pi-H | 3.97 | −0.9 | |
| Co-crystallized ligand | N 2 | OD2 ASP29 (A) | H-donor | 2.87 | −3.7 |
| N 3 | O GLY27 (A) | H-donor | 3.16 | −1.4 | |
| O 4 | OD1 ASP25 (B) | H-donor | 2.70 | −3.6 | |
| O 1 | N ASP29 (A) | H-acceptor | 3.02 | −3.0 | |
| 6-ring | N ASP29 (B) | pi-H | 3.93 | −1.1 | |
| 6-ring | CB ASP29 (B) | pi-H | 4.06 | −0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okafor, S.N.; Meyer, A.; Gadsden, J.; Ahmed, F.; Guzmán, L.; Ahmed, H.; Romero, J.A.F.; Angsantikul, P. Drug Reprofiling to Identify Potential HIV-1 Protease Inhibitors. Molecules 2023, 28, 6330. https://doi.org/10.3390/molecules28176330
Okafor SN, Meyer A, Gadsden J, Ahmed F, Guzmán L, Ahmed H, Romero JAF, Angsantikul P. Drug Reprofiling to Identify Potential HIV-1 Protease Inhibitors. Molecules. 2023; 28(17):6330. https://doi.org/10.3390/molecules28176330
Chicago/Turabian StyleOkafor, Sunday N., Abigail Meyer, Jay Gadsden, Fadi Ahmed, Lilian Guzmán, Hashim Ahmed, José A. Fernández Romero, and Pavimol Angsantikul. 2023. "Drug Reprofiling to Identify Potential HIV-1 Protease Inhibitors" Molecules 28, no. 17: 6330. https://doi.org/10.3390/molecules28176330
APA StyleOkafor, S. N., Meyer, A., Gadsden, J., Ahmed, F., Guzmán, L., Ahmed, H., Romero, J. A. F., & Angsantikul, P. (2023). Drug Reprofiling to Identify Potential HIV-1 Protease Inhibitors. Molecules, 28(17), 6330. https://doi.org/10.3390/molecules28176330