

Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2

 , , , , , , and
, , , , , , and 
Abstract
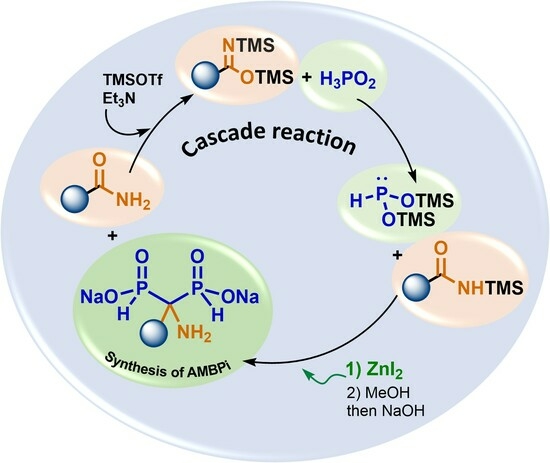
:
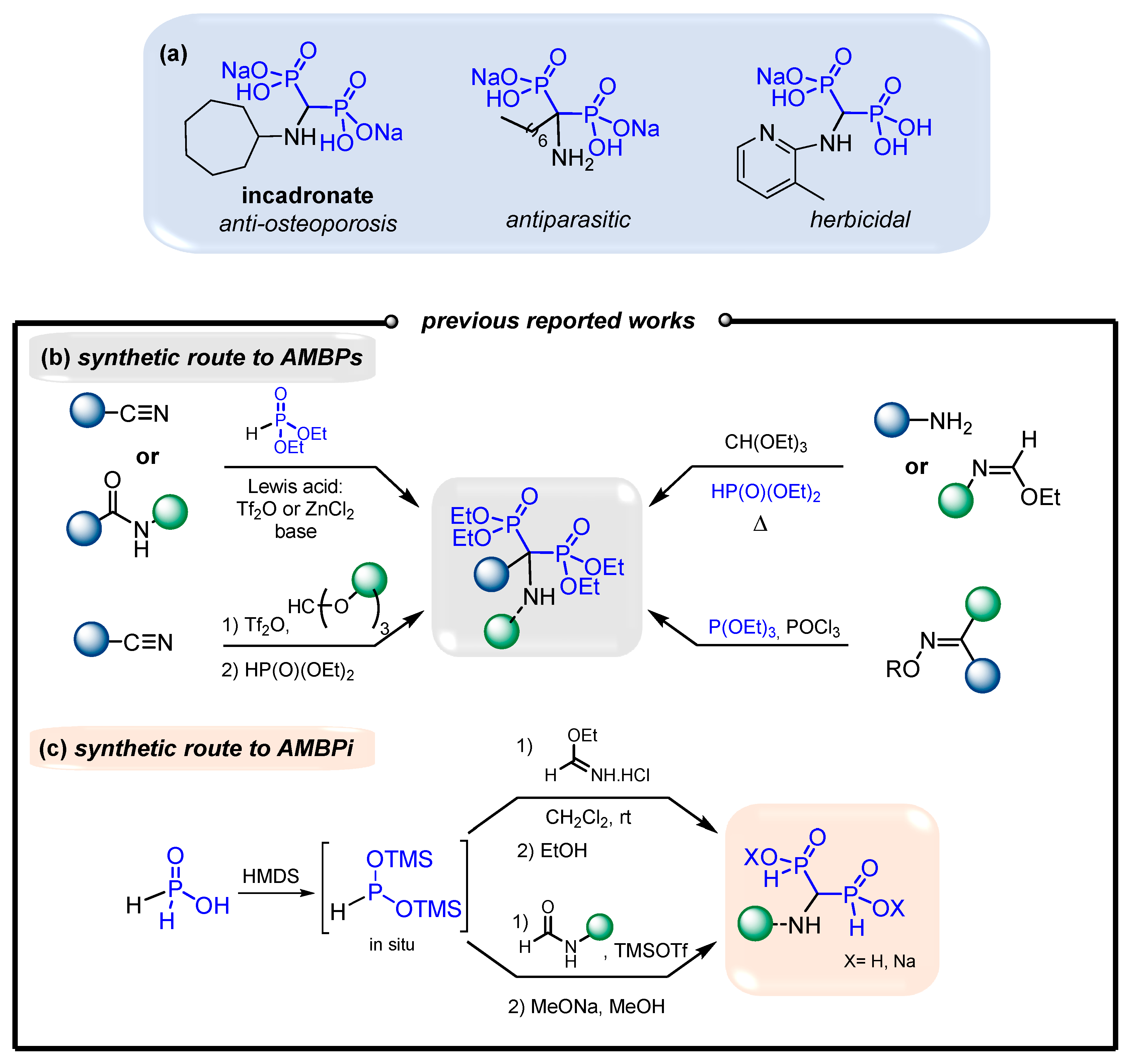
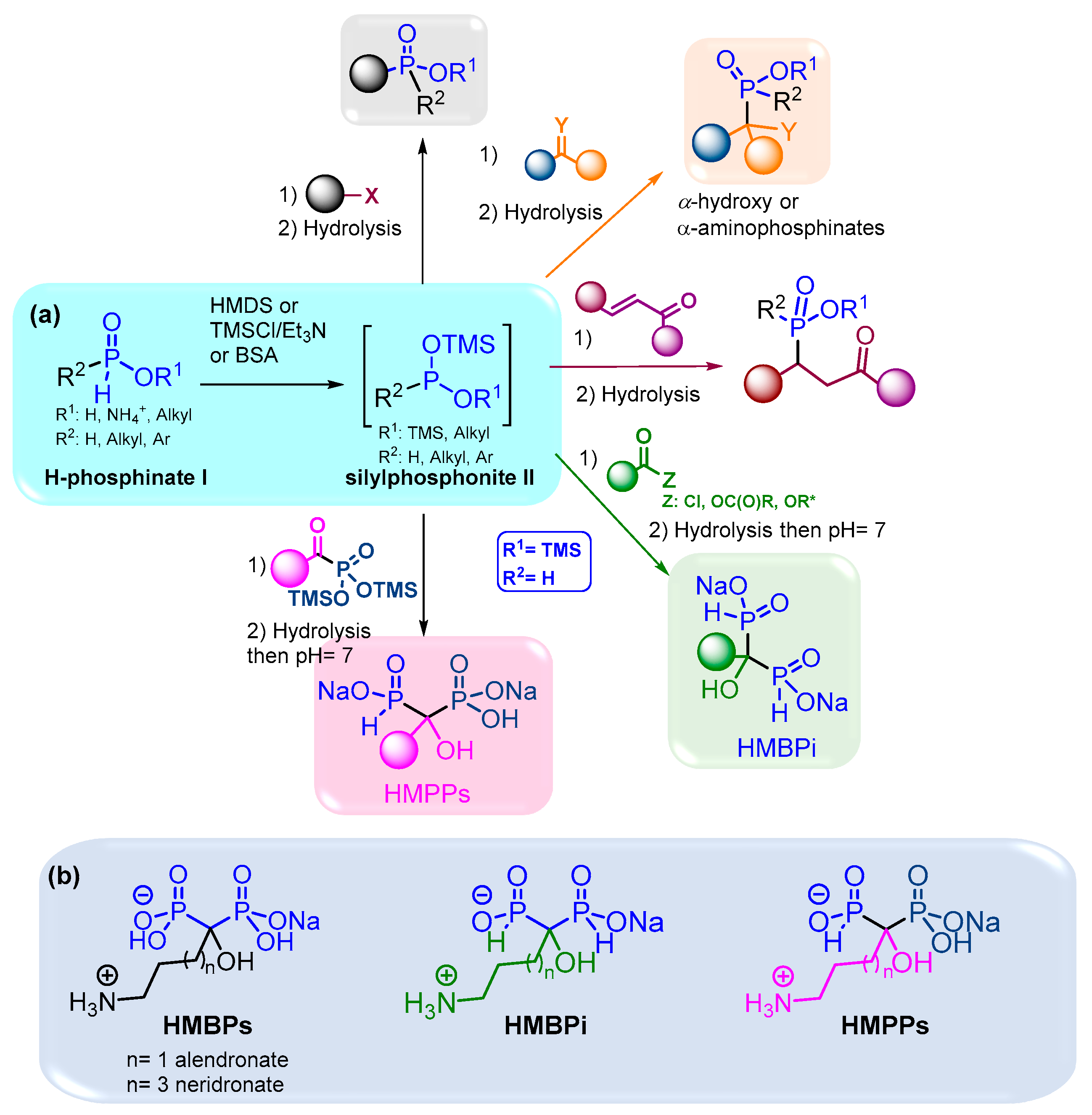
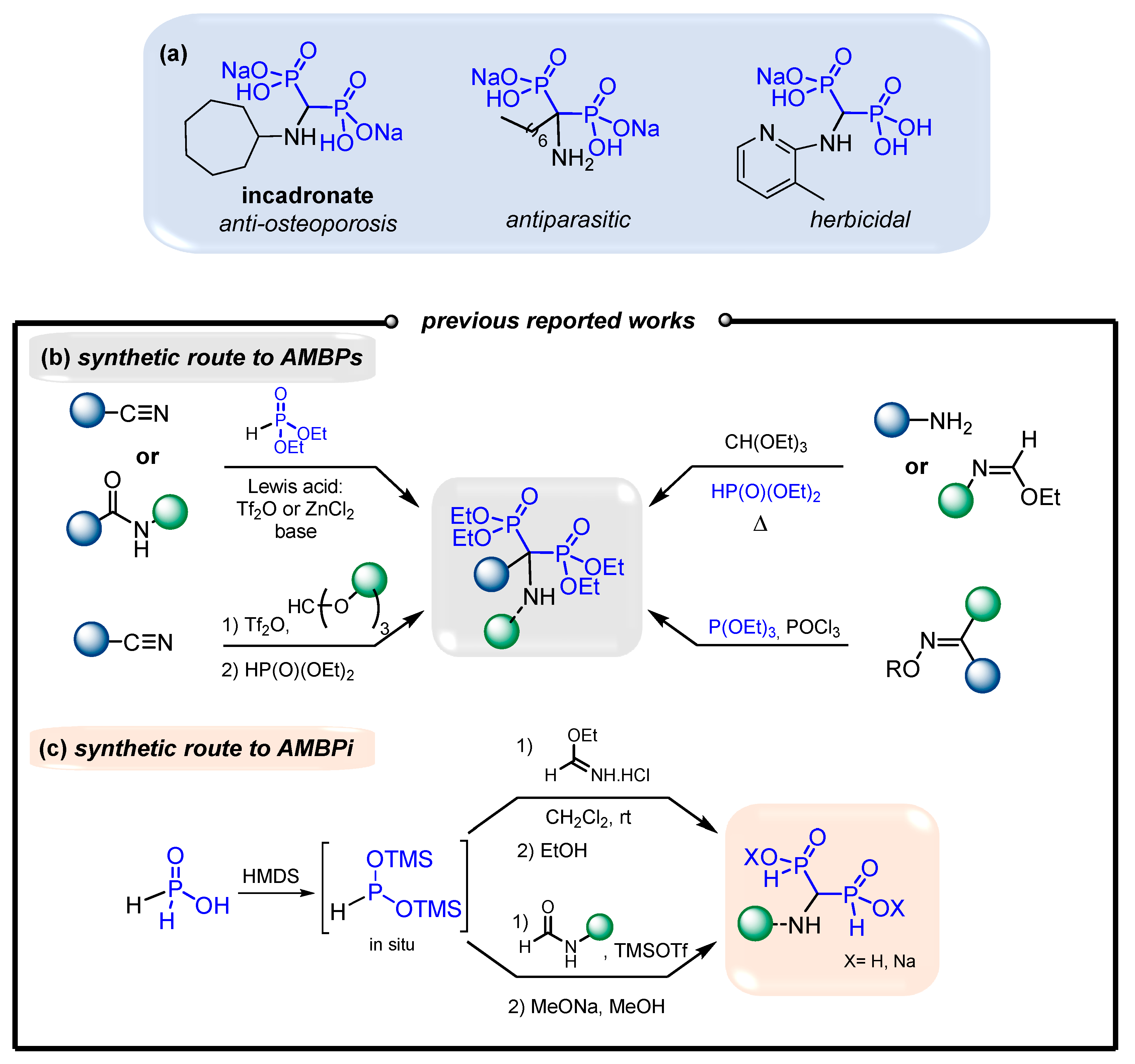
1. Introduction
2. Results and Discussion
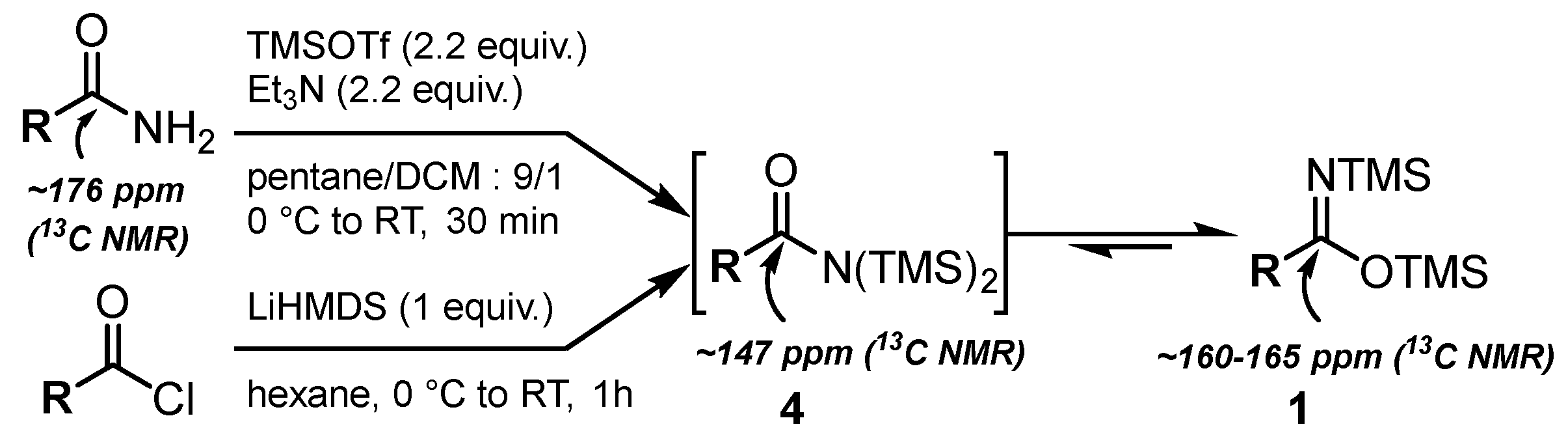
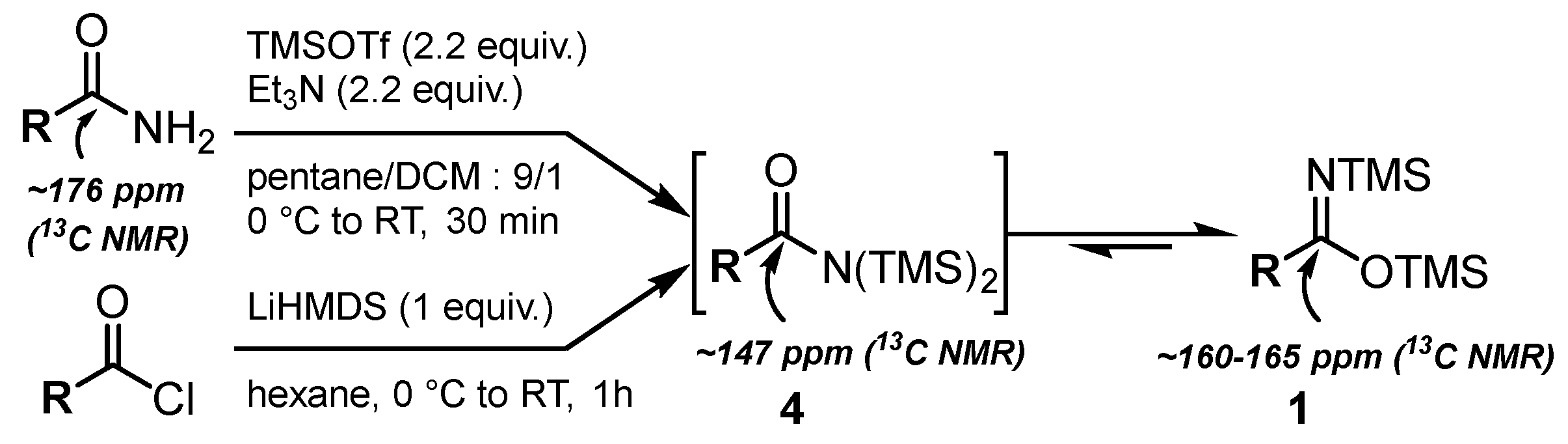
2.1. Synthesis of Bis(silyl)imidates 1
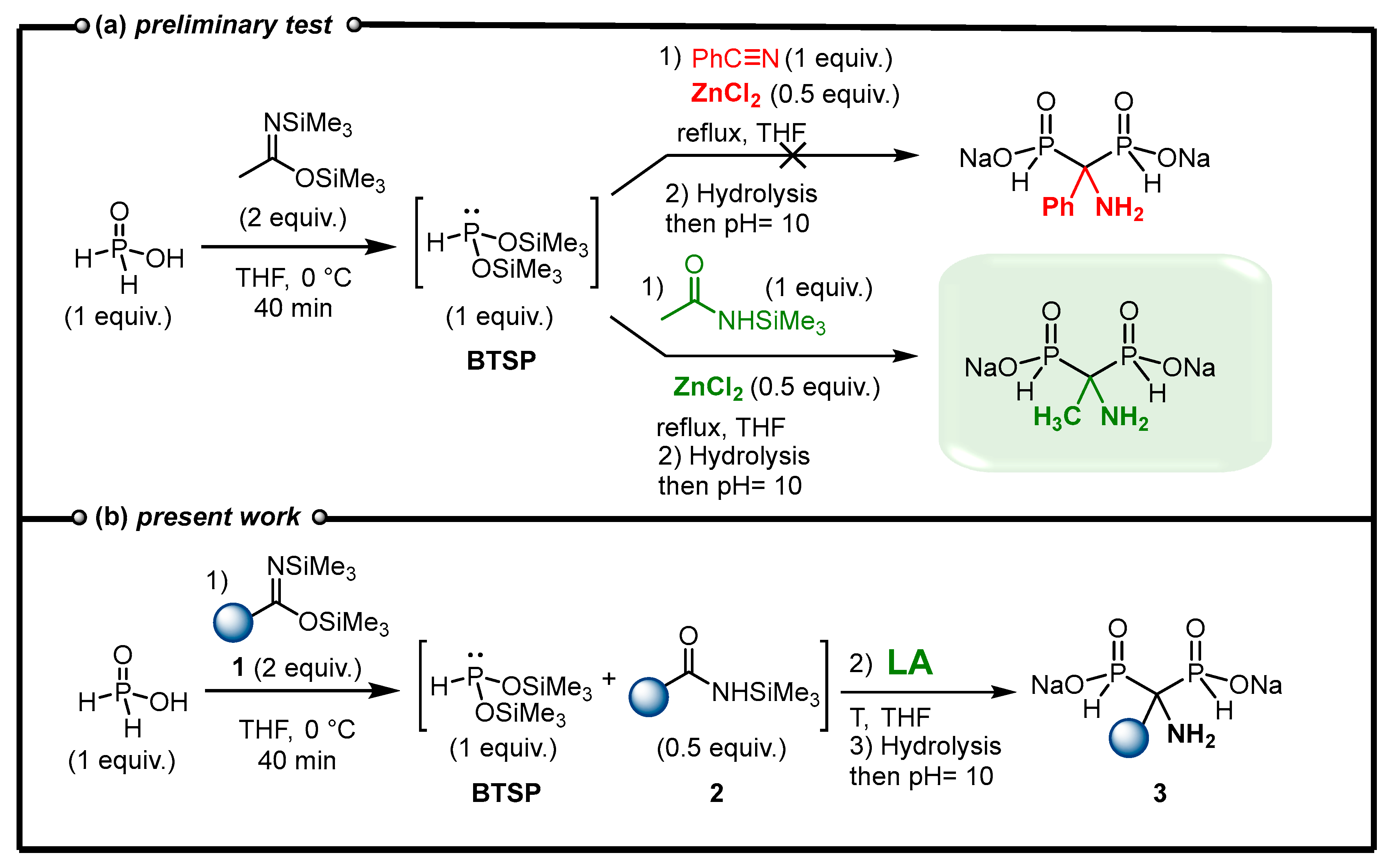
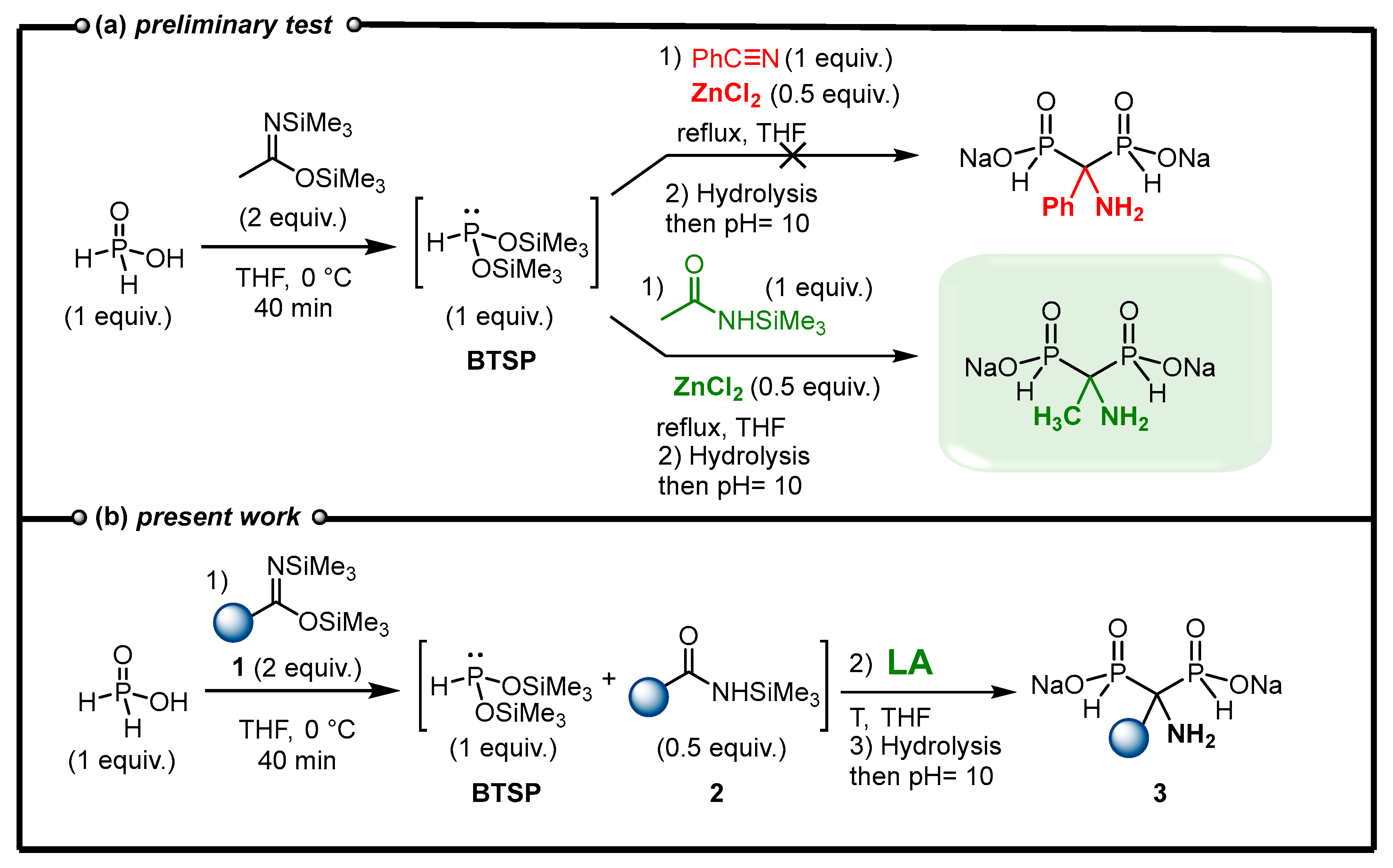
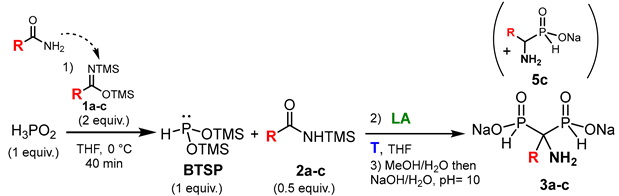
2.2. Optimization of the Reaction between N,O-Bis(trimethylsilyl)imidates and Phosphorous Acid Mediated by Lewis Acid
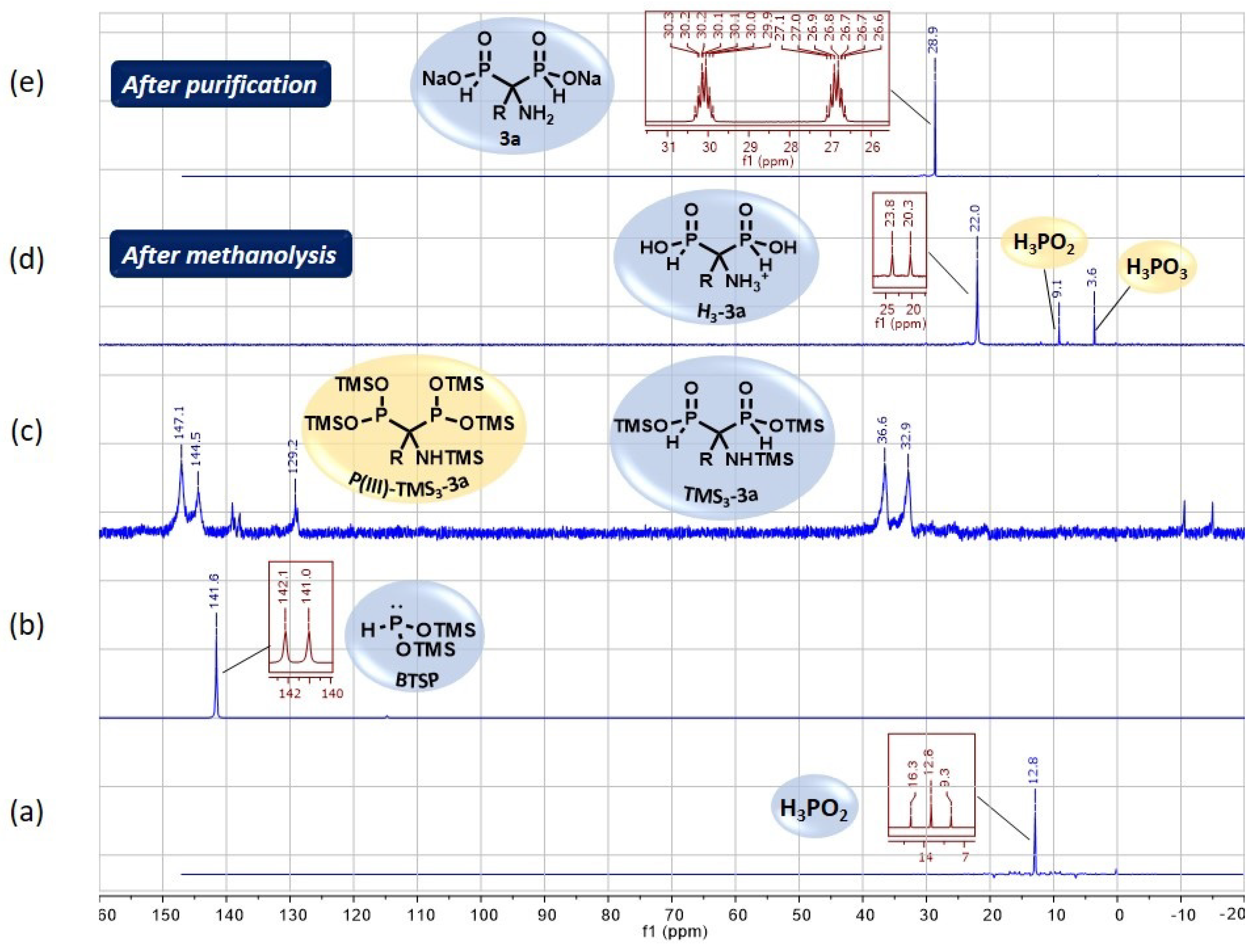
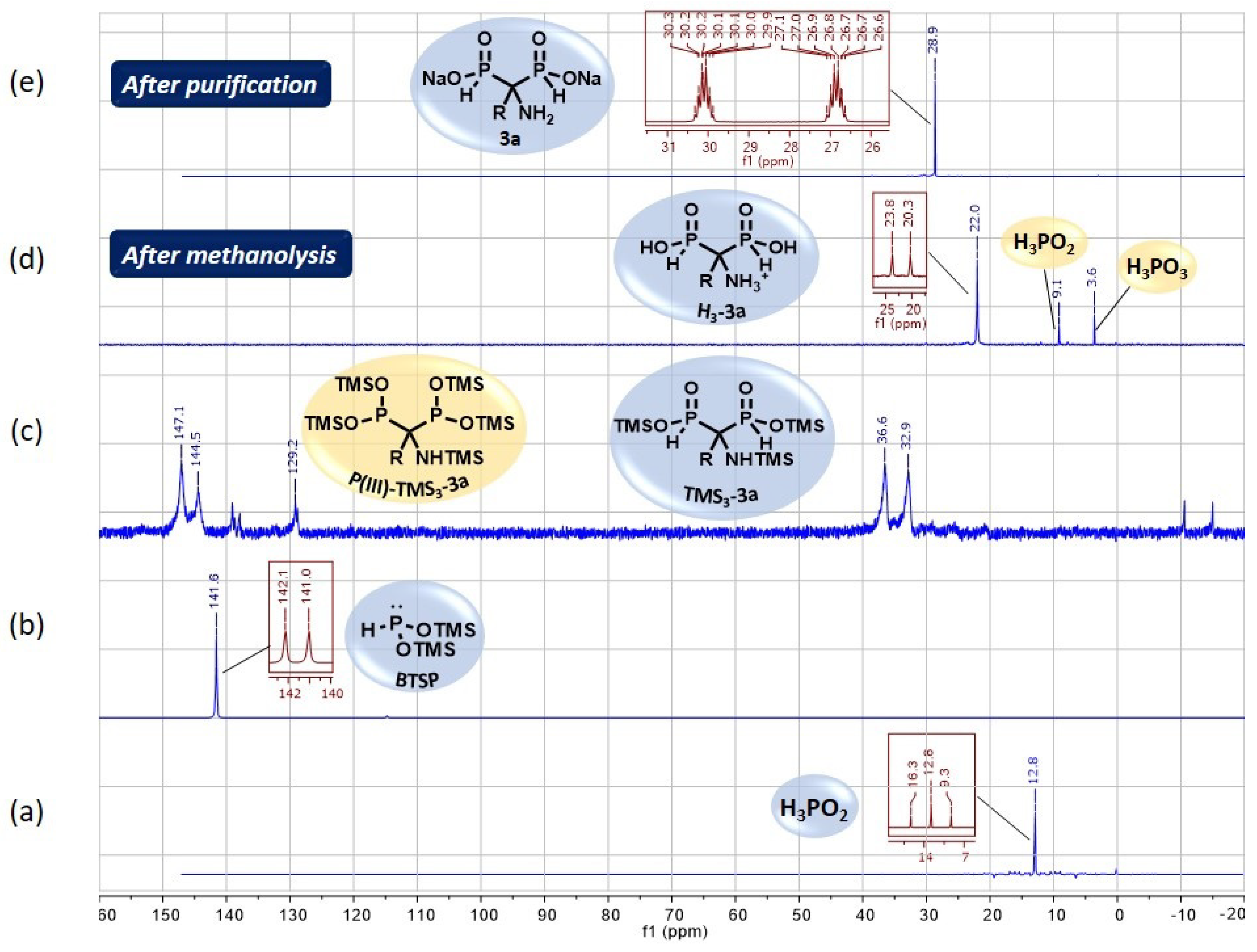
2.3. NMR Monitoring and Purification Details
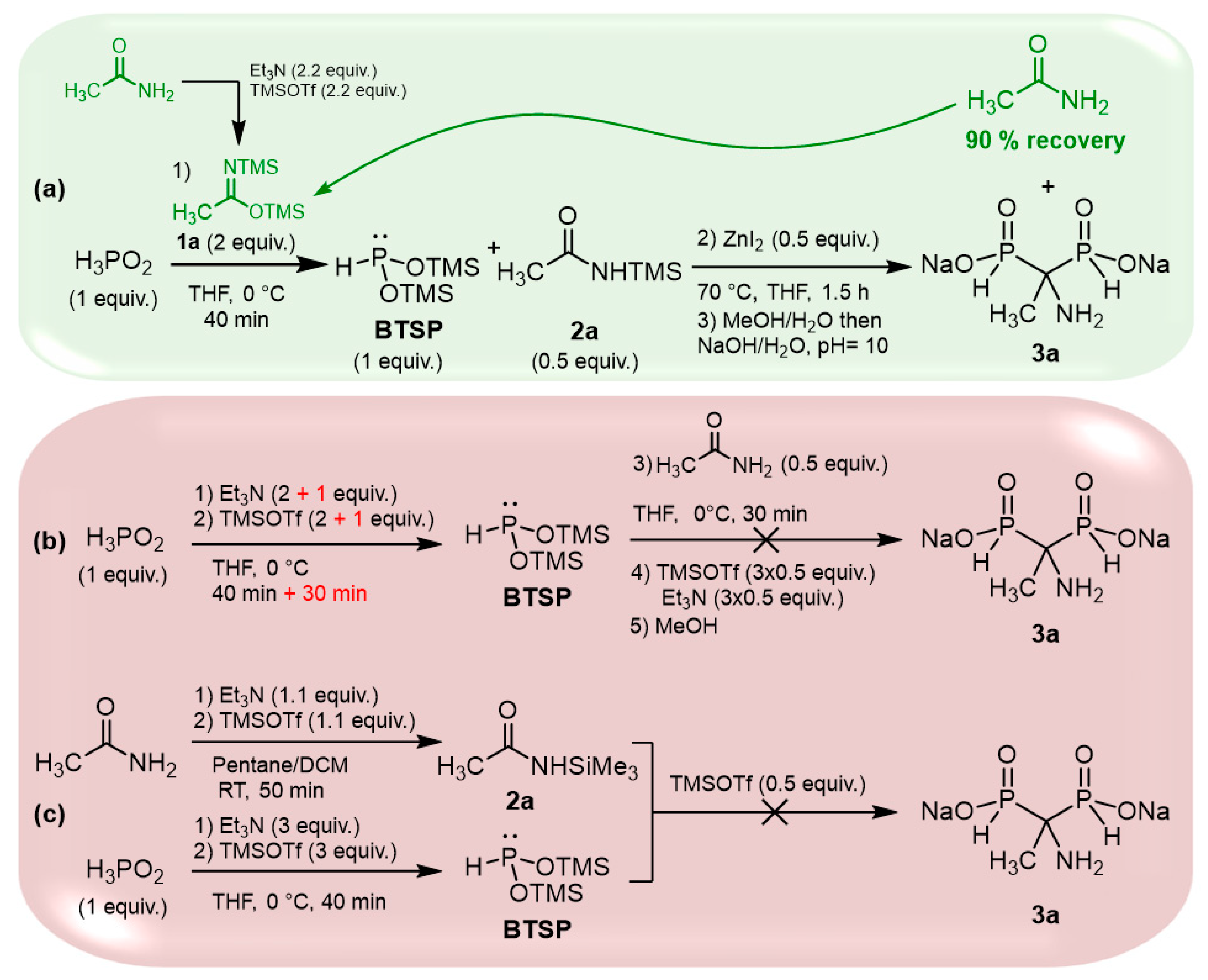
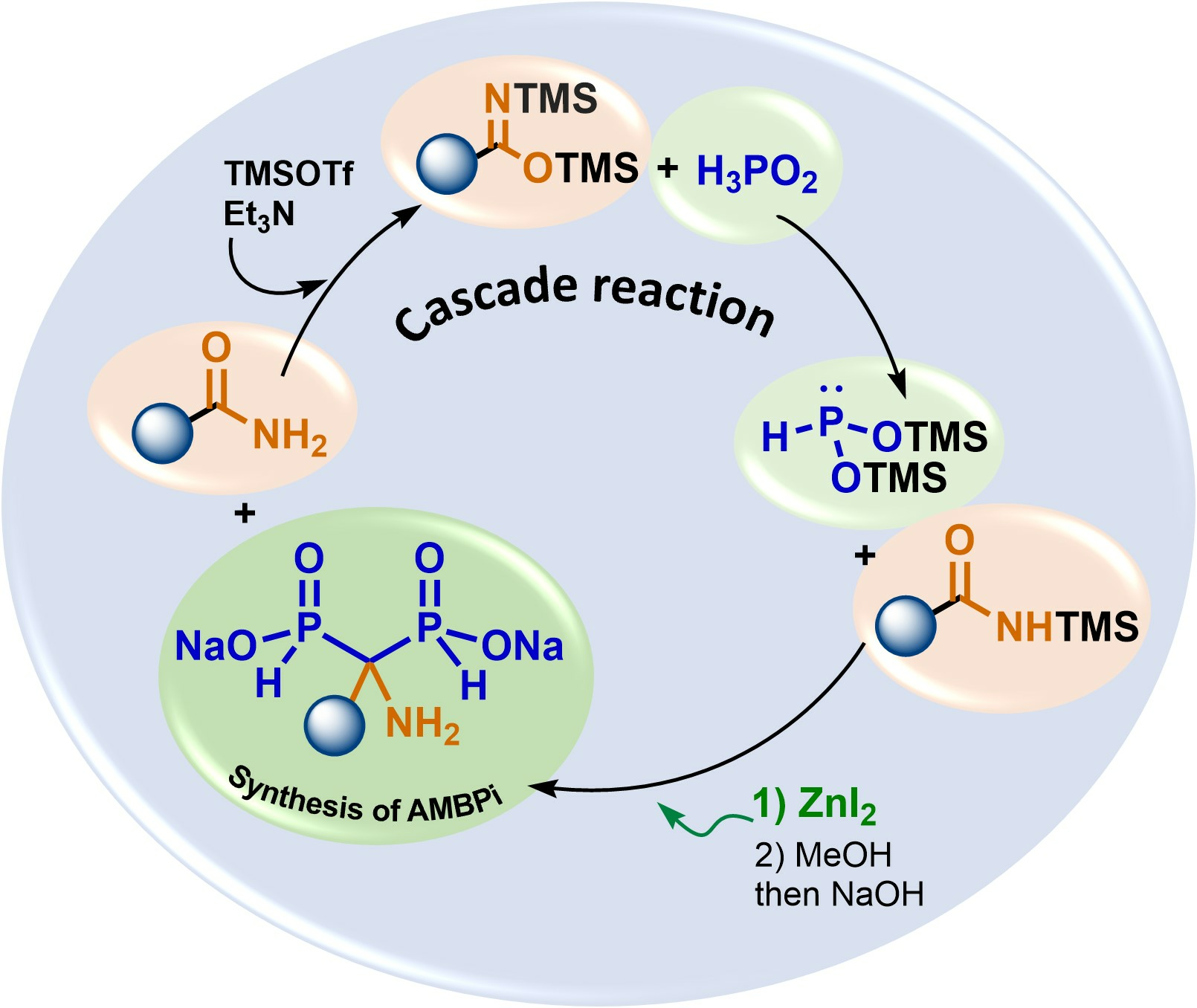
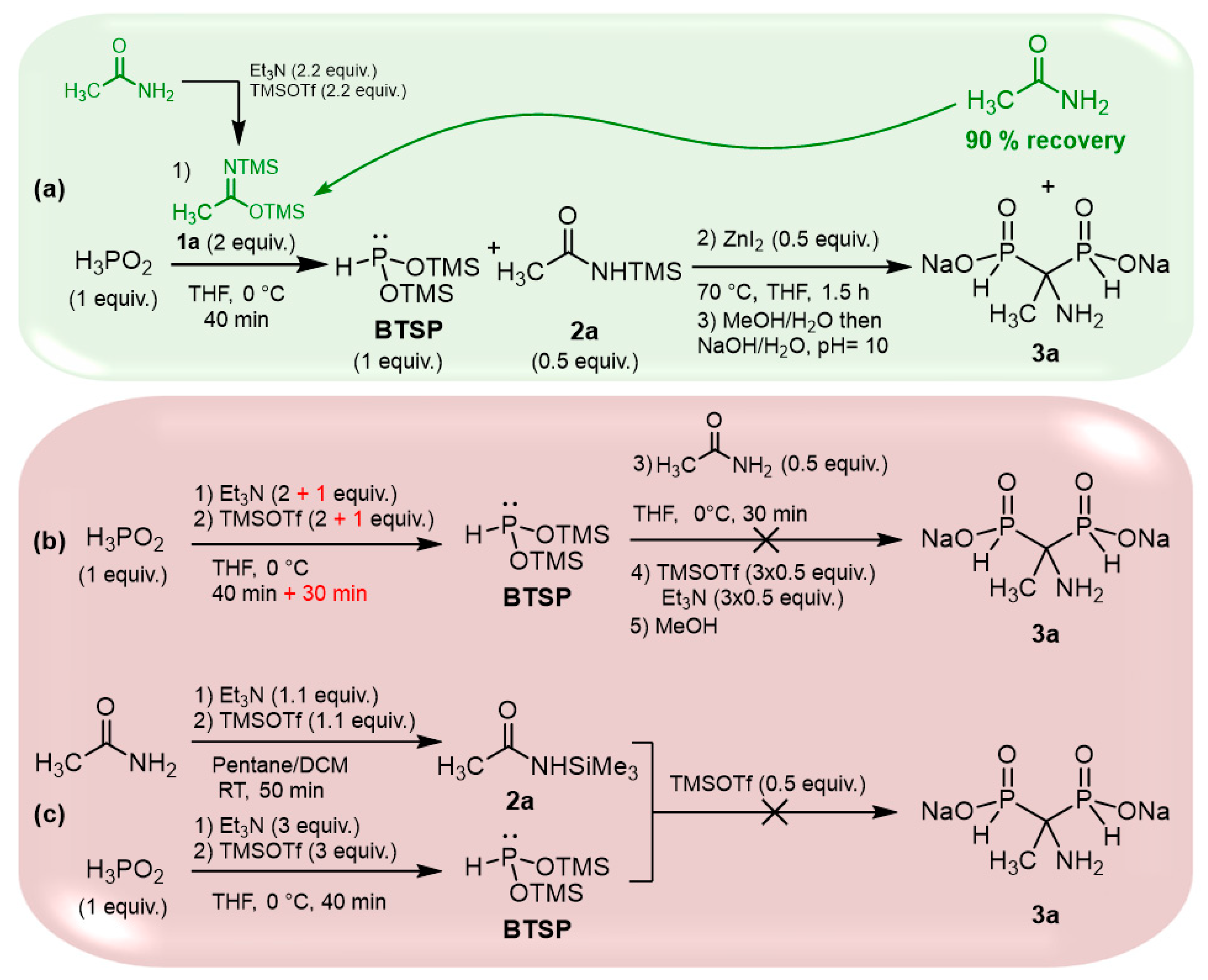
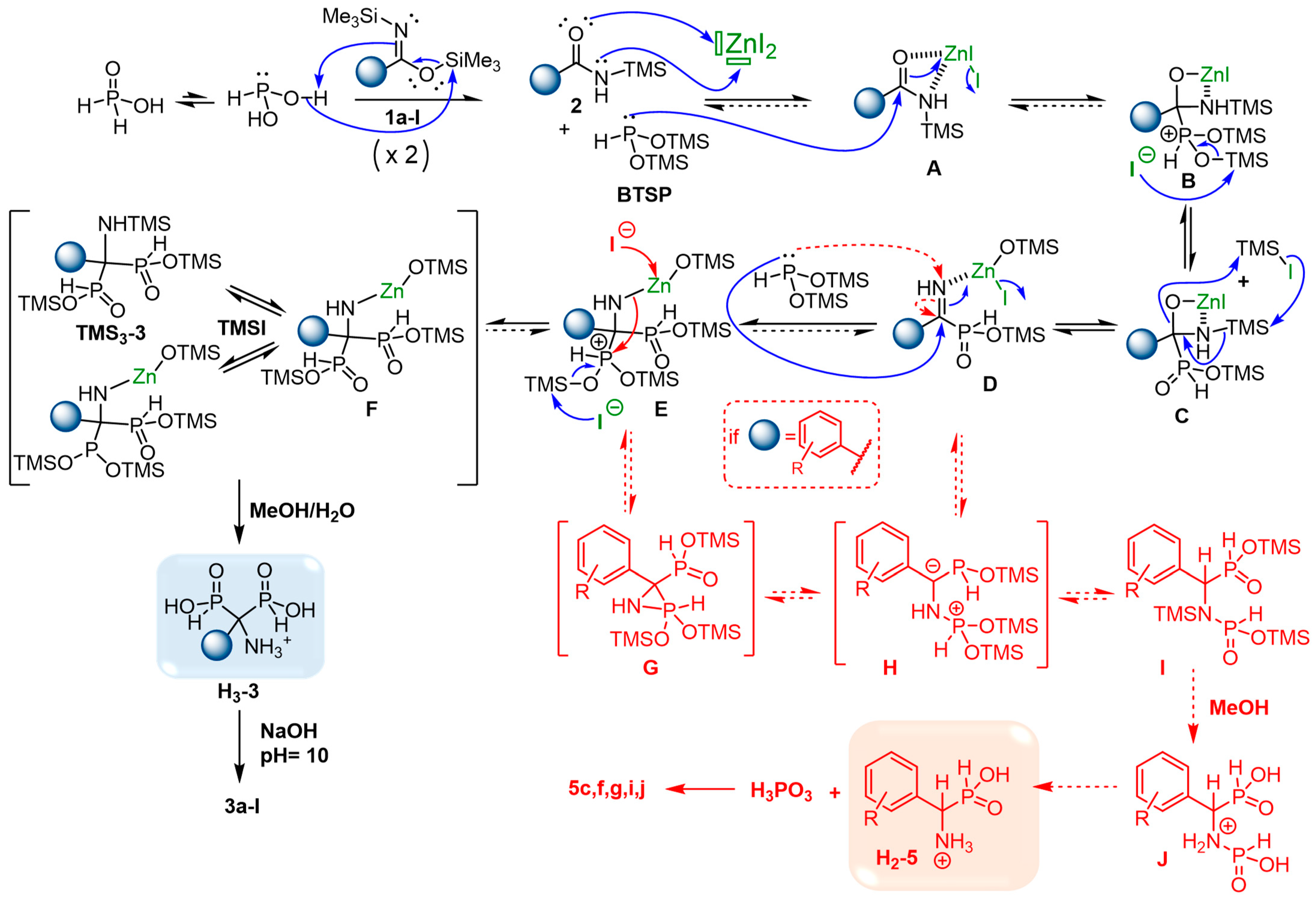
2.4. Viability Considerations of the Cascade Reaction
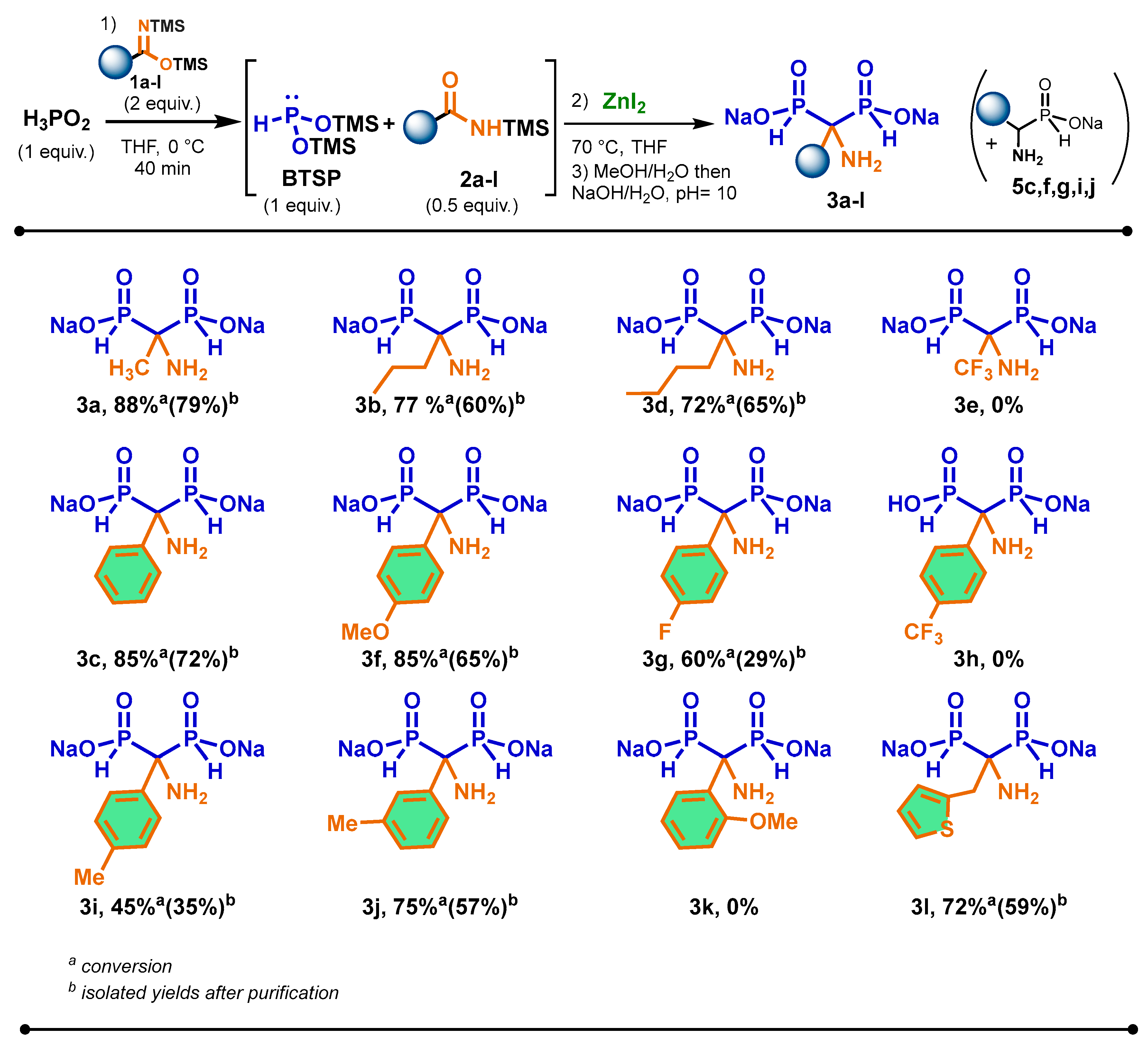
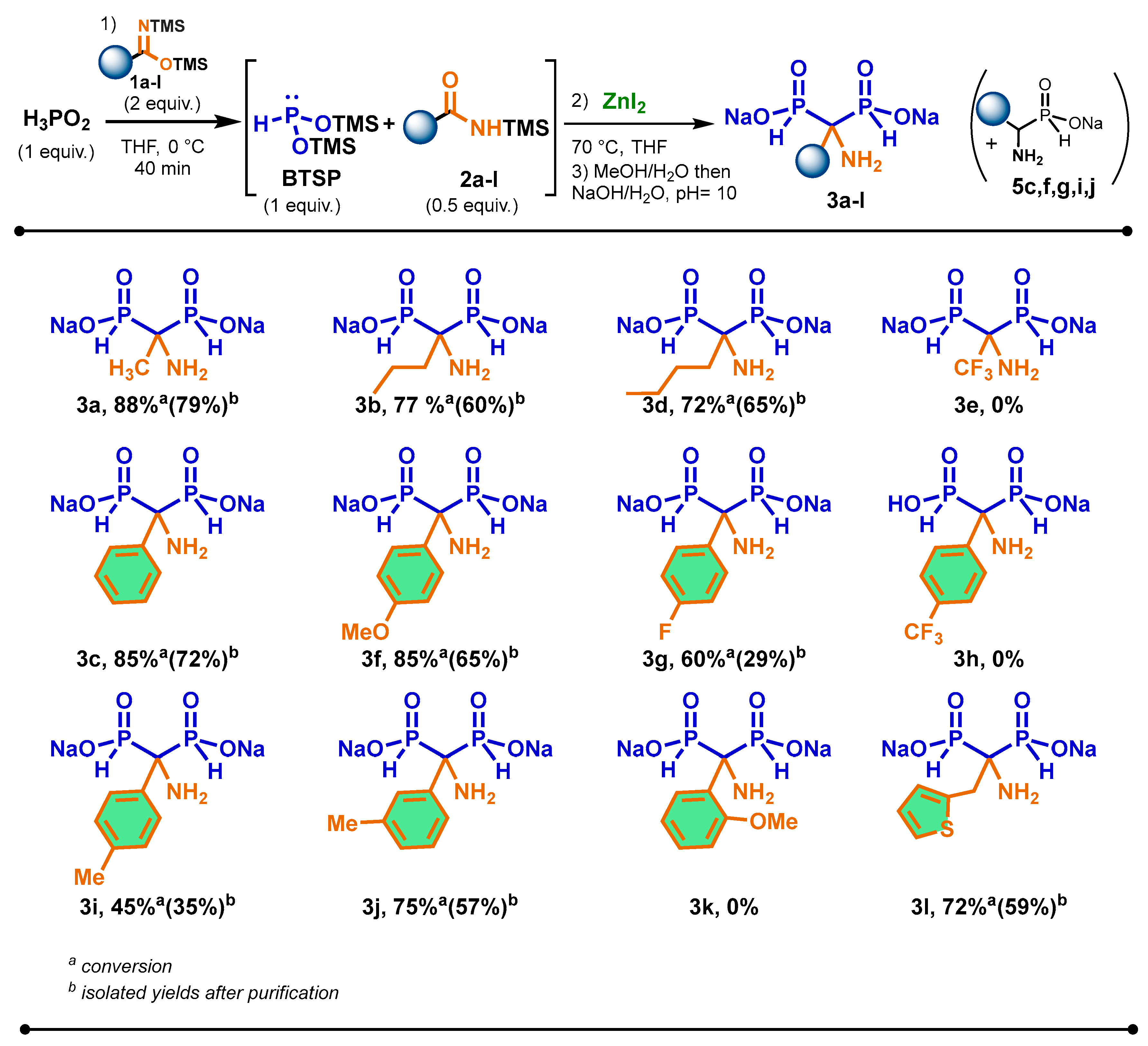
2.5. Scope of the Cascade Reaction
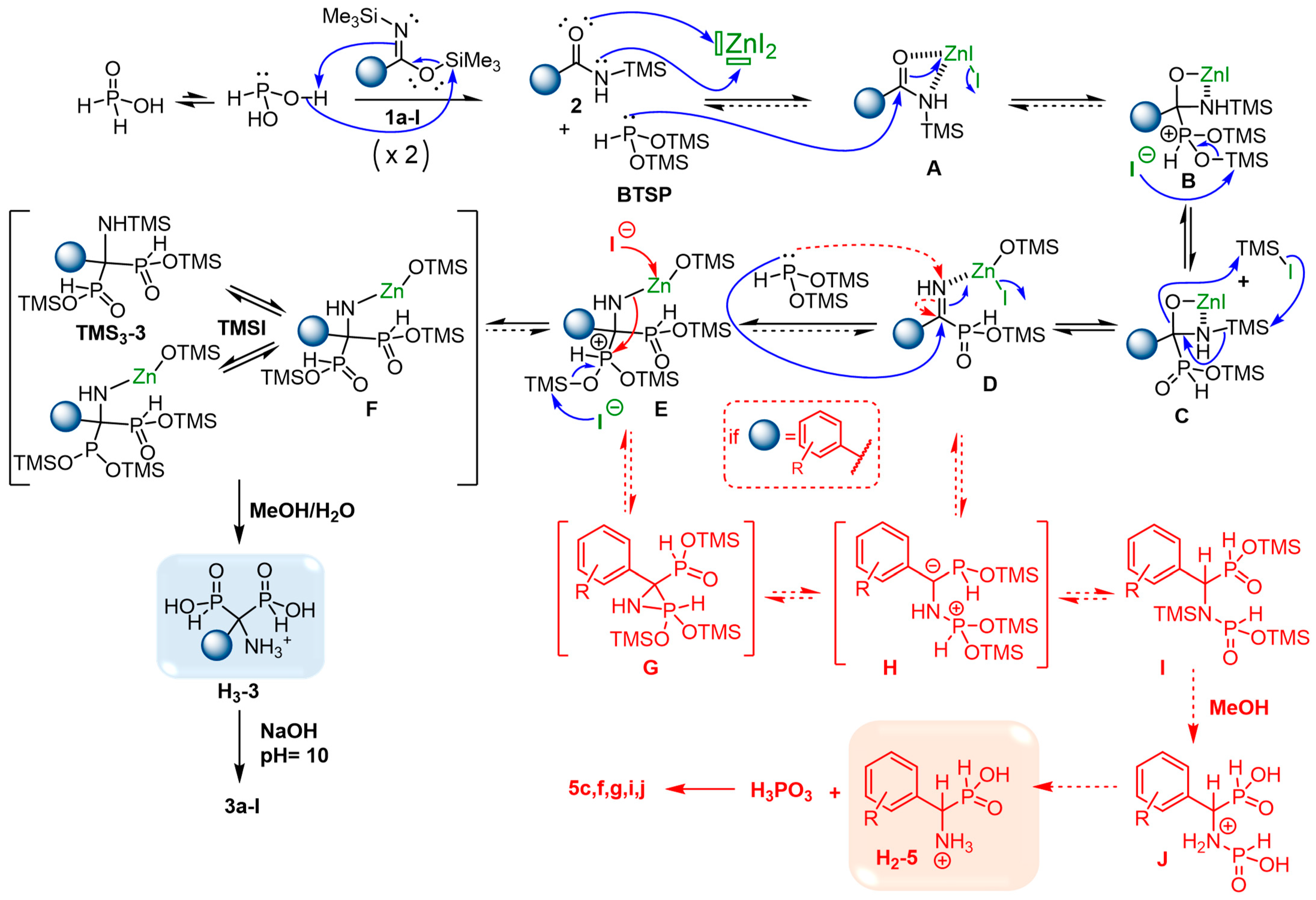
2.6. Plausible Mechanism of the Cascade Reaction
3. Materials and Methods
3.1. General Informations
3.2. General Procedure for the Cascade Synthesis of Aminomethylenebisphosphinates 3a–l
3.3. Spectral Data of Aminomethylenebisphosphinates 3a–l
- 1-aminoethane-1,1-bis(H-phosphinate) disodium salts 3a. White powder; 425 mg, 79% yield. 31P {1H} NMR (162 MHz, D2O) δ 28.5 (s). 31P NMR (162 MHz, D2O) δ 28.5 (dm, 1JP-H = 524.9 Hz). 1H NMR (400 MHz, D2O) δ 6.80 (dt, 1JP-H = 525.3 Hz, 2J = 11.7 Hz, 2H), 1.19 (t, 2JP-H = 15.8 Hz, 3H). 13C NMR (101 MHz, D2O) δ 52.2 (t, 1JP-C = 89.4 Hz), 15.0. MS (ESI-) m/z 171.99 [M − H]−, 193.97 [M − 2H + Na]−, 153.98 [M − H-H2O]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C2H8NO4P2]: 171.9934; found: 171.9934.
- 1-amino-1-propylmethane-1,1-bis(H-phosphinate) disodium salts 3b. White powder; 359 mg, 60% yield. 31P {1H} NMR (162 MHz, D2O) δ 28.1 (s). 31P NMR (162 MHz, D2O) δ 28.1 (dp, 1JP-H = 523.6 Hz, 2J = 13.4 Hz). 1H NMR (400 MHz, D2O) δ 6.84 (dt, 1JP-H = 523.5 Hz, 2J = 12.1 Hz, 2H), 1.72–1.55 (m, 2H), 1.54–1.39 (m, 2H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, D2O) δ 55.3 (t, 1JP-C = 89.0 Hz, 33.1, 16,6 (t, 2JP-C = 6.8 Hz), 14.3. MS (ESI-) m/z 200.02 [M − H]−, 222.00 [M − 2H + Na]−, 182.01 [M − H-H2O]−, 134.04 [M − H-H3PO2]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C4H12NO4P2]: 200.0247; found: 200.0247.
- 1-amino-1-phenylmethane-1,1-bis(H-phosphinate) disodium salts 3c. White powder; 500 mg, 72% yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H = 538.6 Hz, J = 12.1 Hz). 1H NMR (400 MHz, D2O) δ 7.43 (d, 3JP-H = 8.1 Hz, 2H), 7.32 (t, 4JP-H = 7.6 Hz, 2H), 7.23 (t, 1JP-H = 7.6 Hz, 1H), 6.87 (dt, 1JP-H = 539.0 Hz, 2J = 10.3 Hz, 2H). 13C NMR (101 MHz, D2O) δ 164.0, 128.5, 127.0, 126.0, 60.5 (t, 1JP-C = 86.0 Hz. MS (ESI-) m/z 234.00 [M − H]−, 215.99 [M − H-H2O]−, 170.04 [M − H-HPO2]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C7H10NO4P2]: 234.0090; found: 234.0090.
- 1-amino-1-butylmethane-1,1-bis(H-phosphinate) disodium salts 3d. White powder; 417 mg, 65% yield. 31P {1H} NMR (162 MHz, D2O) δ 27.9 (s). 31P NMR (162 MHz, D2O) δ 27.9 (dp, 1JP-H = 523.8 Hz, 2J = 13.2 Hz). 1H NMR (400 MHz, D2O) δ 6.85 (dt, 1JP-H = 524.3 Hz, 2J = 11.9 Hz, 2H), 1.73–1.62 (m, 2H), 1.48–1.40 (m, 2H), 1.28 (hex., J = 7.3 Hz, 2H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, D2O) δ 55.2 (t, 1JP-C = 88.6 Hz), 30.45 (C2), 25.1 (t, 3JP-C = 6.7 Hz), 23.0, 13.1. MS (ESI-) m/z 214.04 [M − H]−, 236.02 [M − 2H + Na]−, 196.03 [M − H-H2O]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C5H14NO4P2]: 214.0403; found: 214.0403.
- 1-amino-1-(4-methoxyphenyl)methane-1,1-bis(H-phosphinate) disodium salts 3f. White powder; 0.483 mg, 65% yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H = 538.7 Hz, 2J = 12.4 Hz). 1H NMR (400 MHz, D2O) 7.47 (d, 3J = 8.9 Hz, 2H), 7.02 (d, 3JP-H = 8.5 Hz, 2H), δ 6.95 (dt, 1JP-H = 537.9 Hz, 2J= 11.1 Hz, 2H), 3.81 (s, 3H). 13C NMR (101 MHz, D2O) δ 157.8 (t, J = 2.4 Hz), 128.2, 127.4 (t, 3JP-C = 4.9 Hz), 114.0, 59.8 (t, 1JP-C = 86.8 Hz), 55.3. MS (ESI-) m/z 264.02 [M − H]−, 286.00 [M − 2H + Na]−, 246.01 [M − H-H2O]−, 200.05 [M − H-HPO2]−. HRMS (ESI-) m/z: [M-H]−. Calcd. for [C8H12NO5P2]: 264.0196; found: 200.0204.
- 1-amino-1-(4-fluorophenyl)methane-1,1-bis(H-phosphinate) disodium salts 3g. White powder; 220 mg, 29% yield. 31P {1H} NMR (162 MHz, D2O) δ 26.4 (s). 31P NMR (162 MHz, D2O) δ 26.4 (dt, 1JP-H = 538.6 Hz, J = 11.1 Hz). 19P NMR (377 MHz, D2O) δ 116.8 (m). 1H NMR (400 MHz, D2O) δ 7.57–7.46 (m, 2H), 7.14 (t, 4JP-H = 8.8 Hz, 2H), 6.96 (dt, 1JP-H = 537.4 Hz, 1JP-H = 10.8 Hz, 2H). 13C NMR (101 MHz, D2O) δ 160.7 (dt, 1JC-F = 243.2 Hz, 4JP-C = 2.8 Hz), 131.6–131.5 (m), 127.8–127.7 (m), 115.2, 115.0, 60.0 (t, 1JP-C = 86.0 Hz). MS (ESI-) m/z 252.00 [M − H]−, 273.98 [M − 2H + Na]−, 233.99 [M − H-H2O]−, 188.03 [M − H-HPO2]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C7H9FNO4P2]: 251.9996; found: 251.9996.
- 1-amino-1-(4-tolyl)methane-1,1-bis(H-phosphinate) disodium salts 3i. White powder; 250 mg, 35% yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H = 539.8 Hz, J = 12.0 Hz). 1H NMR (400 MHz, D2O) δ 7.46–7.40 (m, 2H), 7.26–7.22 (m, 2H), 6.96 (dt, 1JP-H = 538.2 Hz, 2J = 10.8 Hz, 2H), 2.30 (s, 3H). 13C NMR (101 MHz, D2O) δ 137.0, 132.6, 129.1, 126.05 (t, 3JP-C = 4.8 Hz), 60.2 (t, 1JP-C = 86.5 Hz), 20.1. MS (ESI-) m/z 248.02 [M − H]−, 270.00 [M − H-H2O]−, 184.05 [M − H-HPO2]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C8H12FNO4P2]: 248.0247; found: 248.0247.
- 1-amino-1-(3-tolyl)phenyl)methane-1,1-bis(H-phosphinate) disodium salts 3j. White powder; 400 mg, 57% yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H = 542.3 Hz, J= 11.5 Hz). 1H NMR (400 MHz, D2O) δ 7.36 (s, 1H), 7.30−7.29 (m, 2H), 7.15 (s, 1H), 6.96 (dt, 1JP-H = 538.9 Hz, 2J = 11.8 Hz, 2H), 2.32 (s, 3H). 13C NMR (101 MHz, D2O) δ 138.4, 135.8 (t, 4JP-C = 2.3 Hz), 128.4, 127.6, 126.7 (t, 3JP-C = 4.9 Hz), 123.0 (t, 3JP-C = 5.0 Hz), 60.5 (t, 1JP-C = 86.1 Hz), 20.7. MS (ESI-) m/z 248.02 [M − H]−, 270.01 [M − 2H + Na]−, 230.01 [M − H-H2O]−, 184.05 [M − H-HPO2]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C8H12NO4P2]: 248.0247; found: 248.0247.
- 1-amino-1-(2-thienyl)ethane-1,1-bis(H-phosphinate) disodium salts 3l. White powder; 437 mg, 59% yield. 31P {1H} NMR (162 MHz, D2O) δ 26.5 (s). 31P NMR (162 MHz, D2O) δ 26.5 (dm, 1JP-H = 531.6 Hz). 1H NMR (400 MHz, D2O) δ 7.28–7.27 (m, 1H, H6), 6.99–6.97 (m, 2H), 6.83 (dt, 1JP-H = 530.6 Hz, 2J = 11.8 Hz, 2H), 3.26 (t, J = 12.7 Hz, 2H). 13C NMR (101 MHz, D2O) δ 136.9 (t, 3JP-C = 9.1 Hz), 128.5, 126.9, 125.1, 55.1 (t, 1JP-C = 89.3 Hz), 29.8. MS (ESI-) m/z 253.98 [M − H]−, 275.96 [M − 2H + Na]−, 253.97 [M − H-H2O]−. HRMS (ESI-) m/z: [M − H]−. Calcd. for [C6H10NO4P2S]: 253.9811; found: 253.9811.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Virieux, D.; Volle, J.N.; Bakalara, N.; Pirat, J.L. Synthesis and biological applications of phosphinates and derivatives. Top. Curr. Chem. 2015, 360, 39–114. [Google Scholar] [CrossRef] [PubMed]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, H.; Shi, E.; Tang, W. Development and Clinical Application of Phosphorus-Containing Drugs. Med. Drug. Discov. 2020, 8, 100063. [Google Scholar] [CrossRef]
- Montchamp, J.-L. Challenges and solutions in phosphinate chemistry. Pure Appl. Chem. 2019, 91, 113–120. [Google Scholar] [CrossRef]
- Montchamp, J.-L. Recent advances in phosphorus–carbon bond formation: Synthesis of H-phosphinic acid derivatives from hypophosphorous compounds. J. Organomet. Chem. 2005, 690, 2388–2406. [Google Scholar] [CrossRef]
- Dussart, J.; Deschamp, J.; Monteil, M.; Gager, O.; Migianu-Griffoni, E.; Lecouvey, M. A General Protocol for the Synthesis of H --Hydroxyphosphinates. Synthesis 2019, 51, 421–432. [Google Scholar] [CrossRef]
- Guedeney, N.; Dussart, J.; Deschamp, J.; Ouechtati, M.; Migianu-Griffoni, E.; Lecouvey, M. A convenient one-pot synthesis of 1-hydroxymethylene-1,1-bisphosphinic acids. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 323–325. [Google Scholar] [CrossRef]
- Dussart, J.; Guedeney, N.; Deschamp, J.; Monteil, M.; Gager, O.; Legigan, T.; Migianu-Griffoni, E.; Lecouvey, M. A convenient synthetic route towards H-bisphosphinates. Org. Biomol. Chem. 2018, 16, 6969–6979. [Google Scholar] [CrossRef]
- Dussart-Gautheret, J.; Deschamp, J.; Monteil, M.; Gager, O.; Legigan, T.; Migianu-Griffoni, E.; Lecouvey, M. Formation of 1-Hydroxymethylene-1,1-bisphosphinates through the Addition of a Silylated Phosphonite on Various Trivalent Derivatives. J. Org. Chem. 2020, 85, 14559–14569. [Google Scholar] [CrossRef]
- Dussart-Gautheret, J.; Deschamp, J.; Legigan, T.; Monteil, M.; Migianu-Griffoni, E.; Lecouvey, M. One-Pot Synthesis of Phosphinylphosphonate Derivatives and Their Anti-Tumor Evaluations. Molecules 2021, 26, 7609. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.S.; Almeida Paz, F.A.; Braga, S.S. Bisphosphonates, Old Friends of Bones and New Trends in Clinics. J. Med. Chem. 2021, 64, 1260–1282. [Google Scholar] [CrossRef] [PubMed]
- Dussart, J.; Deschamp, J.; Migianu-Griffoni, E.; Lecouvey, M. From Industrial Method to the Use of Silylated P(III) Reagents for the Synthesis of Relevant Phosphonylated Molecules. Org. Process Res. Dev. 2020, 24, 637–651. [Google Scholar] [CrossRef]
- Reszka, A.A.; Rodan, G.A. Nitrogen-Containing Bisphosphonate Mechanism of Action. Mini Rev. Med. Chem. 2004, 4, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Ebetino, F.H.; Hogan, A.M.; Sun, S.; Tsoumpra, M.K.; Duan, X.; Triffitt, J.T.; Kwaasi, A.A.; Dunford, J.E.; Barnett, B.L.; Oppermann, U.; et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011, 49, 20–33. [Google Scholar] [CrossRef]
- Clézardin, P. Bisphosphonates’ antitumor activity: An unravelled side of a multifaceted drug class. Bone 2011, 48, 71–79. [Google Scholar] [CrossRef]
- Rogers, M.J.; Frith, J.C.; Luckman, S.P.; Coxon, F.P.; Benford, H.L.; Mönkkönen, J.; Auriola, S.; Chilton, K.M.; Russell, R.G.G. Molecular mechanisms of action of bisphosphonates. Bone 1999, 24, 73S–79S. [Google Scholar] [CrossRef]
- Lin, J.H. Bisphosphonates: A review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [Google Scholar] [CrossRef]
- Mimura, M.; Hayashida, M.; Nomiyama, K.; Ikegami, S.; Iida, Y.; Tamura, M.; Hiyama, Y.; Ohishi, Y. Synthesis and Evaluation of (Piperidinomethylene)bis(phosphonic acid) Derivatives as Anti-osteoporosis Agents. Chem. Pharm. Bull. 1993, 41, 1971–1986. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Ravaschino, E.L.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef]
- Kotsikorou, E.; Song, Y.; Chan, J.M.W.; Faelens, S.; Tovian, Z.; Broderick, E.; Bakalara, N.; Docampo, R.; Oldfield, E. Bisphosphonate Inhibition of the Exopolyphosphatase Activity of the Trypanosoma brucei Soluble Vacuolar Pyrophosphatase. J. Med. Chem. 2005, 48, 6128–6139. [Google Scholar] [CrossRef]
- Leon, A.; Liu, L.; Yang, Y.; Hudock, M.P.; Hall, P.; Yin, F.; Studer, D.; Puan, K.-J.; Morita, C.T.; Oldfield, E. Isoprenoid Biosynthesis as a Drug Target: Bisphosphonate Inhibition of Escherichia coli K12 Growth and Synergistic Effects of Fosmidomycin. J. Med. Chem. 2006, 49, 7331–7341. [Google Scholar] [CrossRef]
- Occhipinti, A.; Berlicki, L.; Giberti, S.; Dziedziola, G.; Kafarski, P.; Forlani, G. Effectiveness and mode of action of phosphonate inhibitors of plant glutamine synthetase. Pest Manag. Sci. 2010, 66, 51–58. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B.; Forlani, G. Herbicidally active aminomethylenebisphosphonic acids. Heteroat. Chem. 2000, 11, 449–453. [Google Scholar] [CrossRef]
- Simoni, D.; Gebbia, N.; Invidiata, F.P.; Eleopra, M.; Marchetti, P.; Rondanin, R.; Baruchello, R.; Provera, S.; Marchioro, C.; Tolomeo, M.; et al. Design, Synthesis, and Biological Evaluation of Novel Aminobisphosphonates Possessing an in Vivo Antitumor Activity Through a γδ-T Lymphocytes-Mediated Activation Mechanism. J. Med. Chem. 2008, 51, 6800–6807. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kafarski, P. Synthetic Procedures Leading towards Aminobisphosphonates. Molecules 2016, 21, 1474. [Google Scholar] [CrossRef]
- Hong, Y.C.; Ye, J.L.; Huang, P.Q. One-Pot Synthesis of α-Amino Bisphosphonates from Nitriles via Tf2O/HC(OR)3-Mediated Interrupted Ritter-Type Reaction. J. Org. Chem. 2022, 87, 9044–9055. [Google Scholar] [CrossRef]
- Kaboudin, B.; Esfandiari, H.; Moradi, A.; Kazemi, F.; Aoyama, H. ZnCl2-Mediated Double Addition of Dialkylphosphite to Nitriles for the Synthesis of 1-Aminobisphosphonates. J. Org. Chem. 2019, 84, 14943–14948. [Google Scholar] [CrossRef]
- Islas, R.E.; García, J.J. Nickel-Catalyzed Hydrophosphonylation and Hydrogenation of Aromatic Nitriles Assisted by Lewis Acid. ChemCatChem 2019, 11, 1337–1345. [Google Scholar] [CrossRef]
- Wang, A.E.; Chang, Z.; Sun, W.T.; Huang, P.Q. General and chemoselective bisphosphonylation of secondary and tertiary amides. Org. Lett. 2015, 17, 732–735. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis of new functionalized aryl and pyridyl aminomethylenebisphosphonic acids and their derivatives via silicon-assisted methodology. J. Organomet. Chem. 2020, 912, 121177. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Silicon-assisted synthesis of new aminomethylenebisphosphonic acids with quinolines moieties. J. Organomet. Chem. 2020, 917, 121286. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Organosilicon based synthesis of new functionalized aminomethylenediphosphonates with moieties of amino acids. J. Organomet. Chem. 2018, 871, 36–39. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Tris(trimethylsilyl) phosphite as key synthon for convenient synthesis of new organosilicon(phosphorus)-containing N-heterocycles. J. Organomet. Chem. 2018, 867, 149–154. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Alekseyev, R.S.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Terenin, V.I.; Petrosyan, V.S. Synthesis of new functionalized mono- and diphosphonic acids with five-membered aza-heterocycles moieties. Heteroat. Chem. 2017, 28, e21353. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Ershov, I.S.; Petrosyan, V.S. Synthesis of new types of aminomethylenediphosphorus-containing acids and their derivatives. Russ. J. Gen. Chem. 2015, 85, 370–379. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Ershov, I.S.; Petrosyan, V.S. Synthesis of the New Types of N-Unsubstituted Aminomethylenebisorganophosphorus Acids and Their Derivatives. Heteroat. Chem. 2015, 26, 101–105. [Google Scholar] [CrossRef]
- Minaeva, L.I.; Patrikeeva, L.S.; Kabachnik, M.M.; Beletskaya, I.P.; Orlinson, B.S.; Novakov, I.A. Synthesis of novel aminomethylenebisphosphonates and bisphosphonic acids, containing adamantyl fragment. Heteroat. Chem. 2011, 22, 55–58. [Google Scholar] [CrossRef]
- Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar] [CrossRef]
- Kaboudin, B.; Alipour, S. A microwave-assisted solvent- and catalyst-free synthesis of aminomethylene bisphosphonates. Tetrahedron Lett. 2009, 50, 4243–4245. [Google Scholar] [CrossRef]
- Wu, M.; Chen, R.; Huang, Y. Simple, Efficient and One-Pot Method for Synthesis of Aminomethylene gem-Diphosphonic Acid Derivatives from Ketones via Beckmann Rearrangement. Synthesis 2004, 2004, 2441–2444. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Yoshida, Y.; Nakabayashi, N.; Shibuya, S. Simple and Efficient Method for Preparation of Conformationally Constrained Aminomethylene gem-Diphosphonate Derivatives via Beckmann Rearrangement. J. Org. Chem. 1994, 59, 7562–7564. [Google Scholar] [CrossRef]
- David, T.; Procházková, S.; Kotek, J.; Kubíček, V.; Hermann, P.; Lukeš, I. Aminoalkyl-1,1-bis(phosphinic acids): Stability, Acid-Base, and Coordination Properties. Eur. J. Inorg. Chem. 2014, 2014, 4357–4368. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Ershov, I.S.; Petrosyan, V.S. Synthesis and Reactivity of the New Trimethylsilyl Esters of Aminomethylenebisorganophosphorus Acids. Heteroat. Chem. 2013, 24, 355–360. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Ershov, I.S.; Petrosyan, V.S. Synthesis of N-substituted aminomethylenediphosphonites and their derivatives. Russ. J. Gen. Chem. 2013, 83, 1175–1177. [Google Scholar] [CrossRef]
- Samples, M.S.; Yoder, C.H. The structure of bis(organosily1) amides containing the dimethylsilyl and bis(dimethylsilyl) ethylene groups. J. Organomet. Chem. 1987, 332, 69–73. [Google Scholar] [CrossRef]
- Klebe, J.F.; Finkbeiner, H.; White, D.M. Silylations with Bis(trimethylsilyl)acetamide, a Highly Reactive Silyl Donor. J. Am. Chem. Soc. 1966, 88, 3390–3395. [Google Scholar] [CrossRef]
- Knapp, S.B.S.; Gibson, F.S. IODOLACTAMIZATION: 8-exo-iodo-2-azabicyclo[3.3.0]octan-3-one. Org. Synth. 1992, 70, 101. [Google Scholar] [CrossRef]
- Knapp, S.; Levorse, A.T. Synthesis and reactions of iodo lactams. J. Org. Chem. 1988, 53, 4006–4014. [Google Scholar] [CrossRef]
- Hanada, S.; Motoyama, Y.; Nagashima, H. Hydrosilanes Are Not Always Reducing Agents for Carbonyl Compounds but Can Also Induce Dehydration: A Ruthenium-Catalyzed Conversion of Primary Amides to Nitriles. Eur. J. Org. Chem. 2008, 2008, 4097–4100. [Google Scholar] [CrossRef]
- Dwak, B.; Lasocki, Z. Structure and tautomerism of cyclic silylamides I. Disiloxane derivatives of acetamide and benzamides. J. Organomet. Chem. 1983, 246, 151–158. [Google Scholar] [CrossRef]
- Bassindale, A.R.; Posner, T.B. The Structure of Silylated Amides-. N-Methyl-NTrimethylsilyltrifluoroacetamide, a Reassignment of Structure. J. Organomet. Chem. 1979, 175, 273–284. [Google Scholar] [CrossRef]
- Itoh, K.; Katsuda, M.; Ishii, Y. Reactions of Group IV Organometallic Compounds. Part X1X.l Substituent Effects on the Rate of Trimethylsilyl Migration in Substituted N,O-Bis(trimethylsilyl)benzimidates. J. Chem. Soc. B 1970, 302–304. [Google Scholar] [CrossRef]
- Deprèle, S.; Montchamp, J.L. Triethylborane-Initiated Room Temperature Radical Addition of Hypophosphites to Olefins: Synthesis of Monosubstituted Phosphinic Acids and Esters. J. Org. Chem. 2001, 66, 6745–6755. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 2a–c | R | LA | T, °C | Time, Hour | 3a–c/5c, Yield (%) 1 |
| Entry 1 | 2a | Me 5 | ZnCl2 | 70 | 18 | 3a, 90 2 (75) 3 |
| Entry 2 | 2a | Me 5 | ZnI2 | 70 | 1.5 | 3a, 88 2 (83) 3 |
| Entry 3 | 2a | Me 5 | TMSOTf | 0 | 0.5 | 3a, 90 2 (75) 3 |
| Entry 4 | 2a | Me 5 | BF3·OEt2 | 0–70 | 18 | - |
| Entry 5 | 2a | Me 6 | ZnCl2 | 70 | 15 | 3a, 90 2 (75) 3 |
| Entry 6 | 2a | Me 6 | ZnI2 | 70 | 1.5 | 3a, 88 2 (79) 3 |
| Entry 7 | 2a | Me 6 | TMSOTf | 0 | 0.5 | 3a, 90 2 (75) 3 |
| Entry 8 | 2b | Pr | ZnCl2 | 70 | 18 | 3b, 77 2 (60) 3 |
| Entry 9 | 2b | Pr | ZnI2 | 70 | 2 | 3b, 77 2 (60) 3 |
| Entry 10 | 2b | Pr | TMSOTf | 0 | 0.5 | 3b, 6 4 |
| Entry 11 | 2c | Ph | ZnCl2 | 70 | 18 | 5c, 15 4 |
| Entry 12 | 2c | Ph | ZnI2 | 70 | 1 | 3c, 85 2 (72) 3 |
| Entry 13 | 2c | Ph | TMSOTf | 0 | 0.5 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayadi, N.; Descamps, A.; Legigan, T.; Dussart-Gautheret, J.; Monteil, M.; Migianu-Griffoni, E.; Ben Ayed, T.; Deschamp, J.; Lecouvey, M. Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2. Molecules 2023, 28, 6226. https://doi.org/10.3390/molecules28176226
Ayadi N, Descamps A, Legigan T, Dussart-Gautheret J, Monteil M, Migianu-Griffoni E, Ben Ayed T, Deschamp J, Lecouvey M. Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2. Molecules. 2023; 28(17):6226. https://doi.org/10.3390/molecules28176226
Chicago/Turabian StyleAyadi, Nouha, Aurélie Descamps, Thibaut Legigan, Jade Dussart-Gautheret, Maelle Monteil, Evelyne Migianu-Griffoni, Taïcir Ben Ayed, Julia Deschamp, and Marc Lecouvey. 2023. "Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2" Molecules 28, no. 17: 6226. https://doi.org/10.3390/molecules28176226
APA StyleAyadi, N., Descamps, A., Legigan, T., Dussart-Gautheret, J., Monteil, M., Migianu-Griffoni, E., Ben Ayed, T., Deschamp, J., & Lecouvey, M. (2023). Synthesis of Aminobisphosphinates through a Cascade Reaction between Hypophosphorous Acid and Bis(trimethylsilyl)imidates Mediated by ZnI2. Molecules, 28(17), 6226. https://doi.org/10.3390/molecules28176226