
A Molecular Electron Density Theory Study of the Domino Reaction of N-Phenyl Iminoboranes with Benzaldehyde Yielding Fused Bicyclic Compounds




Abstract
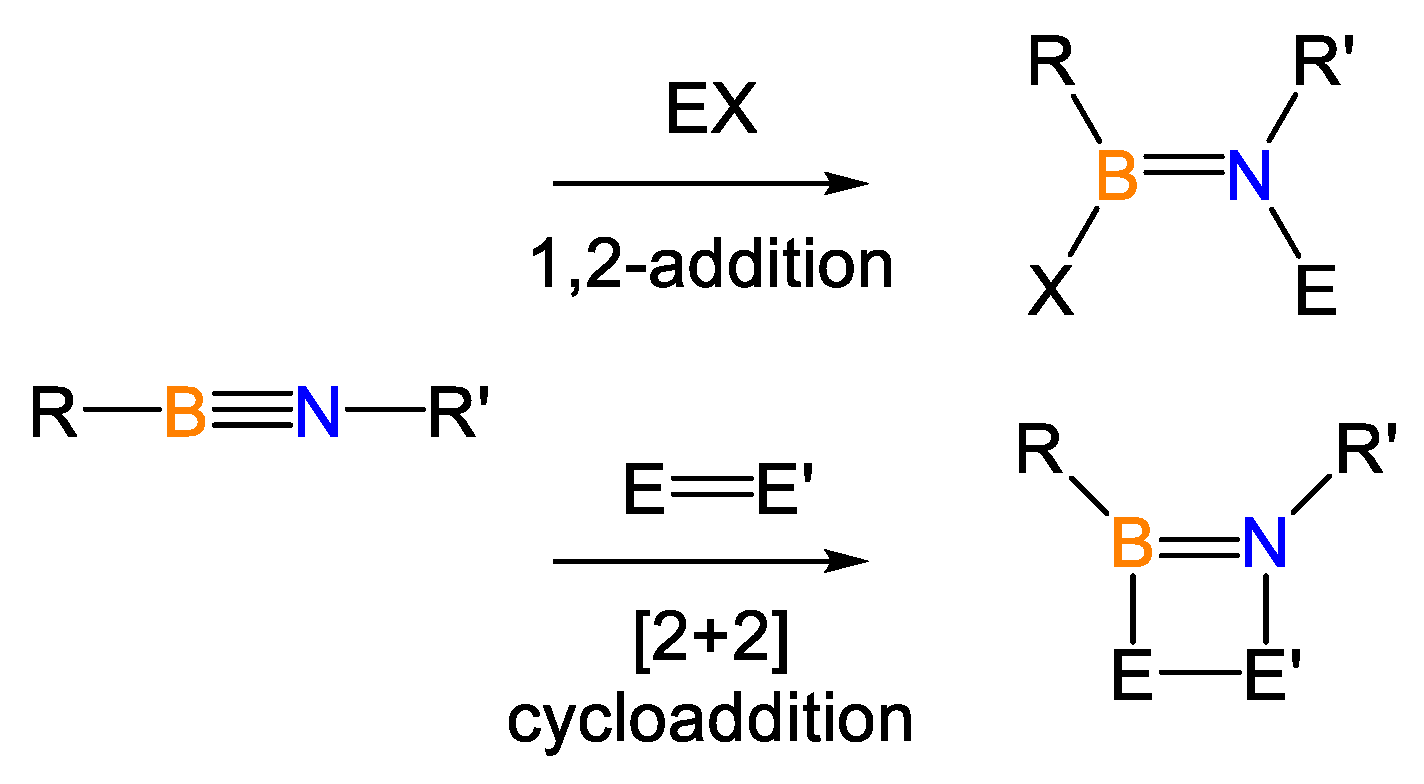
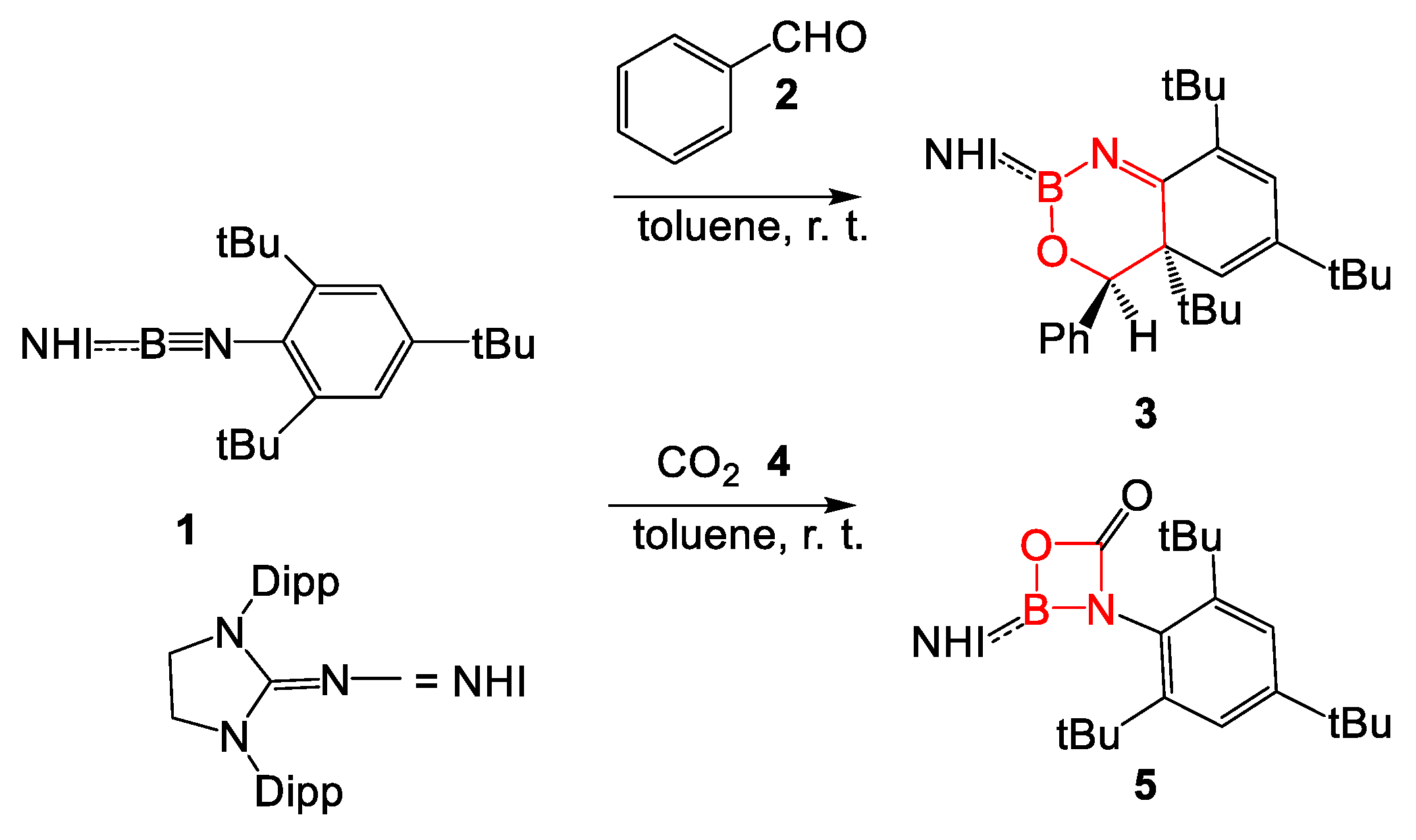
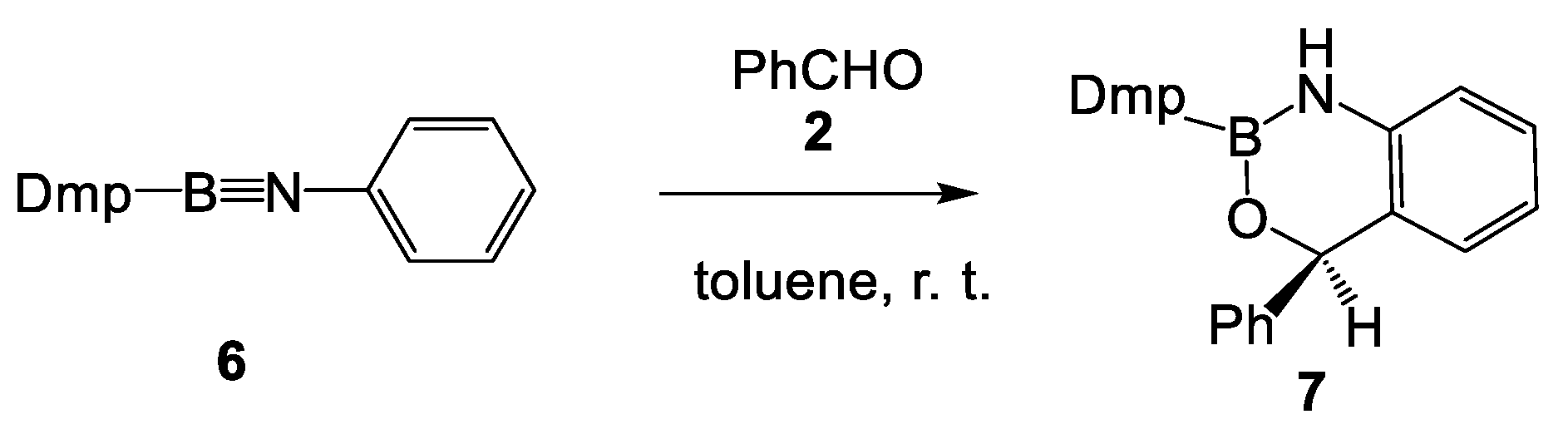
:1. Introduction
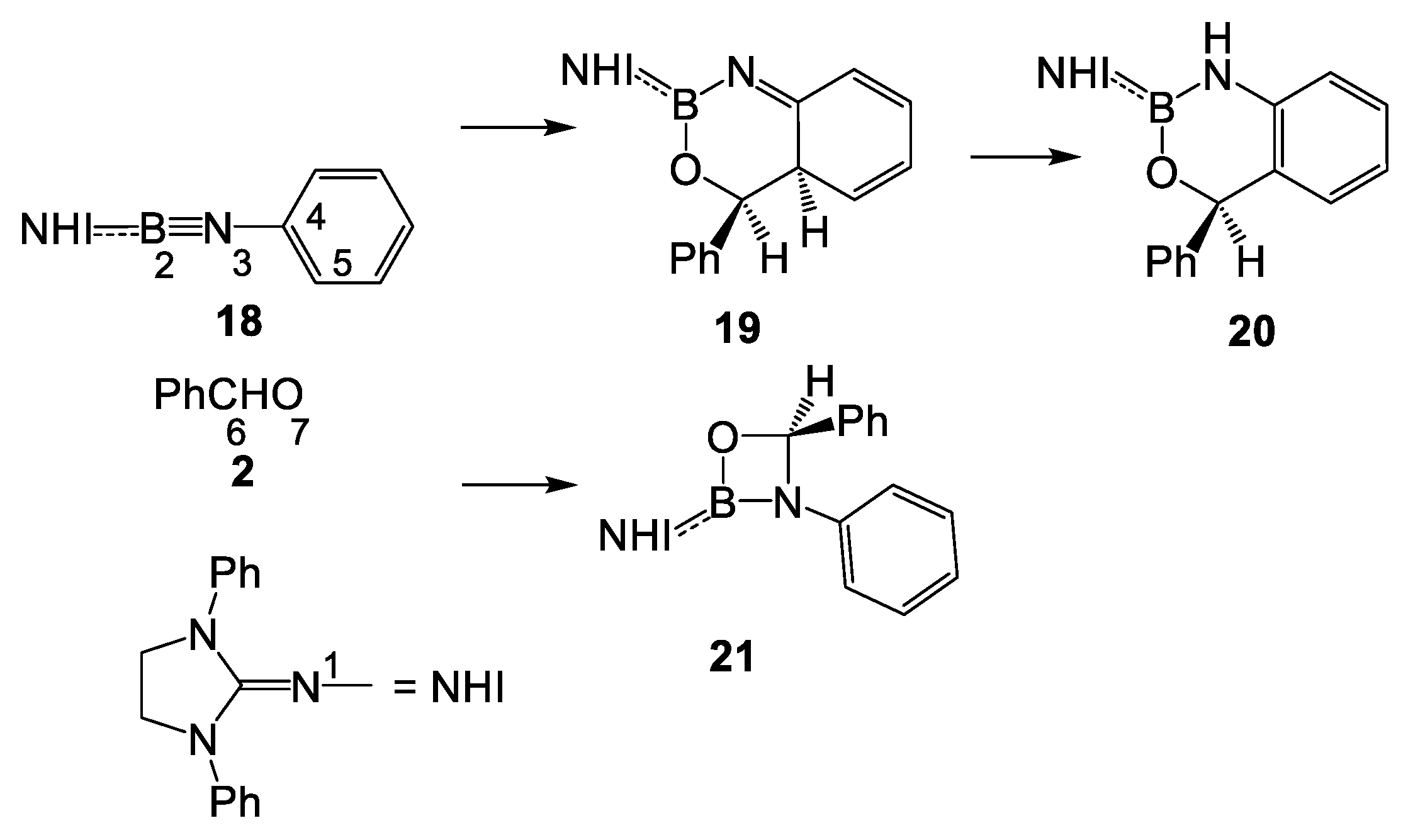
2. Results and Discussion
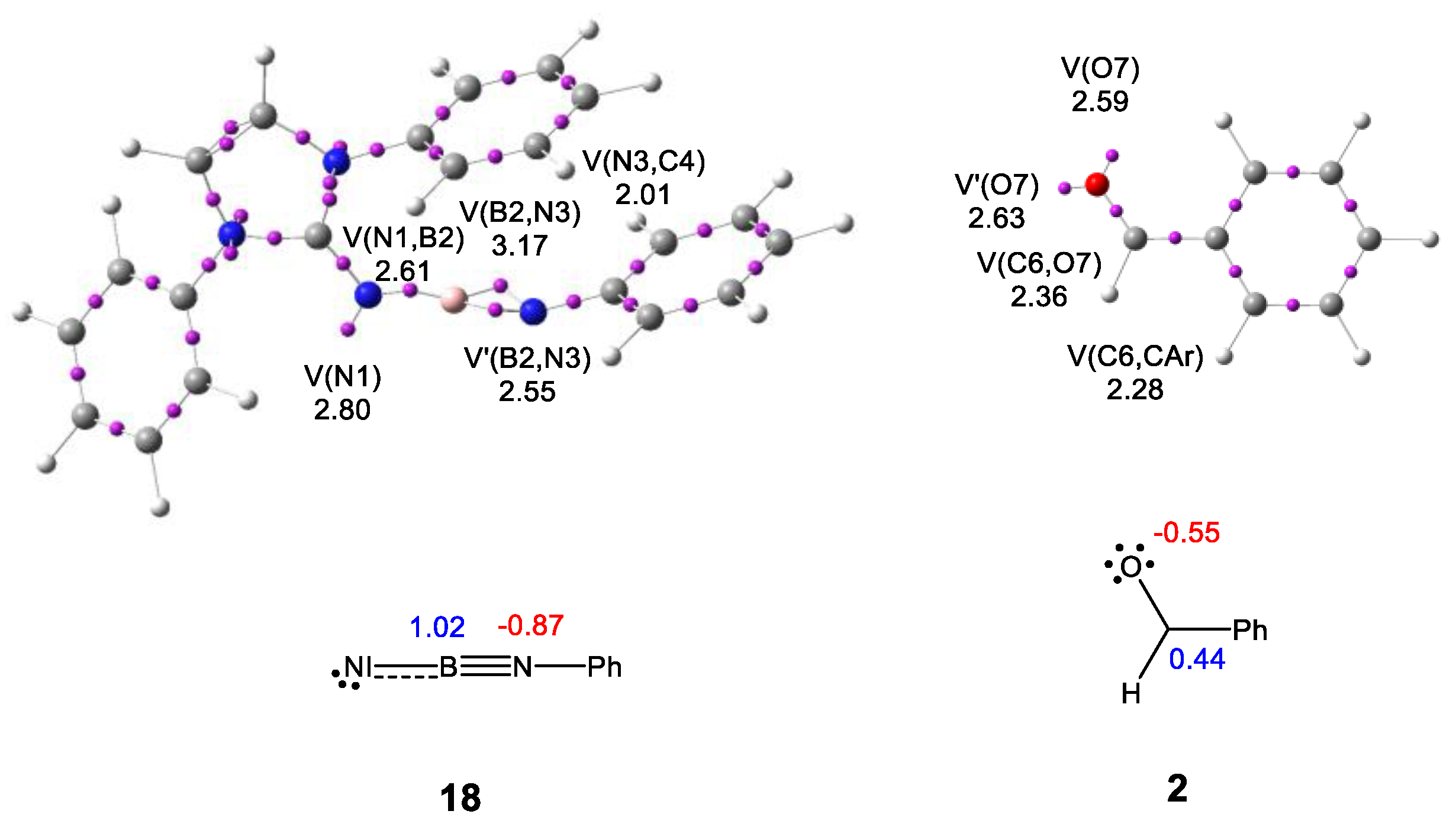
2.1. ELF and AIM Characterization of the Electronic Structure of N-Phenyl Iminoborane 18 and Benzaldehyde 2
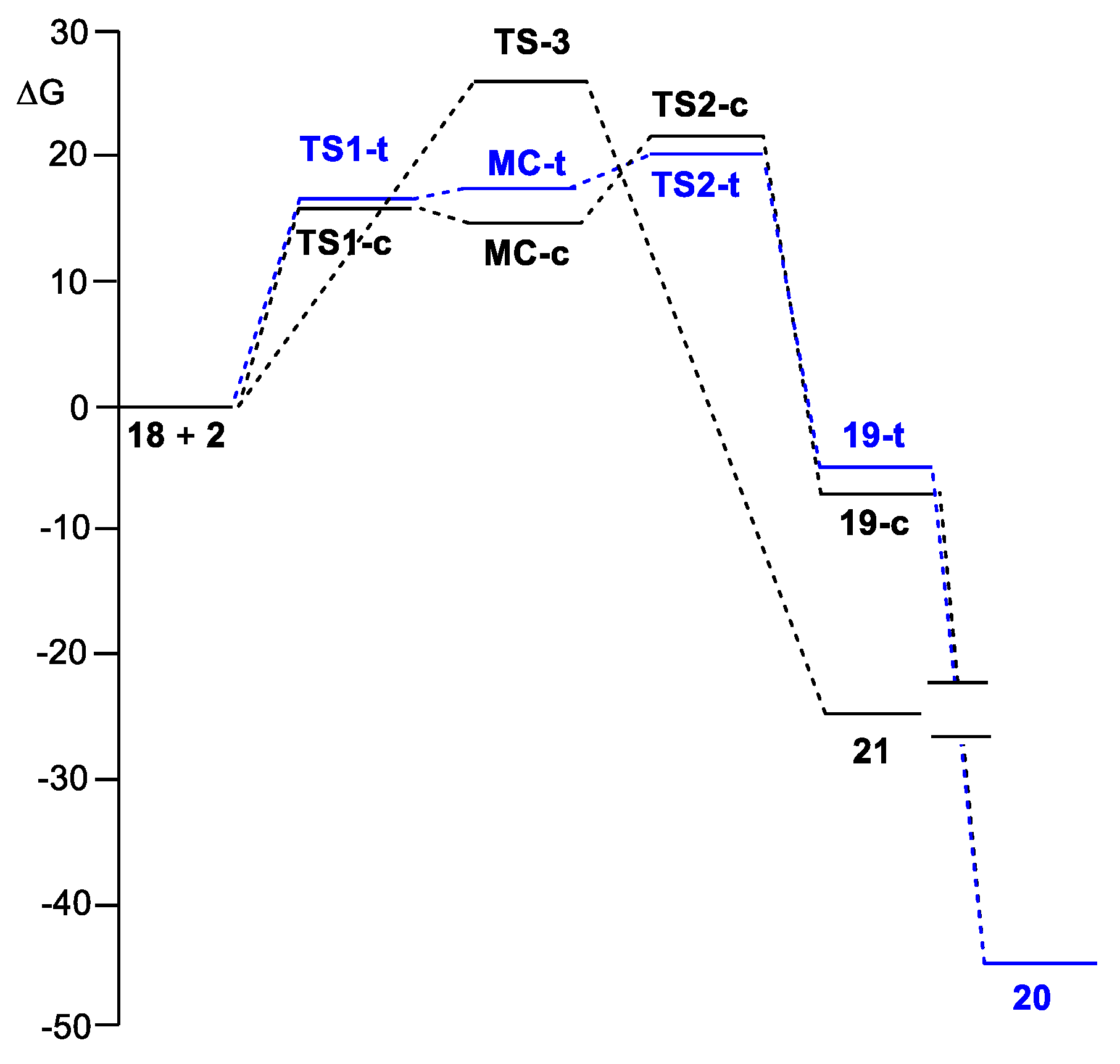
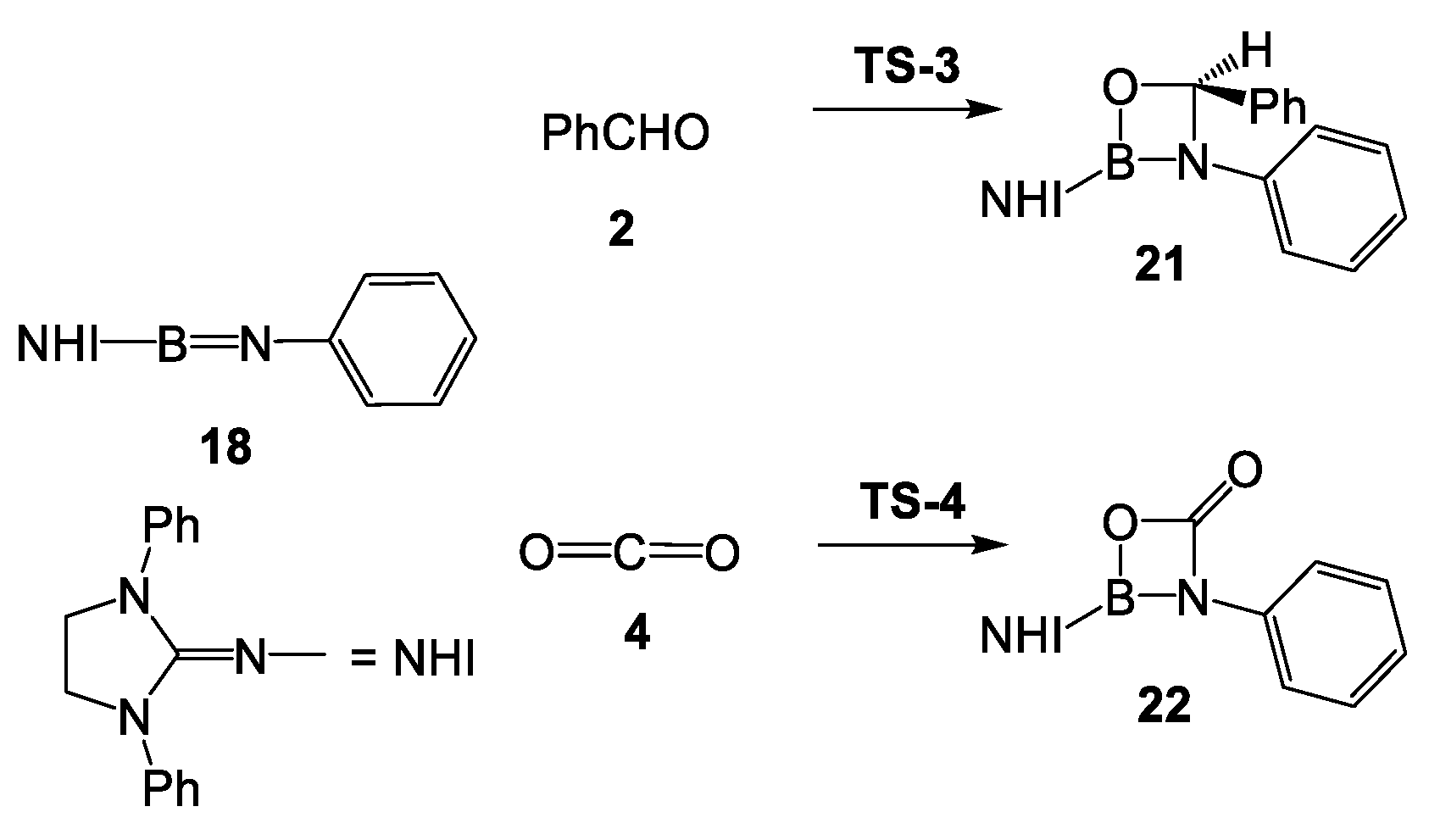
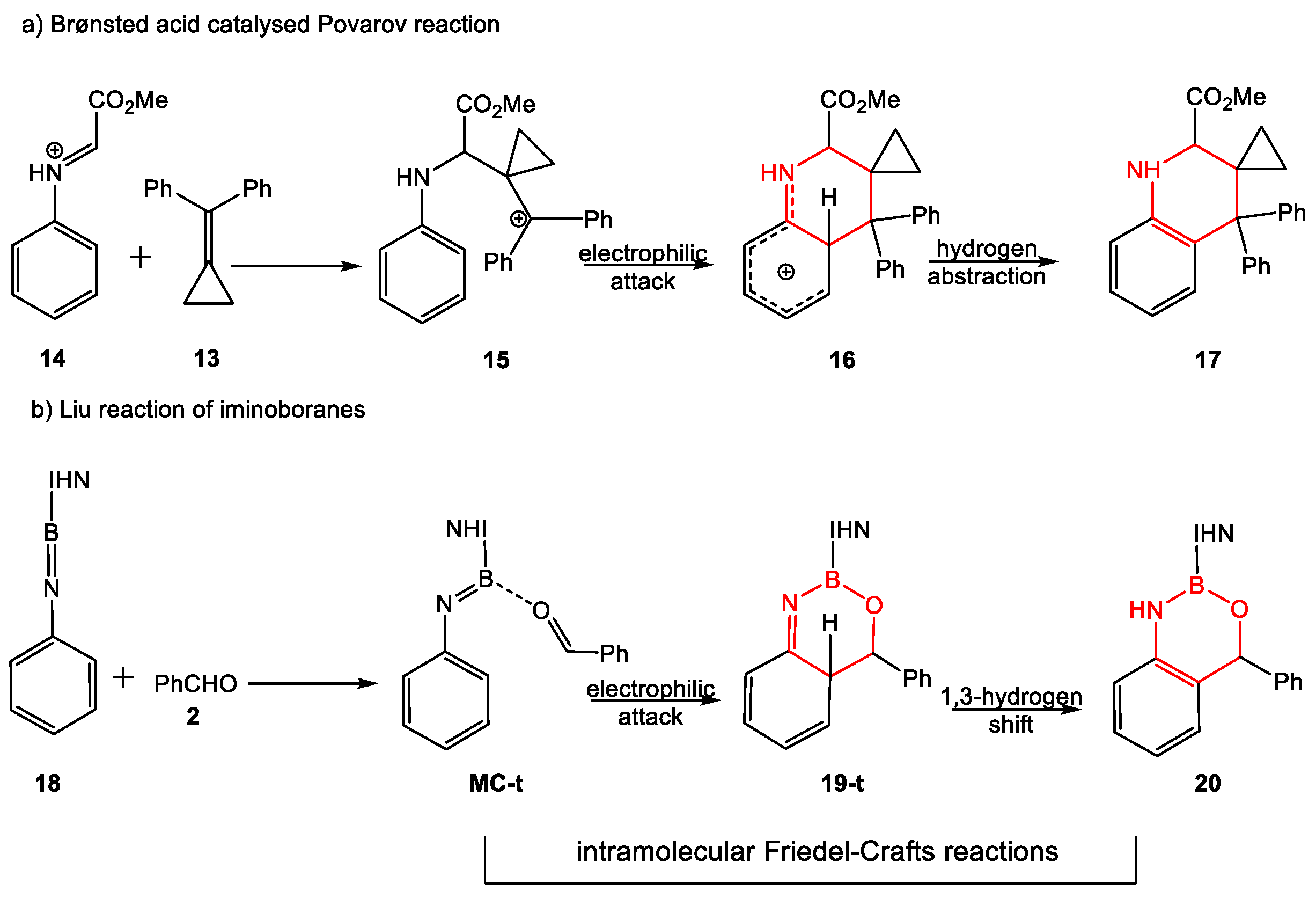
2.2. Study of the Reaction of N-Phenyl Iminoborane 18 with Benzaldehyde 2
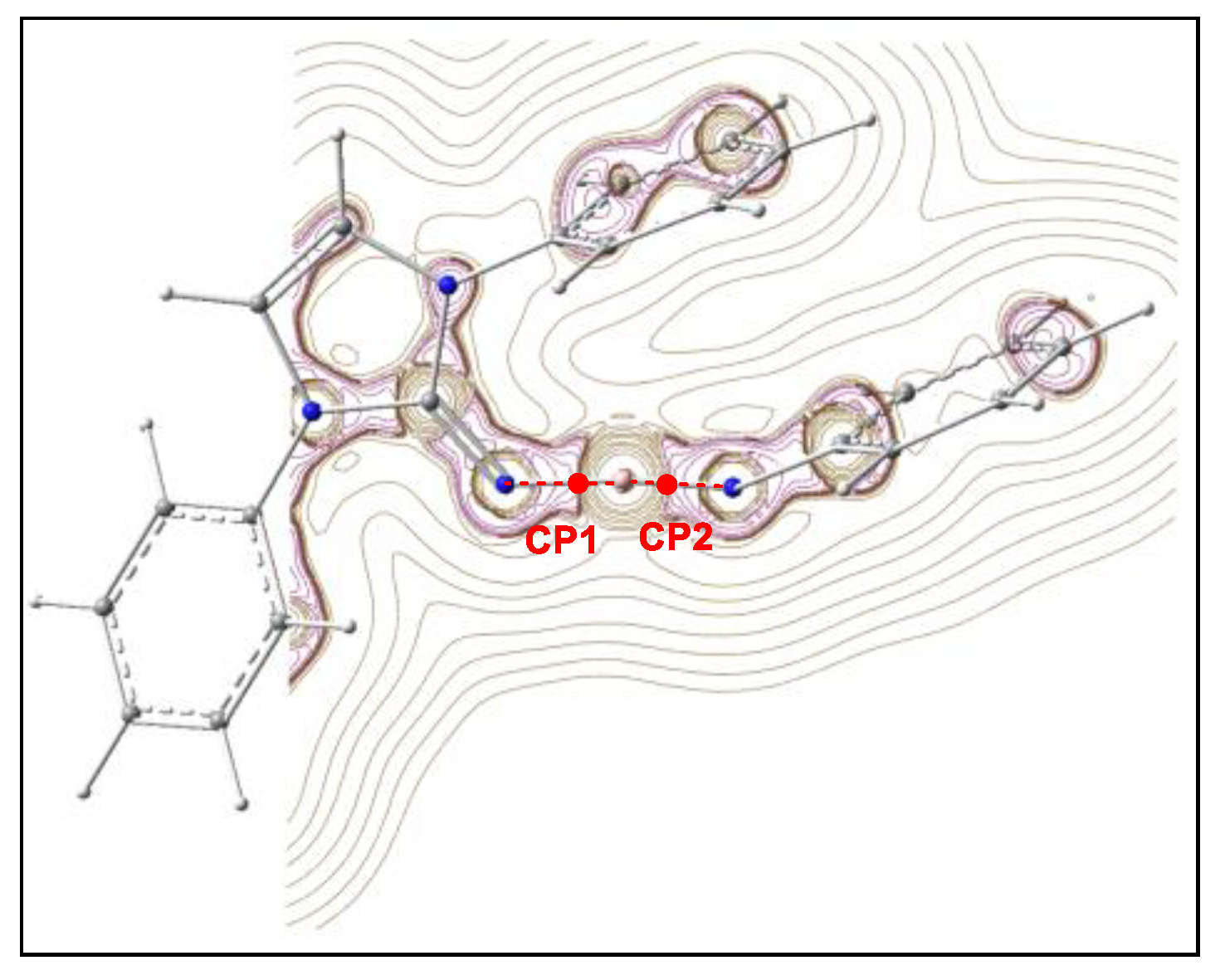
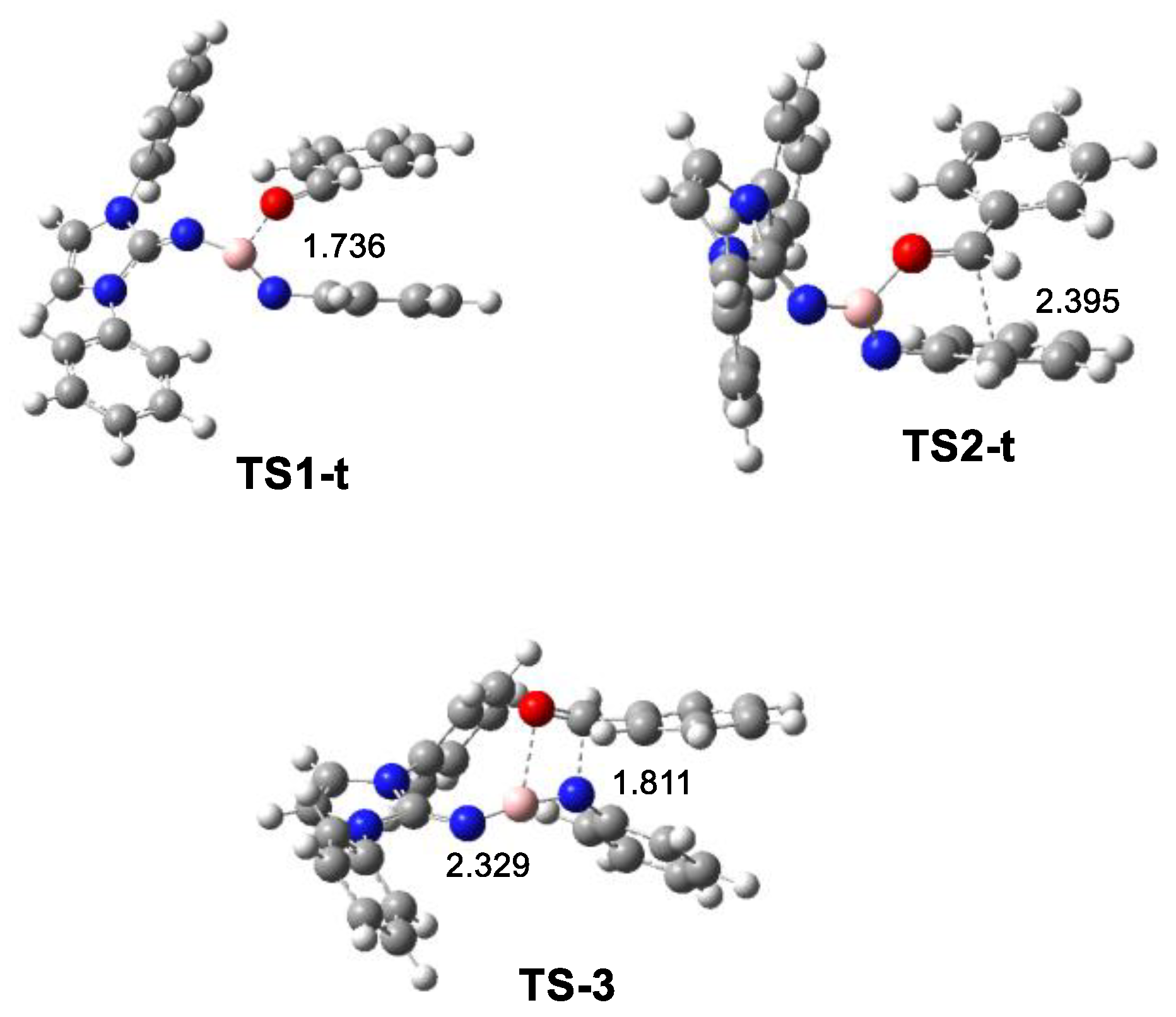
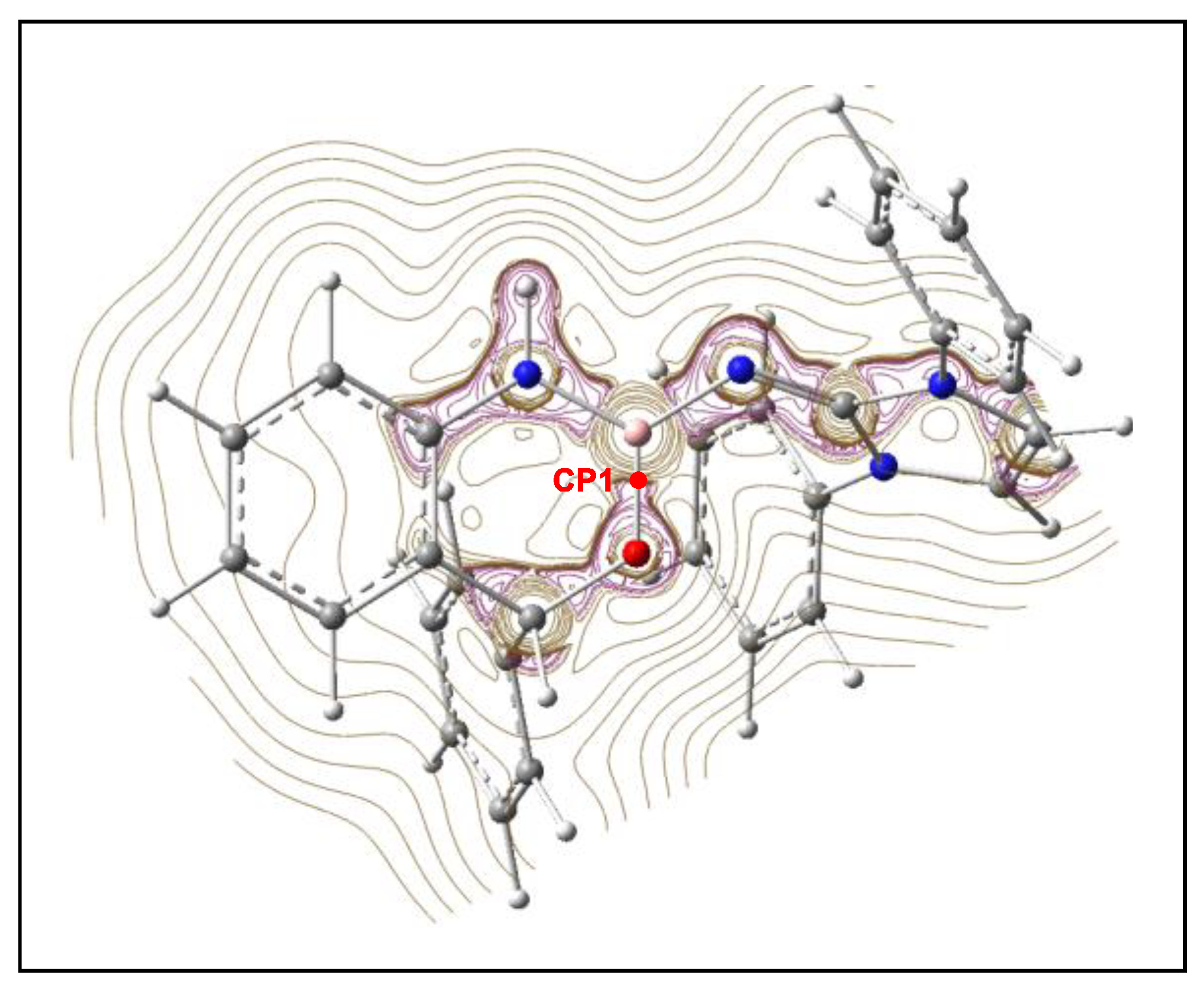
2.3. ELF and QTAIM Analysis of the Stationary Points Associated with the Trans Reaction Path of the Domino Reaction between N-Phenyl Iminoborane 18 and Benzaldehyde 2
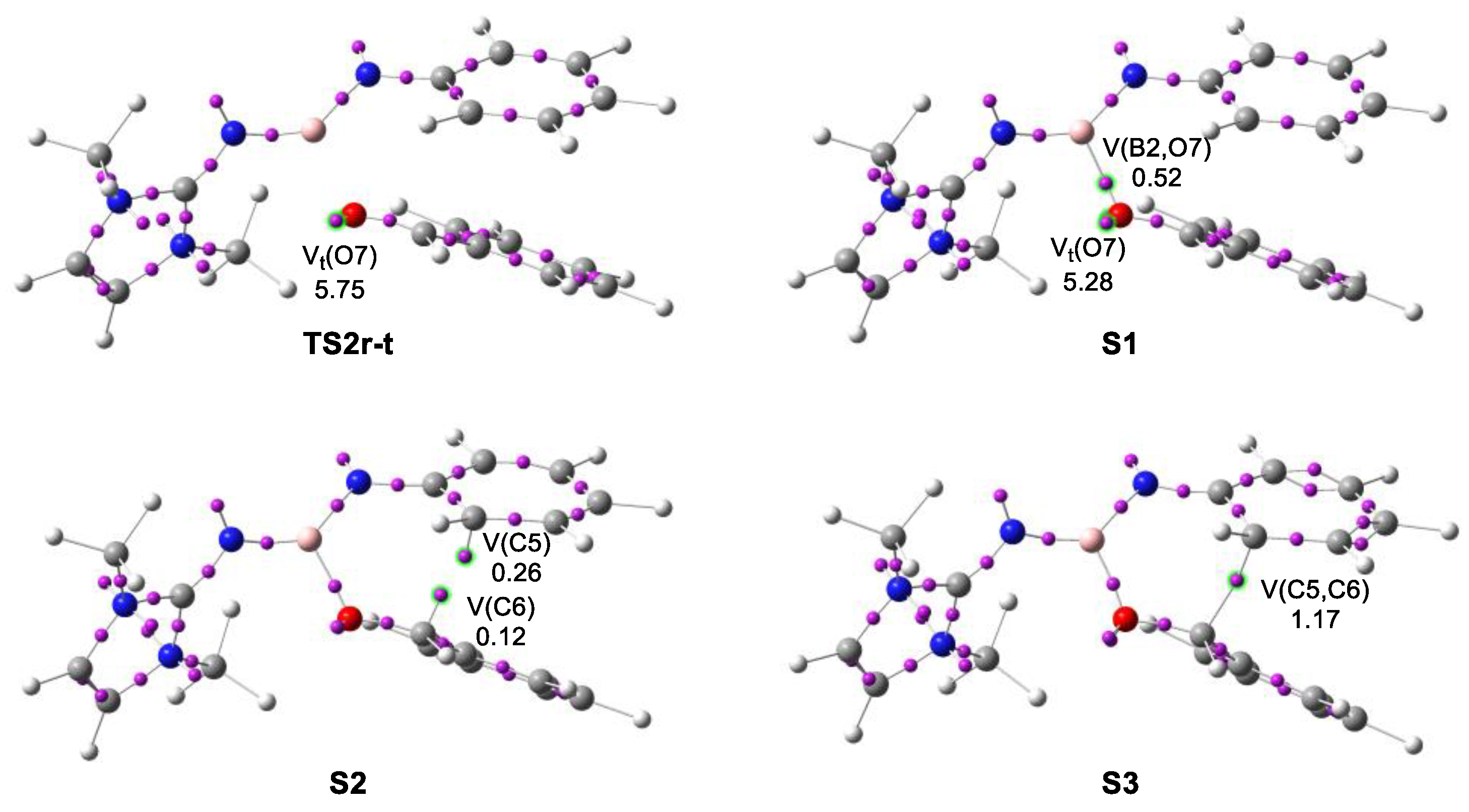
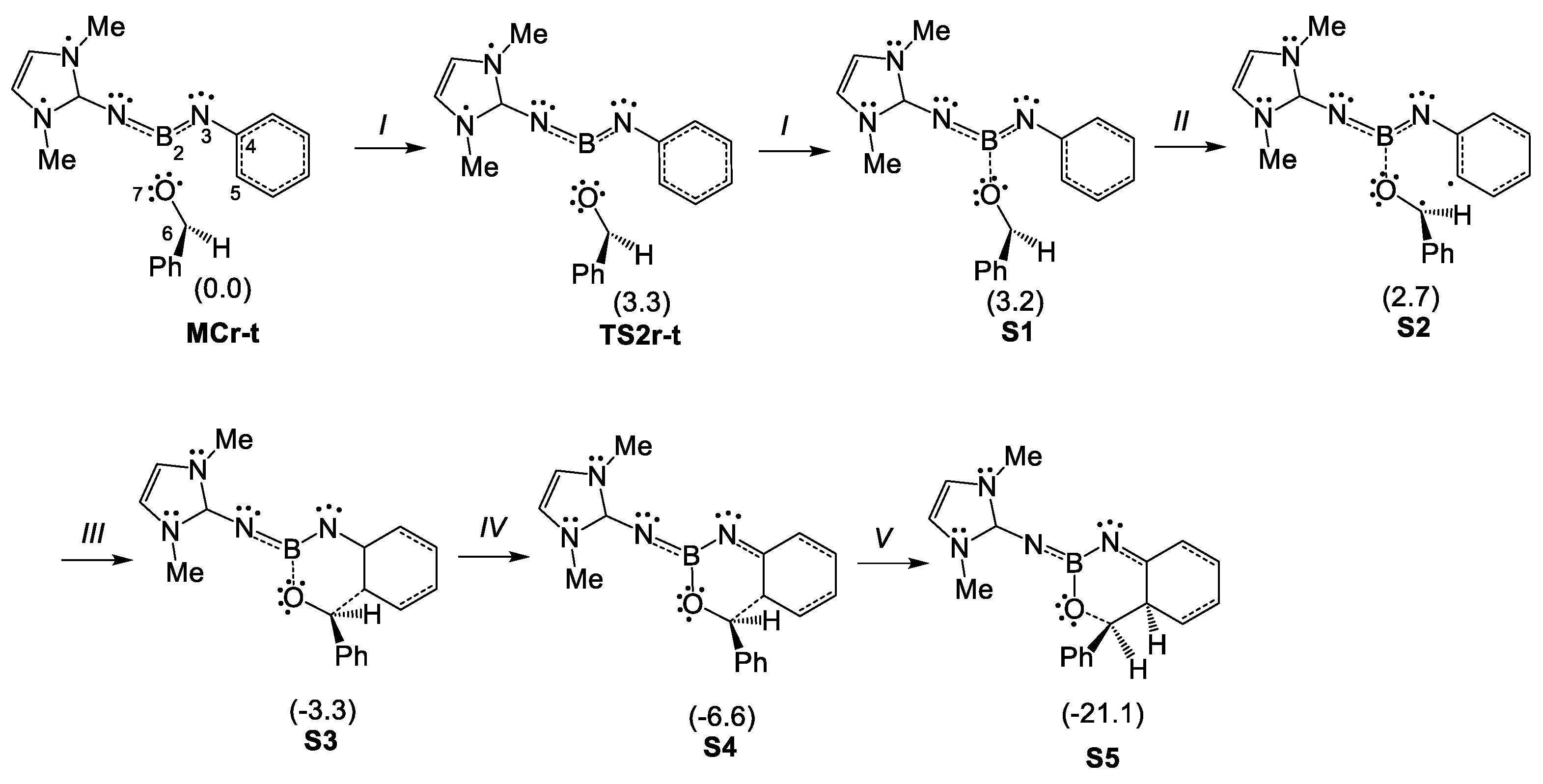
2.4. BET Study of the Molecular Mechanisms of the Electrophilic Attack of Benzaldehyde 2 on the Phenyl Substituent of Imine Borane 18
2.5. REG-IQA Energy Partitioning Analysis of the Activation Energy
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nöth, H. The Chemistry of Amino Imino Boranes. Angew. Chem. Int. Ed. 1988, 27, 1603–1622. [Google Scholar] [CrossRef]
- Paetzold, P. Iminoboranes. Adv. Inorg. Chem. 1987, 31, 123–170. [Google Scholar]
- Braunschweig, H.; Ewing, W.C.; Geetharani, K.; Schäfer, M. The Reactivities of Iminoboranes with Carbenes: BN Isosteres of Carbene–Alkyne Adducts. Angew. Chem. Int. Ed. 2015, 54, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Winner, L.; Bélanger-Chabot, G.; Celik, M.A.; Schäfer, M.; Braunschweig, H. Intriguing migrations in transient iminoborane adducts: Two new pathways to aminoboranes. Chem. Commun. 2018, 54, 9349–9351. [Google Scholar] [CrossRef]
- Winner, L.; Hermann, A.; Bélanger-Chabot, G.; González-Belman, O.F.; Jiménez-Hall, J.O.C.; Kelch, H.; Braunschweig, H. Cleavage of BN triple bonds by main group reagents. Chem. Commun. 2018, 54, 8210–8213. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zhang, X.; Hu, C.; Chu, H.; Li, Q.; Ruiz, D.A.; Liu, L.L.; Tung, C.-H.; Kong, L. Unveiling Hetero-Enyne Reactivity of Aryliminoboranes: Dearomative Hetero-Diels-Alder-Like Reactions. Angew. Chem. Int. Ed. 2022, 61, e202205814. [Google Scholar] [CrossRef]
- Brückner, R.; Huisgen, R. 2,2-bis(trifluoromethyl)ethylene-1,1-dicarbonitrile and styrenes the concertedness of the [2 + 4] cycloaddition. Tetrahedron Lett. 1990, 31, 7133–7136. [Google Scholar] [CrossRef]
- Brückner, R.; Huisgen, R.; Schmid, J. 2,2-bis(trifluoromethyl)ethylene-1,1-dicarbonitrile and styrenes a dichotomy of cycloaddition pathways. Tetrahedron Lett. 1990, 31, 7129–7132. [Google Scholar] [CrossRef]
- Middleton, W.J. 1,1-Dicyano-2,2-bis(trifluoromethyl)ethylene. J. Org. Chem. 1965, 30, 1402–1407. [Google Scholar] [CrossRef]
- Povarov, L.S. α,β-Unsaturated ethers and their analogues in reactions of diene synthesis. Russ. Chem. Rev. 1967, 36, 656–670. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Sáez, J.A.; Mekelleche, S.M. Understanding the mechanism of the Povarov reaction. A DFT study RSC Adv. 2014, 4, 25268–25278. [Google Scholar] [CrossRef]
- Rios-Gutierrez, M.; Layeb, H.; Domingo, L.R. A DFT Study of the Mechanisms of Bronsted Acid Catalysed Povarov Reactions. Tetrahedron 2015, 71, 9339–9345. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Tang, Y.H.; Tal, Y.; Biegler-König, F.W. Properties of toms and bonds in hydrocarbon molecules. J. Am. Chem. Soc. 1982, 104, 946–952. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, NY, USA, 1994. [Google Scholar]
- Gatti, C. Chemical Bonding in Crystals: New Directions. Z. Krist. 2005, 220, 399–457. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Silvi, B. The Synaptic Order: A Key Concept to Understand Multicenter Bonding. J. Mol. Struct. 2002, 614, 3–10. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Gervasio, G.; Bianchi, R.; Marabello, D. About the Topological Classification of the Metal-Metal Bond. Chem. Phys. Lett. 2004, 387, 481–484. [Google Scholar] [CrossRef]
- Mierzwa, G.; Gordon, A.J.; Berski, S. The nature of multiple boron-nitrogen bonds studied using electron localization function (ELF), electron density (AIM), and natural bond orbital (NBO) methods. J. Mol. Model. 2020, 26, 136. [Google Scholar] [CrossRef]
- Andrés, J.; Domingo, L.R.; Picher, M.T.; Safont, V.S. Comparative theoretical study of transition structures, barrier heights, and reaction energies for the intramolecular tautomerization in acetaldehyde vinyl alcohol and acetaldimine/vinylamine systems. Int. J. Quant. Chem. 1998, 66, 9–24. [Google Scholar] [CrossRef]
- Emamian, S.R.; Domingo, L.R.; Tayyari, S.F. Tautomerism in pyridazin-3(2H)-one: A theoretical study using implicit/explicit solvation models. J. Mol. Graph. Model. 2014, 49, 47–54. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Berski, S.; Latajka, Z.; Gordon, A.J. Theoretical insights and quantitative rediction of the nature of boron–chalcogen (O, S, Se, Te) interactions using the electron density and the electron localisation function (ELF). New J. Chem. 2011, 35, 89–96. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the Electronic Reorganization along the Non-polar [3 + 2] cycloaddition reactions of carbonyl ylides. J. Org. Chem. 2011, 76, 373–379. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Falcioni, F.; Domingo, L.R.; Popelier, P.L.A. A Combined BET and IQA-REG Study of the Activation Energy of non-polar zw-type [3+2] Cycloaddition Reactions. Phys. Chem. Chem. Phys. 2023, 25, 10853–10865. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Phys. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hehre, M.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions–Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, 6th ed.; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Douglas, D.H.; Peucker, T.K. Algorithms for the reduction of the number of points required to represent a digitized line or its caricature. Cartographica 1973, 10, 112–122. [Google Scholar] [CrossRef]
- Ramer, U. An iterative procedure for the polygonal approximation of plane curves. Comput. Graph. Image Process. 1972, 1, 244–256. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Popelier, P.L.A. The ANANKE relative energy gradient (REG) method to automate IQA analysis over configurational change. Theor. Chem. Acc. 2017, 136, 86. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019; Available online: aim.tkgristmill.com (accessed on 8 August 2023).
- Falcioni, F.; Duarte Jose, L.; Popelier, L.A.P. REG.py, Version 0.1; Computer Software; 2023. Available online: https://github.com/popelier-group/REG (accessed on 8 August 2023).
- Wilson, A.L.; Popelier, P.L.A. Exponential Relationships Capturing Atomistic Short-Range Repulsion from the Interacting Quantum Atoms (IQA) Method. J. Phys. Chem. A 2016, 120, 9647–9659. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Kosov, D.S. Atom–atom partitioning of intramolecular and intermolecular Coulomb energy. J. Chem. Phys. 2001, 114, 6539–6547. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Joubert, L.; Kosov, D.S. Convergence of the Electrostatic Interaction Based on Topological Atoms. J. Phys. Chem. A 2001, 105, 8254–8261. [Google Scholar] [CrossRef]
- Martín Pendás, A.; Francisco, E.M.; Blanco, A.; Gatti, C. Bond Paths as Privileged Exchange Channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CP1 (N1–B2) | CP2 (B2–N3) | |
|---|---|---|
| Density ρ(r) | 0.2284 | 0.2648 |
| Laplacian ∇2ρ(r) | 0.4435 | 0.9547 |
| G(r) | 0.3463 | 0.5161 |
| V(r) | −0.5817 | −0.7936 |
| H(r) | −0.2354 | −0.2775 |
| G(r)/ρ(r) | 1.5160 | 1.9484 |
| H(r)/ρ(r) | −1.0306 | −1.0473 |
| |V(r)|/G(r) | 1.6797 | 1.5376 |
| ΔH | ΔG | |
|---|---|---|
| TS1-c | 0.3 | 15.8 |
| TS1-t | 1.7 | 16.7 |
| MC-c | 0.7 | 14.9 |
| MC-t | 2.3 | 17.2 |
| TS2-c | 4.0 | 21.1 |
| TS2-t | 2.7 | 19.8 |
| 19-c | −22.6 | −6.8 |
| 19-t | −21.3 | −4.6 |
| 20 | −62.8 | −45.1 |
| TS-3 | 11.1 | 26.4 |
| 21 | −40.1 | −24.4 |
| TS-4 | 7.8 | 19.4 |
| 22 | −27.5 | −14.1 |
| 18 | 2 | TS1-t | MC-t | TS2-t | 19-t | 20 | |
|---|---|---|---|---|---|---|---|
| V(N1,B2) | 2.61 | 2.74 | 2.44 | 2.43 | 2.53 | 2.48 | |
| V(N1) | 2.80 | 2.74 | 2.74 | 2.75 | 2.78 | 2.85 | |
| V(B2,N3) | 3.17 | 1.45 | 1.33 | 2.78 | 2.27 | 2.72 | |
| 2.55 | 1.80 | 1.82 | |||||
| V(N3,H) | 2.14 | ||||||
| V(N3) | 2.48 | 2.55 | 2.72 | 2.62 | 1.19 | ||
| V(N3,C4) | 2.01 | 2.00 | 2.01 | 2.18 | 2.75 | 1.91 | |
| V(C4,C5) | 2.75 | 2.67 | 2.65 | 2.53 | 2.07 | 2.79 | |
| V(C5,C6) | 1.9 | 2.08 | |||||
| V(C6,O7) | 2.36 | 2.10 | 2.05 | 1.82 | 1.36 | 1.33 | |
| V(O7) | 2.63 | 2.81 | 2.89 | 3.17 | 4.28 | 4.3 | |
| V(O7) | 2.59 | 2.66 | 2.66 | 2.65 | |||
| V(B2,O7) | 2.01 | 2.02 |
| MC-t | 19-t | 20 | |||
|---|---|---|---|---|---|
| CP1 (B2–O7) | CP1 (B2–O7) | CP2 (C5–C6) | CP1 (B2–O7) | CP2 (C5–C6) | |
| Density ρ(r) | 0.0938 | 0.1833 | 0.2374 | 0.1902 | 0.2533 |
| Laplacian ∇2ρ(r) | 0.3036 | 0.8081 | −0.5244 | 0.8242 | −0.6088 |
| G(r) | 0.1316 | 0.3405 | 0.0547 | 0.3538 | 0.0571 |
| V(r) | −0.1872 | −0.4791 | −0.2406 | −0.5015 | −0.2664 |
| H(r) | −0.0557 | −0.1386 | −0.1858 | −0.1476 | −0.2093 |
| G(r)/ρ(r) | 1.4030 | 1.8576 | 0.2304 | 1.8601 | 0.2254 |
| H(r)/ρ(r) | −0.5938 | −0.7561 | −0.7826 | −0.7760 | −0.8263 |
| |V(r)|/G(r) | 1.4228 | 1.4070 | 4.3983 | 1.4176 | 4.6626 |
| Structures | MCr-t | TS2r-t | S1 | S2 | S3 | S4 | S5 |
|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | ||
| d(B2–O7) | 1.657 | 1.630 | 1.586 | 1.542 | 1.471 | 1.456 | 1.418 |
| d(C5–C6) | 2.829 | 2.461 | 2.39 | 2.284 | 1.995 | 1.906 | 1.575 |
| ΔE | 0.0 | 3.3 | 3.2 | 2.7 | −3.3 | −6.6 | −21.1 |
| V(N1,B2) | 2.66 | 2.43 | 2.58 | 2.56 | 2.56 | 2.55 | 2.55 |
| V(B2,N3) | 3.03 | 2.74 | 2.68 | 2.61 | 2.45 | 2.41 | 2.28 |
| V(N3) | 2.66 | 2.77 | 2.77 | 2.75 | 2.66 | 2.63 | 2.61 |
| V(N3,C4) | 2.02 | 2.19 | 2.23 | 2.30 | 2.53 | 2.59 | 2.74 |
| V(C4,C5) | 2.65 | 2.76 | 2.78 | 2.55 | 2.29 | 2.23 | 2.08 |
| V(C6,O7) | 2.16 | 1.89 | 1.83 | 1.73 | 1.52 | 1.49 | 1.37 |
| V(O7) | 5.41 * | 5.75 * | 5.28 * | 5.10 * | 4.74 * | 4.61 | 4.40 |
| V(B2,O7) | 0.52 | 0.81 | 1.39 | 1.53 | 1.88 | ||
| V(C5) | 0.26 | ||||||
| V(C6) | 0.12 | ||||||
| V(C5,C6) | 1.17 | 1.34 | 1.81 | ||||
| Energy Term | REG | ||
|---|---|---|---|
| Vinter(C6,O7) | 72.42 | 50.74 | 21.68 |
| Vinter(B2,N3) | 60.39 | 47.69 | 12.71 |
| Vinter(B2,C4) | 30.40 | 30.36 | 0.04 |
| Vinter(N3,C6) | 21.61 | 21.67 | −0.06 |
| Vinter(N1,O7) | 18.28 | 20.43 | −2.16 |
| Vinter(C4,O7) | −22.44 | −21.79 | −0.65 |
| Eintra(C6) | −31.07 | ||
| Vinter(C5,C6) | −38.14 | −2.30 | −35.83 |
| Vinter(N3,C4) | −56.85 | −35.99 | −20.85 |
| Vinter(B2,O7) | −103.97 | −92.58 | −11.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Aurell, M.J.; Ríos-Gutiérrez, M. A Molecular Electron Density Theory Study of the Domino Reaction of N-Phenyl Iminoboranes with Benzaldehyde Yielding Fused Bicyclic Compounds. Molecules 2023, 28, 6211. https://doi.org/10.3390/molecules28176211
Domingo LR, Aurell MJ, Ríos-Gutiérrez M. A Molecular Electron Density Theory Study of the Domino Reaction of N-Phenyl Iminoboranes with Benzaldehyde Yielding Fused Bicyclic Compounds. Molecules. 2023; 28(17):6211. https://doi.org/10.3390/molecules28176211
Chicago/Turabian StyleDomingo, Luis R., María José Aurell, and Mar Ríos-Gutiérrez. 2023. "A Molecular Electron Density Theory Study of the Domino Reaction of N-Phenyl Iminoboranes with Benzaldehyde Yielding Fused Bicyclic Compounds" Molecules 28, no. 17: 6211. https://doi.org/10.3390/molecules28176211
APA StyleDomingo, L. R., Aurell, M. J., & Ríos-Gutiérrez, M. (2023). A Molecular Electron Density Theory Study of the Domino Reaction of N-Phenyl Iminoboranes with Benzaldehyde Yielding Fused Bicyclic Compounds. Molecules, 28(17), 6211. https://doi.org/10.3390/molecules28176211