
Recent Advances in C–H Functionalisation through Indirect Hydrogen Atom Transfer †


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. HAT Background and Mechanism
1.2. Indirect HAT
2. C–H Functionalisation Using HAT Chemistry
2.1. Nitrogen-Based HAT Reagents
2.1.1. Quinuclidine and DABCO-Style HAT Reagents
2.1.2. Amide HAT Reagents
2.1.3. Azidyl Radical as a HAT Reagent
2.2. Sulfur-Based HAT Agents
2.2.1. Thiols and Thioacid HAT Reagents
2.2.2. BINOL-Derived Thiophosphoric Acids
2.3. Oxygen-Based HAT Reagents
2.3.1. Pyridinium N-Oxide HAT Reagents
2.3.2. Peroxide HAT Reagents
2.3.3. Miscellaneous Oxygen HAT Reagents
2.4. Carbon and Boron HAT Agents
2.4.1. Carbon HAT Reagents
2.4.2. Boron HAT Reagents
3. Conclusions and Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| (TRIPS)2 | Bis(2,4,6-triisopropylphenyl) disulfide |
| 4CzIPN | 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene |
| Acr | Acridinium |
| Ar | Aryl |
| Bn | Benzyl |
| Boc | tert-butyloxycarbonyl |
| BOX | Bis(oxazoline) (ligands) |
| Bz | Benzoyl |
| CAD | Catalytic Acceptorless Dehydrogenation |
| Cbz | Carboxybenzyl |
| CT | Chain transfer |
| Cz | Carbazolyl |
| DABCO | Diazabicyclooctane |
| DCE | 1,2-Dichloroethane |
| DFT | Density-functional theory |
| DMA | Dimethylacetamide |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| dr | Diastereomeric ratio |
| Dtbbpy | Di–tert-butylbipyridyl |
| EAC | Electron acceptor catalyst |
| EDA | Electron–donor–acceptor |
| ee | Enantiomeric excess |
| er | Enantiomeric ratio |
| EWG | Electron-withdrawing group |
| HAT | Hydrogen atom transfer |
| HFIP | Hexafluoroisopropanol |
| IBX | 2-Iodoxybenzoic acid |
| iPr | Isopropyl |
| LED | Light-emitting diode |
| LSF | Late-stage functionalisation |
| MesAcr | Mesityl acridinium |
| MLCT | Metal–ligand charge transfer |
| OA | Oxidation addition |
| PCET | Proton-coupled electron transfer |
| PFTB | Perfluoro-tert-butanol |
| PRC | Polarity reversal catalysis |
| PTH | N-phenylphenothiazine |
| Pyf | Tetrafluoropyridinyl |
| RE | Reductive elimination |
| SCE | Saturated calomel electrode |
| SCS | Spin-centred-shift |
| SET | Single electron transfer |
| SFL | Sulfolane |
| SOMO | Singly occupied molecular orbital |
| TBAB | Tetrabutylammonium bromide |
| tBu | tert-butyl |
| TEDA2+ | Selectfluor Radical Dication |
| TIPS | Triisopropylsilane |
| TMS | Trimethylsilyl |
| TMS | Trimethylsilyl group |
| TPI | Thiophosphoric imide |
References
- Lam, N.Y.S.; Wu, K.; Yu, J.-Q. Advancing the Logic of Chemical Synthesis: C–H Activation as Strategic and Tactical Disconnections for C–C Bond Construction. Angew. Chem. Int. Ed. 2021, 60, 15767–15790. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.C.K.; Rovis, T. Complementary Strategies for Directed C(sp3)–H Functionalisation: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed. 2018, 57, 62–101. [Google Scholar] [CrossRef] [PubMed]
- Abrams, D.J.; Provencher, P.A.; Sorensen, E.J. Recent applications of C–H functionalisation in complex natural product synthesis. Chem. Soc. Rev. 2018, 47, 8925–8967. [Google Scholar] [CrossRef]
- Yoshioka, S.; Nagatomo, M.; Inoue, M. Application of Two Direct C(sp3)–H Functionalisations for Total Synthesis of (+)-Lactacystin. Org. Lett. 2015, 17, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Falcone, N.A.; Bosse, A.T.; Park, H.; Yu, J.-Q.; Davies, H.M.L.; Sorensen, E.J. A C–H Functionalization Strategy Enables an Enantioselective Formal Synthesis of (–)-Aflatoxin B2. Org. Lett. 2021, 23, 9393–9397. [Google Scholar] [CrossRef] [PubMed]
- Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L.L.; Musaev, D.G.; Wencel-Delord, J.; Roberts, C.A.; et al. C–H activation. Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar] [CrossRef]
- Capaldo, L.; Quadri, L.L.; Ravelli, D. Photocatalytic hydrogen atom transfer: The philosopher’s stone for late-stage functionalisation? Green Chem. 2020, 22, 3376–3396. [Google Scholar] [CrossRef]
- Barham, J.P.; John, M.P.; Murphy, J.A. Contra-thermodynamic Hydrogen Atom Abstraction in the Selective C–H Functionalisation of Trialkylamine N-CH3 Groups. J. Am. Chem. Soc. 2016, 138, 15482–15487. [Google Scholar] [CrossRef]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalisation of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef]
- Candish, L.; Collins, K.D.; Cook, G.C.; Douglas, J.J.; Gómez-Suárez, A.; Jolit, A.; Keess, S. Photocatalysis in the Life Science Industry. Chem. Rev. 2022, 122, 2907–2980. [Google Scholar] [CrossRef]
- Karimov, R.R.; Hartwig, J.F. Transition-Metal-Catalyzed Selective Functionalisation of C(sp3)–H Bonds in Natural Products. Angew. Chem. Int. Ed. 2018, 57, 4234–4241. [Google Scholar] [CrossRef]
- Britton, L.; Docherty, J.; Sklyaruk, J.; Cooney, J.; Nichol, G.S.; Dominey, A.; Thomas, S.P. Iron-catalysed Alkene and Heteroarene H/D Exchange by Reversible Protonation of Iron-hydride Intermediates. Chem. Sci. 2022, 13, 10291–10298. [Google Scholar] [CrossRef]
- Britton, L.; Skrodzki, M.; Nichol, G.S.; Dominey, A.P.; Pawluć, P.; Docherty, J.H.; Thomas, S.P. Manganese-Catalyzed C(sp2)–H Borylation of Furan and Thiophene Derivatives. ACS Catal. 2021, 11, 6857–6864. [Google Scholar] [CrossRef]
- Britton, L.; Docherty, J.H.; Dominey, A.P.; Thomas, S.P. Iron-Catalysed C(sp2)–H Borylation Enabled by Carboxylate Activation. Molecules. 2020, 25, 905. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, Z.; Wu, K.; Ma, J.; Zhou, Y.-G.; Yu, Z. Recent advances in transition-metal-catalyzed carbene insertion to C–H bonds. Chem. Soc. Rev. 2022, 51, 2759–2852. [Google Scholar] [CrossRef]
- Han, F.; Choi, P.H.; Ye, C.-X.; Grell, Y.; Xie, X.; Ivlev, S.I.; Chen, S.; Meggers, E. Cyclometalated Chiral-at-Ruthenium Catalyst for Enantioselective Ring-Closing C(sp3)–H Carbene Insertion to Access Chiral Flavanones. ACS Catal. 2022, 12, 10304–10312. [Google Scholar] [CrossRef]
- Peil, S.; Gutiérrez González, A.; Leutzsch, M.; Fürstner, A. C–H Insertion via Ruthenium Catalyzed gem-Hydrogenation of 1,3-Enynes. J. Am. Chem. Soc. 2022, 144, 4158–4167. [Google Scholar] [CrossRef]
- Romero, E.; Jones, B.S.; Hogg, B.N.; Rué Casamajo, A.; Hayes, M.A.; Flitsch, S.L.; Turner, N.J.; Schnepel, C. Enzymatic Late-Stage Modifications: Better Late Than Never. Angew. Chem. Int. Ed. 2021, 60, 16824–16855. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. [Google Scholar] [CrossRef]
- Golden, D.L.; Suh, S.-E.; Stahl, S.S. Radical C(sp3)–H functionalisation and cross-coupling reactions. Nat. Rev. Chem. 2022, 6, 405–427. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S. If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Quevedo, R.E.; Kohrt, J.T.; Oderinde, M.S.; Reilly, U.; White, M.C. Late-stage oxidative C(sp3)–H methylation. Nature. 2020, 580, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Luan, Y.-X.; Lam, N.Y.S.; Li, J.-F.; Li, Y.; Ye, M.; Yu, J.-Q. A directive Ni catalyst overrides conventional site selectivity in pyridine C–H alkenylation. Nat. Chem. 2021, 13, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Lo, J.C.; Edwards, J.T.; Baran, P.S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. [Google Scholar] [CrossRef] [PubMed]
- Crespi, S.; Fagnoni, M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790–9833. [Google Scholar] [CrossRef] [PubMed]
- Leifert, D.; Studer, A. The Persistent Radical Effect in Organic Synthesis. Angew. Chem. Int. Ed. 2020, 59, 74–108. [Google Scholar] [CrossRef]
- Blakemore, D.C.; Castro, L.; Churcher, I.; Rees, D.C.; Thomas, A.W.; Wilson, D.M.; Wood, A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018, 10, 383–394. [Google Scholar] [CrossRef]
- Le Vaillant, F.; Waser, J. Alkynylation of radicals: Spotlight on the “Third Way” to transfer triple bonds. Chem. Sci. 2019, 10, 8909–8923. [Google Scholar] [CrossRef]
- Bietti, M. Activation and Deactivation Strategies Promoted by Medium Effects for Selective Aliphatic C–H Bond Functionalisation. Angew. Chem. Int. Ed. 2018, 57, 16618–16637. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, M.; Salamone, M.; Bietti, M. Electronic control over site-selectivity in hydrogen atom transfer (HAT) based C(sp3)–H functionalisation promoted by electrophilic reagents. Chem. Soc. Rev. 2022, 51, 2171–2223. [Google Scholar] [CrossRef]
- Bonciolini, S.; Noël, T.; Capaldo, L. Synthetic Applications of Photocatalyzed Halogen-radical mediated Hydrogen Atom Transfer for C–H Bond Functionalisation. Eur. J. Org. Chem. 2022, 2022, e202200417. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Li, J.; Li, C.-J. Photocatalytic C(sp3) radical generation via C–H, C–C, and C–X bond cleavage. Chem. Sci. 2022, 13, 5465–5504. [Google Scholar] [CrossRef] [PubMed]
- Holmberg-Douglas, N.; Nicewicz, D.A. Photoredox-Catalyzed C–H Functionalisation Reactions. Chem. Rev. 2022, 122, 1925–2016. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wang, S.; An, Q.; Liu, L.; Wang, H.; Li, Y.; Feng, K.; Zuo, Z. Resurgence and advancement of photochemical hydrogen atom transfer processes in selective alkane functionalisations. Chem. Sci. 2023, 14, 6841–6859. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Lin, Y.-M.; Gong, L. The Merger of Photocatalyzed Hydrogen Atom Transfer with Transition Metal Catalysis for C–H Functionalisation of Alkanes and Cycloalkanes. Eur. J. Org. Chem. 2021, 2021, 5545–5556. [Google Scholar] [CrossRef]
- Zhang, S.; Findlater, M. Electrochemically Driven Hydrogen Atom Transfer Catalysis: A Tool for C(sp3)/Si–H Functionalisation and Hydrofunctionalisation of Alkenes. ACS Catal. 2023, 13, 8731–8751. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Rubio, Á.; Martínez-Balart, P.; Álvarez-Constantino, A.M.; Fañanás-Mastral, M. C–C bond formation via photocatalytic direct functionalisation of simple alkanes. Chem. Commun. 2023, 59, 9424–9444. [Google Scholar] [CrossRef]
- Turner, O.J.; Murphy, J.A.; Hirst, D.J.; Talbot, E.P.A. Hydrogen Atom Transfer-Mediated Cyclisations of Nitriles. Chem. Eur. J. 2018, 24, 18658–18662. [Google Scholar] [CrossRef]
- Turner, O.J.; Hirst, D.J.; Murphy, J.A. Hydrogen Atom Transfer-Mediated Domino Cyclisation Reaction to Access (Spiro)Quinazolinones. Chem. Eur. J. 2020, 26, 3026–3029. [Google Scholar] [CrossRef]
- Obradors, C.; Martinez, R.M.; Shenvi, R.A. Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers. J. Am. Chem. Soc. 2016, 138, 4962–4971. [Google Scholar] [CrossRef]
- Matos, J.L.M.; Vásquez-Céspedes, S.; Gu, J.; Oguma, T.; Shenvi, R.A. Branch-Selective Addition of Unactivated Olefins into Imines and Aldehydes. J. Am. Chem. Soc. 2018, 140, 16976–16981. [Google Scholar] [CrossRef] [PubMed]
- Crossley, S.W.M.; Obradors, C.; Martinez, R.M.; Shenvi, R.A. Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalisations of Olefins. Chem. Rev. 2016, 116, 8912–9000. [Google Scholar] [CrossRef] [PubMed]
- Green, S.A.; Matos, J.L.M.; Yagi, A.; Shenvi, R.A. Branch-Selective Hydroarylation: Iodoarene–Olefin Cross-Coupling. J. Am. Chem. Soc. 2016, 138, 12779–12782. [Google Scholar] [CrossRef]
- Iwasaki, K.; Wan, K.K.; Oppedisano, A.; Crossley, S.W.M.; Shenvi, R.A. Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc. 2014, 136, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Green, S.A.; Crossley, S.W.M.; Matos, J.L.M.; Vásquez-Céspedes, S.; Shevick, S.L.; Shenvi, R.A. The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer. Acc. Chem. Res. 2018, 51, 2628–2640. [Google Scholar] [CrossRef] [PubMed]
- Green, S.A.; Huffman, T.R.; McCourt, R.O.; van der Puyl, V.; Shenvi, R.A. Hydroalkylation of Olefins to form Quaternary Carbons. J. Am. Chem. Soc. 2019, 141, 7709–7714. [Google Scholar] [CrossRef]
- Wu, J.; Ma, Z. Metal-hydride hydrogen atom transfer (MHAT) reactions in natural product synthesis. Org. Chem. Front. 2021, 8, 7050–7076. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D.; Fagnoni, M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924. [Google Scholar] [CrossRef]
- Gu, Y.; Yin, H.; Wakeling, M.; An, J.; Martin, R. Defunctionalisation of sp3 C–Heteroatom and sp3 C–C Bonds Enabled by Photoexcited Triplet Ketone Catalysts. ACS Catal. 2022, 12, 1031–1036. [Google Scholar] [CrossRef]
- Xu, S.; Ping, Y.; Li, W.; Guo, H.; Su, Y.; Li, Z.; Wang, M.; Kong, W. Enantioselective C(sp3)–H Functionalisation of Oxacycles via Photo-HAT/Nickel Dual Catalysis. J. Am. Chem. Soc. 2023, 145, 5231–5241. [Google Scholar] [CrossRef]
- Sanjosé-Orduna, J.; Silva, R.C.; Raymenants, F.; Reus, B.; Thaens, J.; de Oliveira, K.T.; Noël, T. Dual role of benzophenone enables a fast and scalable C-4 selective alkylation of pyridines in flow. Chem. Sci. 2022, 13, 12527–12532. [Google Scholar] [CrossRef]
- Shen, Y.; Gu, Y.; Martin, R. sp3 C–H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc. 2018, 140, 12200–12209. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.W.; Yuan, M.; Polites, V.C.; Gutierrez, O.; Molander, G.A. Photochemical C–H Activation Enables Nickel-Catalyzed Olefin Dicarbofunctionalisation. J. Am. Chem. Soc. 2021, 143, 3901–3910. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.; Prakash, G.; Teja, C.; Lahiri, G.K.; Maiti, D. Metal-free photoinduced hydrogen atom transfer assisted C(sp3)–H thioarylation. Green Chem. 2023, 25, 3431–3436. [Google Scholar] [CrossRef]
- Masuda, Y.; Tsuda, H.; Murakami, M. C1 Oxidation/C2 Reduction Isomerisation of Unprotected Aldoses Induced by Light/Ketone. Angew. Chem. Int. Ed. 2020, 59, 2755–2759. [Google Scholar] [CrossRef]
- Dewanji, A.; Krach, P.E.; Rueping, M. The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C–H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem. Int. Ed. 2019, 58, 3566–3570. [Google Scholar] [CrossRef]
- Xia, J.-B.; Zhu, C.; Chen, C. Visible Light-Promoted Metal-Free C–H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Difluorination. J. Am. Chem. Soc. 2013, 135, 17494–17500. [Google Scholar] [CrossRef]
- Pulcinella, A.; Bonciolini, S.; Lukas, F.; Sorato, A.; Noël, T. Photocatalytic Alkylation of C(sp3)–H Bonds Using Sulfonylhydrazones. Angew. Chem. Int. Ed. 2023, 62, e202215374. [Google Scholar] [CrossRef]
- Sarver, P.J.; Bacauanu, V.; Schultz, D.M.; DiRocco, D.A.; Lam, Y.-h.; Sherer, E.C.; MacMillan, D.W.C. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem. 2020, 12, 459–467. [Google Scholar] [CrossRef]
- Dong, Y.-J.; Zhu, B.; Liang, Y.-J.; Guan, W.; Su, Z.-M. Origin and Regioselectivity of Direct Hydrogen Atom Transfer Mechanism of C(sp3)–H Arylation by [W10O32]4–/Ni Metallaphotoredox Catalysis. Inorg. Chem. 2021, 60, 18706–18714. [Google Scholar] [CrossRef]
- Capaldo, L.; Bonciolini, S.; Pulcinella, A.; Nuño, M.; Noël, T. Modular allylation of C(sp3)–H bonds by combining decatungstate photocatalysis and HWE olefination in flow. Chem. Sci. 2022, 13, 7325–7331. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Wen, Z.; Laudadio, G.; Capaldo, L.; Lammers, R.; Rincón, J.A.; García-Losada, P.; Mateos, C.; Frederick, M.O.; Broersma, R.; et al. Accelerated and Scalable C(sp3)–H Amination via Decatungstate Photocatalysis Using a Flow Photoreactor Equipped with High-Intensity LEDs. ACS Cent. Sci. 2022, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Mazzarella, D.; Pulcinella, A.; Bovy, L.; Broersma, R.; Noël, T. Rapid and Direct Photocatalytic C(sp3)–H Acylation and Arylation in Flow. Angew. Chem. Int. Ed. 2021, 60, 21277–21282. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Maheshwari, A.; Sambiagio, C.; Deng, Y.; Laudadio, G.; Van Aken, K.; Sun, Y.; Gemoets, H.P.L.; Noël, T. Optimisation of a Decatungstate-Catalyzed C(sp3)–H Alkylation Using a Continuous Oscillatory Millistructured Photoreactor. Org. Process Res. Dev. 2020, 24, 2356–2361. [Google Scholar] [CrossRef]
- Wan, T.; Capaldo, L.; Laudadio, G.; Nyuchev, A.V.; Rincón, J.A.; García-Losada, P.; Mateos, C.; Frederick, M.O.; Nuño, M.; Noël, T. Decatungstate-Mediated C(sp3)–H Heteroarylation via Radical-Polar Crossover in Batch and Flow. Angew. Chem. Int. Ed. 2021, 60, 17893–17897. [Google Scholar] [CrossRef]
- Laudadio, G.; Deng, Y.; van der Wal, K.; Ravelli, D.; Nuño, M.; Fagnoni, M.; Guthrie, D.; Sun, Y.; Noël, T. C(sp(3))–H functionalisations of light hydrocarbons using decatungstate photocatalysis in flow. Science 2020, 369, 92–96. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D. Decatungstate as Direct Hydrogen Atom Transfer Photocatalyst for SOMOphilic Alkynylation. Org. Lett. 2021, 23, 2243–2247. [Google Scholar] [CrossRef]
- Jorea, A.; Bassetti, B.; Gervasoni, K.; Protti, S.; Palmieri, A.; Ravelli, D. More Chips to Nitroolefins: Decatungstate Photocatalysed Hydroalkylation Under Batch and Flow Conditions. Adv. Synth. Catal. 2023, 365, 722–727. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Shih, Y.-L.; Wu, Y.-K.; Ryu, I. Site-Selective C(sp3)–H Alkenylation Using Decatungstate Anion as Photocatalyst. Adv. Synth. Catal. 2022, 364, 1039–1043. [Google Scholar] [CrossRef]
- Fukuyama, T.; Yamada, K.; Nishikawa, T.; Ravelli, D.; Fagnoni, M.; Ryu, I. Site-selectivity in TBADT-photocatalyzed C(sp3)–H Functionalisation of Saturated Alcohols and Alkanes. Chem. Lett. 2018, 47, 207–209. [Google Scholar] [CrossRef]
- Ravelli, D.; Fagnoni, M.; Fukuyama, T.; Nishikawa, T.; Ryu, I. Site-Selective C–H Functionalisation by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal. 2018, 8, 701–713. [Google Scholar] [CrossRef]
- Paolillo, J.M.; Duke, A.D.; Gogarnoiu, E.S.; Wise, D.E.; Parasram, M. Anaerobic Hydroxylation of C(sp3)–H Bonds Enabled by the Synergistic Nature of Photoexcited Nitroarenes. J. Am. Chem. Soc. 2023, 145, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Horn, E.J.; Rosen, B.R.; Chen, Y.; Tang, J.; Chen, K.; Eastgate, M.D.; Baran, P.S. Scalable and sustainable electrochemical allylic C–H oxidation. Nature 2016, 533, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.M. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. [Google Scholar] [CrossRef]
- Lai, W.; Li, C.; Chen, H.; Shaik, S. Hydrogen-Abstraction Reactivity Patterns from A to Y: The Valence Bond Way. Angew. Chem. Int. Ed. 2012, 51, 5556–5578. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.D.; Murphy, J.A. Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents. Chem. Soc. Rev. 2021, 50, 9540–9685. [Google Scholar] [CrossRef]
- Salamone, M.; Bietti, M. Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C–H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects. Acc. Chem. Res. 2015, 48, 2895–2903. [Google Scholar] [CrossRef]
- Zavitsas, A.A.; Pinto, J.A. Meaning of the polar effect in hydrogen abstractions by free radicals. Reactions of the tert-butoxy radical. J. Am. Chem. Soc. 1972, 94, 7390–7396. [Google Scholar] [CrossRef]
- Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, USA, 2002; pp. 18, 38, 41, 68, 69, 80, 146, 169, 170, 172, 209, 240–269, 291, 293, 298. [Google Scholar]
- Salamone, M.; Galeotti, M.; Romero-Montalvo, E.; van Santen, J.A.; Groff, B.D.; Mayer, J.M.; DiLabio, G.A.; Bietti, M. Bimodal Evans–Polanyi Relationships in Hydrogen Atom Transfer from C(sp3)–H Bonds to the Cumyloxyl Radical. A Combined Time-Resolved Kinetic and Computational Study. J. Am. Chem. Soc. 2021, 143, 11759–11776. [Google Scholar] [CrossRef]
- Dénès, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef]
- Ide, T.; Barham, J.P.; Fujita, M.; Kawato, Y.; Egami, H.; Hamashima, Y. Regio- and chemoselective Csp3–H arylation of benzylamines by single electron transfer/hydrogen atom transfer synergistic catalysis. Chem. Sci. 2018, 9, 8453–8460. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.J.; Zhu, Q.; Miller, D.C.; Gu, C.J.; Knowles, R.R. Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 2016, 539, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.P. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: Concepts and applications in organic chemistry. Chem. Soc. Rev. 1999, 28, 25–35. [Google Scholar] [CrossRef]
- Giese, B. Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew. Chem. Int. Ed. 1983, 22, 753–764. [Google Scholar] [CrossRef]
- Tedder, J.M. Which Factors Determine the Reactivity and Regioselectivity of Free Radical Substitution and Addition Reactions? Angew. Chem. Int. Ed. 1982, 21, 401–410. [Google Scholar] [CrossRef]
- Ruffoni, A.; Mykura, R.C.; Bietti, M.; Leonori, D. The interplay of polar effects in controlling the selectivity of radical reactions. Nat. Synth. 2022, 1, 682–695. [Google Scholar] [CrossRef]
- Chan, B.; Easton, C.J.; Radom, L. Outcome-Changing Effect of Polarity Reversal in Hydrogen-Atom-Abstraction Reactions. J. Phys. Chem. A 2015, 119, 3843–3847. [Google Scholar] [CrossRef]
- Galeotti, M.; Trasatti, C.; Sisti, S.; Salamone, M.; Bietti, M. Factors Governing Reactivity and Selectivity in Hydrogen Atom Transfer from C(sp3)–H Bonds of Nitrogen-Containing Heterocycles to the Cumyloxyl Radical. J. Org. Chem. 2022, 87, 7456–7463. [Google Scholar] [CrossRef]
- Liu, F.; Ma, S.; Lu, Z.; Nangia, A.; Duan, M.; Yu, Y.; Xu, G.; Mei, Y.; Bietti, M.; Houk, K.N. Hydrogen Abstraction by Alkoxyl Radicals: Computational Studies of Thermodynamic and Polarity Effects on Reactivities and Selectivities. J. Am. Chem. Soc. 2022, 144, 6802–6812. [Google Scholar] [CrossRef]
- Martin, T.; Galeotti, M.; Salamone, M.; Liu, F.; Yu, Y.; Duan, M.; Houk, K.N.; Bietti, M. Deciphering Reactivity and Selectivity Patterns in Aliphatic C–H Bond Oxygenation of Cyclopentane and Cyclohexane Derivatives. J. Org. Chem. 2021, 86, 9925–9937. [Google Scholar] [CrossRef]
- Dantignana, V.; Milan, M.; Cussó, O.; Company, A.; Bietti, M.; Costas, M. Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Salamone, M.; Martin, T.; Milan, M.; Costas, M.; Bietti, M. Electronic and Torsional Effects on Hydrogen Atom Transfer from Aliphatic C–H Bonds: A Kinetic Evaluation via Reaction with the Cumyloxyl Radical. J. Org. Chem. 2017, 82, 13542–13549. [Google Scholar] [CrossRef] [PubMed]
- Bietti, M.; Forcina, V.; Lanzalunga, O.; Lapi, A.; Martin, T.; Mazzonna, M.; Salamone, M. Kinetic Study of the Reaction of the Phthalimide-N-oxyl Radical with Amides: Structural and Medium Effects on the Hydrogen Atom Transfer Reactivity and Selectivity. J. Org. Chem. 2016, 81, 11924–11931. [Google Scholar] [CrossRef] [PubMed]
- Salamone, M.; Mangiacapra, L.; Carboni, G.; Bietti, M. Hydrogen atom transfer from tertiary alkanamides to the cumyloxyl radical. The role of substrate structure on alkali and alkaline earth metal ion induced C–H bond deactivation. Tetrahedron 2016, 72, 7757–7763. [Google Scholar] [CrossRef]
- Salamone, M.; Carboni, G.; Bietti, M. Fine Control over Site and Substrate Selectivity in Hydrogen Atom Transfer-Based Functionalisation of Aliphatic C–H Bonds. J. Org. Chem. 2016, 81, 9269–9278. [Google Scholar] [CrossRef]
- Salamone, M.; DiLabio, G.A.; Bietti, M. Hydrogen Atom Abstraction Reactions from Tertiary Amines by Benzyloxyl and Cumyloxyl Radicals: Influence of Structure on the Rate-Determining Formation of a Hydrogen-Bonded Prereaction Complex. J. Org. Chem. 2011, 76, 6264–6270. [Google Scholar] [CrossRef]
- Salamone, M.; Mangiacapra, L.; Bietti, M. Kinetic Solvent Effects on the Reactions of the Cumyloxyl Radical with Tertiary Amides. Control over the Hydrogen Atom Transfer Reactivity and Selectivity through Solvent Polarity and Hydrogen Bonding. J. Org. Chem. 2015, 80, 1149–1154. [Google Scholar] [CrossRef]
- Salamone, M.; Carboni, G.; Mangiacapra, L.; Bietti, M. Binding to Redox-Inactive Alkali and Alkaline Earth Metal Ions Strongly Deactivates the C–H Bonds of Tertiary Amides toward Hydrogen Atom Transfer to Reactive Oxygen Centered Radicals. J. Org. Chem. 2015, 80, 9214–9223. [Google Scholar] [CrossRef]
- Salamone, M.; Ortega, V.B.; Bietti, M. Enhanced Reactivity in Hydrogen Atom Transfer from Tertiary Sites of Cyclohexanes and Decalins via Strain Release: Equatorial C–H Activation vs Axial C–H Deactivation. J. Org. Chem. 2015, 80, 4710–4715. [Google Scholar] [CrossRef]
- Salamone, M.; Basili, F.; Mele, R.; Cianfanelli, M.; Bietti, M. Reactions of the Cumyloxyl Radical with Secondary Amides. The Influence of Steric and Stereoelectronic Effects on the Hydrogen Atom Transfer Reactivity and Selectivity. Org. Lett. 2014, 16, 6444–6447. [Google Scholar] [CrossRef]
- Milan, M.; Salamone, M.; Bietti, M. Hydrogen Atom Transfer from 1,n-Alkanediamines to the Cumyloxyl Radical. Modulating C–H Deactivation Through Acid–Base Interactions and Solvent Effects. J. Org. Chem. 2014, 79, 5710–5716. [Google Scholar] [CrossRef] [PubMed]
- Kushch, O.V.; Hordieieva, I.O.; Kompanets, M.O.; Zosenko, O.O.; Opeida, I.A.; Shendrik, A.N. Hydrogen Atom Transfer from Benzyl Alcohols to N-Oxyl Radicals. Reactivity Parameters. J. Org. Chem. 2021, 86, 3792–3799. [Google Scholar] [CrossRef] [PubMed]
- Ruffoni, A.; Hampton, C.; Simonetti, M.; Leonori, D. Photoexcited nitroarenes for the oxidative cleavage of alkenes. Nature. 2022, 610, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hampton, C.; Simonetti, M.; Leonori, D. Olefin Dihydroxylation Using Nitroarenes as Photoresponsive Oxidants. Angew. Chem. Int. Ed. 2023, 62, e202214508. [Google Scholar] [CrossRef]
- Gaster, E.; Kozuch, S.; Pappo, D. Selective Aerobic Oxidation of Methylarenes to Benzaldehydes Catalyzed by N-Hydroxyphthalimide and Cobalt(II) Acetate in Hexafluoropropan-2-ol. Angew. Chem. Int. Ed. 2017, 56, 5912–5915. [Google Scholar] [CrossRef] [PubMed]
- Bietti, M.; Salamone, M. Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. The Role of Hydrogen Bonding. Org. Lett. 2010, 12, 3654–3657. [Google Scholar] [CrossRef] [PubMed]
- Bietti, M.; Martella, R.; Salamone, M. Understanding Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. Org. Lett. 2011, 13, 6110–6113. [Google Scholar] [CrossRef]
- Salamone, M.; Giammarioli, I.; Bietti, M. Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. The Importance of Solvent Hydrogen-Bond Interactions with the Substrate and the Abstracting Radical. J. Org. Chem. 2011, 76, 4645–4651. [Google Scholar] [CrossRef]
- Paul, V.; Roberts, B.P. Homolytic reactions of ligated boranes. Part 8. Electron spin resonance studies of radicals derived from ligated alkylboranes. J. Chem. Soc. Perkin Trans. II 1988, 1183–1193. [Google Scholar] [CrossRef]
- Paul, V.; Roberts, B.P.; Willis, C.R. Homolytic reactions of ligated boranes. Part 12. Amine–alkylboranes as polarity reversal catalysts for hydrogen-atom abstraction by t-butoxyl radicals. J. Chem. Soc. Perkin Trans. II 1989, 1953–1961. [Google Scholar] [CrossRef]
- Sheldon, R.A. Synthesis and uses of alkyl hydroperoxides and dialkyl peroxides. In Peroxides; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1983; p. 169. [Google Scholar]
- Okada, K.; Okamoto, K.; Oda, M. A new and practical method of decarboxylation: Photosensitised decarboxylation of N-acyloxyphthalimides via electron-transfer mechanism. J. Am. Chem. Soc. 1988, 110, 8736–8738. [Google Scholar] [CrossRef]
- Toriyama, F.; Cornella, J.; Wimmer, L.; Chen, T.G.; Dixon, D.D.; Creech, G.; Baran, P.S. Redox-Active Esters in Fe-Catalyzed C-C Coupling. J. Am. Chem. Soc. 2016, 138, 11132–11135. [Google Scholar] [CrossRef] [PubMed]
- Cornella, J.; Edwards, J.T.; Qin, T.; Kawamura, S.; Wang, J.; Pan, C.-M.; Gianatassio, R.; Schmidt, M.; Eastgate, M.D.; Baran, P.S. Practical Ni-Catalyzed Aryl–Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc. 2016, 138, 2174–2177. [Google Scholar] [CrossRef] [PubMed]
- Huihui, K.M.M.; Caputo, J.A.; Melchor, Z.; Olivares, A.M.; Spiewak, A.M.; Johnson, K.A.; DiBenedetto, T.A.; Kim, S.; Ackerman, L.K.G.; Weix, D.J. Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc. 2016, 138, 5016–5019. [Google Scholar] [CrossRef] [PubMed]
- Leibler, I.N.-M.; Tekle-Smith, M.A.; Doyle, A.G. A general strategy for C(sp3)–H functionalisation with nucleophiles using methyl radical as a hydrogen atom abstractor. Nat. Commun. 2021, 12, 6950. [Google Scholar] [CrossRef]
- Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.-H.; Chen, J.; Starr, J.T.; Baran, P.S. Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, J.D.; MacMillan, D.W.C. The direct arylation of allylic sp3 C–H bonds via organic and photoredox catalysis. Nature. 2015, 519, 74–77. [Google Scholar] [CrossRef]
- Madani, A.; Anghileri, L.; Heydenreich, M.; Möller, H.M.; Pieber, B. Benzylic Fluorination Induced by a Charge-Transfer Complex with a Solvent-Dependent Selectivity Switch. Org. Lett. 2022, 24, 5376–5380. [Google Scholar] [CrossRef]
- Cismesia, M.A.; Yoon, T.P. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. [Google Scholar] [CrossRef]
- Ye, T.; Zhang, F.-L.; Xia, H.-M.; Zhou, X.; Yu, Z.-X.; Wang, Y.-F. Stereoselective hydrogen atom transfer to acyclic radicals: A switch enabling diastereodivergent borylative radical cascades. Nat. Commun 2022, 13, 426. [Google Scholar] [CrossRef]
- Katz, R.B.; Mistry, J.; Mitchell, M.B. An Improved Method for the Mono-Hydroxymethylation of Pyridines. A Modification of the Minisci Procedure. Synth. Commun. 1989, 19, 317–325. [Google Scholar] [CrossRef]
- Chan, W.C.; Vinod, J.K.; Koide, K. Acetal Addition to Electron-Deficient Alkenes with Hydrogen Atom Transfer as a Radical Chain Propagation Step. J. Org. Chem. 2021, 86, 3674–3682. [Google Scholar] [CrossRef]
- Curran, D.P.; Kim, D.; Liu, H.T.; Shen, W. Translocation of radical sites by intramolecular 1,5-hydrogen atom transfer. J. Am. Chem. Soc. 1988, 110, 5900–5902. [Google Scholar] [CrossRef]
- Takahira, Y.; Chen, M.; Kawamata, Y.; Mykhailiuk, P.; Nakamura, H.; Peters, B.K.; Reisberg, S.H.; Li, C.; Chen, L.; Hoshikawa, T.; et al. Electrochemical C(sp3)–H Fluorination. Synlett 2019, 30, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.S.J.; Phipps, R.J. Recent Advances in Minisci-Type Reactions. Angew. Chem. Int. Ed. 2019, 58, 13666–13699. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.; MacMillan, D.W.C. Photoredox α-Vinylation of α-Amino Acids and N-Aryl Amines. J. Am. Chem. Soc. 2014, 136, 11602–11605. [Google Scholar] [CrossRef]
- Jeffrey, J.L.; Petronijević, F.R.; MacMillan, D.W.C. Selective Radical–Radical Cross-Couplings: Design of a Formal β-Mannich Reaction. J. Am. Chem. Soc. 2015, 137, 8404–8407. [Google Scholar] [CrossRef]
- McManus, J.B.; Onuska, N.P.R.; Nicewicz, D.A. Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 9056–9060. [Google Scholar] [CrossRef]
- Liu, K.; Studer, A. Formal β-C–H Arylation of Aldehydes and Ketones by Cooperative Nickel and Photoredox Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202206533. [Google Scholar] [CrossRef]
- Landwehr, E.M.; Baker, M.A.; Oguma, T.; Burdge, H.E.; Kawajiri, T.; Shenvi, R.A. Concise syntheses of GB22, GB13, and himgaline by cross-coupling and complete reduction. Science. 2022, 375, 1270–1274. [Google Scholar] [CrossRef]
- Shen, Y.; Funez-Ardoiz, I.; Schoenebeck, F.; Rovis, T. Site-Selective α-C–H Functionalisation of Trialkylamines via Reversible Hydrogen Atom Transfer Catalysis. J. Am. Chem. Soc. 2021, 143, 18952–18959. [Google Scholar] [CrossRef] [PubMed]
- Pratley, C.; Fenner, S.; Murphy, J.A. Nitrogen-Centered Radicals in Functionalisation of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev. 2022, 122, 8181–8260. [Google Scholar] [CrossRef] [PubMed]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-Z.; Bordwell, F.G. Gas-Phase and Solution-Phase Homolytic Bond Dissociation Energies of H–N+ Bonds in the Conjugate Acids of Nitrogen Bases. J. Org. Chem. 1996, 61, 4778–4783. [Google Scholar] [CrossRef]
- Twilton, J.; Christensen, M.; DiRocco, D.A.; Ruck, R.T.; Davies, I.W.; MacMillan, D.W.C. Selective Hydrogen Atom Abstraction through Induced Bond Polarisation: Direct α-Arylation of Alcohols through Photoredox, HAT, and Nickel Catalysis. Angew. Chem. Int. Ed. 2018, 57, 5369–5373. [Google Scholar] [CrossRef]
- Jeffrey, J.L.; Terrett, J.A.; MacMillan, D.W. O-H hydrogen bonding promotes H-atom transfer from α C-H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. [Google Scholar] [CrossRef]
- Le, C.; Liang, Y.; Evans, R.W.; Li, X.; MacMillan, D.W.C. Selective sp3 C-H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [Google Scholar] [CrossRef]
- Shaw, M.H.; Shurtleff, V.W.; Terrett, J.A.; Cuthbertson, J.D.; MacMillan, D.W.C. Native functionality in triple catalytic cross-coupling: sp3 C-H bonds as latent nucleophiles. Science 2016, 352, 1304–1308. [Google Scholar] [CrossRef]
- Yang, H.-B.; Feceu, A.; Martin, D.B.C. Catalyst-Controlled C–H Functionalisation of Adamantanes Using Selective H-Atom Transfer. ACS Catal. 2019, 9, 5708–5715. [Google Scholar] [CrossRef]
- Romano, C.; Talavera, L.; Gómez-Bengoa, E.; Martin, R. Conformational Flexibility as a Tool for Enabling Site-Selective Functionalisation of Unactivated sp3 C–O Bonds in Cyclic Acetals. J. Am. Chem. Soc. 2022, 144, 11558–11563. [Google Scholar] [CrossRef]
- Juliá, F.; Constantin, T.; Leonori, D. Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis. Chem. Rev. 2022, 122, 2292–2352. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Rueping, M. Direct Cross-Coupling of Allylic C(sp3)–H Bonds with Aryl- and Vinylbromides by Combined Nickel and Visible-Light Catalysis. Angew. Chem. Int. Ed. 2018, 57, 10333–10337. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.I.; Gygi, D.; Qin, Y.; Zhu, Q.; Johnson, E.J.; Chen, Y.-S.; Nocera, D.G. Taming the Chlorine Radical: Enforcing Steric Control over Chlorine-Radical-Mediated C–H Activation. J. Am. Chem. Soc. 2022, 144, 1464–1472. [Google Scholar] [CrossRef]
- Gygi, D.; Gonzalez, M.I.; Hwang, S.J.; Xia, K.T.; Qin, Y.; Johnson, E.J.; Gygi, F.; Chen, Y.-S.; Nocera, D.G. Capturing the Complete Reaction Profile of a C–H Bond Activation. J. Am. Chem. Soc. 2021, 143, 6060–6064. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-Q.; Wu, Y.; Wang, R.; Song, H.; Liu, Y.; Wang, Q. Photoredox/Hydrogen Atom Transfer Cocatalyzed C–H Difluoroallylation of Amides, Ethers, and Alkyl Aldehydes. Org. Lett. 2021, 23, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; MacMillan, D.W.C. Direct Aldehyde C–H Arylation and Alkylation via the Combination of Nickel, Hydrogen Atom Transfer, and Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 11353–11356. [Google Scholar] [CrossRef] [PubMed]
- Ryu, I.; Sonoda, N. Free-Radical Carbonylations: Then and Now. Angew. Chem. Int. Ed. 1996, 35, 1050–1066. [Google Scholar] [CrossRef]
- Luo, C.; Zhou, Y.; Chen, H.; Wang, T.; Zhang, Z.-B.; Han, P.; Jing, L.-H. Photoredox Metal-Free Allylic Defluorinative Silylation of α-Trifluoromethylstyrenes with Hydrosilanes. Org. Lett. 2022, 24, 4286–4291. [Google Scholar] [CrossRef]
- Zhou, R.; Goh, Y.Y.; Liu, H.; Tao, H.; Li, L.; Wu, J. Visible-Light-Mediated Metal-Free Hydrosilylation of Alkenes through Selective Hydrogen Atom Transfer for Si–H Activation. Angew. Chem. Int. Ed. 2017, 56, 16621–16625. [Google Scholar] [CrossRef]
- Milligan, J.A.; Phelan, J.P.; Polites, V.C.; Kelly, C.B.; Molander, G.A. Radical/Polar Annulation Reactions (RPARs) Enable the Modular Construction of Cyclopropanes. Org. Lett. 2018, 20, 6840–6844. [Google Scholar] [CrossRef]
- Askey, H.E.; Grayson, J.D.; Tibbetts, J.D.; Turner-Dore, J.C.; Holmes, J.M.; Kociok-Kohn, G.; Wrigley, G.L.; Cresswell, A.J. Photocatalytic Hydroaminoalkylation of Styrenes with Unprotected Primary Alkylamines. J. Am. Chem. Soc. 2021, 143, 15936–15945. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Guo, L.; Shi, C.; Zhu, Y.; Yang, C.; Xia, W. Transition Metal-Free Radical α-Oxy C–H Cyclobutylation via Photoinduced Hydrogen Atom Transfer. Adv. Synth. Catal. 2022, 364, 2140–2145. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, X.; Liu, R.; Wu, J. Quinuclidine and its derivatives as hydrogen-atom-transfer catalysts in photoinduced reactions. Chin. Chem. Lett. 2021, 32, 1847–1856. [Google Scholar] [CrossRef]
- Sun, T.; Jin, R.; Yang, Y.; Jia, Y.; Hu, S.; Jin, Y.; Wang, Q.; Li, Z.; Zhang, Y.; Wu, J.; et al. Direct α-C–H Alkylation of Structurally Diverse Alcohols via Combined Tavaborole and Photoredox Catalysis. Org. Lett. 2022, 24, 7637–7642. [Google Scholar] [CrossRef]
- Sakai, K.; Oisaki, K.; Kanai, M. Identification of Bond-Weakening Spirosilane Catalyst for Photoredox α-C–H Alkylation of Alcohols. Adv. Synth. Catal. 2020, 362, 337–343. [Google Scholar] [CrossRef]
- Sakai, K.; Oisaki, K.; Kanai, M. A Bond-Weakening Borinate Catalyst that Improves the Scope of the Photoredox α-C–H Alkylation of Alcohols. Synthesis 2020, 52, 2171–2189. [Google Scholar] [CrossRef]
- Dimakos, V.; Su, H.Y.; Garrett, G.E.; Taylor, M.S. Site-Selective and Stereoselective C–H Alkylations of Carbohydrates via Combined Diarylborinic Acid and Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 5149–5153. [Google Scholar] [CrossRef]
- Dimakos, V.; Gorelik, D.; Su, H.Y.; Garrett, G.E.; Hughes, G.; Shibayama, H.; Taylor, M.S. Site-selective redox isomerisations of furanosides using a combined arylboronic acid/photoredox catalyst system. Chem. Sci. 2020, 11, 1531–1537. [Google Scholar] [CrossRef]
- Merkens, K.; Sanosa, N.; Funes-Ardoiz, I.; Gómez-Suárez, A. Accessing α-Amino Ketyl Radicals from β-Amino Alcohols via Chemoselective Hydrogen Atom Transfer Catalysis. ACS Catal. 2022, 12, 13186–13192. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Ye, J.; Kalvet, I.; Schoenebeck, F.; Rovis, T. Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics. Nat. Chem. 2018, 10, 1037–1041. [Google Scholar] [CrossRef]
- Sakai, K.; Oisaki, K.; Kanai, M. A Germanium Catalyst Accelerates the Photoredox α-C(sp3)–H Alkylation of Primary Amines. Org. Lett. 2022, 24, 3325–3330. [Google Scholar] [CrossRef]
- Alder, R.W.; Arrowsmith, R.J.; Casson, A.; Sessions, R.B.; Heilbronner, E.; Kovac, B.; Huber, H.; Taagepera, M. Proton affinities and ionisation energies of bicyclic amines and diamines. Effects of ring strain and of 3-electron .sigma. bonding. J. Am. Chem. Soc. 1981, 103, 6137–6142. [Google Scholar] [CrossRef]
- Matsumoto, A.; Yamamoto, M.; Maruoka, K. Cationic DABCO-Based Catalyst for Site-Selective C–H Alkylation via Photoinduced Hydrogen-Atom Transfer. ACS Catal. 2022, 12, 2045–2051. [Google Scholar] [CrossRef]
- Aguilar Troyano, F.J.; Merkens, K.; Gómez-Suárez, A. Selectfluor® Radical Dication (TEDA2+.)—A Versatile Species in Modern Synthetic Organic Chemistry. Asian J. Org. Chem. 2020, 9, 992–1007. [Google Scholar] [CrossRef]
- Xiang, M.; Xin, Z.-K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Exploring the Reducing Ability of Organic Dye (Acr+-Mes) for Fluorination and Oxidation of Benzylic C(sp3)–H Bonds under Visible Light Irradiation. Org. Lett. 2017, 19, 3009–3012. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Liu, J.; Liang, X.-A.; Wang, S.; Lei, A. Visible light-induced direct α C–H functionalisation of alcohols. Nat. Commun. 2019, 10, 467. [Google Scholar] [CrossRef]
- Liang, X.-A.; Niu, L.; Wang, S.; Liu, J.; Lei, A. Visible-Light-Induced C(sp3)–H Oxidative Arylation with Heteroarenes. Org. Lett. 2019, 21, 2441–2444. [Google Scholar] [CrossRef]
- Zhao, H.; Jin, J. Visible Light-Promoted Aliphatic C–H Arylation Using Selectfluor as a Hydrogen Atom Transfer Reagent. Org. Lett. 2019, 21, 6179–6184. [Google Scholar] [CrossRef]
- Danahy, K.E.; Cooper, J.C.; Van Humbeck, J.F. Benzylic Fluorination of Aza-Heterocycles Induced by Single-Electron Transfer to Selectfluor. Angew. Chem. Int. Ed. 2018, 57, 5134–5138. [Google Scholar] [CrossRef]
- Ventre, S.; Petronijevic, F.R.; MacMillan, D.W.C. Decarboxylative Fluorination of Aliphatic Carboxylic Acids via Photoredox Catalysis. J. Am. Chem. Soc. 2015, 137, 5654–5657. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-D.; Wang, Y.; Xue, X.-S.; Cheng, J.-P. A Systematic Evaluation of the N–F Bond Strength of Electrophilic N–F Reagents: Hints for Atomic Fluorine Donating Ability. J. Org. Chem. 2017, 82, 4129–4135. [Google Scholar] [CrossRef]
- Capilato, J.N.; Pitts, C.R.; Rowshanpour, R.; Dudding, T.; Lectka, T. Site-Selective Photochemical Fluorination of Ketals: Unanticipated Outcomes in Selectivity and Stability. J. Org. Chem. 2020, 85, 2855–2864. [Google Scholar] [CrossRef]
- Ghorbani, F.; Harry, S.A.; Capilato, J.N.; Pitts, C.R.; Joram, J.; Peters, G.N.; Tovar, J.D.; Smajlagic, I.; Siegler, M.A.; Dudding, T.; et al. Carbonyl-Directed Aliphatic Fluorination: A Special Type of Hydrogen Atom Transfer Beats Out Norrish II. J. Am. Chem. Soc. 2020, 142, 14710–14724. [Google Scholar] [CrossRef] [PubMed]
- Harry, S.A.; Xiang, M.R.; Holt, E.; Zhu, A.; Ghorbani, F.; Patel, D.; Lectka, T. Hydroxy-directed fluorination of remote unactivated C(sp3)–H bonds: A new age of diastereoselective radical fluorination. Chem. Sci. 2022, 13, 7007–7013. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Wang, L.; Nicewicz, D.A. Synthesis and Characterisation of Acridinium Dyes for Photoredox Catalysis. Synlett 2019, 30, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Pangu, A.; Lévesque, F.; Roth, H.G.; Oliver, S.F.; Campeau, L.-C.; Nicewicz, D.; DiRocco, D.A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81, 7244–7249. [Google Scholar] [CrossRef]
- Bortolamei, N.; Isse, A.A.; Gennaro, A. Estimation of standard reduction potentials of alkyl radicals involved in atom transfer radical polymerisation. Electrochim. Acta 2010, 55, 8312–8318. [Google Scholar] [CrossRef]
- Davies, J.; Lyonnet, J.R.; Zimin, D.P.; Martin, R. The road to industrialisation of fine chemical carboxylation reactions. Chem 2021, 7, 2927–2942. [Google Scholar] [CrossRef]
- Lamy, E.; Nadjo, L.; Saveant, J.M. Standard potential and kinetic parameters of the electrochemical reduction of carbon dioxide in dimethylformamide. J. Electroanal. Chem. Interf. Electrochem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- Chmiel, A.F.; Williams, O.P.; Chernowsky, C.P.; Yeung, C.S.; Wickens, Z.K. Non-innocent Radical Ion Intermediates in Photoredox Catalysis: Parallel Reduction Modes Enable Coupling of Diverse Aryl Chlorides. J. Am. Chem. Soc. 2021, 143, 10882–10889. [Google Scholar] [CrossRef] [PubMed]
- Hendy, C.M.; Smith, G.C.; Xu, Z.; Lian, T.; Jui, N.T. Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2•−). J. Am. Chem. Soc. 2021, 143, 8987–8992. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gao, Y.; Zhou, C.; Li, G. Visible-Light-Driven Reductive Carboarylation of Styrenes with CO2 and Aryl Halides. J. Am. Chem. Soc. 2020, 142, 8122–8129. [Google Scholar] [CrossRef] [PubMed]
- Grills, D.C.; Lymar, S.V. Radiolytic formation of the carbon dioxide radical anion in acetonitrile revealed by transient IR spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 10011–10017. [Google Scholar] [CrossRef]
- Till, N.A.; Tian, L.; Dong, Z.; Scholes, G.D.; MacMillan, D.W.C. Mechanistic Analysis of Metallaphotoredox C–N Coupling: Photocatalysis Initiates and Perpetuates Ni(I)/Ni(III) Coupling Activity. J. Am. Chem. Soc. 2020, 142, 15830–15841. [Google Scholar] [CrossRef]
- Xu, P.; Wang, S.; Xu, H.; Liu, Y.-Q.; Li, R.-B.; Liu, W.-W.; Wang, X.-Y.; Zou, M.-L.; Zhou, Y.; Guo, D.; et al. Dicarboxylation of Alkenes with CO2 and Formate via Photoredox Catalysis. ACS Catal. 2023, 13, 2149–2155. [Google Scholar] [CrossRef]
- Mangaonkar, S.R.; Hayashi, H.; Takano, H.; Kanna, W.; Maeda, S.; Mita, T. Photoredox/HAT-Catalyzed Dearomative Nucleophilic Addition of the CO2 Radical Anion to (Hetero)Aromatics. ACS Catal. 2023, 13, 2482–2488. [Google Scholar] [CrossRef]
- Guo, W.; Wang, Q.; Zhu, J. Visible light photoredox-catalysed remote C–H functionalisation enabled by 1,5-hydrogen atom transfer (1,5-HAT). Chem. Soc. Rev. 2021, 50, 7359–7377. [Google Scholar] [CrossRef]
- Mao, R.; Bera, S.; Turla, A.C.; Hu, X. Copper-Catalyzed Intermolecular Functionalisation of Unactivated C(sp3)–H Bonds and Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2021, 143, 14667–14675. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Nechab, M.; Morlet-Savary, F.; Dietlin, C.; Graff, B.; Gigmes, D.; Fouassier, J.-P.; Lalevée, J. Stable copper acetylacetonate-based oxidizing agents in redox (NIR photoactivated) polymerisation: An opportunity for the one pot grafting from approach and an example on a 3D printed object. Polym. Chem. 2018, 9, 2173–2182. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Morlet-Savary, F.; Dietlin, C.; Fouassier, J.P.; Lalevée, J. A New Highly Efficient Amine-Free and Peroxide-Free Redox System for Free Radical Polymerisation under Air with Possible Light Activation. Macromolecules 2016, 49, 6296–6309. [Google Scholar] [CrossRef]
- Vasilopoulos, A.; Golden, D.L.; Buss, J.A.; Stahl, S.S. Copper-Catalyzed C–H Fluorination/Functionalisation Sequence Enabling Benzylic C–H Cross Coupling with Diverse Nucleophiles. Org. Lett. 2020, 22, 5753–5757. [Google Scholar] [CrossRef] [PubMed]
- Pitts, C.R.; Bloom, S.; Woltornist, R.; Auvenshine, D.J.; Ryzhkov, L.R.; Siegler, M.A.; Lectka, T. Direct, Catalytic Monofluorination of sp3 C–H Bonds: A Radical-Based Mechanism with Ionic Selectivity. J. Am. Chem. Soc. 2014, 136, 9780–9791. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, H.; Lv, Y.; Tan, X.; Shen, H.; Yu, H.-Z.; Li, C. Radical Carbofluorination of Unactivated Alkenes with Fluoride Ions. J. Am. Chem. Soc. 2018, 140, 6169–6175. [Google Scholar] [CrossRef]
- Na, C.G.; Ravelli, D.; Alexanian, E.J. Direct Decarboxylative Functionalisation of Carboxylic Acids via O–H Hydrogen Atom Transfer. J. Am. Chem. Soc. 2020, 142, 44–49. [Google Scholar] [CrossRef]
- Quinn, R.K.; Könst, Z.A.; Michalak, S.E.; Schmidt, Y.; Szklarski, A.R.; Flores, A.R.; Nam, S.; Horne, D.A.; Vanderwal, C.D.; Alexanian, E.J. Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. [Google Scholar] [CrossRef]
- Tierney, M.M.; Crespi, S.; Ravelli, D.; Alexanian, E.J. Identifying Amidyl Radicals for Intermolecular C–H Functionalisations. J. Org. Chem. 2019, 84, 12983–12991. [Google Scholar] [CrossRef]
- Williamson, J.B.; Na, C.G.; Johnson, R.R.; Daniel, W.F.M.; Alexanian, E.J.; Leibfarth, F.A. Chemo- and Regioselective Functionalisation of Isotactic Polypropylene: A Mechanistic and Structure–Property Study. J. Am. Chem. Soc. 2019, 141, 12815–12823. [Google Scholar] [CrossRef]
- Schmidt, V.A.; Quinn, R.K.; Brusoe, A.T.; Alexanian, E.J. Site-Selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light. J. Am. Chem. Soc. 2014, 136, 14389–14392. [Google Scholar] [CrossRef]
- Liang, L.; Guo, G.; Li, C.; Wang, S.-L.; Wang, Y.-H.; Guo, H.-M.; Niu, H.-Y. Copper-Catalyzed Intermolecular Alkynylation and Allylation of Unactivated C(sp3)–H Bonds via Hydrogen Atom Transfer. Org. Lett. 2021, 23, 8575–8579. [Google Scholar] [CrossRef]
- Fazekas, T.J.; Alty, J.W.; Neidhart, E.K.; Miller, A.S.; Leibfarth, F.A.; Alexanian, E.J. Diversification of aliphatic C-H bonds in small molecules and polyolefins through radical chain transfer. Science 2022, 375, 545–550. [Google Scholar] [CrossRef]
- Carestia, A.M.; Ravelli, D.; Alexanian, E.J. Reagent-dictated site selectivity in intermolecular aliphatic C–H functionalisations using nitrogen-centered radicals. Chem. Sci. 2018, 9, 5360–5365. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.S.; Alexanian, E.J. Heteroarylation of unactivated C–H bonds suitable for late-stage functionalisation. Chem. Sci. 2022, 13, 11878–11882. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Zhang, P.; Xu, J.; Ma, W.; Tu, D.; Lu, C.-S.; Yan, H. Direct B–H Functionalisation of Icosahedral Carboranes via Hydrogen Atom Transfer. J. Am. Chem. Soc. 2023, 145, 7638–7647. [Google Scholar] [CrossRef] [PubMed]
- Ohmatsu, K.; Suzuki, R.; Furukawa, Y.; Sato, M.; Ooi, T. Zwitterionic 1,2,3-Triazolium Amidate as a Catalyst for Photoinduced Hydrogen-Atom Transfer Radical Alkylation. ACS Catal. 2020, 10, 2627–2632. [Google Scholar] [CrossRef]
- Czaplyski, W.L.; Na, C.G.; Alexanian, E.J. C–H Xanthylation: A Synthetic Platform for Alkane Functionalisation. J. Am. Chem. Soc. 2016, 138, 13854–13857. [Google Scholar] [CrossRef]
- Minami, K.; Ohmatsu, K.; Ooi, T. Hydrogen-Atom-Transfer-Mediated Acceptorless Dehydrogenative Cross-Coupling Enabled by Multiple Catalytic Functions of Zwitterionic Triazolium Amidate. ACS Catal. 2022, 12, 1971–1976. [Google Scholar] [CrossRef]
- Ohmatsu, K.; Fujita, H.; Suzuki, R.; Ooi, T. Hydrogen-Atom Transfer Catalysis for C–H Alkylation of Benzylic Fluorides. Org. Lett. 2022, 24, 3134–3137. [Google Scholar] [CrossRef]
- Ohmatsu, K.; Suzuki, R.; Fujita, H.; Ooi, T. Zwitterionic Diphenylphosphinyl Amidate as a Powerful Photoinduced Hydrogen-Atom-Transfer Catalyst for C–H Alkylation of Simple Alkanes. J. Org. Chem. 2023, 88, 6553–6556. [Google Scholar] [CrossRef]
- Shee, M.; Singh, N.D.P. Chemical versatility of azide radical: Journey from a transient species to synthetic accessibility in organic transformations. Chem. Soc. Rev. 2022, 51, 2255–2312. [Google Scholar] [CrossRef]
- Constantin, T.; Górski, B.; Tilby, M.J.; Chelli, S.; Juliá, F.; Llaveria, J.; Gillen, K.J.; Zipse, H.; Lakhdar, S.; Leonori, D. Halogen-atom and group transfer reactivity enabled by hydrogen tunneling. Science 2022, 377, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Groves, J.T. Taming Azide Radicals for Catalytic C–H Azidation. ACS Catal. 2016, 6, 751–759. [Google Scholar] [CrossRef]
- Ryder, A.S.H.; Cunningham, W.B.; Ballantyne, G.; Mules, T.; Kinsella, A.G.; Turner-Dore, J.; Alder, C.M.; Edwards, L.J.; McKay, B.S.J.; Grayson, M.N.; et al. Photocatalytic α-Tertiary Amine Synthesis via C–H Alkylation of Unmasked Primary Amines. Angew. Chem. Int. Ed. 2020, 59, 14986–14991. [Google Scholar] [CrossRef]
- Grayson, J.D.; Cresswell, A.J. γ-Amino phosphonates via the photocatalytic α-C–H alkylation of primary amines. Tetrahedron 2021, 81, 131896. [Google Scholar] [CrossRef]
- Sim, J.; Ryou, B.; Choi, M.; Lee, C.; Park, C.-M. Electrochemical C(sp3)–H Functionalisation of γ-Lactams Based on Hydrogen Atom Transfer. Org. Lett. 2022, 24, 4264–4269. [Google Scholar] [CrossRef]
- Wiss, J.; Fleury, C.; Onken, U. Safety Improvement of Chemical Processes Involving Azides by Online Monitoring of the Hydrazoic Acid Concentration. Org. Process Res. Dev. 2006, 10, 349–353. [Google Scholar] [CrossRef]
- Sneha, M.; Thornton, G.L.; Lewis-Borrell, L.; Ryder, A.S.H.; Espley, S.G.; Clark, I.P.; Cresswell, A.J.; Grayson, M.N.; Orr-Ewing, A.J. Photoredox-HAT Catalysis for Primary Amine α-C–H Alkylation: Mechanistic Insight with Transient Absorption Spectroscopy. ACS Catal. 2023, 13, 8004–8013. [Google Scholar] [CrossRef]
- Chinn, A.J.; Sedillo, K.; Doyle, A.G. Phosphine/Photoredox Catalyzed Anti-Markovnikov Hydroamination of Olefins with Primary Sulfonamides via α-Scission from Phosphoranyl Radicals. J. Am. Chem. Soc. 2021, 143, 18331–18338. [Google Scholar] [CrossRef]
- Guo, X.; Wenger, O.S. Reductive Amination by Photoredox Catalysis and Polarity-Matched Hydrogen Atom Transfer. Angew. Chem. Int. Ed. 2018, 57, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Pu, C.-D.; Shen, Y.-M.; Wu, C.-D.; Peng, X.-G. Visible-Light Photocatalytic Synthesis of Amines from Imines via Transfer Hydrogenation Using Quantum Dots as Catalysts. J. Org. Chem. 2018, 83, 11886–11895. [Google Scholar] [CrossRef]
- Yue, W.-J.; Day, C.S.; Brenes Rucinski, A.J.; Martin, R. Catalytic Hydrodifluoroalkylation of Unactivated Olefins. Org. Lett. 2022, 24, 5109–5114. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, L.; Ji, Y.; Zou, G.; Shen, H.; Nicewicz, D.A.; Chen, J.; Huang, Y. Direct Synthesis of Bicyclic Acetals via Visible Light Catalysis. iScience 2020, 23, 101395. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Mendoza, A. Modular Enantioselective Synthesis of cis-Cyclopropanes through Self-Sensitised Stereoselective Photodecarboxylation with Benzothiazolines. ACS Catal. 2021, 11, 13312–13319. [Google Scholar] [CrossRef] [PubMed]
- Qvortrup, K.; Rankic, D.A.; MacMillan, D.W.C. A General Strategy for Organocatalytic Activation of C–H Bonds via Photoredox Catalysis: Direct Arylation of Benzylic Ethers. J. Am. Chem. Soc. 2014, 136, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P.; Salvesen, K. Equilibrium deuterium isotope effects on the ionisation of thiol acids. J. Am. Chem. Soc. 1971, 93, 4433–4436. [Google Scholar] [CrossRef]
- Studer, A. The Persistent Radical Effect in Organic Synthesis. Chem. Eur. J. 2001, 7, 1159–1164. [Google Scholar] [CrossRef]
- Kobayashi, F.; Fujita, M.; Ide, T.; Ito, Y.; Yamashita, K.; Egami, H.; Hamashima, Y. Dual-Role Catalysis by Thiobenzoic Acid in Cα–H Arylation under Photoirradiation. ACS Catal. 2021, 11, 82–87. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Mazzarella, D.; Melchiorre, P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, Y.; Cao, K.; Zhang, X.; Jiang, H.; Li, J. Synthetic reactions driven by electron-donor-acceptor (EDA) complexes. Beilstein J. Org. Chem. 2021, 17, 771–799. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lim, C.-H.; Miyake, G.M. Visible-Light-Promoted C–S Cross-Coupling via Intermolecular Charge Transfer. J. Am. Chem. Soc. 2017, 139, 13616–13619. [Google Scholar] [CrossRef]
- Vu, M.D.; Das, M.; Guo, A.; Ang, Z.-E.; D̵okić, M.; Soo, H.S.; Liu, X.-W. Visible-Light Photoredox Enables Ketone Carbonyl Alkylation for Easy Access to Tertiary Alcohols. ACS Catal. 2019, 9, 9009–9014. [Google Scholar] [CrossRef]
- Walczyk, K.R.; Popkirov, G.S.; Schindler, R.N. Investigation of the Redox Couple Benzophenone/Benzophenone Anion Radical in Acetonitrile and N,N-Dimethylformamide by Electrochemical and Spectroelectrochemical Methods. Bunsenges. Phys. Chem. 1995, 99, 1028–1036. [Google Scholar] [CrossRef]
- Flamigni, L.; Barbieri, A.; Sabatini, C.; Ventura, B.; Barigelletti, F. Photochemistry and Photophysics of Coordination Compounds: Iridium. In Photochemistry and Photophysics of Coordination Compounds II; Balzani, V., Campagna, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; p. 193. [Google Scholar]
- Zhou, R.; Li, J.; Cheo, H.W.; Chua, R.; Zhan, G.; Hou, Z.; Wu, J. Visible-light-mediated deuteration of silanes with deuterium oxide. Chem. Sci. 2019, 10, 7340–7344. [Google Scholar] [CrossRef]
- Zheng, W.; Xu, Y.; Luo, H.; Feng, Y.; Zhang, J.; Lin, L. Light-Promoted Arylsilylation of Alkenes with Hydrosilanes. Org. Lett. 2022, 24, 7145–7150. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Chiba, S. Leveraging of Sulfur Anions in Photoinduced Molecular Transformations. JACS Au 2021, 1, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, C.; Wang, Y.; Huang, X.; Zhao, X.; Qiao, B.; Jiang, Z. Catalytic Asymmetric Reductive Azaarylation of Olefins via Enantioselective Radical Coupling. J. Am. Chem. Soc. 2022, 144, 7805–7814. [Google Scholar] [CrossRef] [PubMed]
- Queen, A.E.; Selmani, A.; Schoenebeck, F. Hydrogermylation of Alkenes via Organophotoredox-Initiated HAT Catalysis. Org. Lett. 2022, 24, 406–409. [Google Scholar] [CrossRef]
- Fricke, C.; Schoenebeck, F. Organogermanes as Orthogonal Coupling Partners in Synthesis and Catalysis. Acc. Chem. Res. 2020, 53, 2715–2725. [Google Scholar] [CrossRef]
- Jia, J.; Kancherla, R.; Rueping, M.; Huang, L. Allylic C(sp3)–H alkylation via synergistic organo- and photoredox catalyzed radical addition to imines. Chem. Sci. 2020, 11, 4954–4959. [Google Scholar] [CrossRef]
- Nakashima, T.; Ohmatsu, K.; Ooi, T. Mannich-type allylic C–H functionalisation of enol silyl ethers under photoredox–thiol hybrid catalysis. Org. Biomol. Chem. 2021, 19, 141–145. [Google Scholar] [CrossRef]
- Shen, Y.; Rovis, T. Late-Stage N-Me Selective Arylation of Trialkylamines Enabled by Ni/Photoredox Dual Catalysis. J. Am. Chem. Soc. 2021, 143, 16364–16369. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Barham, J.P. Site-Selective C(sp3)–H Functionalisations Mediated by Hydrogen Atom Transfer Reactions via α-Amino/α-Amido Radicals. Synthesis 2022, 54, 1461–1477. [Google Scholar]
- Zhang, Y.-A.; Gu, X.; Wendlandt, A.E. A Change from Kinetic to Thermodynamic Control Enables trans-Selective Stereochemical Editing of Vicinal Diols. J. Am. Chem. Soc. 2022, 144, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Escoubet, S.; Gastaldi, S.; Vanthuyne, N.; Gil, G.; Siri, D.; Bertrand, M.P. Thiyl Radical Mediated Racemisation of Benzylic Amines. Eur. J. Org. Chem. 2006, 2006, 3242–3250. [Google Scholar] [CrossRef]
- Panferova, L.I.; Zubkov, M.O.; Kokorekin, V.A.; Levin, V.V.; Dilman, A.D. Using the Thiyl Radical for Aliphatic Hydrogen-Atom Transfer: Thiolation of Unactivated C–H Bonds. Angew. Chem. Int. Ed. 2021, 60, 2849–2854. [Google Scholar] [CrossRef]
- Wang, H.; Jui, N.T. Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc. 2018, 140, 163–166. [Google Scholar] [CrossRef]
- Vogt, D.B.; Seath, C.P.; Wang, H.; Jui, N.T. Selective C–F Functionalisation of Unactivated Trifluoromethylarenes. J. Am. Chem. Soc. 2019, 141, 13203–13211. [Google Scholar] [CrossRef]
- Jacobsen, E.; Roberts, J.L.; Sawyer, D.T. Electrochemical oxidation of formate in dimethylsulfoxide at gold and platinum electrodes. J. Electroanal. Chem. Interf. Electrochem. 1968, 16, 351–360. [Google Scholar] [CrossRef]
- Alektiar, S.N.; Wickens, Z.K. Photoinduced Hydrocarboxylation via Thiol-Catalyzed Delivery of Formate Across Activated Alkenes. J. Am. Chem. Soc. 2021, 143, 13022–13028. [Google Scholar] [CrossRef]
- Xu, P.; Wang, X.-Y.; Wang, Z.; Zhao, J.; Cao, X.-D.; Xiong, X.-C.; Yuan, Y.-C.; Zhu, S.; Guo, D.; Zhu, X. Defluorinative Alkylation of Trifluoromethylbenzimidazoles Enabled by Spin-Center Shift: A Synergistic Photocatalysis/Thiol Catalysis Process with CO2•−. Org. Lett. 2022, 24, 4075–4080. [Google Scholar] [CrossRef]
- Alektiar, S.N.; Han, J.; Dang, Y.; Rubel, C.Z.; Wickens, Z.K. Radical Hydrocarboxylation of Unactivated Alkenes via Photocatalytic Formate Activation. J. Am. Chem. Soc. 2023, 145, 10991–10997. [Google Scholar] [CrossRef]
- Williams, O.P.; Chmiel, A.F.; Mikhael, M.; Bates, D.M.; Yeung, C.S.; Wickens, Z.K. Practical and General Alcohol Deoxygenation Protocol. Angew. Chem. Int. Ed. 2023, 62, e202300178. [Google Scholar] [CrossRef]
- Mikhael, M.; Alektiar, S.N.; Yeung, C.S.; Wickens, Z.K. Translating planar heterocycles into three-dimensional analogs via photoinduced hydrocarboxylation. Angew. Chem. Int. Ed. 2023, 62, e202303264. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.W.; Polites, V.C.; Patel, S.; Lipson, J.E.; Majhi, J.; Molander, G.A. Photochemical C–F Activation Enables Defluorinative Alkylation of Trifluoroacetates and -Acetamides. J. Am. Chem. Soc. 2021, 143, 19648–19654. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, B.; Majhi, J.; Granados, A.; Sharique, M.; Martin, R.T.; Gutierrez, O.; Molander, G.A. Transition metal-free photochemical C–F activation for the preparation of difluorinated-oxindole derivatives. Chem. Sci. 2023, 14, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.-H.; Bellotti, P.; Heusel, C.; Glorius, F. Photoredox-Catalyzed Defluorinative Functionalisations of Polyfluorinated Aliphatic Amides and Esters. Angew. Chem. Int. Ed. 2022, 61, e202115456. [Google Scholar] [CrossRef] [PubMed]
- Fuse, H.; Mitsunuma, H.; Kanai, M. Catalytic Acceptorless Dehydrogenation of Aliphatic Alcohols. J. Am. Chem. Soc. 2020, 142, 4493–4499. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Mitsunuma, H.; Kanai, M. Catalytic Allylation of Aldehydes Using Unactivated Alkenes. J. Am. Chem. Soc. 2020, 142, 12374–12381. [Google Scholar] [CrossRef]
- Margrey, K.A.; Nicewicz, D.A. A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalisation Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1997–2006. [Google Scholar] [CrossRef]
- Fuse, H.; Nakao, H.; Saga, Y.; Fukatsu, A.; Kondo, M.; Masaoka, S.; Mitsunuma, H.; Kanai, M. Photocatalytic redox-neutral hydroxyalkylation of N-heteroaromatics with aldehydes. Chem. Sci. 2020, 11, 12206–12211. [Google Scholar] [CrossRef]
- Jin, J.; MacMillan, D.W. Alcohols as alkylating agents in heteroarene C-H functionalisation. Nature. 2015, 525, 87–90. [Google Scholar] [CrossRef]
- Ji, X.; Liu, Q.; Wang, Z.; Wang, P.; Deng, G.-J.; Huang, H. LiBr-promoted photoredox neutral Minisci hydroxyalkylations of quinolines with aldehydes. Green Chem. 2020, 22, 8233–8237. [Google Scholar] [CrossRef]
- Bieszczad, B.; Perego, L.A.; Melchiorre, P. Photochemical C–H Hydroxyalkylation of Quinolines and Isoquinolines. Angew. Chem. Int. Ed. 2019, 58, 16878–16883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-L.; Li, B.; Houk, K.N.; Wang, Y.-F. Application of the Spin-Center Shift in Organic Synthesis. JACS Au 2022, 2, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-M.; Bellotti, P.; Chen, P.-P.; Houk, K.N.; Glorius, F. Allylic C(sp3)–H arylation of olefins via ternary catalysis. Nat. Synth. 2022, 1, 59–68. [Google Scholar] [CrossRef]
- Berman, R.S.; Kochi, J.K. Kinetics and mechanism of oxygen atom transfer from nitro compounds mediated by nickel(0) complexes. Inorg. Chem. 1980, 19, 248–254. [Google Scholar] [CrossRef]
- Percec, V.; Bae, J.-Y.; Zhao, M.; Hill, D.H. Aryl Mesylates in Metal-Catalyzed Homocoupling and Cross-Coupling Reactions. 1. Functional Symmetrical Biaryls from Phenols via Nickel-Catalyzed Homocoupling of Their Mesylates. J. Org. Chem. 1995, 60, 176–185. [Google Scholar] [CrossRef]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef]
- Ting, S.I.; Williams, W.L.; Doyle, A.G. Oxidative Addition of Aryl Halides to a Ni(I)-Bipyridine Complex. J. Am. Chem. Soc. 2022, 144, 5575–5582. [Google Scholar] [CrossRef]
- McManus, J.B.; Griffin, J.D.; White, A.R.; Nicewicz, D.A. Homobenzylic Oxygenation Enabled by Dual Organic Photoredox and Cobalt Catalysis. J. Am. Chem. Soc. 2020, 142, 10325–10330. [Google Scholar] [CrossRef]
- Fischer, C.; Kerzig, C.; Zilate, B.; Wenger, O.S.; Sparr, C. Modulation of Acridinium Organophotoredox Catalysts Guided by Photophysical Studies. ACS Catal. 2020, 10, 210–215. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, E.; Zanini, M.; Melchiorre, P. Photochemical Organocatalytic Benzylation of Allylic C–H Bonds. J. Am. Chem. Soc. 2022, 144, 1113–1118. [Google Scholar] [CrossRef]
- Correia, J.T.M.; Fernandes, V.A.; Matsuo, B.T.; Delgado, J.A.C.; de Souza, W.C.; Paixão, M.W. Photoinduced deaminative strategies: Katritzky salts as alkyl radical precursors. Chem. Commun. 2020, 56, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Fuse, H.; Irie, Y.; Fuki, M.; Kobori, Y.; Kato, K.; Yamakata, A.; Higashi, M.; Mitsunuma, H.; Kanai, M. Identification of a Self-Photosensitizing Hydrogen Atom Transfer Organocatalyst System. J. Am. Chem. Soc. 2022, 144, 6566–6574. [Google Scholar] [CrossRef]
- Le Saux, E.; Georgiou, E.; Dmitriev, I.A.; Hartley, W.C.; Melchiorre, P. Photochemical Organocatalytic Functionalisation of Pyridines via Pyridinyl Radicals. J. Am. Chem. Soc. 2023, 145, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Li, J.; Liu, W.; Li, C.-J. Diacetyl as a “traceless” visible light photosensitiser in metal-free cross-dehydrogenative coupling reactions. Chem. Sci. 2019, 10, 5018–5024. [Google Scholar] [CrossRef]
- Proctor, R.S.J.; Chuentragool, P.; Colgan, A.C.; Phipps, R.J. Hydrogen Atom Transfer-Driven Enantioselective Minisci Reaction of Amides. J. Am. Chem. Soc. 2021, 143, 4928–4934. [Google Scholar] [CrossRef]
- Wang, B.; Ascenzi Pettenuzzo, C.; Singh, J.; McCabe, G.E.; Clark, L.; Young, R.; Pu, J.; Deng, Y. Photoinduced Site-Selective Functionalisation of Aliphatic C–H Bonds by Pyridine N-oxide Based HAT Catalysts. ACS Catal. 2022, 12, 10441–10448. [Google Scholar] [CrossRef]
- Schlegel, M.; Qian, S.; Nicewicz, D.A. Aliphatic C–H Functionalisation Using Pyridine N-Oxides as H-Atom Abstraction Agents. ACS Catal. 2022, 12, 10499–10505. [Google Scholar] [CrossRef]
- Markham, J.P.; Wang, B.; Stevens, E.D.; Burris, S.C.; Deng, Y. ortho-Alkylation of Pyridine N-Oxides with Alkynes by Photocatalysis: Pyridine N-Oxide as a Redox Auxiliary. Chem. Eur. J. 2019, 25, 6638–6644. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, J.; Bankhead, B.; Markham, J.P.; Zeller, M. Photoinduced oxidative cyclopropanation of ene-ynamides: Synthesis of 3-aza[n.1.0]bicycles via vinyl radicals. Chem. Commun. 2021, 57, 5254–5257. [Google Scholar] [CrossRef]
- Xu, J.-h.; Wu, W.-b.; Wu, J. Photoinduced Divergent Alkylation/Acylation of Pyridine N-Oxides with Alkynes under Anaerobic and Aerobic Conditions. Org. Lett. 2019, 21, 5321–5325. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, F.; Zhang, M.-T. Metal-Free Electrocatalyst for Water Oxidation Initiated by Hydrogen Atom Transfer. ACS Catal. 2021, 11, 68–73. [Google Scholar] [CrossRef]
- Chmurzynski, L.; Warnke, Z. Acid-Base Equilibria of Substituted Pyridine N-Oxides in N,N-Dimethylformamide and Dimethyl Sulfoxide. Aust. J. Chem. 1993, 46, 185–194. [Google Scholar] [CrossRef]
- Ciszewski, Ł.W.; Gryko, D. Pyridine N-oxides as HAT reagents for photochemical C–H functionalisation of electron-deficient heteroarenes. Chem. Commun. 2022, 58, 10576–10579. [Google Scholar] [CrossRef]
- Katsuki, T.; Martin, V. Asymmetric Epoxidation of Allylic Alcohols: The Katsuki–Sharpless Epoxidation Reaction. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 4–5. [Google Scholar]
- Martin, V.S.; Woodard, S.S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless, K.B. Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity? J. Am. Chem. Soc. 1981, 103, 6237–6240. [Google Scholar] [CrossRef]
- Rossiter, B.E.; Katsuki, T.; Sharpless, K.B. Asymmetric epoxidation provides shortest routes to four chiral epoxy alcohols which are key intermediates in syntheses of methymycin, erythromycin, leukotriene C-1, and disparlure. J. Am. Chem. Soc. 1981, 103, 464–465. [Google Scholar] [CrossRef]
- Goor, G.; Glenneberg, J.; Jacobi, S.; Dadabhoy, J.; Candido, E. Hydrogen Peroxide. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 397–411. [Google Scholar]
- Vasilopoulos, A.; Krska, S.W.; Stahl, S.S. C(sp(3))–H methylation enabled by peroxide photosensitisation and Ni-mediated radical coupling. Science 2021, 372, 398–403. [Google Scholar] [CrossRef]
- Murakami, M.; Ishida, N. β-Scission of Alkoxy Radicals in Synthetic Transformations. Chem. Lett. 2017, 46, 1692–1700. [Google Scholar] [CrossRef]
- Avila, D.V.; Brown, C.E.; Ingold, K.U.; Lusztyk, J. Solvent effects on the competitive .beta.-scission and hydrogen atom abstraction reactions of the cumyloxyl radical. Resolution of a long-standing problem. J. Am. Chem. Soc. 1993, 115, 466–470. [Google Scholar] [CrossRef]
- Baciocchi, E.; Bietti, M.; Salamone, M.; Steenken, S. Spectral Properties and Absolute Rate Constants for β-Scission of Ring-Substituted Cumyloxyl Radicals. A Laser Flash Photolysis Study. J. Org. Chem. 2002, 67, 2266–2270. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A. The methylation effect in medicinal chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Fu, J. Methyl-containing pharmaceuticals: Methylation in drug design. Bioorg. Med. Chem. Lett. 2018, 28, 3283–3289. [Google Scholar] [CrossRef]
- Colgan, A.C.; Proctor, R.S.J.; Gibson, D.C.; Chuentragool, P.; Lahdenperä, A.S.K.; Ermanis, K.; Phipps, R.J. Hydrogen Atom Transfer Driven Enantioselective Minisci Reaction of Alcohols. Angew. Chem. Int. Ed. 2022, 61, e202200266. [Google Scholar] [CrossRef]
- Proctor, R.S.J.; Davis, H.J.; Phipps, R.J. Catalytic enantioselective Minisci-type addition to heteroarenes. Science 2018, 360, 419–422. [Google Scholar] [CrossRef]
- Chen, X.; Lian, Z.; Kramer, S. Enantioselective Intermolecular Radical Amidation and Amination of Benzylic C–H Bonds via Dual Copper and Photocatalysis. Angew. Chem. Int. Ed. 2023, 62, e202217638. [Google Scholar] [CrossRef]
- Dai, L.; Chen, Y.-Y.; Xiao, L.-J.; Zhou, Q.-L. Intermolecular Enantioselective Benzylic C(sp3)–H Amination by Cationic Copper Catalysis. Angew. Chem. Int. Ed. 2023, 62, e202304427. [Google Scholar] [CrossRef]
- Golden, D.L.; Zhang, C.; Chen, S.-J.; Vasilopoulos, A.; Guzei, I.A.; Stahl, S.S. Benzylic C–H Esterification with Limiting C–H Substrate Enabled by Photochemical Redox Buffering of the Cu Catalyst. J. Am. Chem. Soc. 2023, 145, 9434–9440. [Google Scholar] [CrossRef]
- Su, Y.-L.; Dong, K.; Zheng, H.; Doyle, M.P. Generation of Diazomethyl Radicals by Hydrogen Atom Abstraction and Their Cycloaddition with Alkenes. Angew. Chem. Int. Ed. 2021, 60, 18484–18488. [Google Scholar] [CrossRef]
- Das, M.; Zamani, L.; Bratcher, C.; Musacchio, P.Z. Azolation of Benzylic C–H Bonds via Photoredox-Catalyzed Carbocation Generation. J. Am. Chem. Soc. 2023, 145, 3861–3868. [Google Scholar] [CrossRef]
- Barthelemy, A.-L.; Tuccio, B.; Magnier, E.; Dagousset, G. Alkoxyl Radicals Generated under Photoredox Catalysis: A Strategy for anti-Markovnikov Alkoxylation Reactions. Angew. Chem. Int. Ed. 2018, 57, 13790–13794. [Google Scholar] [CrossRef]
- Bao, X.; Wang, Q.; Zhu, J. Remote C(sp3)–H Arylation and Vinylation of N-Alkoxypyridinium Salts to δ-Aryl and δ-Vinyl Alcohols. Chem. Eur. J. 2019, 25, 11630–11634. [Google Scholar] [CrossRef] [PubMed]
- Rammal, F.; Gao, D.; Boujnah, S.; Hussein, A.A.; Lalevée, J.; Gaumont, A.-C.; Morlet-Savary, F.; Lakhdar, S. Photochemical C–H Silylation and Hydroxymethylation of Pyridines and Related Structures: Synthetic Scope and Mechanisms. ACS Catal. 2020, 10, 13710–13717. [Google Scholar] [CrossRef]
- Kim, I.; Min, M.; Kang, D.; Kim, K.; Hong, S. Direct Phosphonation of Quinolinones and Coumarins Driven by the Photochemical Activity of Substrates and Products. Org. Lett. 2017, 19, 1394–1397. [Google Scholar] [CrossRef]
- Zheng, M.; Hou, J.; Zhan, L.-W.; Huang, Y.; Chen, L.; Hua, L.-L.; Li, Y.; Tang, W.-Y.; Li, B.-D. Visible-Light-Driven, Metal-Free Divergent Difunctionalisation of Alkenes Using Alkyl Formates. ACS Catal. 2021, 11, 542–553. [Google Scholar] [CrossRef]
- Kim, I.; Park, B.; Kang, G.; Kim, J.; Jung, H.; Lee, H.; Baik, M.-H.; Hong, S. Visible-Light-Induced Pyridylation of Remote C(sp3)–H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem. Int. Ed. 2018, 57, 15517–15522. [Google Scholar] [CrossRef]
- Kim, I.; Kang, G.; Lee, K.; Park, B.; Kang, D.; Jung, H.; He, Y.-T.; Baik, M.-H.; Hong, S. Site-Selective Functionalisation of Pyridinium Derivatives via Visible-Light-Driven Photocatalysis with Quinolinone. J. Am. Chem. Soc. 2019, 141, 9239–9248. [Google Scholar] [CrossRef]
- Fitzpatrick, N.A.; Zamani, L.; Das, M.; Yayla, H.G.; Lall, M.S.; Musacchio, P.Z. A SN1 mechanistic approach to the Williamson ether reaction via photoredox catalysis applied to benzylic C(sp3)–H bonds. Tetrahedron 2022, 125, 132986. [Google Scholar] [CrossRef]
- Zhang, Y.; Fitzpatrick, N.A.; Das, M.; Bedre, I.P.; Yayla, H.G.; Lall, M.S.; Musacchio, P.Z. A photoredox-catalyzed approach for formal hydride abstraction to enable Csp3–H functionalisation with nucleophilic partners (F, C, O, N, and Br/Cl). Chem. Catal. 2022, 2, 292–308. [Google Scholar] [CrossRef]
- Fan, L.-F.; Liu, R.; Ruan, X.-Y.; Wang, P.-S.; Gong, L.-Z. Asymmetric 1,2-oxidative alkylation of conjugated dienes via aliphatic C–H bond activation. Nat. Synth. 2022, 1, 946–955. [Google Scholar] [CrossRef]
- Citterio, A.; Arnoldi, A.; Minisci, F. Nucleophilic character of alkyl radicals. 18. Absolute rate constants for the addition of primary alkyl radicals to conjugated olefins and 1,4-benzoquinone. J. Org. Chem. 1979, 44, 2674–2682. [Google Scholar] [CrossRef]
- Jin, J.; MacMillan, D.W.C. Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C–H Functionalisation and the Minisci Reaction. Angew. Chem. Int. Ed. 2015, 54, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- McCallum, T.; Jouanno, L.-A.; Cannillo, A.; Barriault, L. Persulfate-Enabled Direct C–H Alkylation of Heteroarenes with Unactivated Ethers. Synlett 2016, 27, 1282–1286. [Google Scholar]
- Ueda, M.; Kamikawa, K.; Fukuyama, T.; Wang, Y.-T.; Wu, Y.-K.; Ryu, I. Site-Selective Alkenylation of Unactivated C(sp3)–H Bonds Mediated by Compact Sulfate Radical. Angew. Chem. Int. Ed. 2021, 60, 3545–3550. [Google Scholar] [CrossRef]
- Kim, J.H.; Constantin, T.; Simonetti, M.; Llaveria, J.; Sheikh, N.S.; Leonori, D. A radical approach for the selective C–H borylation of azines. Nature 2021, 595, 677–683. [Google Scholar] [CrossRef]
- Wu, Z.-X.; Hu, G.-W.; Luan, Y.-X. Development of N-Hydroxy Catalysts for C–H Functionalisation via Hydrogen Atom Transfer: Challenges and Opportunities. ACS Catal. 2022, 12, 11716–11733. [Google Scholar] [CrossRef]
- Weidmann, V.; Maison, W. Allylic Oxidations of Olefins to Enones. Synthesis 2013, 45, 2201–2221. [Google Scholar]
- Recupero, F.; Punta, C. Free Radical Functionalisation of Organic Compounds Catalyzed by N-Hydroxyphthalimide. Chem. Rev. 2007, 107, 3800–3842. [Google Scholar] [CrossRef]
- Amorati, R.; Lucarini, M.; Mugnaini, V.; Pedulli, G.F.; Minisci, F.; Recupero, F.; Fontana, F.; Astolfi, P.; Greci, L. Hydroxylamines as Oxidation Catalysts: Thermochemical and Kinetic Studies. J. Org. Chem. 2003, 68, 1747–1754. [Google Scholar] [CrossRef]
- Nutting, J.E.; Rafiee, M.; Stahl, S.S. Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev. 2018, 118, 4834–4885. [Google Scholar] [CrossRef]
- Ueda, C.; Noyama, M.; Ohmori, H.; Masui, M. Reactivity of Phthalimide-N--oxyl: A Kinetic Study. Chem. Pharm. Bull. 1987, 35, 1372–1377. [Google Scholar] [CrossRef]
- Hoque, M.A.; Twilton, J.; Zhu, J.; Graaf, M.D.; Harper, K.C.; Tuca, E.; DiLabio, G.A.; Stahl, S.S. Electrochemical PINOylation of Methylarenes: Improving the Scope and Utility of Benzylic Oxidation through Mediated Electrolysis. J. Am. Chem. Soc. 2022, 144, 15295–15302. [Google Scholar] [CrossRef] [PubMed]
- Sanyo, H. Effects of Cyclodextrins on the Fluorescence of Diphenyl Phosphate in Aqueous Solution. Bull. Chem. Soc. Jpn. 1986, 59, 2979–2982. [Google Scholar] [CrossRef]
- Wakaki, T.; Sakai, K.; Enomoto, T.; Kondo, M.; Masaoka, S.; Oisaki, K.; Kanai, M. C(sp3)–H Cyanation Promoted by Visible-Light Photoredox/Phosphate Hybrid Catalysis. Chem. Eur. J. 2018, 24, 8051–8055. [Google Scholar] [CrossRef]
- Li, D.-S.; Liu, T.; Hong, Y.; Cao, C.-L.; Wu, J.; Deng, H.-P. Stop-Flow Microtubing Reactor-Assisted Visible Light-Induced Hydrogen-Evolution Cross Coupling of Heteroarenes with C(sp3)–H Bonds. ACS Catal. 2022, 12, 4473–4480. [Google Scholar] [CrossRef]
- Hamilton, D.S.; Nicewicz, D.A. Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols. J. Am. Chem. Soc. 2012, 134, 18577–18580. [Google Scholar] [CrossRef]
- Sarkar, S.; Cheung, K.P.S.; Gevorgyan, V. C–H functionalisation reactions enabled by hydrogen atom transfer to carbon-centered radicals. Chem. Sci. 2020, 11, 12974–12993. [Google Scholar] [CrossRef]
- Narayanam, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Wang, C.-S.; Dixneuf, P.H.; Soulé, J.-F. Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Reckenthäler, M.; Griesbeck, A.G. Photoredox Catalysis for Organic Syntheses. Adv. Synth. Catal. 2013, 355, 2727–2744. [Google Scholar] [CrossRef]
- De Vleeschouwer, F.; Van Speybroeck, V.; Waroquier, M.; Geerlings, P.; De Proft, F. Electrophilicity and Nucleophilicity Index for Radicals. Org. Lett. 2007, 9, 2721–2724. [Google Scholar] [CrossRef] [PubMed]
- Kubisiak, M.; Zelga, K.; Bury, W.; Justyniak, I.; Budny-Godlewski, K.; Ochal, Z.; Lewiński, J. Development of zinc alkyl/air systems as radical initiators for organic reactions. Chem. Sci. 2015, 6, 3102–3108. [Google Scholar] [CrossRef]
- Akindele, T.; Yamada, K.-i.; Tomioka, K. Dimethylzinc-Initiated Radical Reactions. Acc. Chem. Res. 2009, 42, 345–355. [Google Scholar] [CrossRef]
- Clerici, A.; Cannella, R.; Panzeri, W.; Pastori, N.; Regolini, E.; Porta, O. TiCl3/PhN2+-mediated radical addition of ethers to aldimines generated in situ under aqueous conditions. Tetrahedron Lett. 2005, 46, 8351–8354. [Google Scholar] [CrossRef]
- Ye, B.; Zhao, J.; Zhao, K.; McKenna, J.M.; Toste, F.D. Chiral Diaryliodonium Phosphate Enables Light Driven Diastereoselective α-C(sp3)–H Acetalisation. J. Am. Chem. Soc. 2018, 140, 8350–8356. [Google Scholar] [CrossRef]
- Liu, Z.; Li, M.; Deng, G.; Wei, W.; Feng, P.; Zi, Q.; Li, T.; Zhang, H.; Yang, X.; Walsh, P.J. Transition-metal-free C(sp3)–H/C(sp3)–H dehydrogenative coupling of saturated heterocycles with N-benzyl imines. Chem. Sci. 2020, 11, 7619–7625. [Google Scholar] [CrossRef]
- Uchikura, T.; Hara, Y.; Tsubono, K.; Akiyama, T. Visible-Light-Driven C–S Bond Formation Based on Electron Donor–Acceptor Excitation and Hydrogen Atom Transfer Combined System. ACS Org. Inorg. Au 2021, 1, 23–28. [Google Scholar] [CrossRef]
- Li, T.; Liang, K.; Tang, J.; Ding, Y.; Tong, X.; Xia, C. A photoexcited halogen-bonded EDA complex of the thiophenolate anion with iodobenzene for C(sp3)–H activation and thiolation. Chem. Sci. 2021, 12, 15655–15661. [Google Scholar] [CrossRef] [PubMed]
- Uchikura, T.; Tsubono, K.; Hara, Y.; Akiyama, T. Dual-Role Halogen-Bonding-Assisted EDA-SET/HAT Photoreaction System with Phenol Catalyst and Aryl Iodide: Visible-Light-Driven Carbon–Carbon Bond Formation. J. Org. Chem. 2022, 87, 15499–15510. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Poole, D.L.; Murphy, J.A. The role of organic electron donors in the initiation of BHAS base-induced coupling reactions between haloarenes and arenes. Sci. China Chem. 2019, 62, 1425–1438. [Google Scholar] [CrossRef]
- Sun, C.-L.; Shi, Z.-J. Transition-Metal-Free Coupling Reactions. Chem. Rev. 2014, 114, 9219–9280. [Google Scholar] [CrossRef]
- Gurry, M.; Aldabbagh, F. A new era for homolytic aromatic substitution: Replacing Bu3SnH with efficient light-induced chain reactions. Org. Biomol. Chem. 2016, 14, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Pak Shing Cheung, K.; Fang, J.; Mukherjee, K.; Mihranyan, A.; Gevorgyan, V. Asymmetric intermolecular allylic C-H amination of alkenes with aliphatic amines. Science 2022, 378, 1207–1213. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, Y.; Rovis, T. Photoinduced Nickel-Catalyzed Selective N-Demethylation of Trialkylamines Using C(sp2)–Bromides as HAT Reagents. J. Am. Chem. Soc. 2023, 145, 3294–3300. [Google Scholar] [CrossRef]
- Zuo, Z.; MacMillan, D.W.C. Decarboxylative Arylation of α-Amino Acids via Photoredox Catalysis: A One-Step Conversion of Biomass to Drug Pharmacophore. J. Am. Chem. Soc. 2014, 136, 5257–5260. [Google Scholar] [CrossRef]
- Qin, T.; Cornella, J.; Li, C.; Malins, L.R.; Edwards, J.T.; Kawamura, S.; Maxwell, B.D.; Eastgate, M.D.; Baran, P.S. A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352, 801–805. [Google Scholar] [CrossRef]
- Capaldo, L.; Noël, T.; Ravelli, D. Photocatalytic generation of ligated boryl radicals from tertiary amine-borane complexes: An emerging tool in organic synthesis. Chem Catal. 2022, 2, 957–966. [Google Scholar] [CrossRef]
- Lei, G.; Xu, M.; Chang, R.; Funes-Ardoiz, I.; Ye, J. Hydroalkylation of Unactivated Olefins via Visible-Light-Driven Dual Hydrogen Atom Transfer Catalysis. J. Am. Chem. Soc. 2021, 143, 11251–11261. [Google Scholar] [CrossRef]
- Gonçalves, C.R.; Lemmerer, M.; Teskey, C.J.; Adler, P.; Kaiser, D.; Maryasin, B.; González, L.; Maulide, N. Unified Approach to the Chemoselective α-Functionalisation of Amides with Heteroatom Nucleophiles. J. Am. Chem. Soc. 2019, 141, 18437–18443. [Google Scholar] [CrossRef]
- Hamada, T.; Chieffi, A.; Åhman, J.; Buchwald, S.L. An Improved Catalyst for the Asymmetric Arylation of Ketone Enolates. J. Am. Chem. Soc. 2002, 124, 1261–1268. [Google Scholar] [CrossRef]
- He, Z.-T.; Hartwig, J.F. Palladium-catalyzed α-arylation for the addition of small rings to aromatic compounds. Nat. Commun. 2019, 10, 4083. [Google Scholar] [CrossRef]
- Baran, P.S.; Maimone, T.J.; Richter, J.M. Total synthesis of marine natural products without using protecting groups. Nature. 2007, 446, 404–408. [Google Scholar] [CrossRef]
- Baran, P.S.; Richter, J.M. Enantioselective Total Syntheses of Welwitindolinone A and Fischerindoles I and G. J. Am. Chem. Soc. 2005, 127, 15394–15396. [Google Scholar] [CrossRef]
- Baran, P.S.; DeMartino, M.P. Intermolecular Oxidative Enolate Heterocoupling. Angew. Chem. Int. Ed. 2006, 45, 7083–7086. [Google Scholar] [CrossRef]
- Guo, F.; Clift, M.D.; Thomson, R.J. Oxidative Coupling of Enolates, Enol Silanes, and Enamines: Methods and Natural Product Synthesis. Eur. J. Org. Chem. 2012, 2012, 4881–4896. [Google Scholar] [CrossRef]
- Zhang, Z.-Q.; Sang, Y.-Q.; Wang, C.-Q.; Dai, P.; Xue, X.-S.; Piper, J.L.; Peng, Z.-H.; Ma, J.-A.; Zhang, F.-G.; Wu, J. Difluoromethylation of Unactivated Alkenes Using Freon-22 through Tertiary Amine-Borane-Triggered Halogen Atom Transfer. J. Am. Chem. Soc. 2022, 144, 14288–14296. [Google Scholar] [CrossRef]

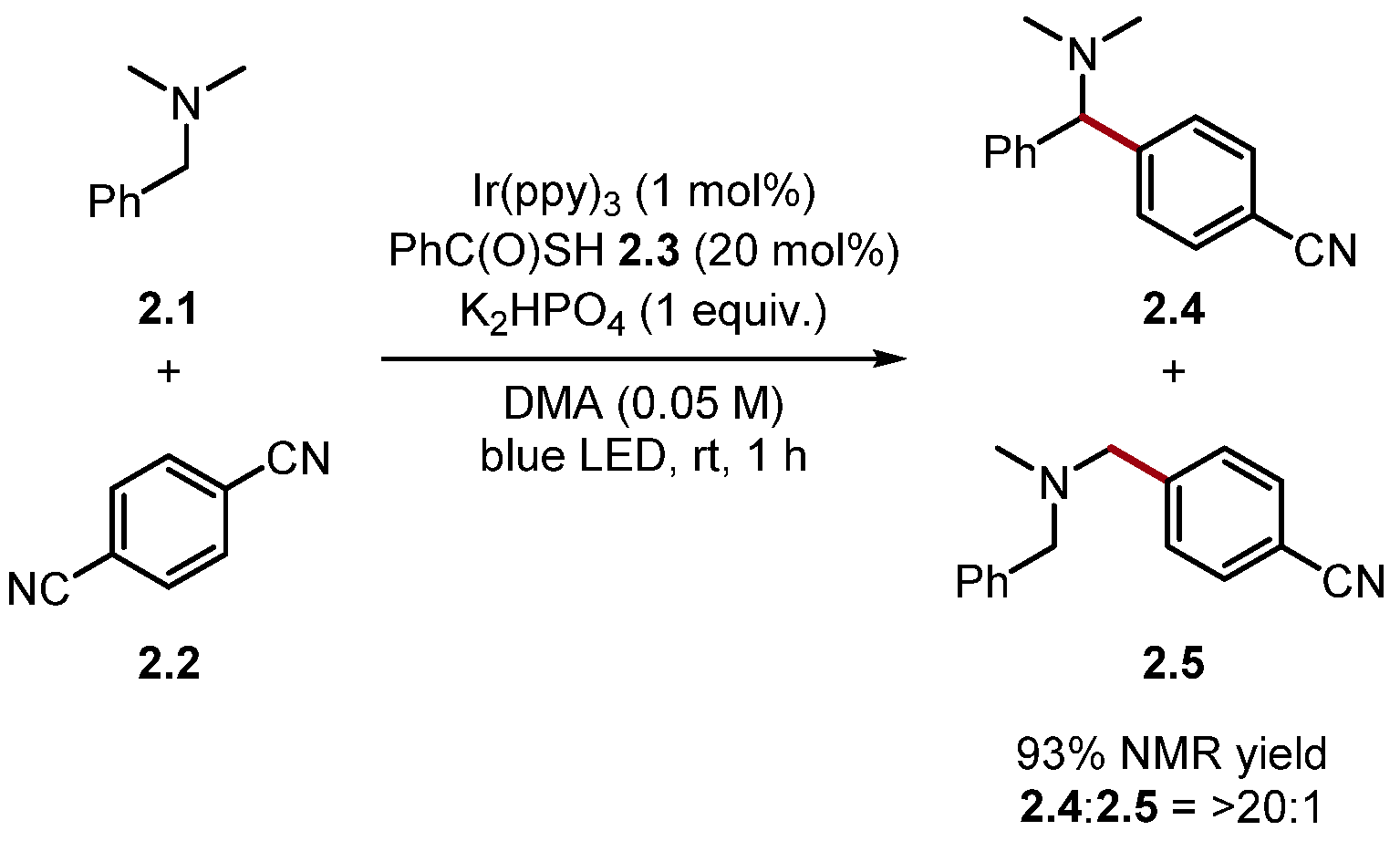
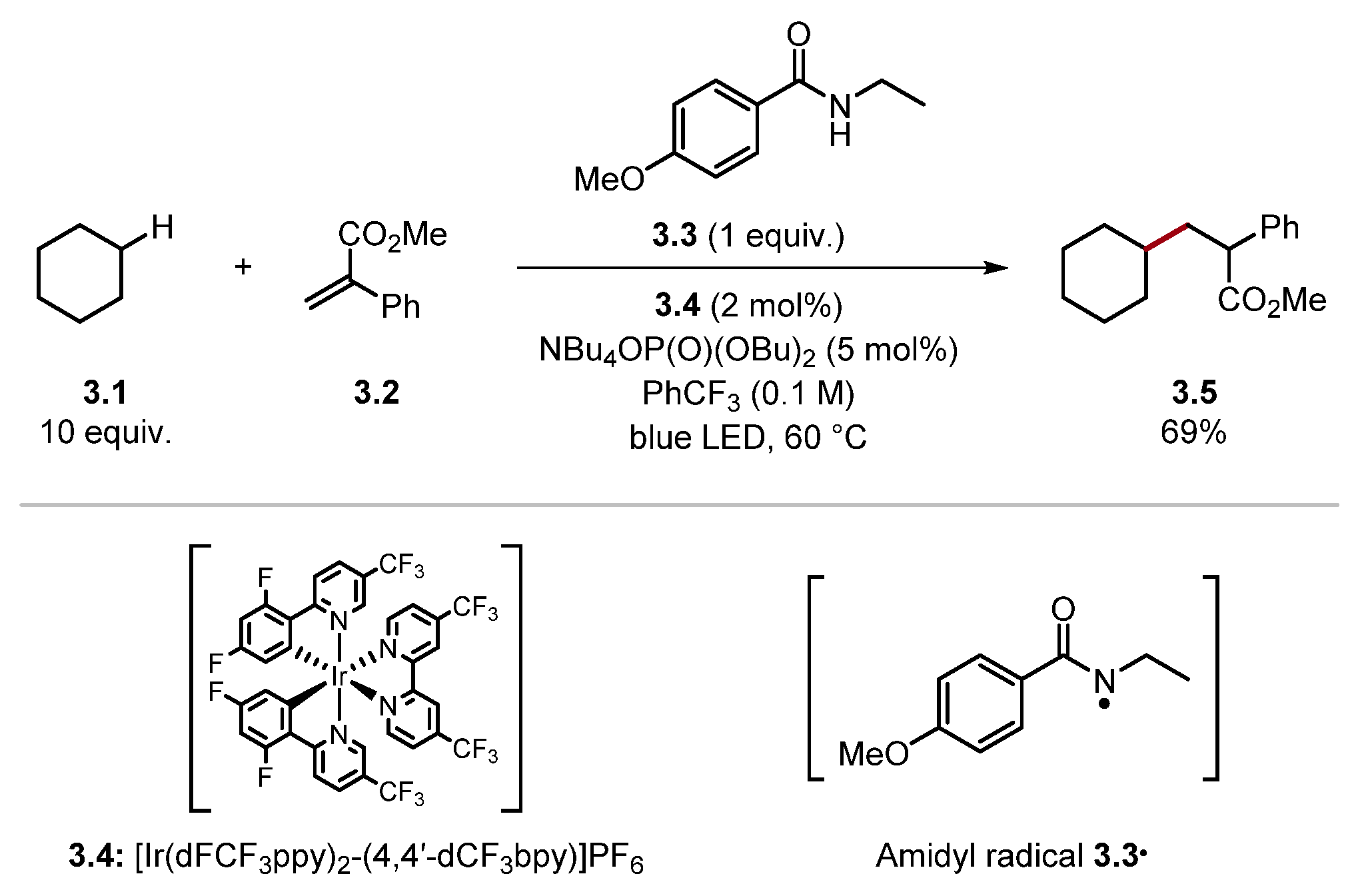

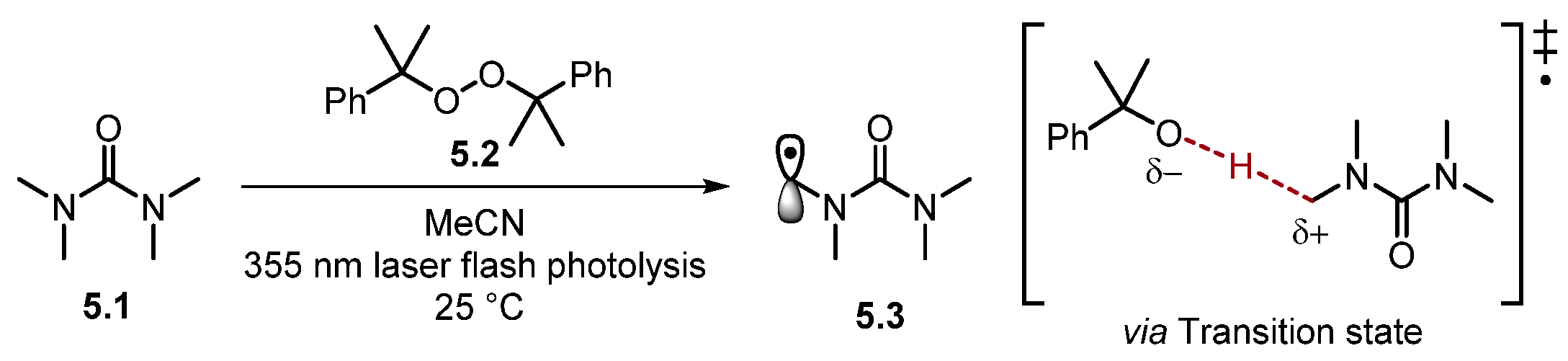
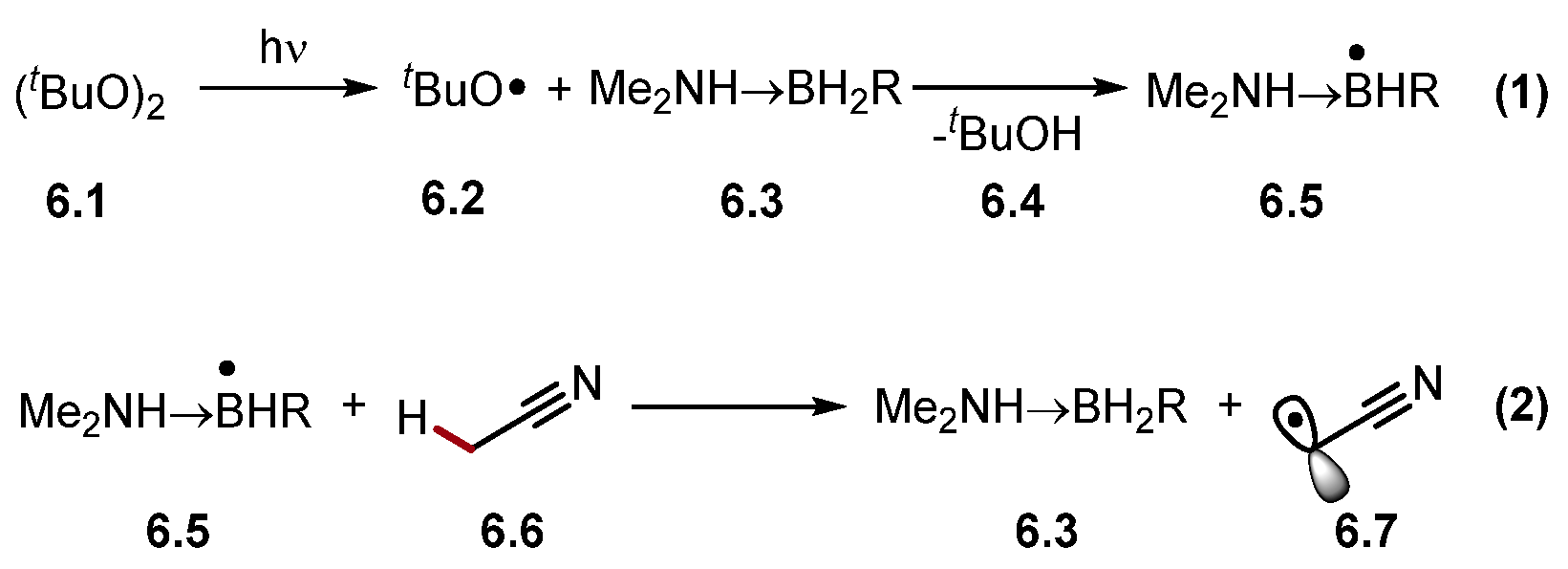

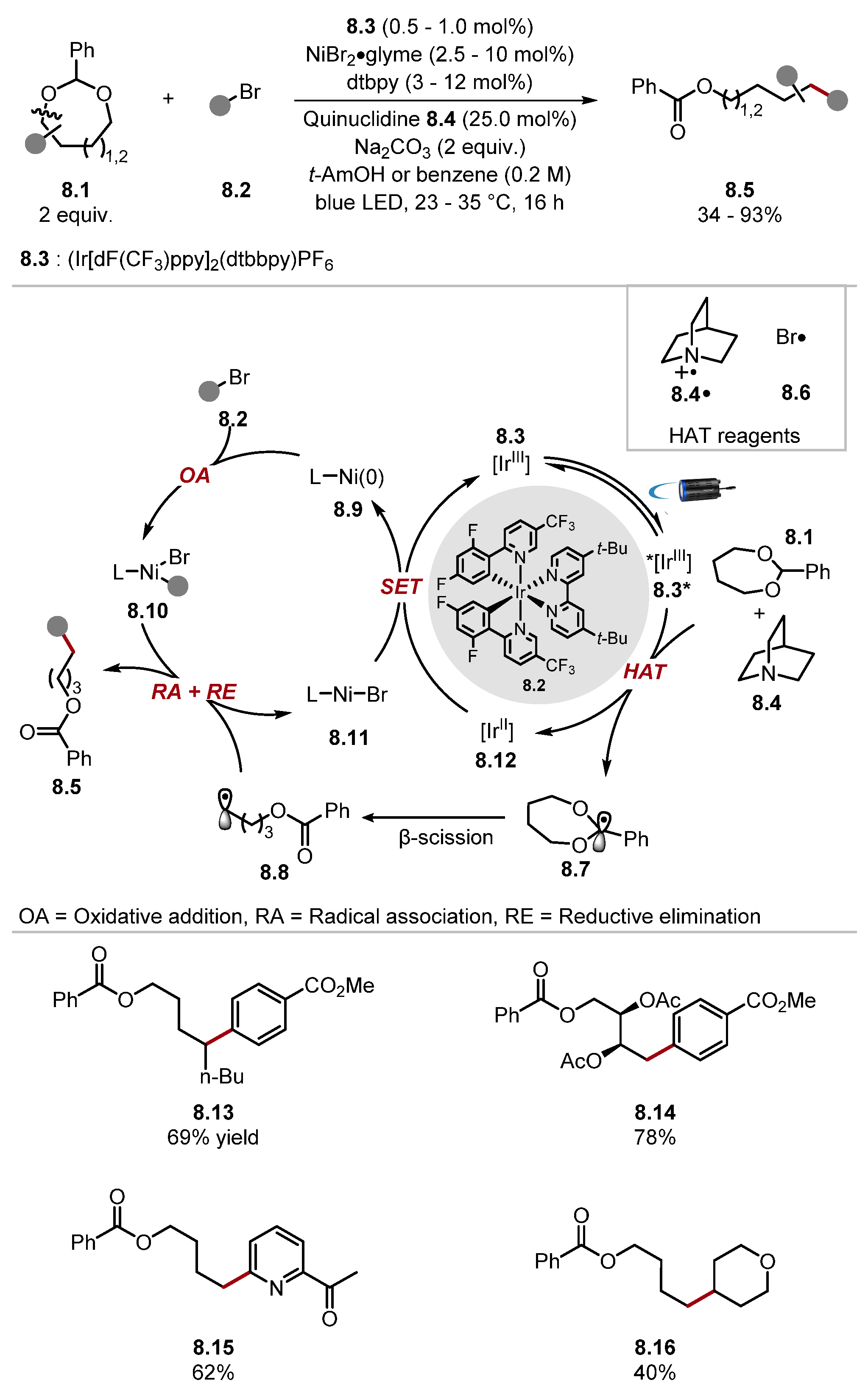
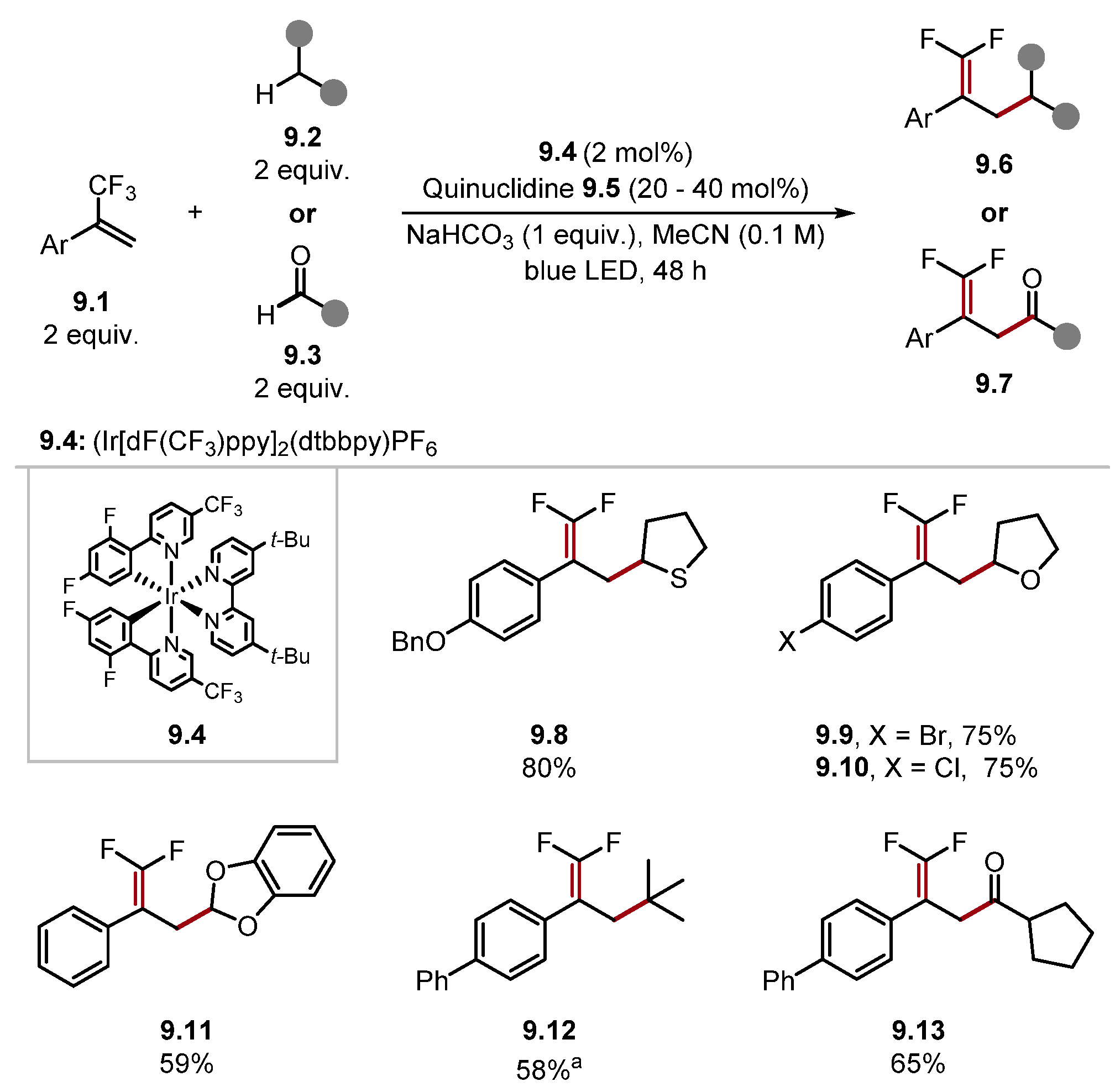
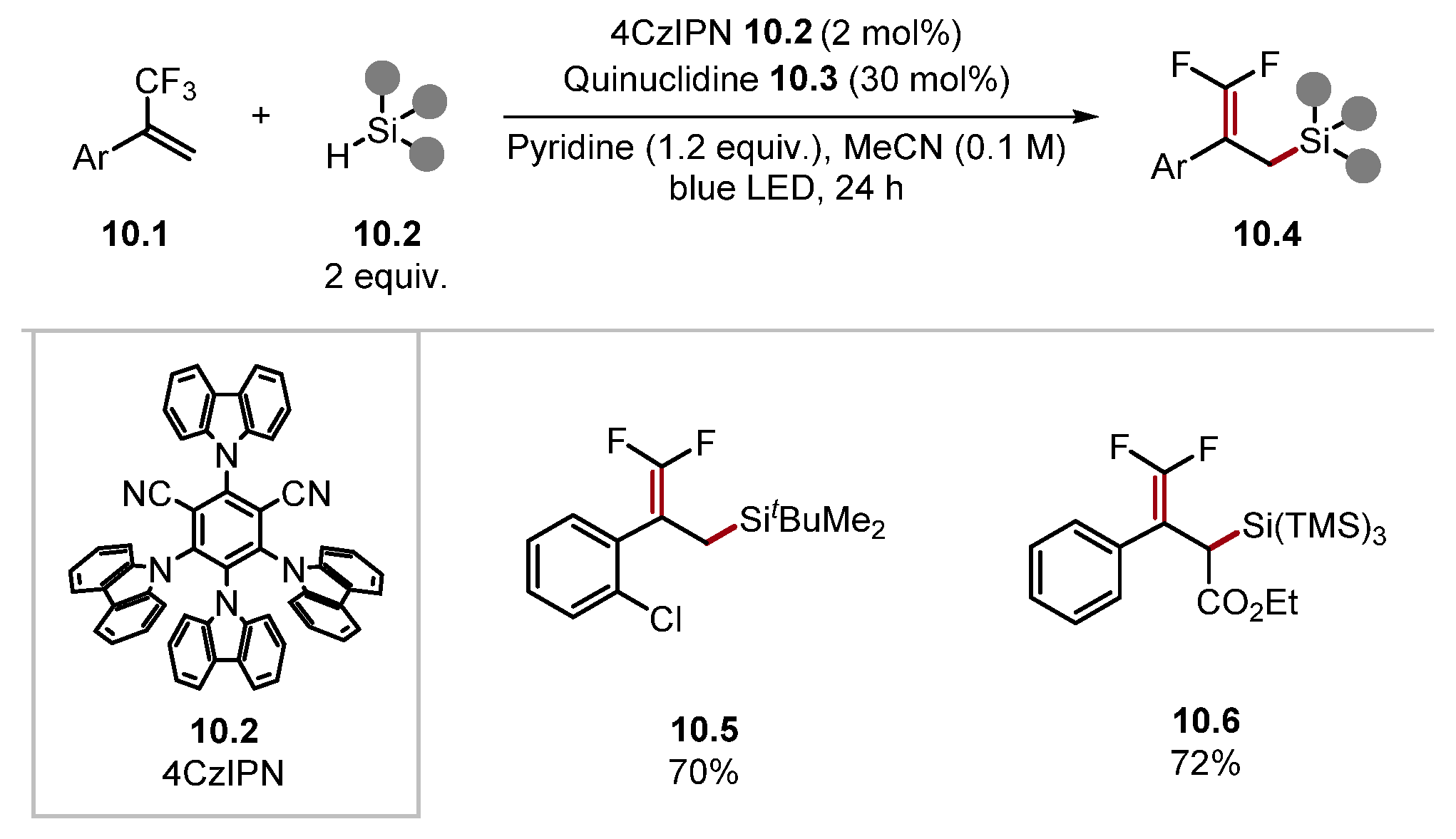
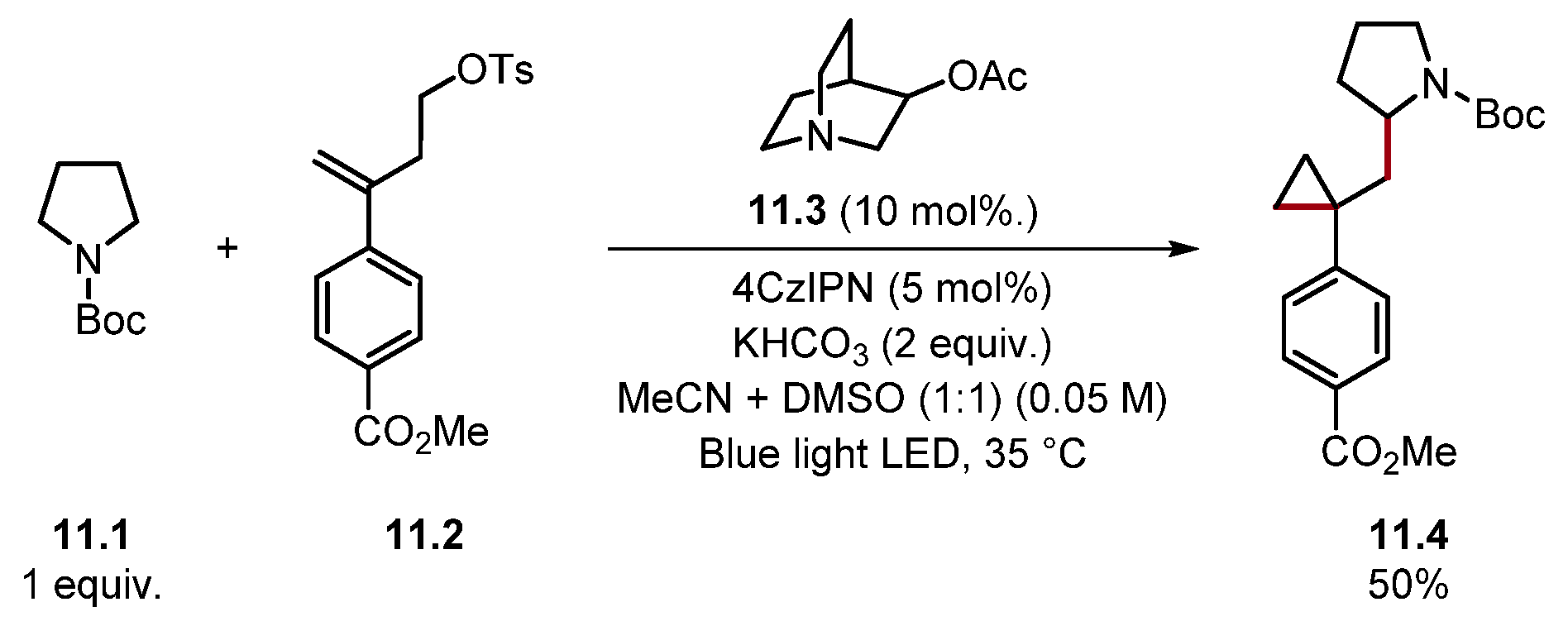



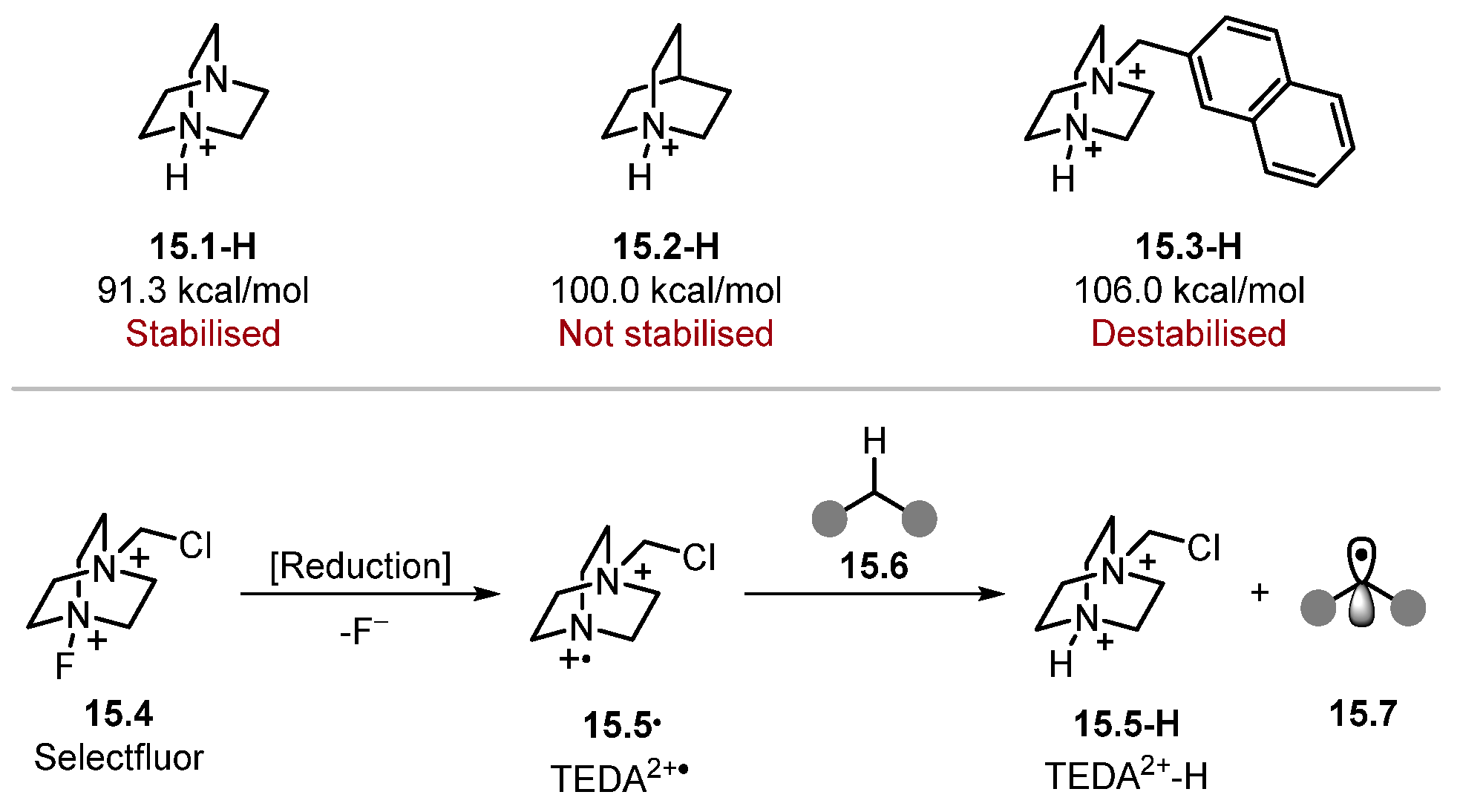
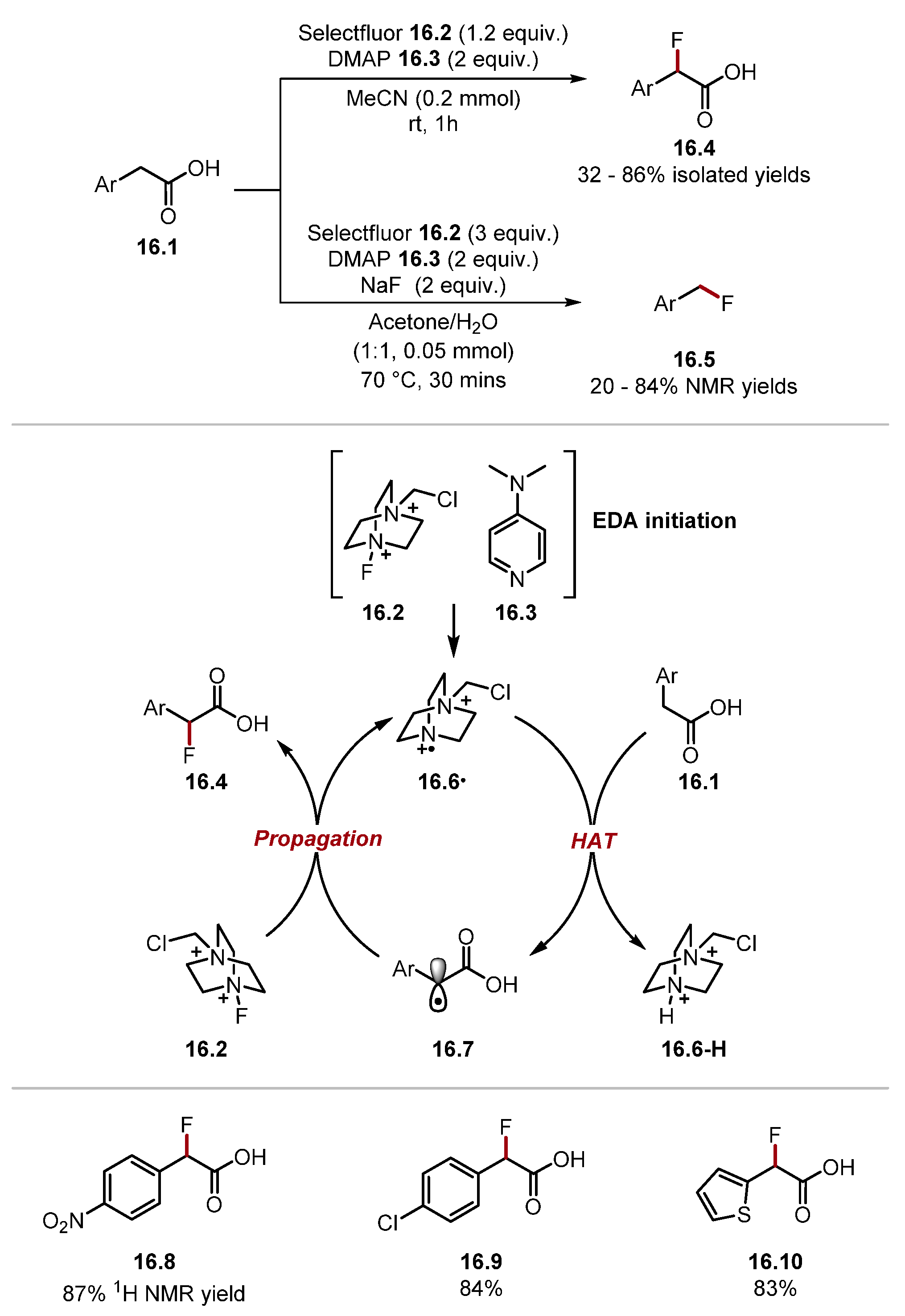
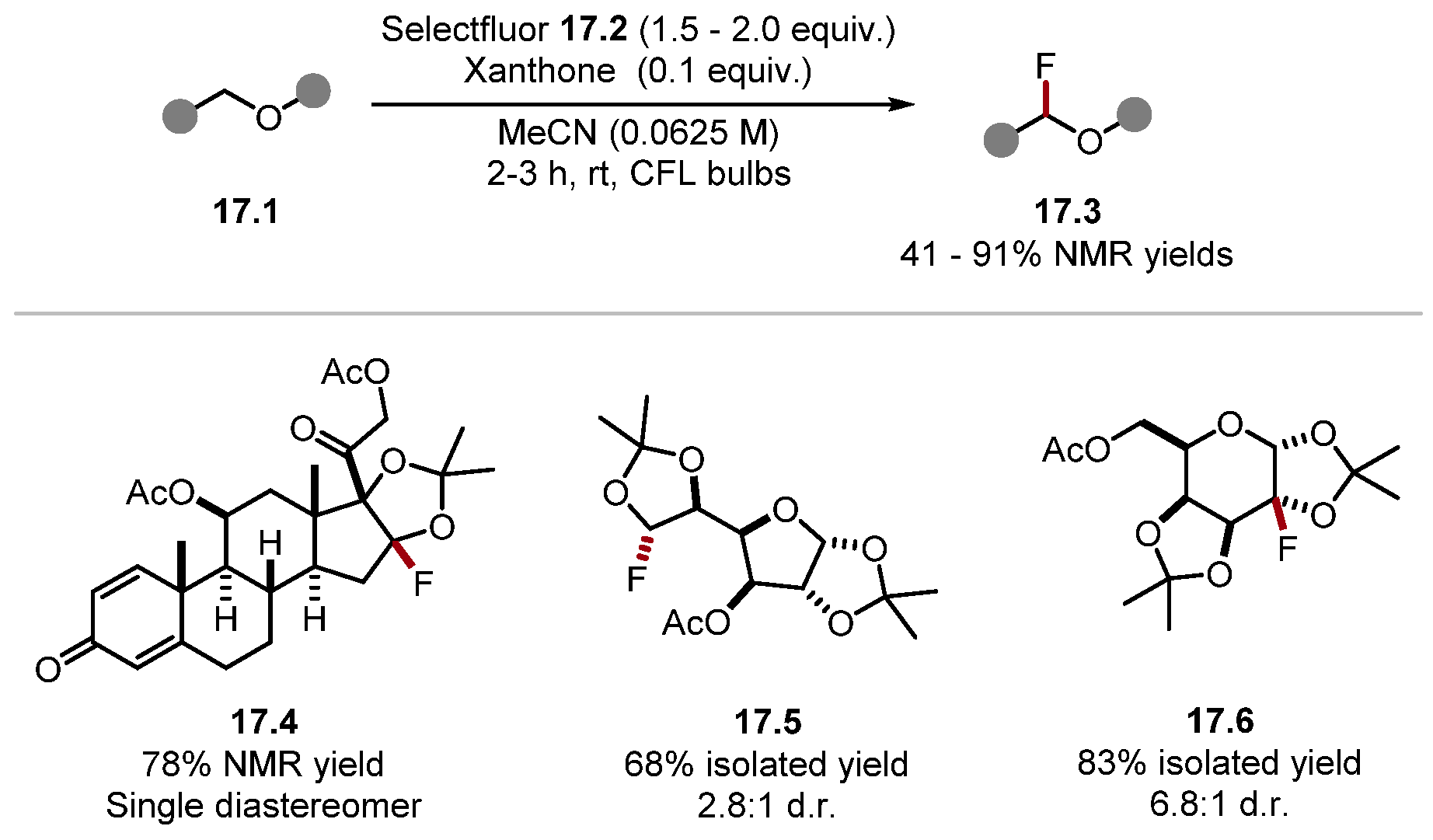
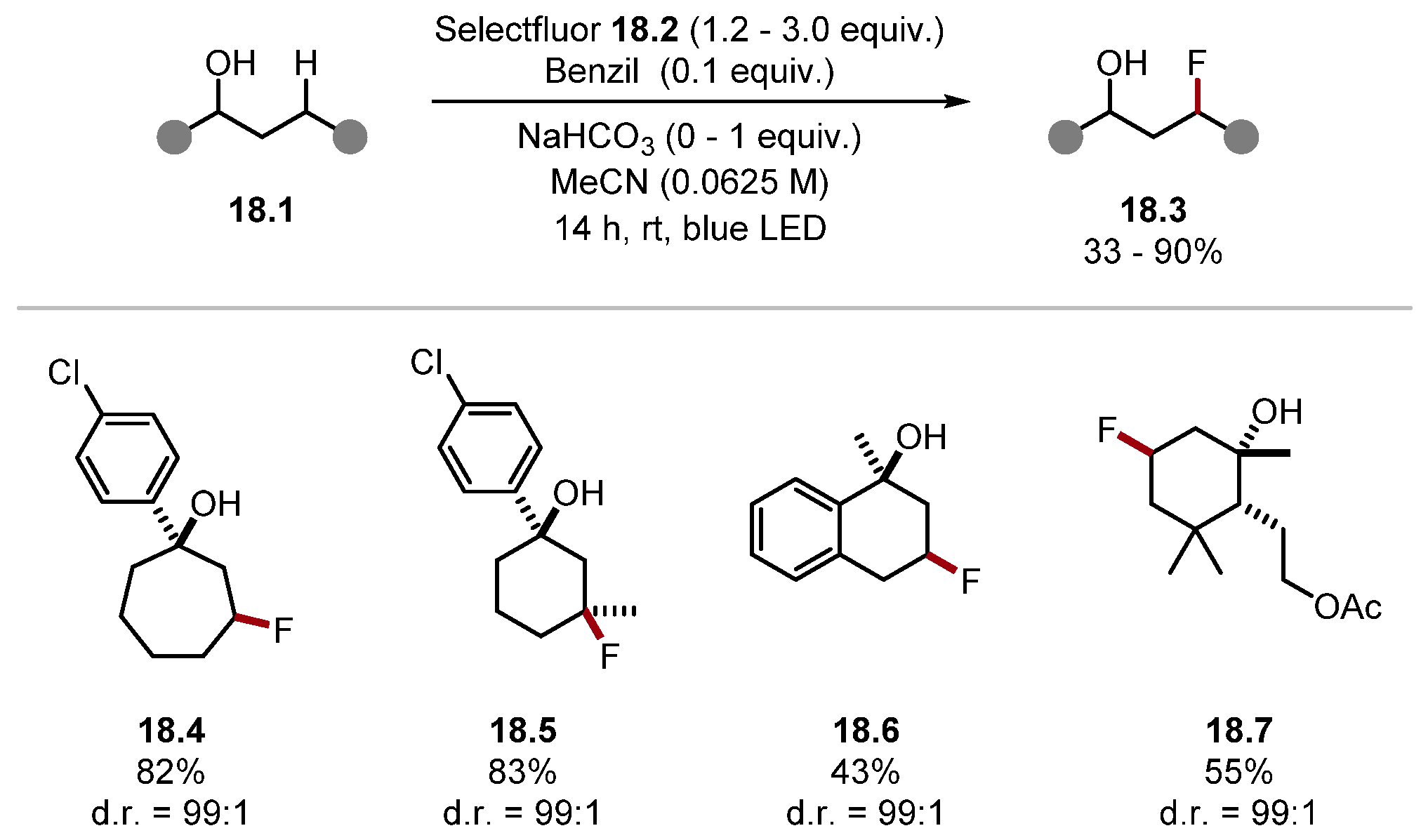
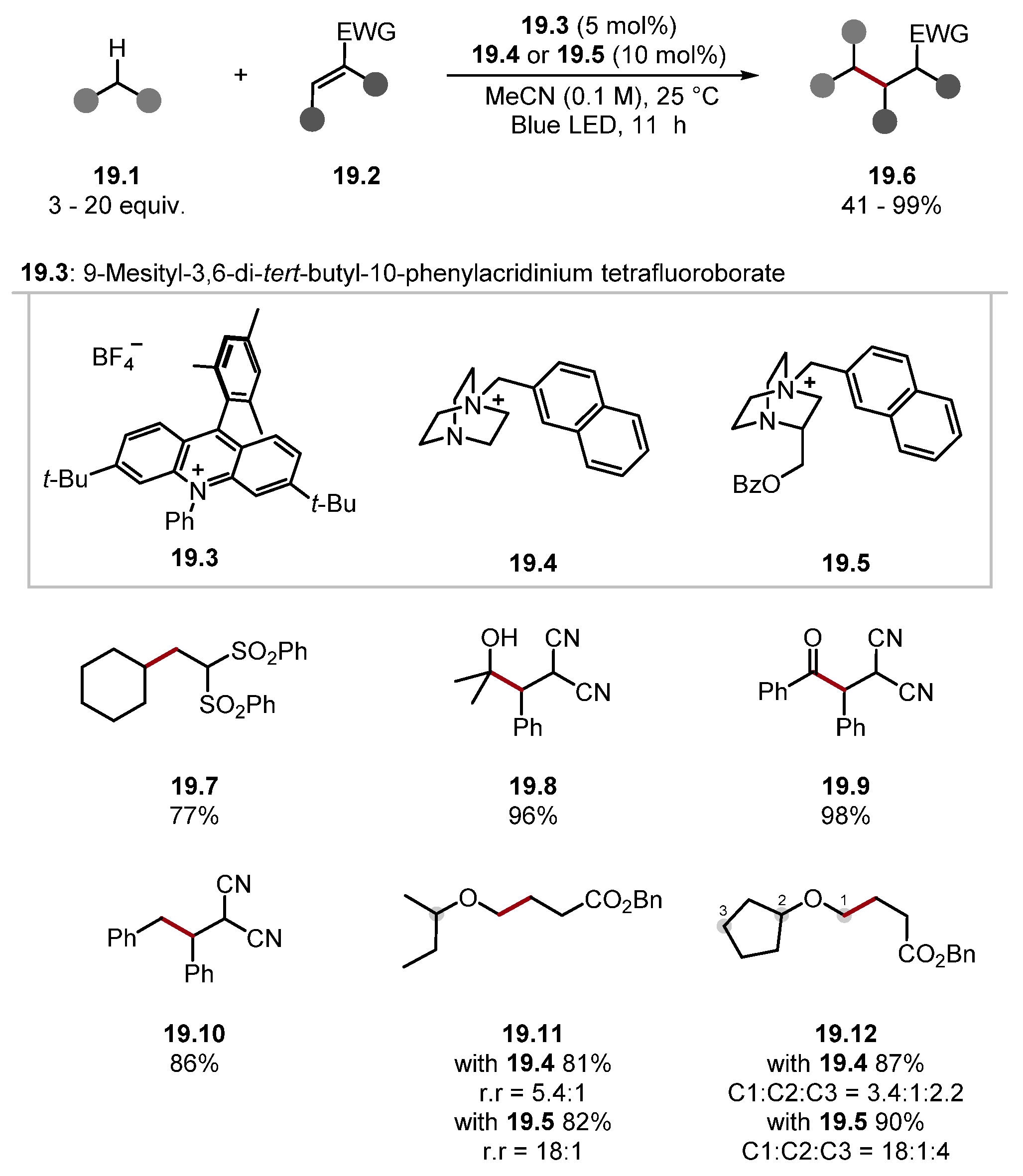
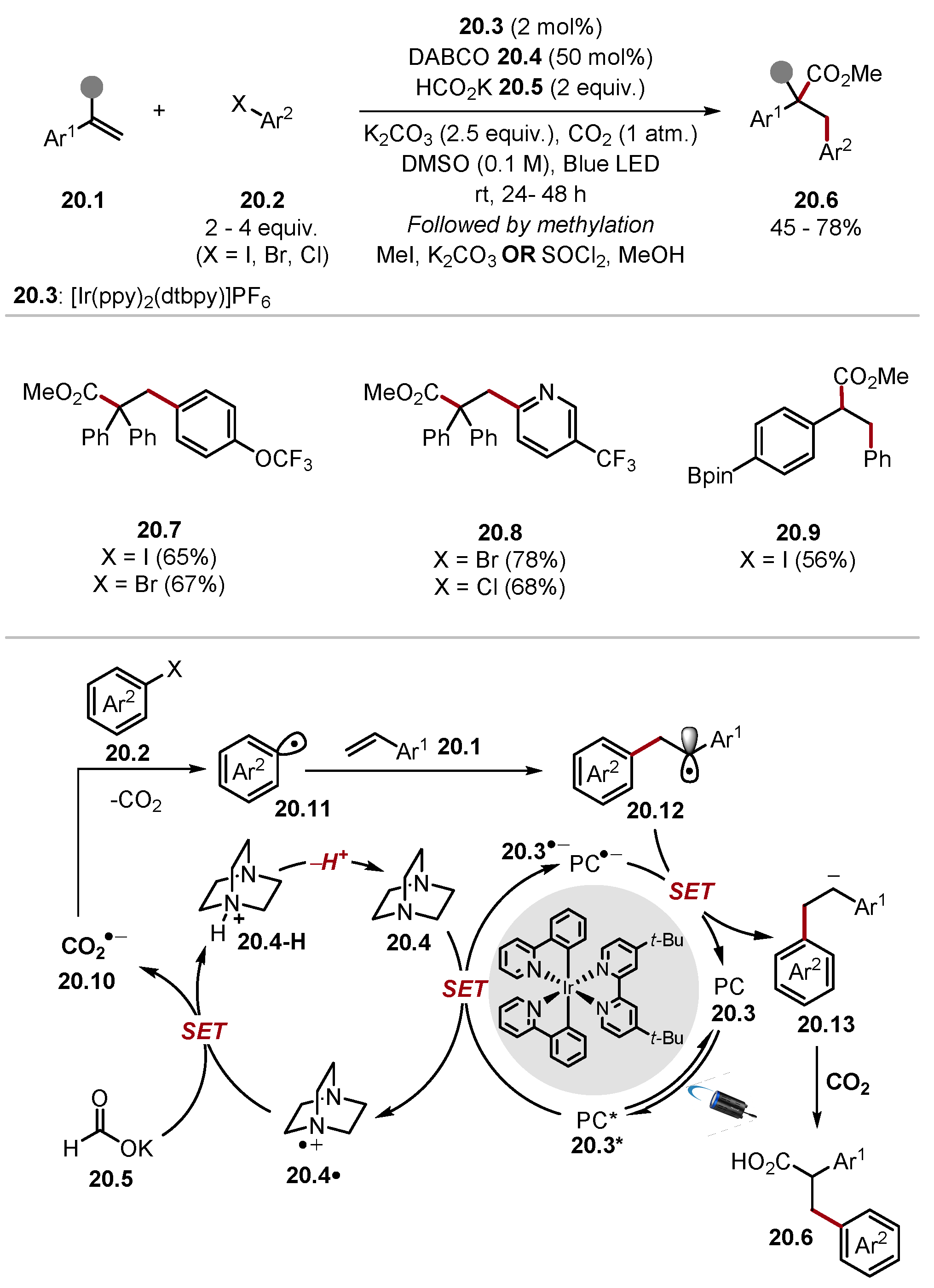
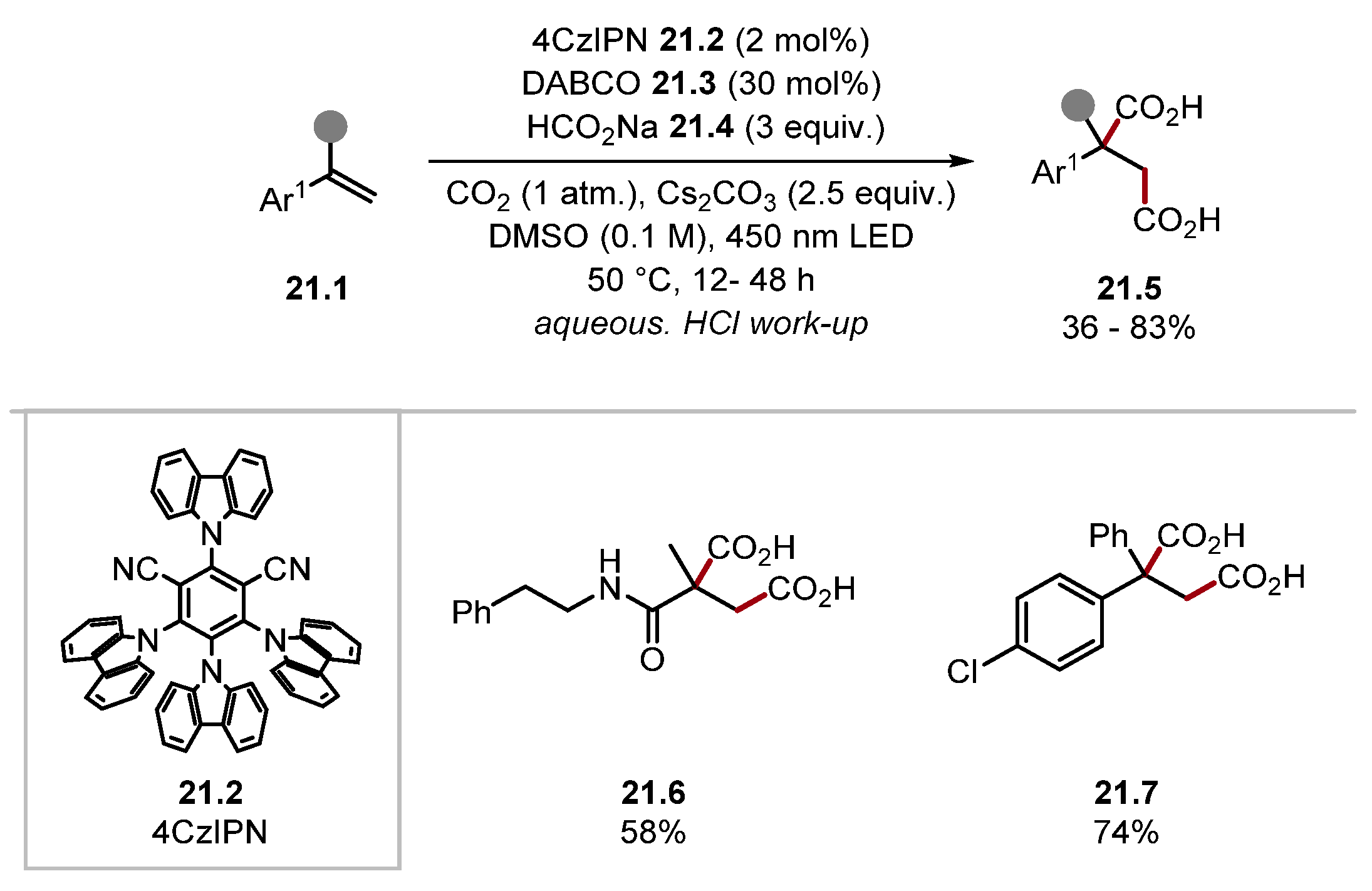
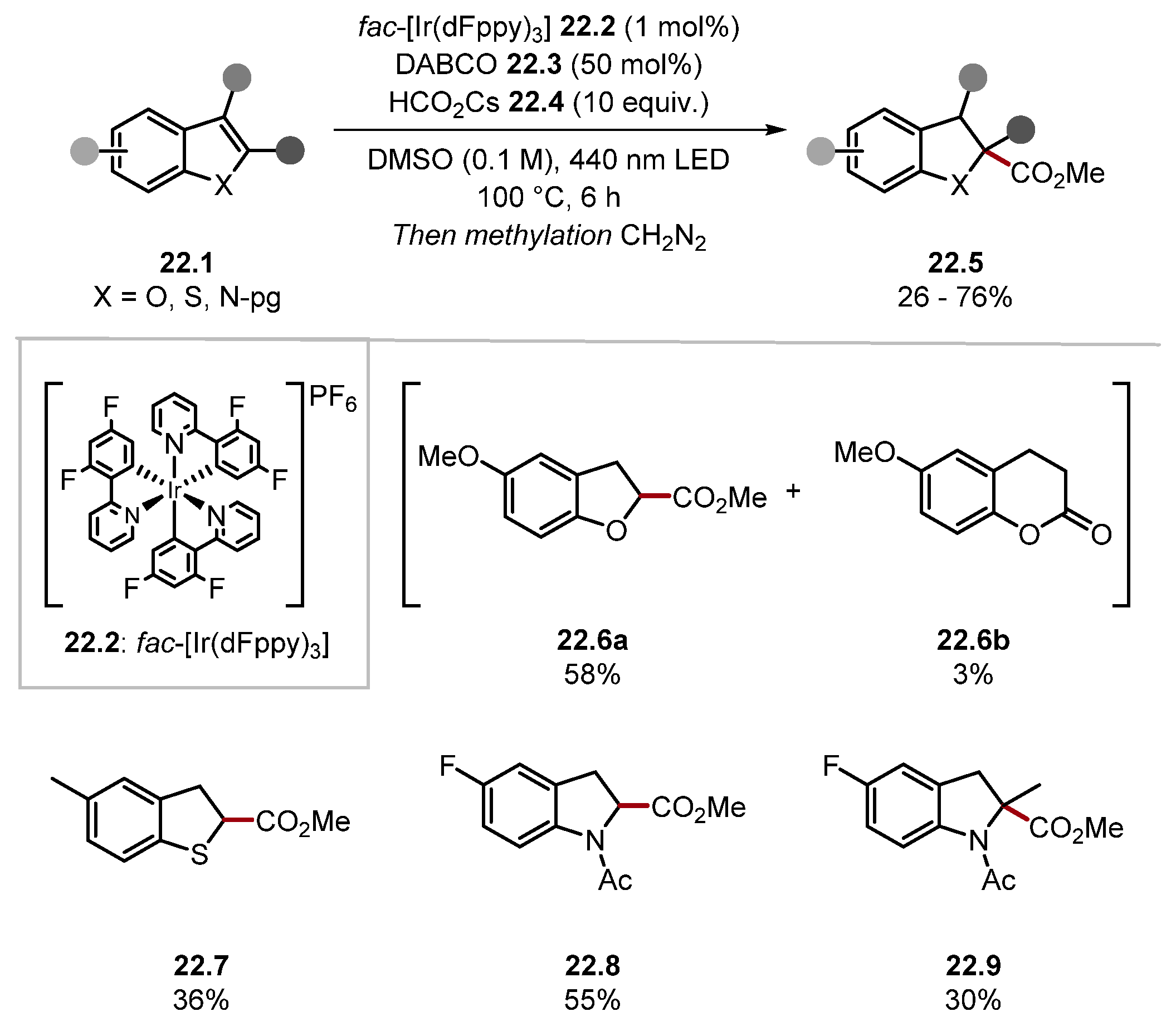
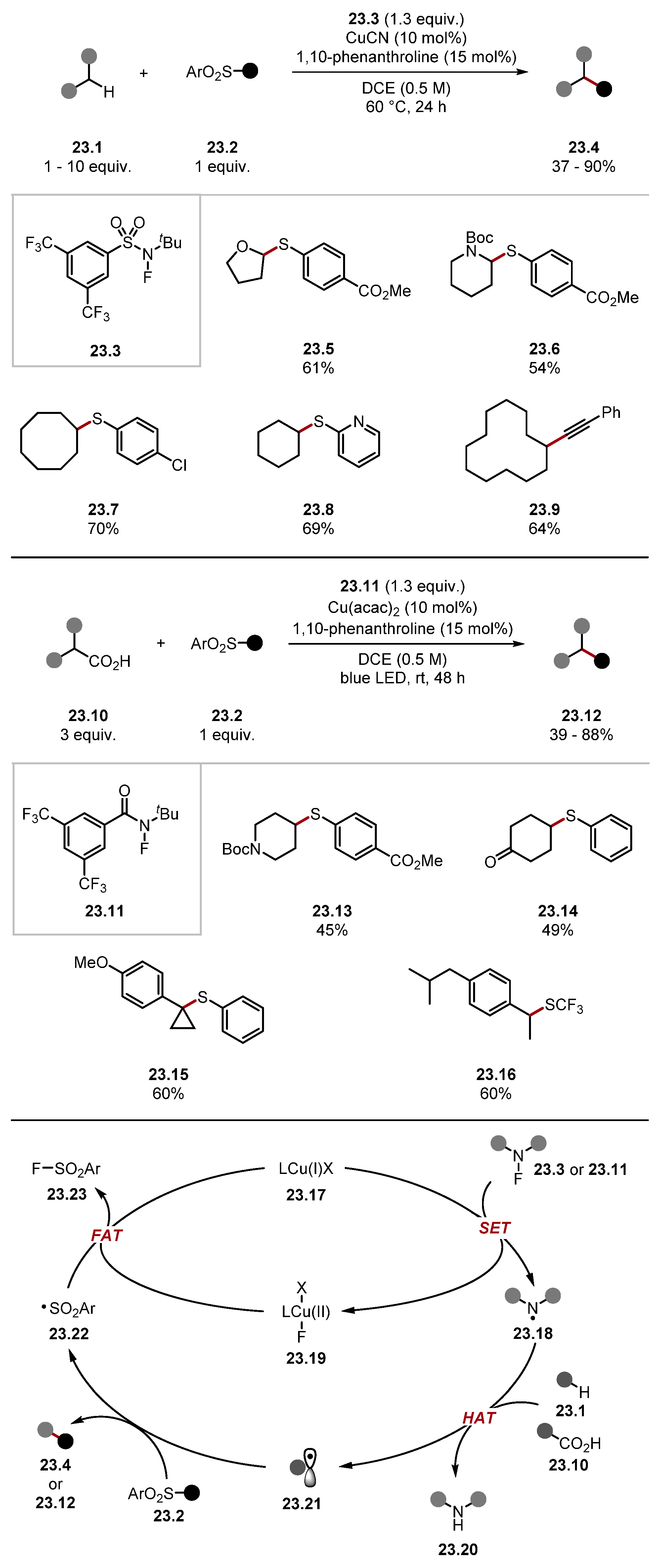

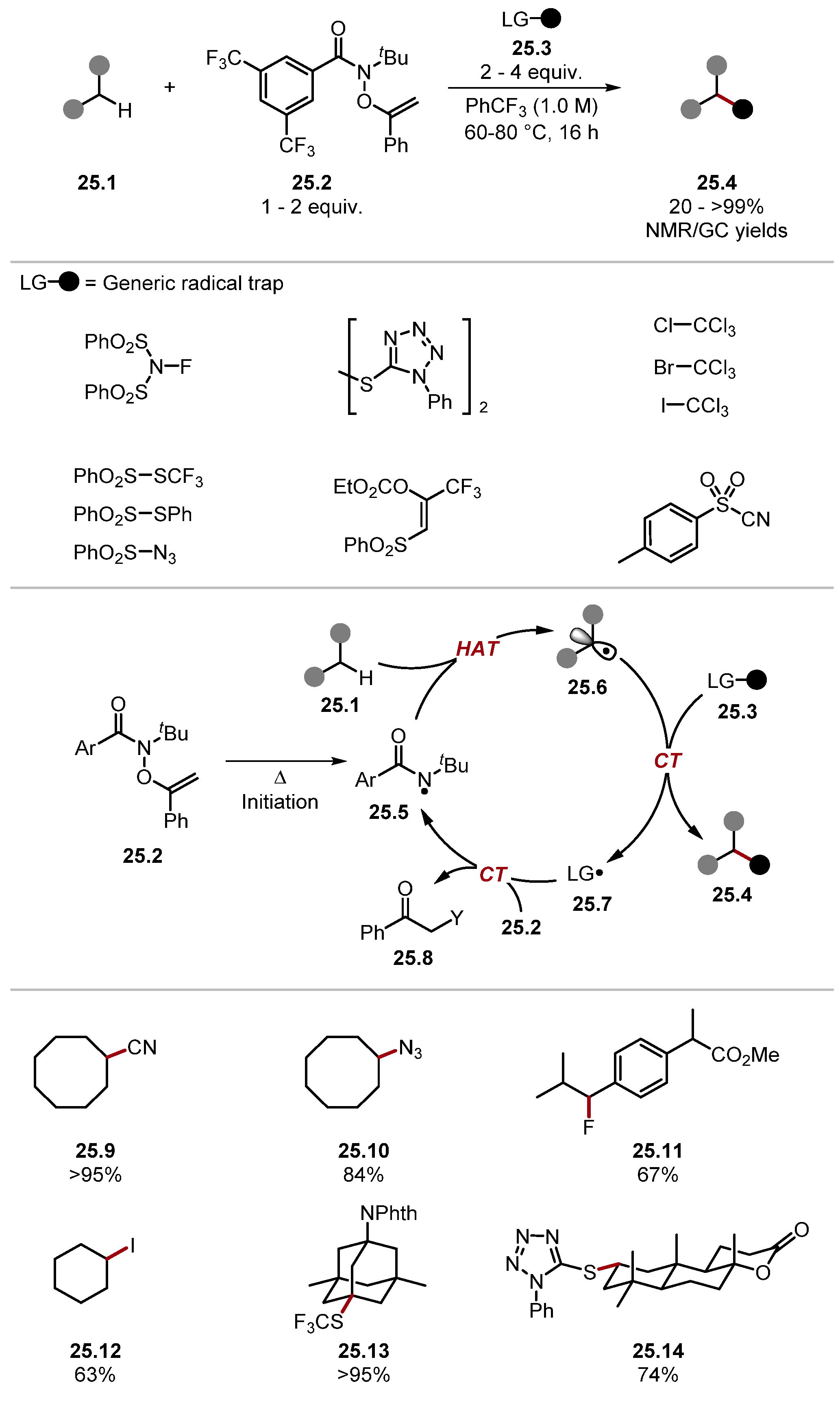
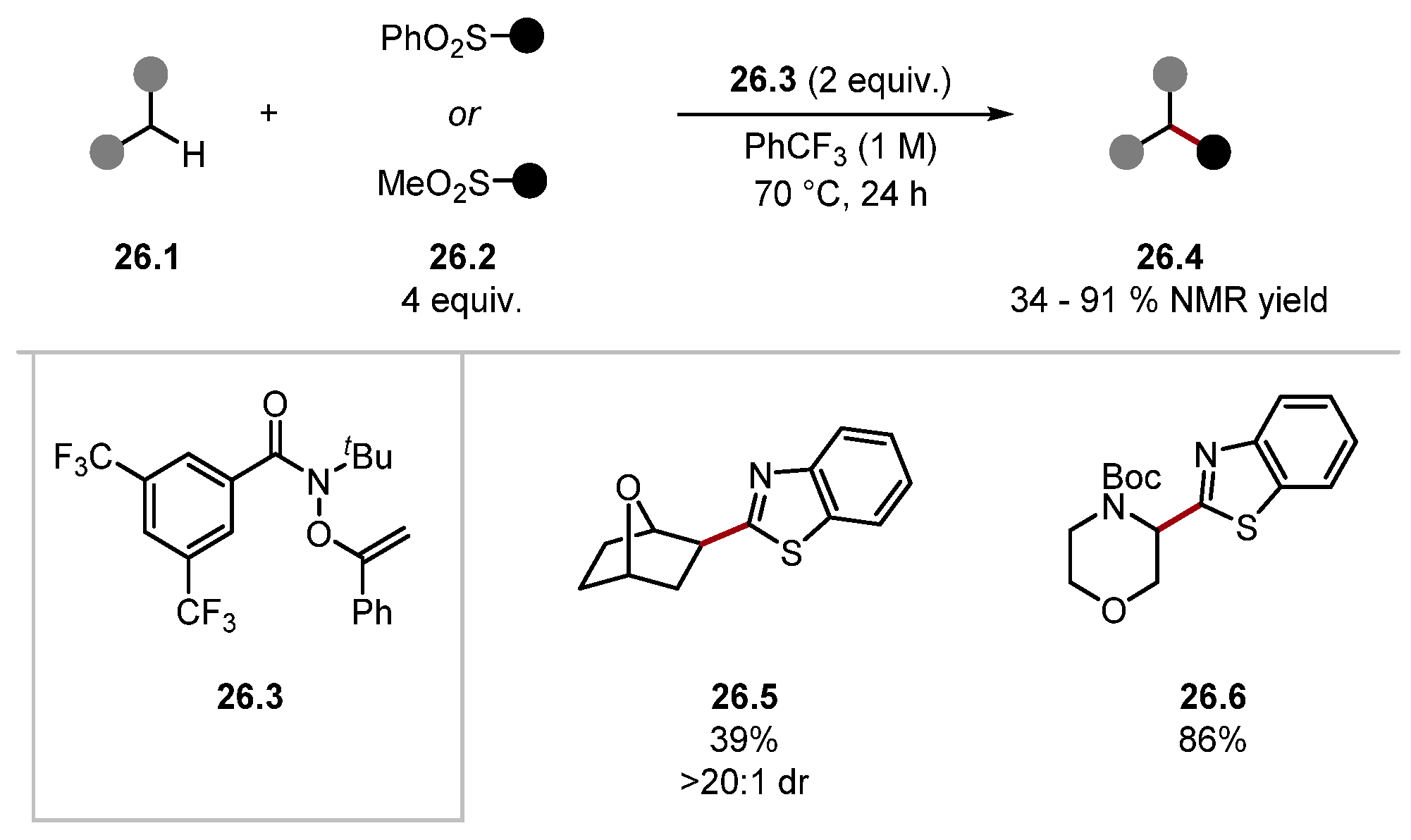

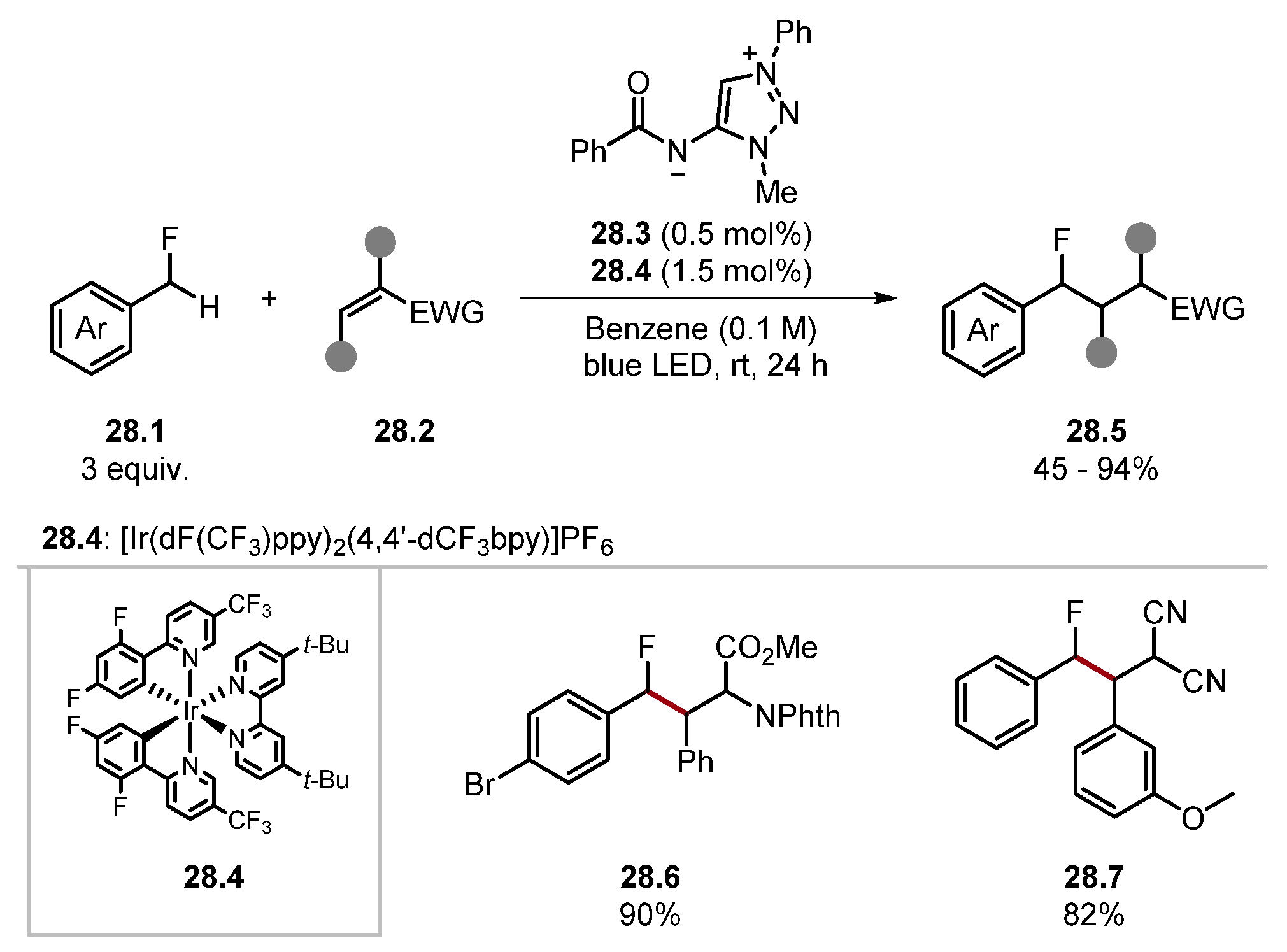
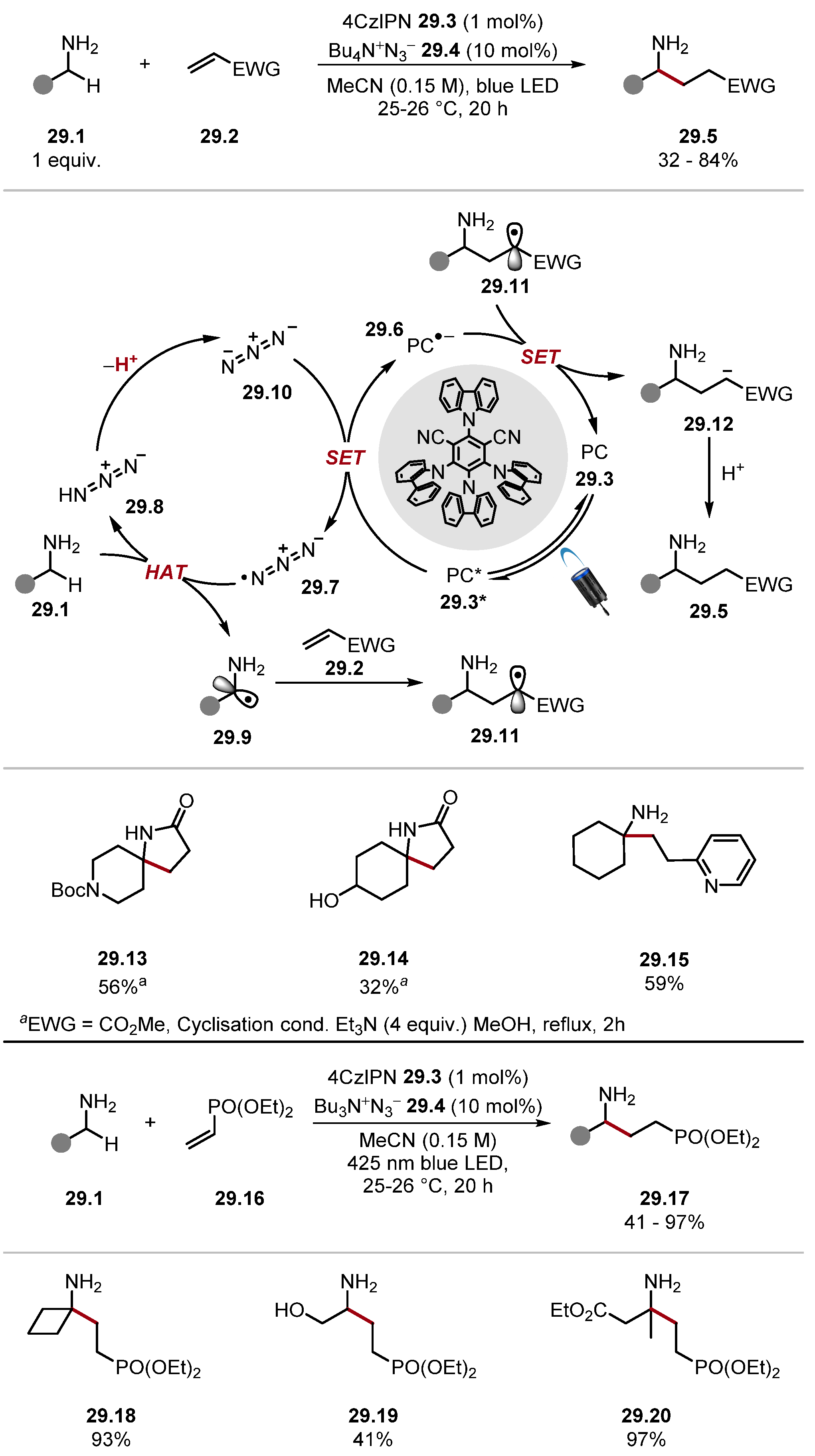
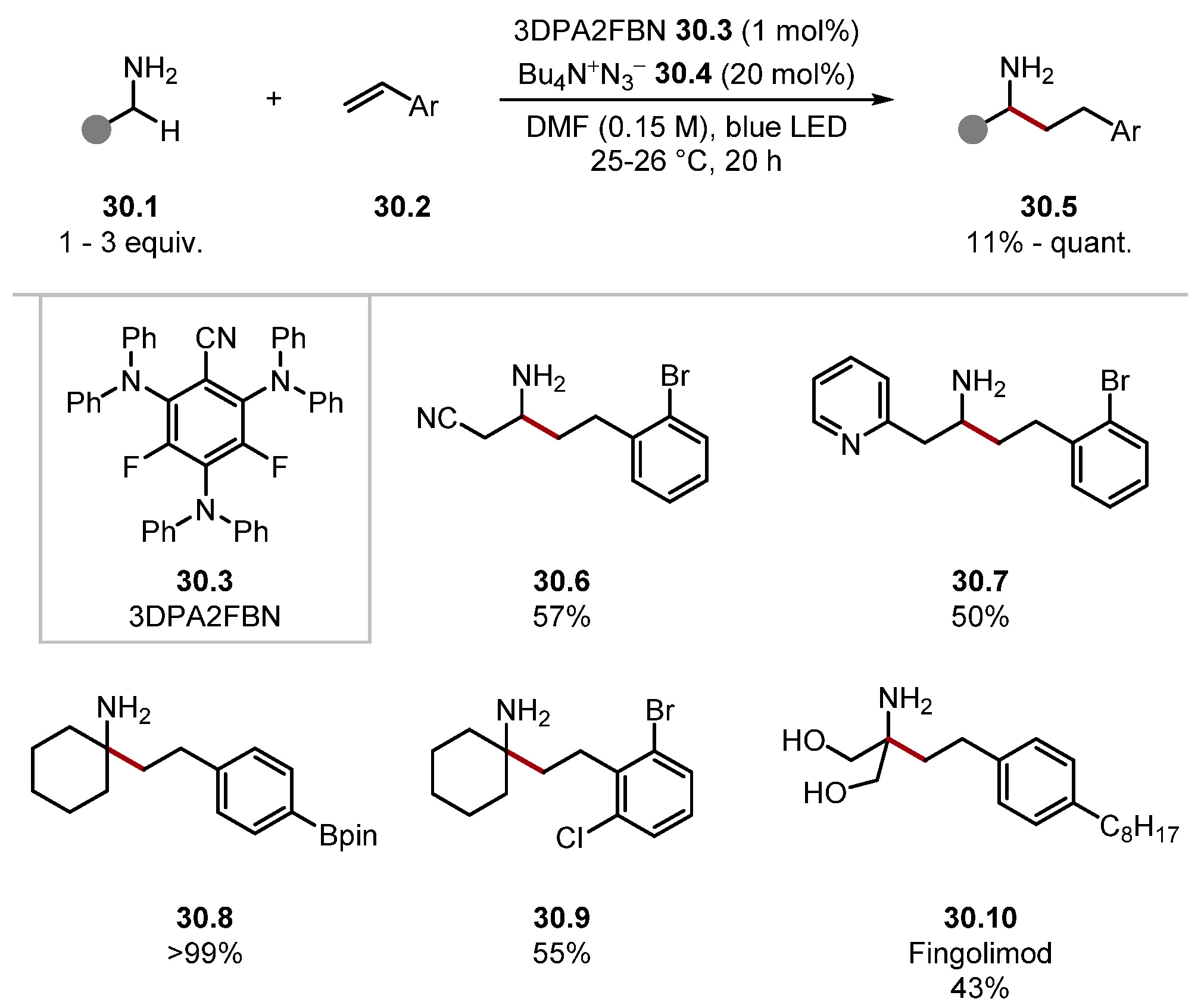
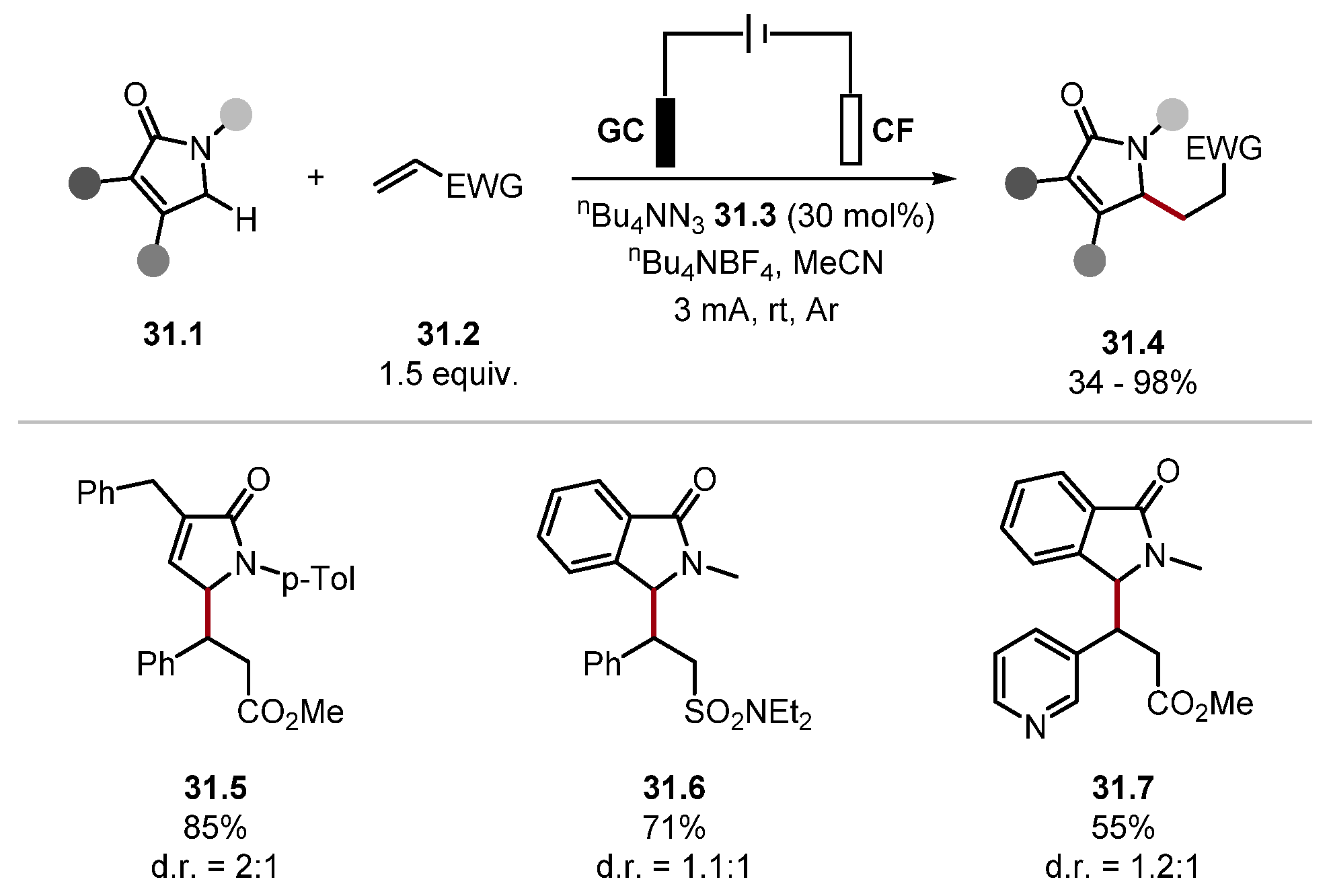

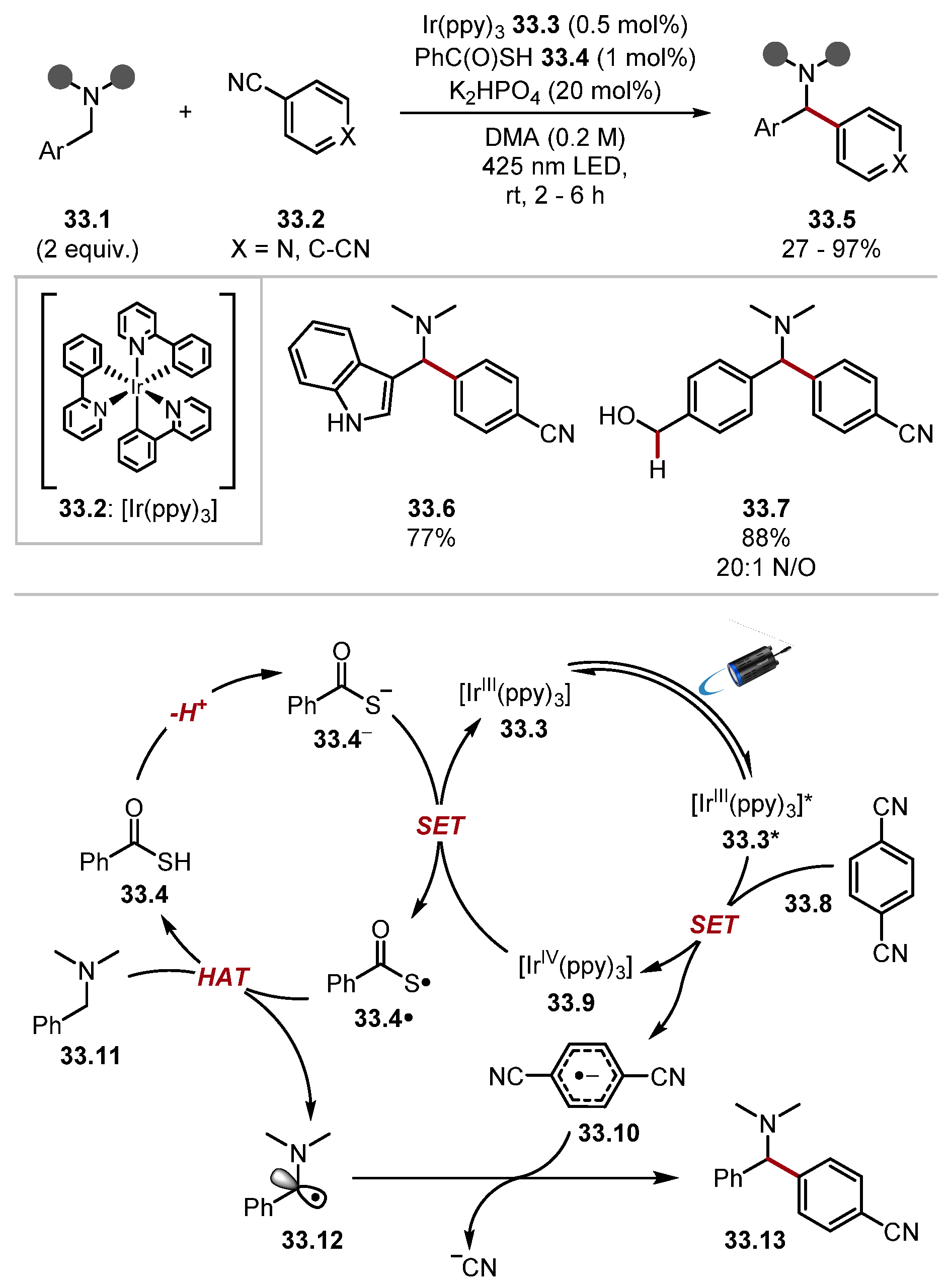
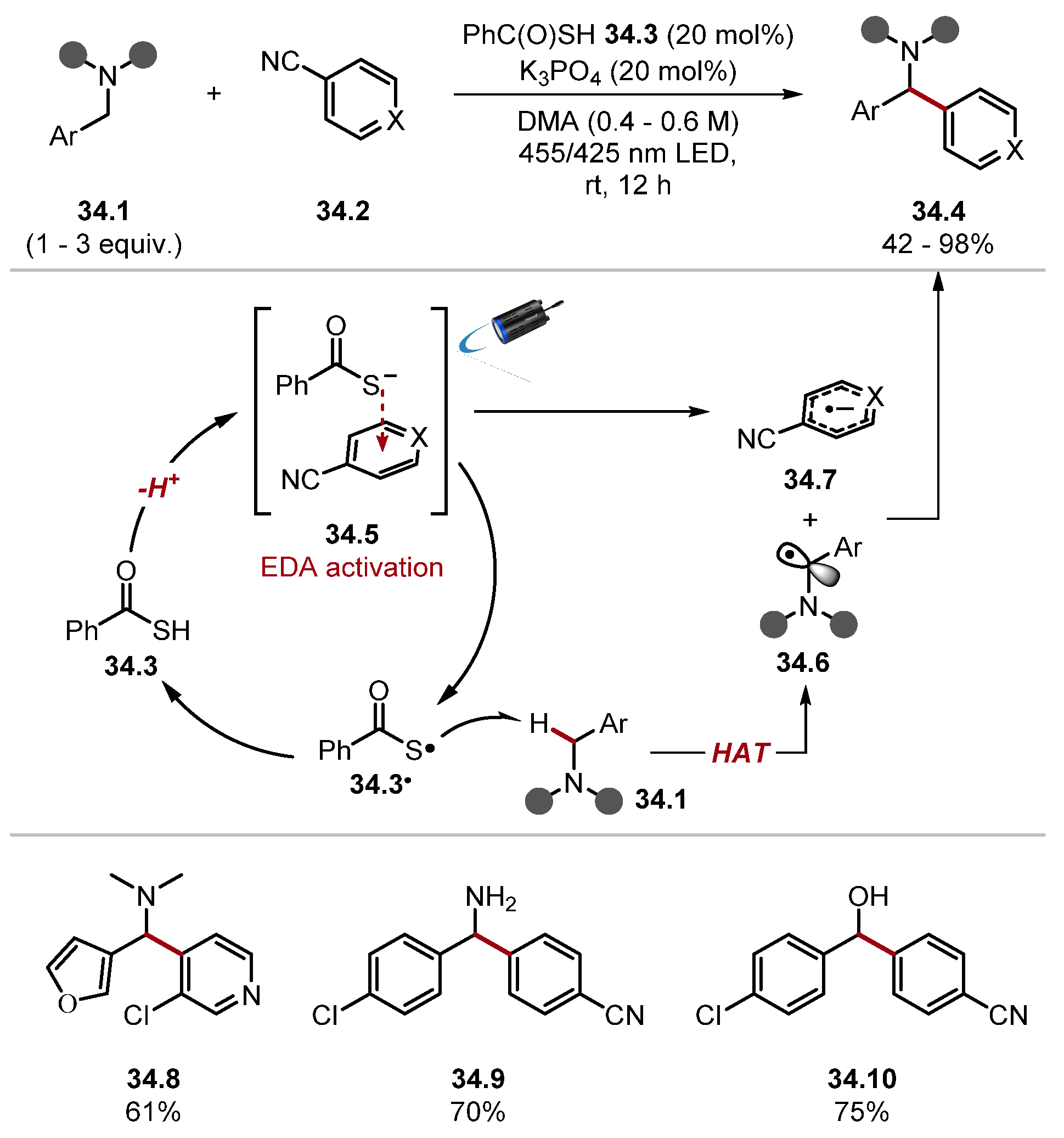
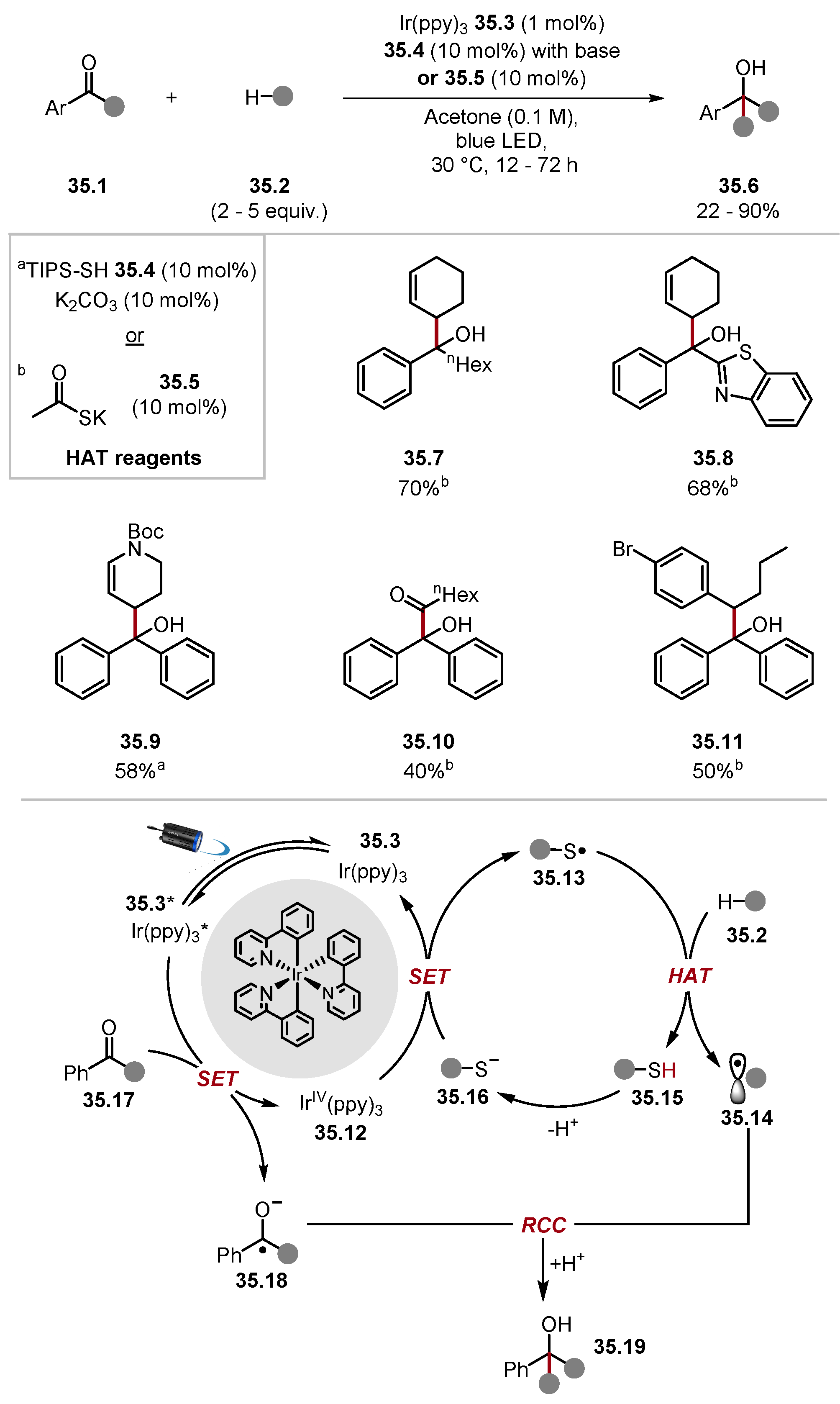
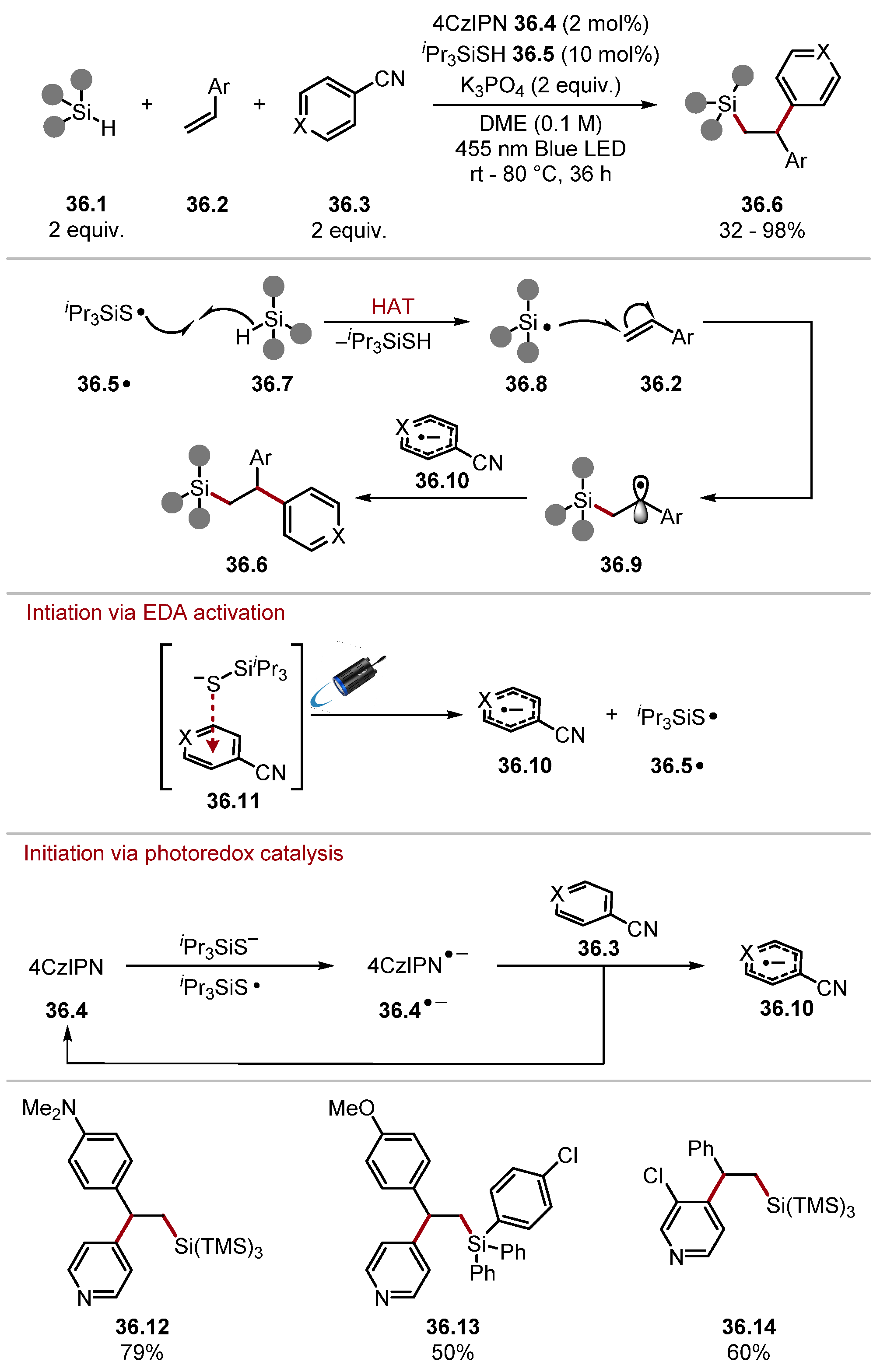

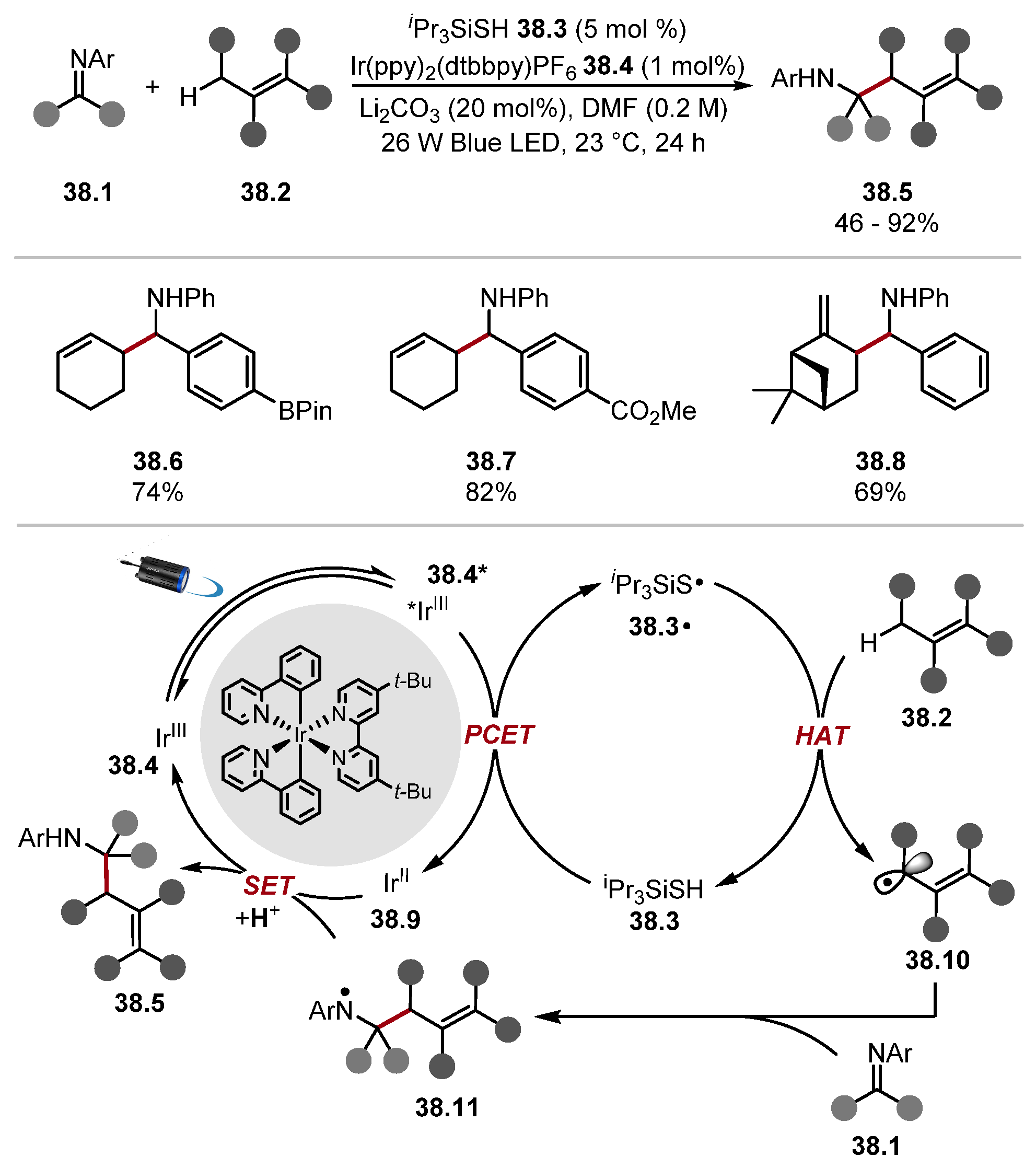
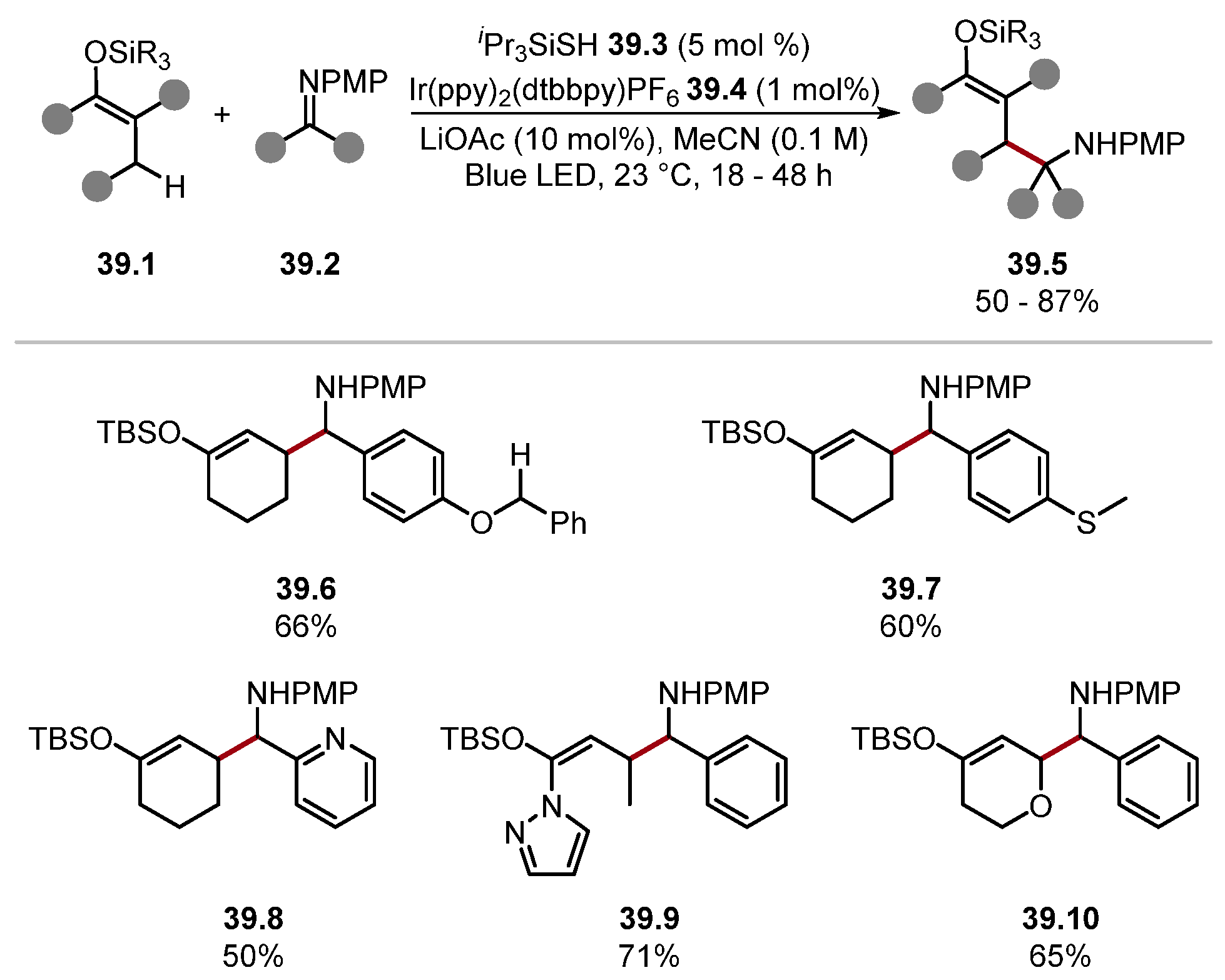
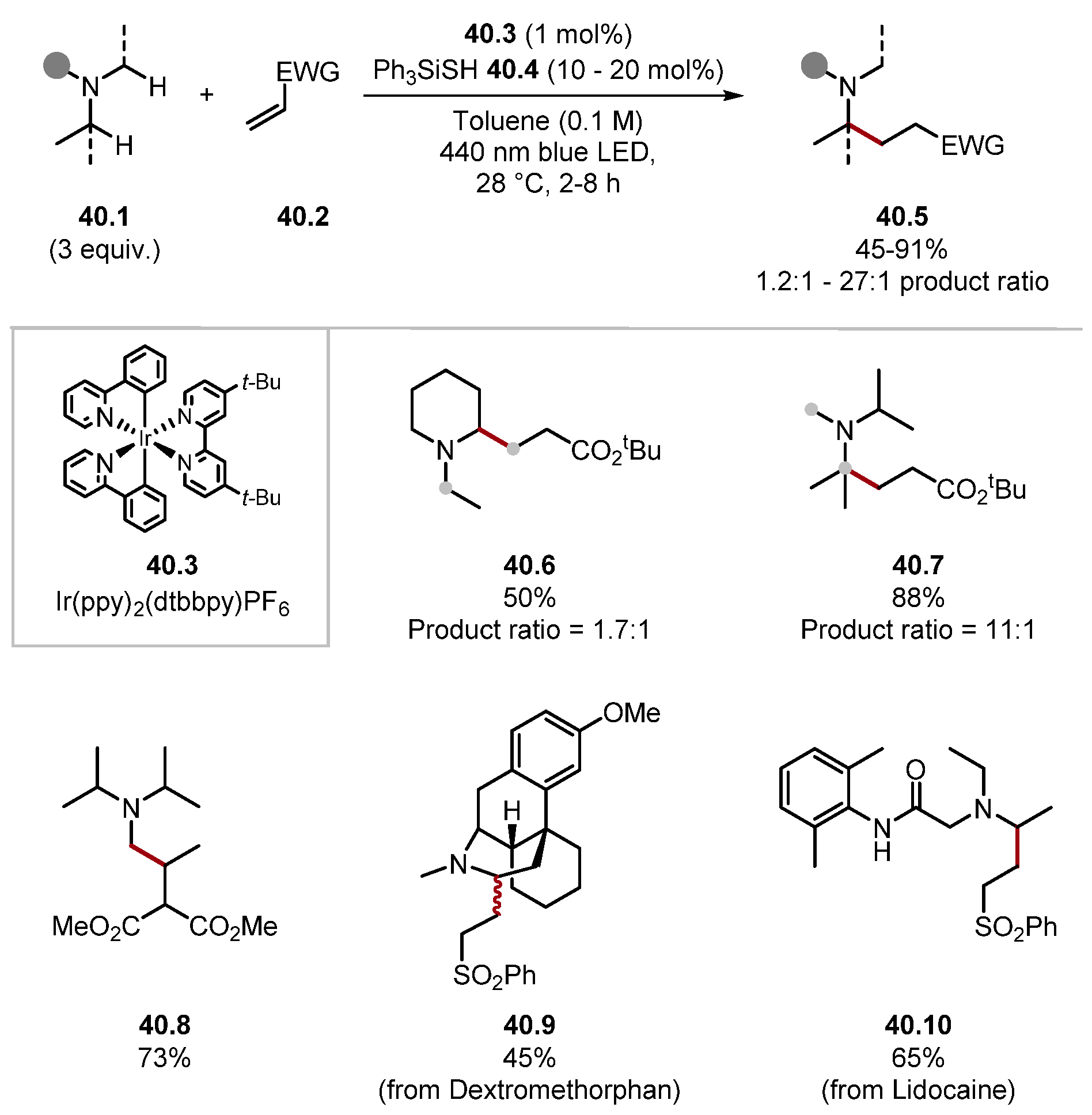
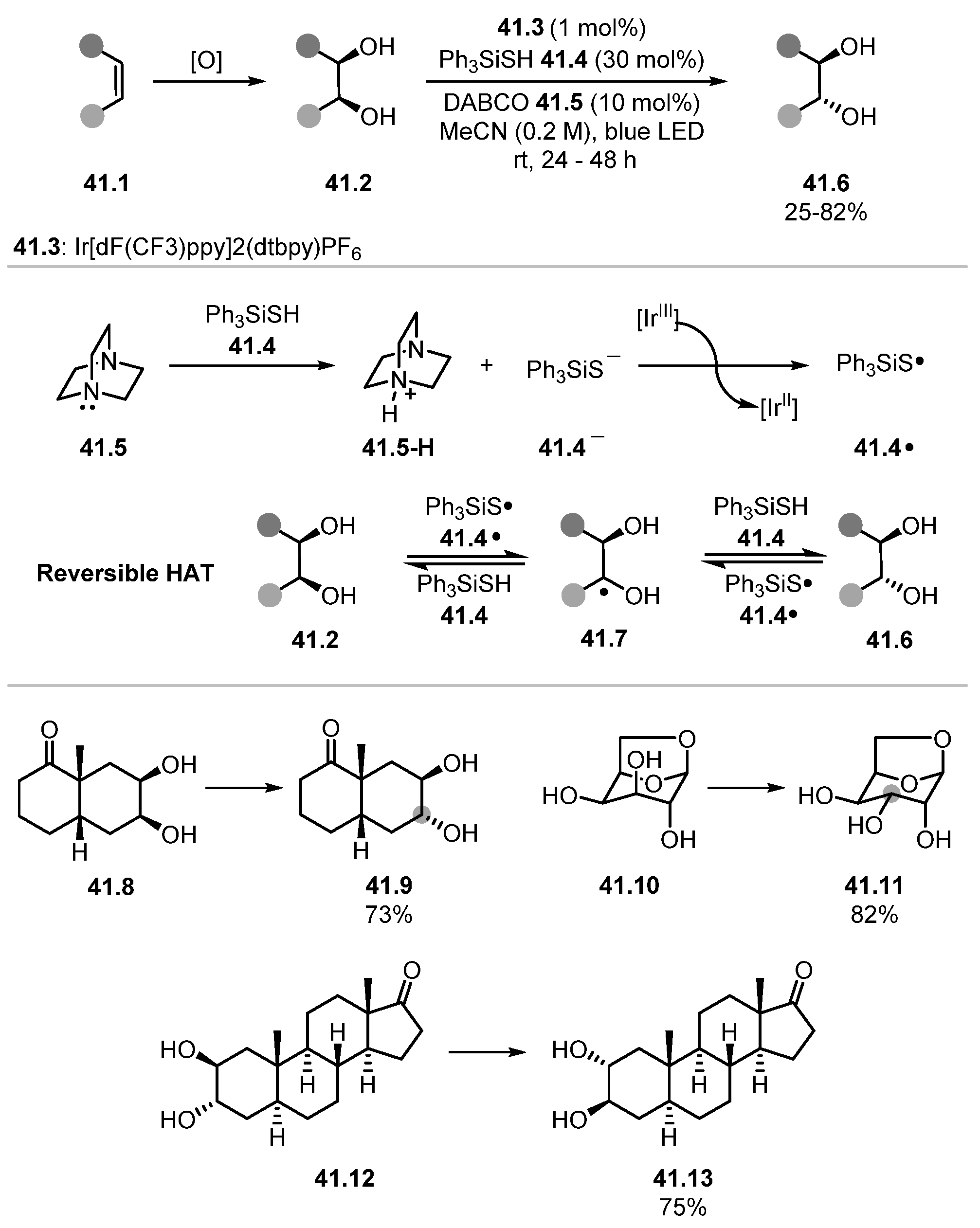
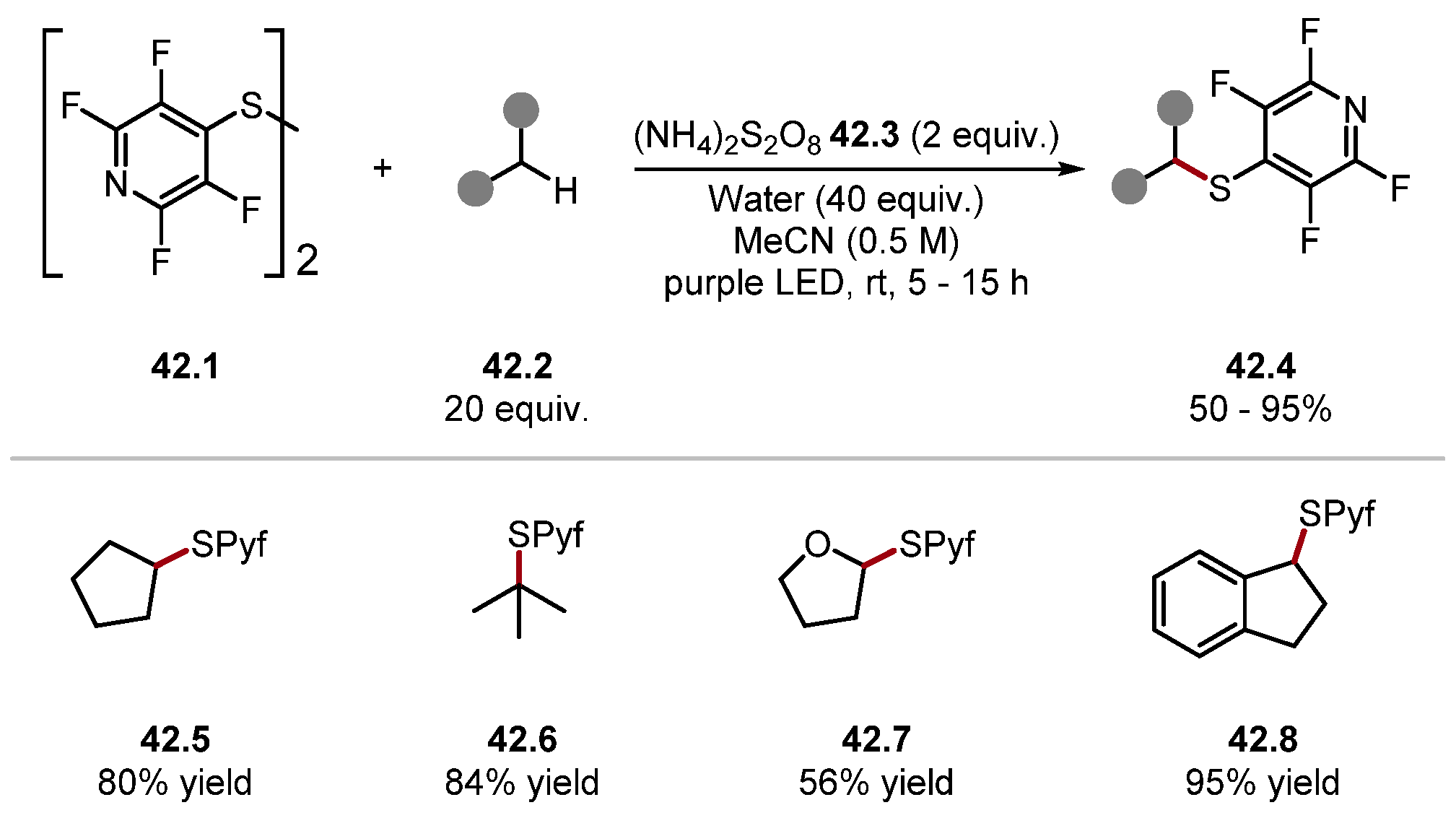
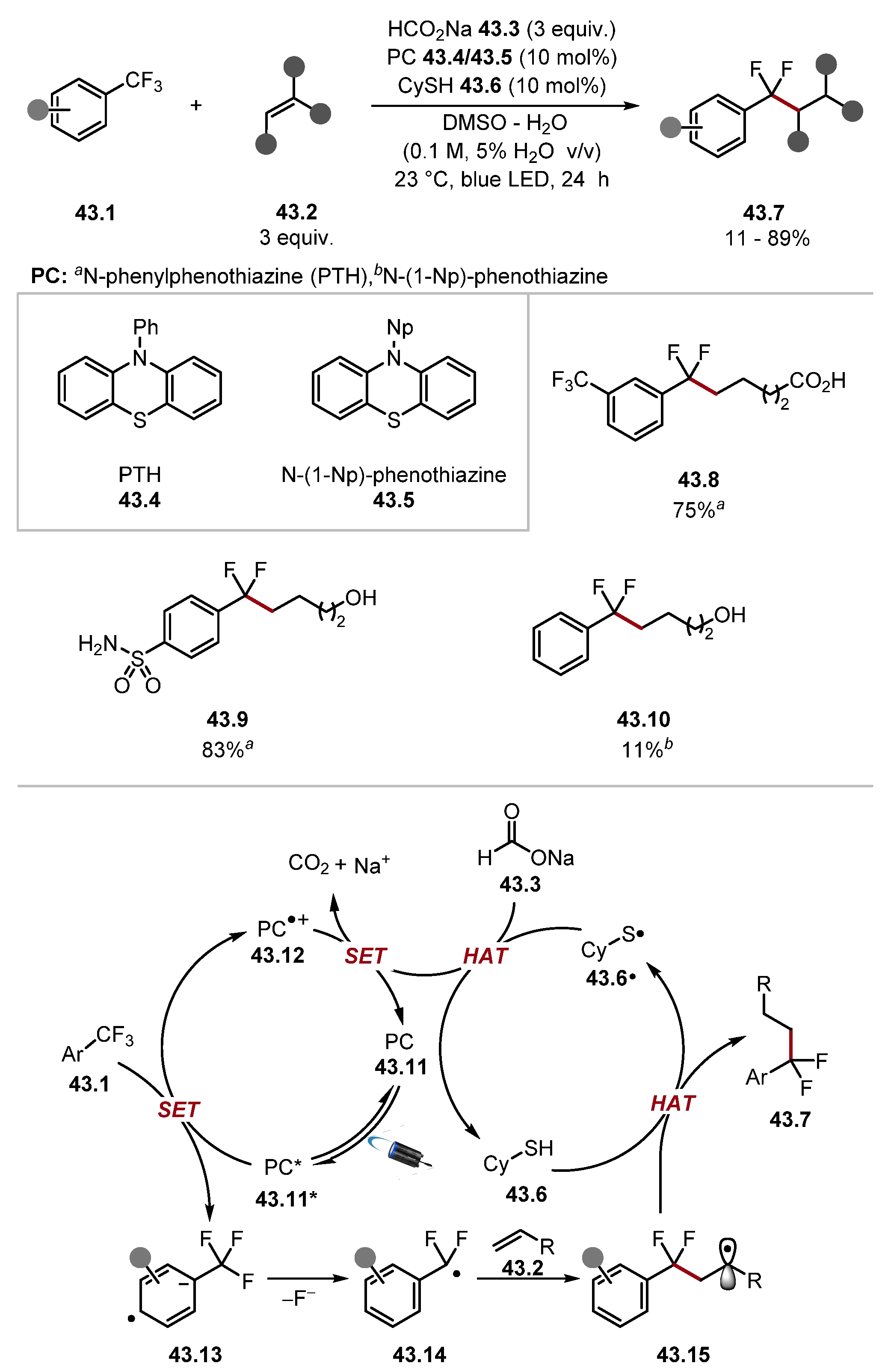

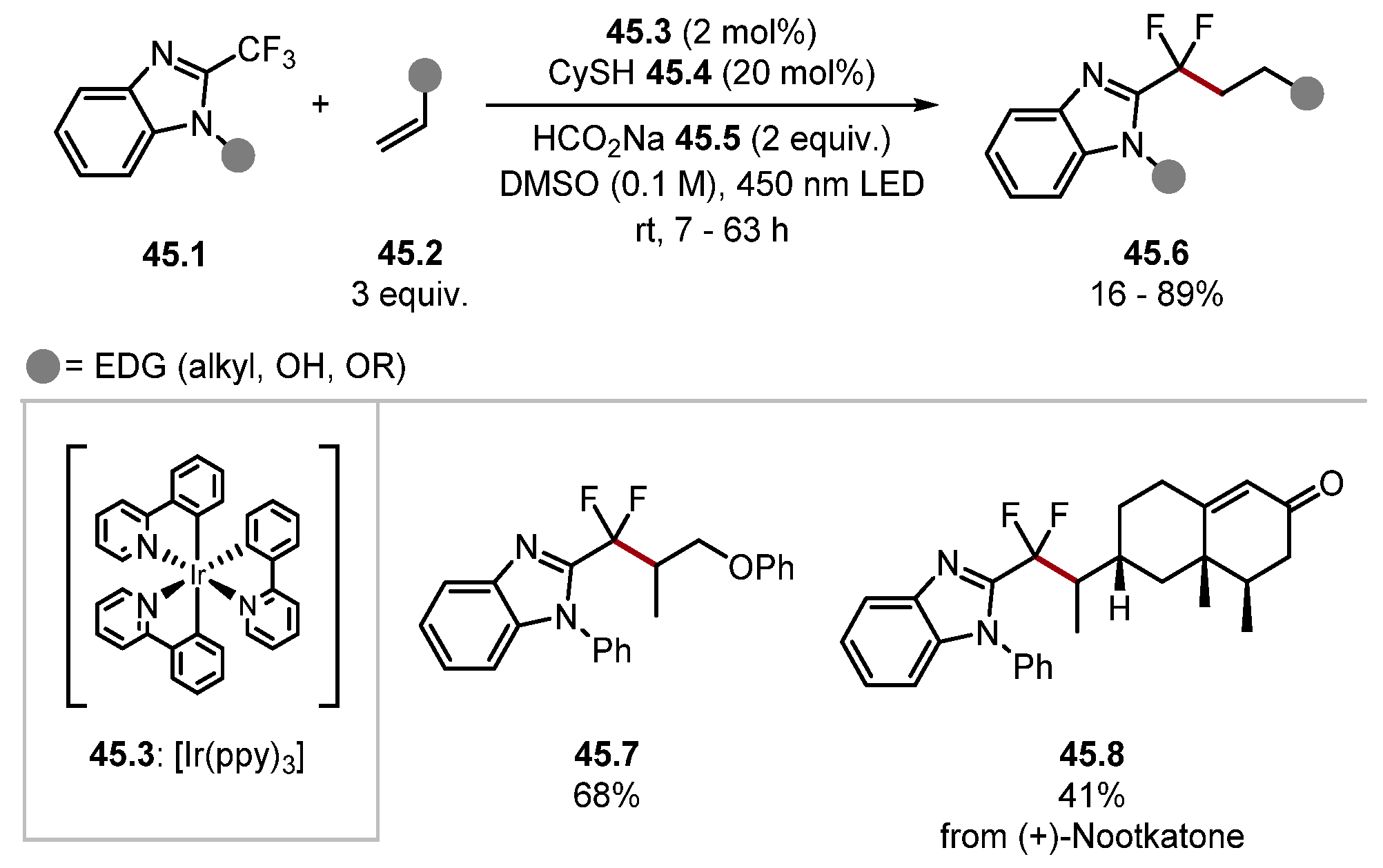
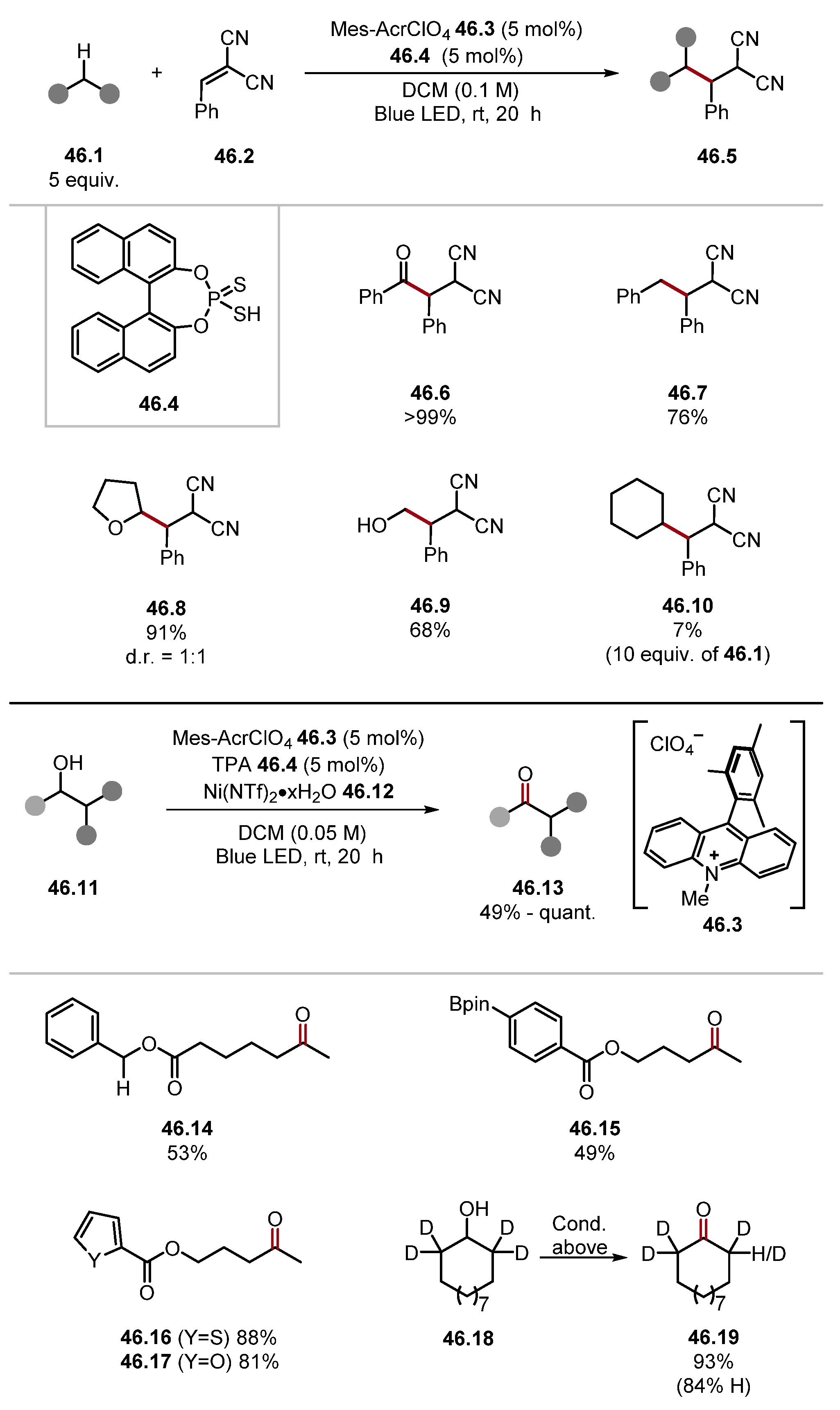
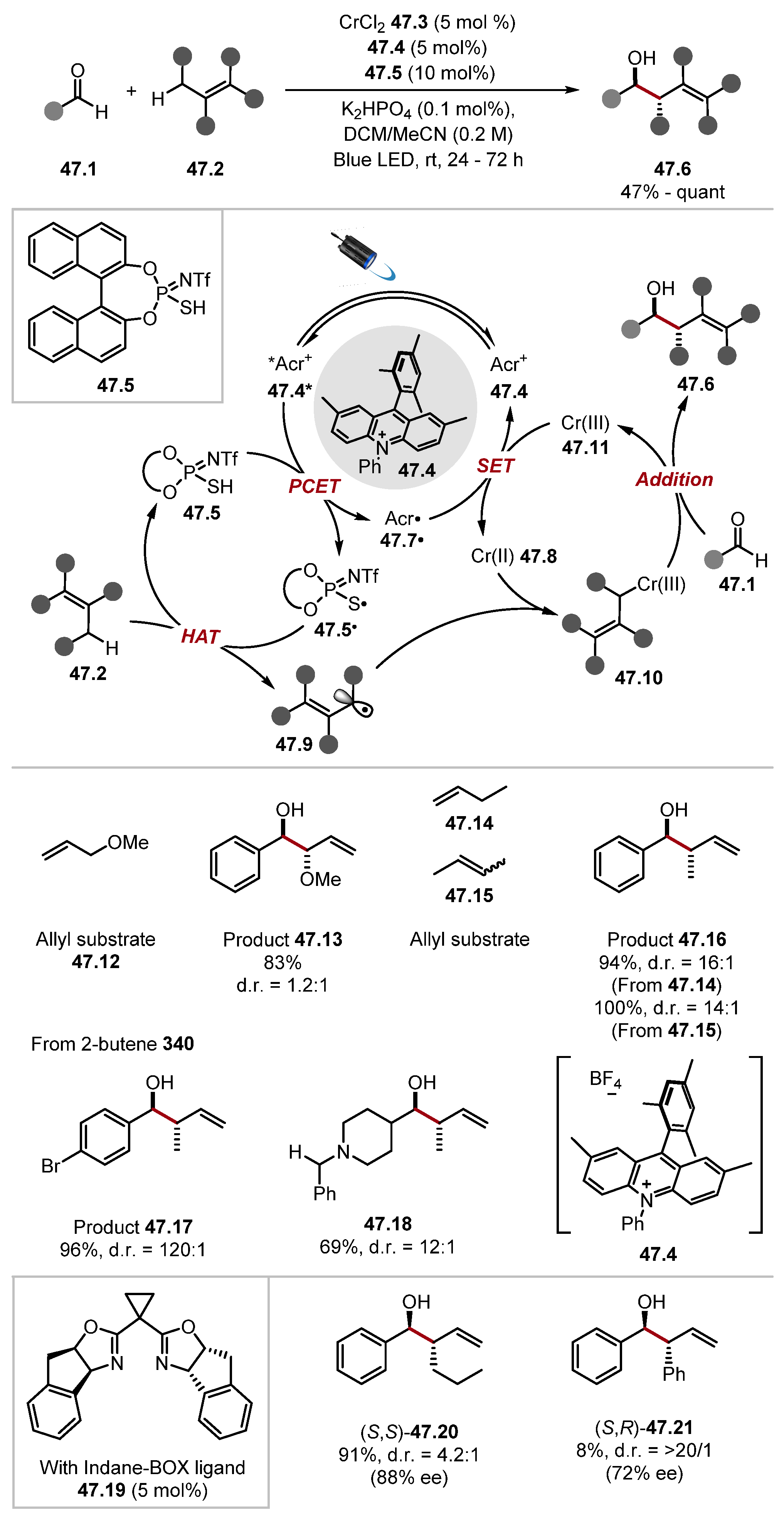
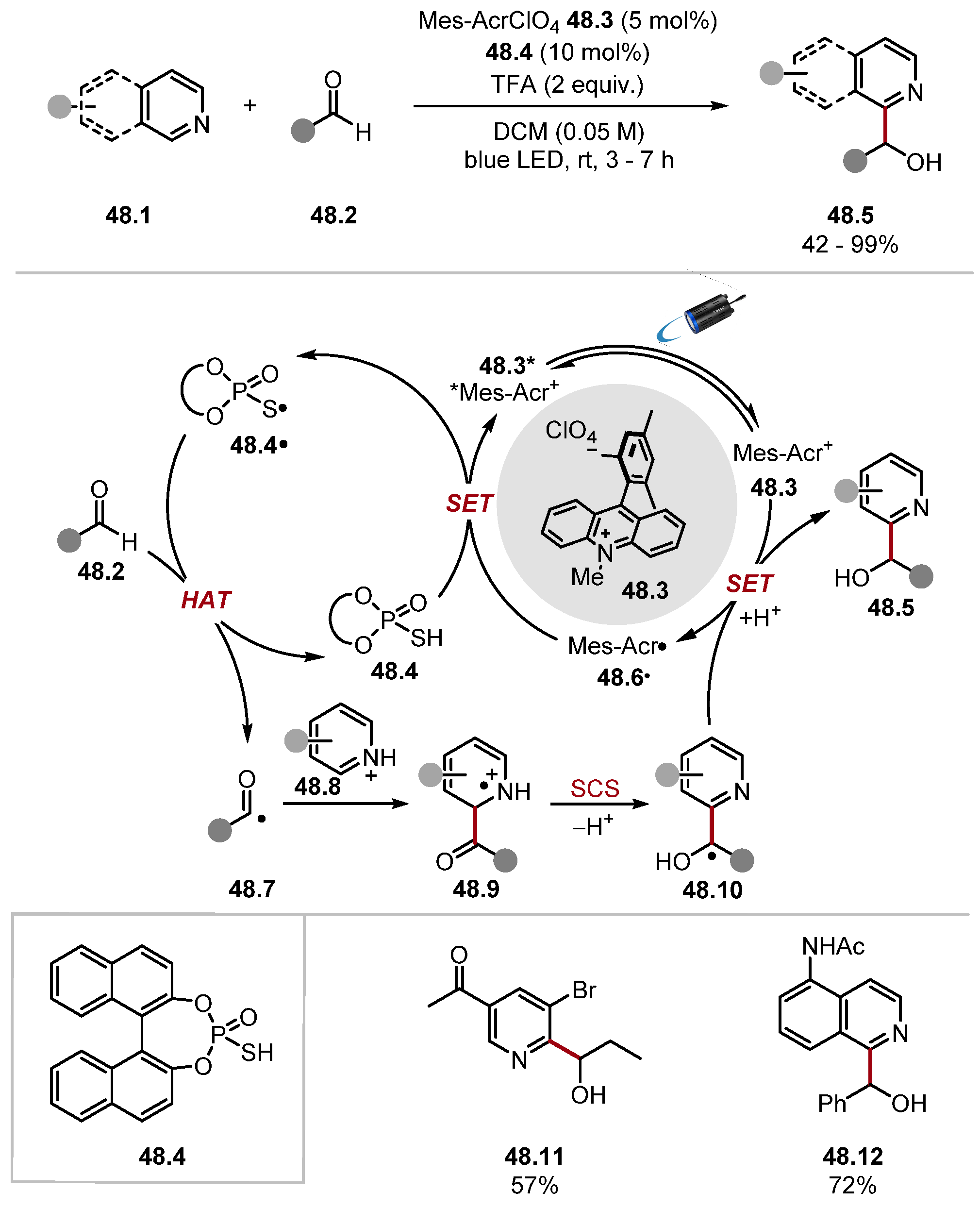
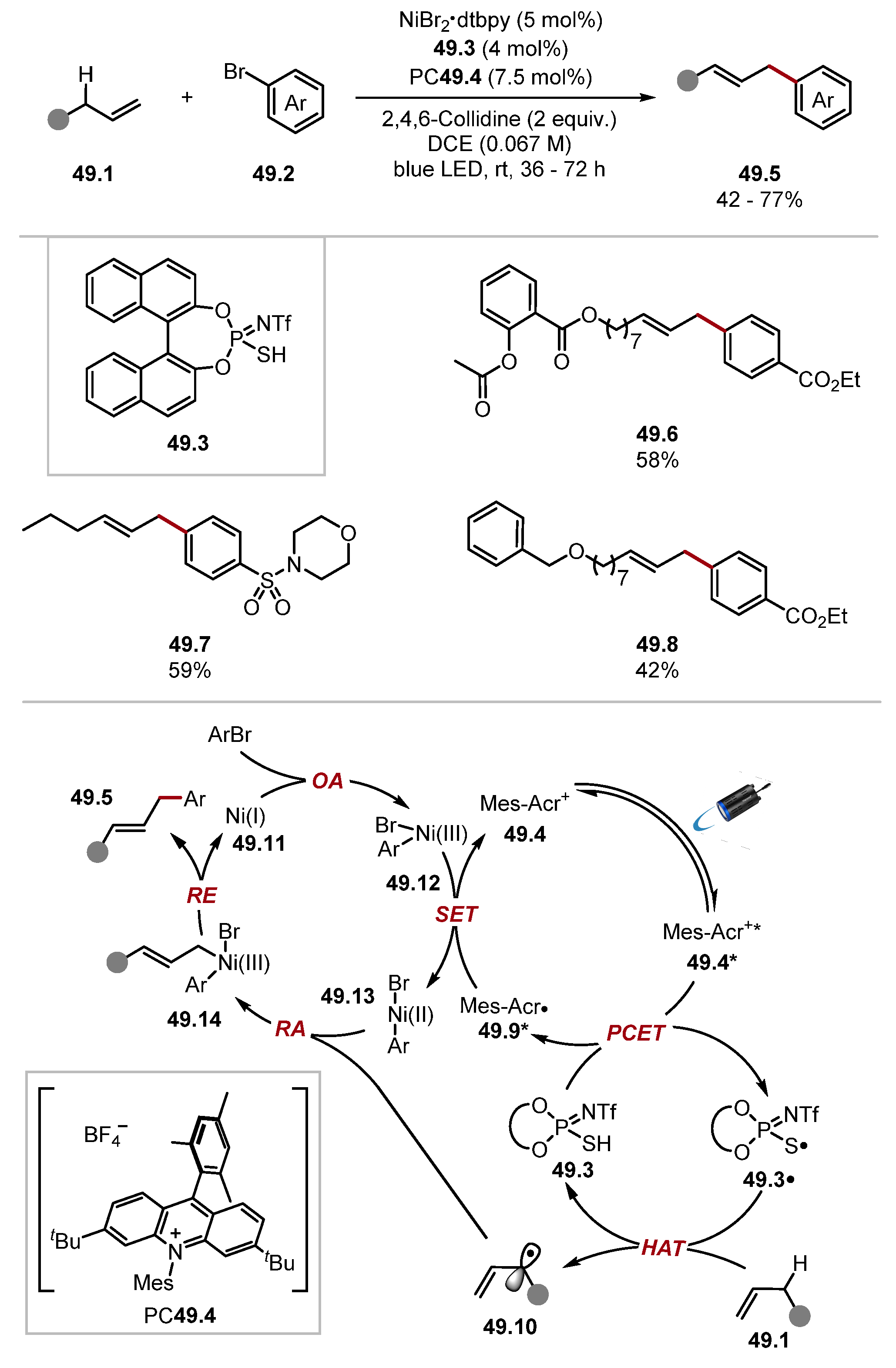



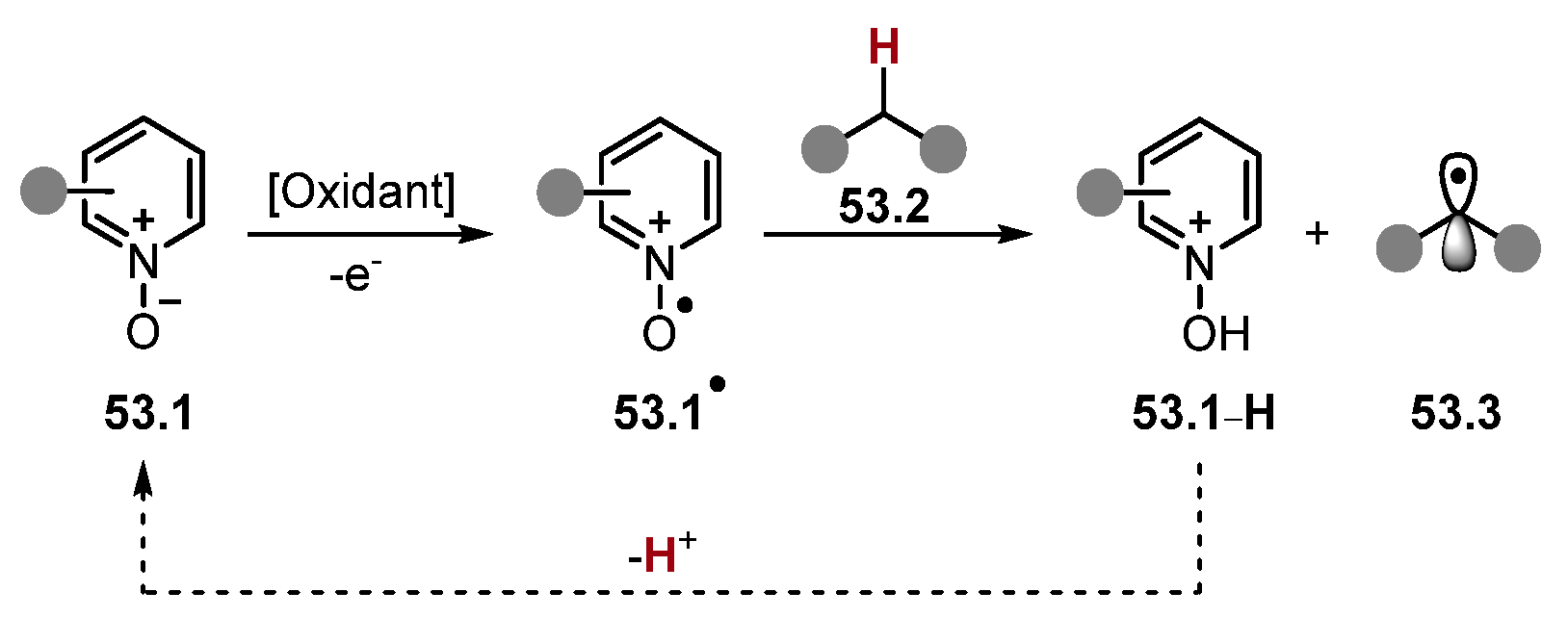
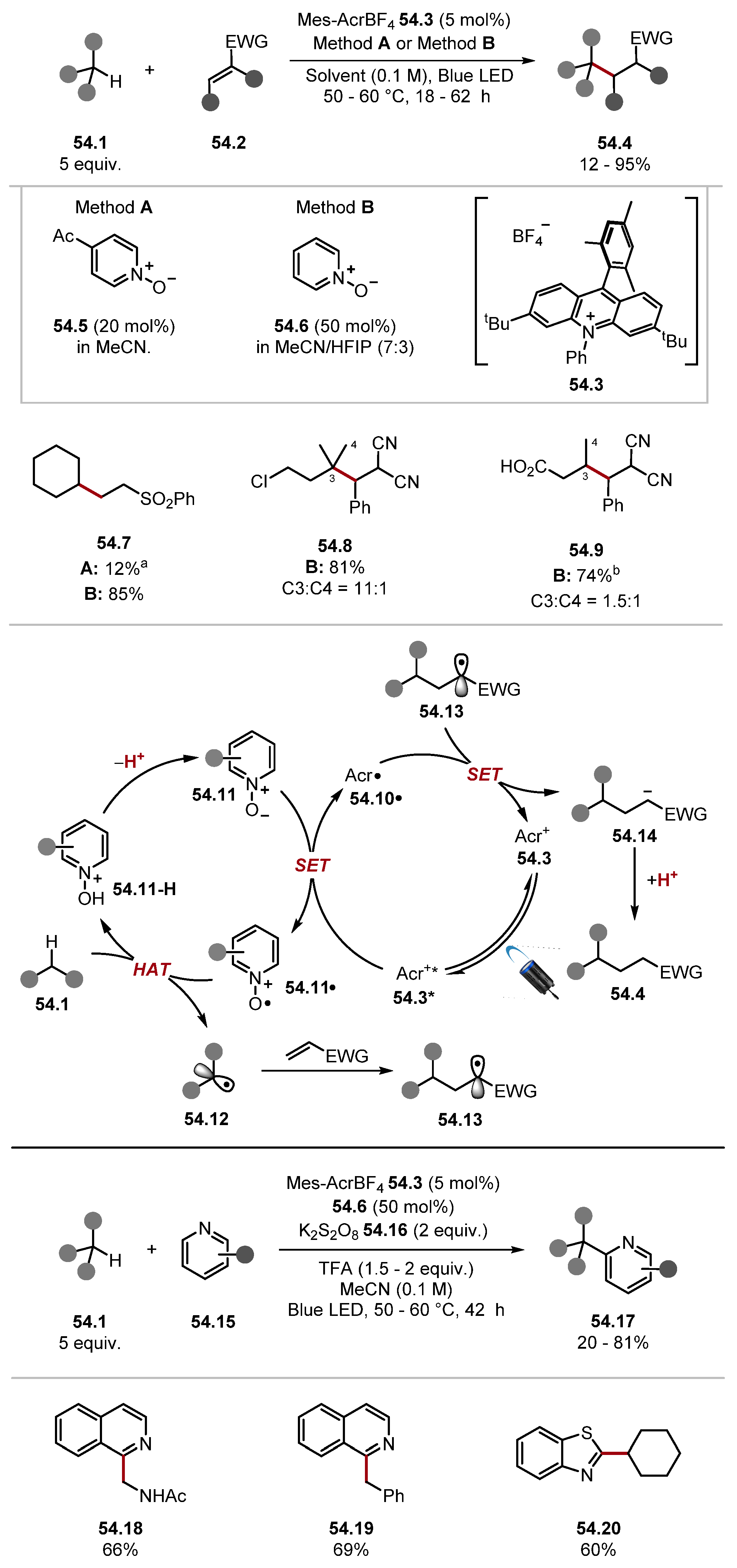

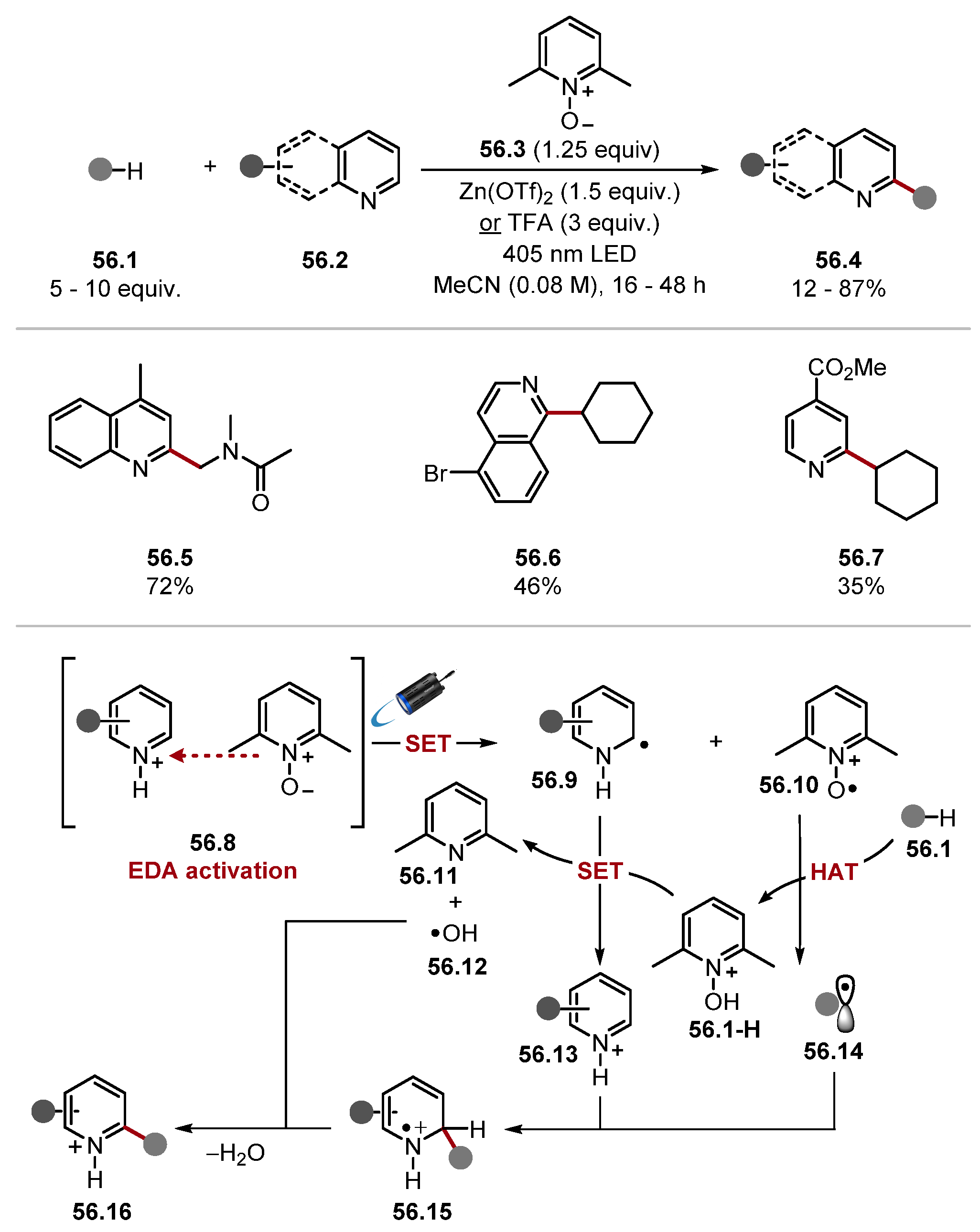

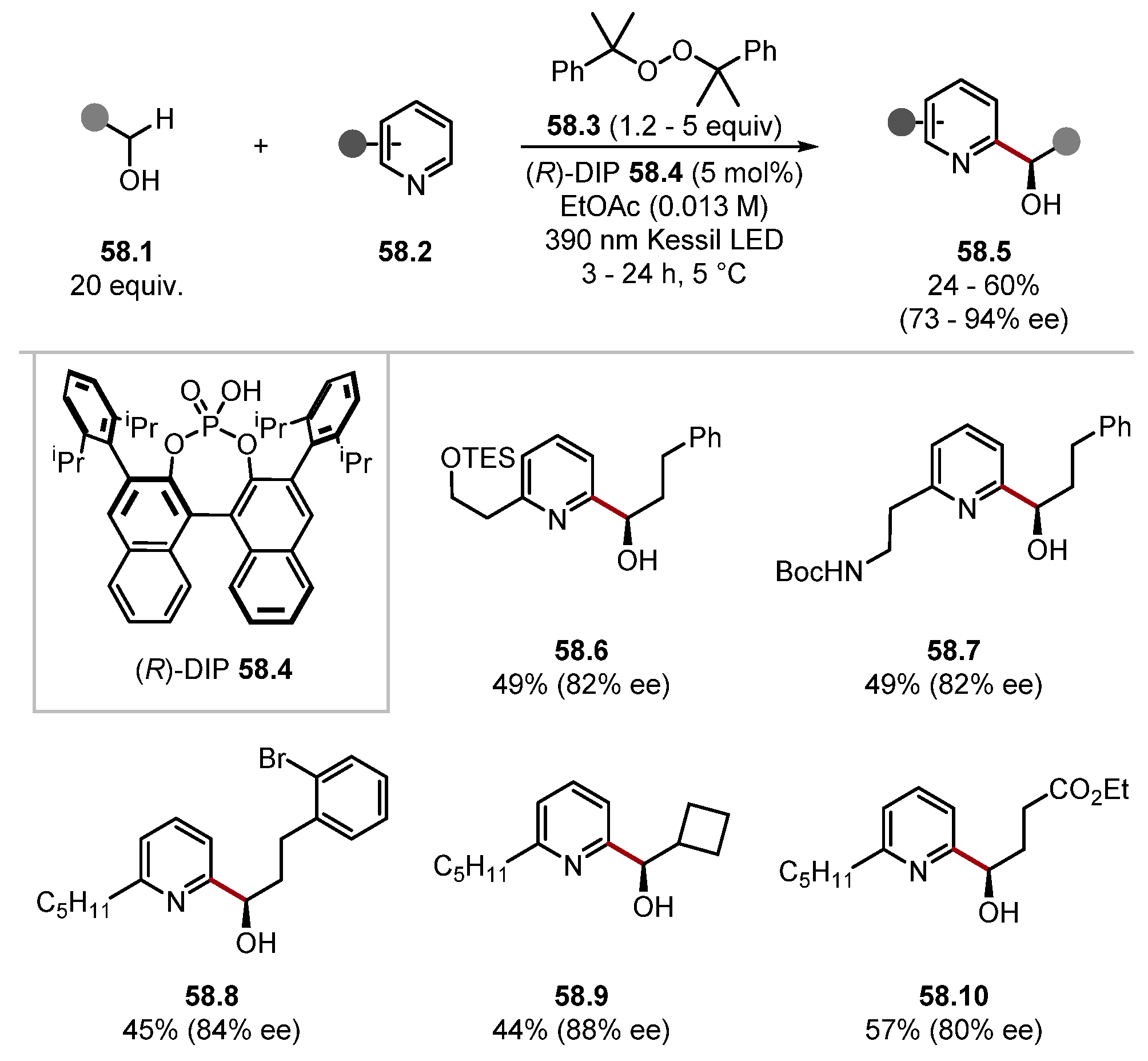


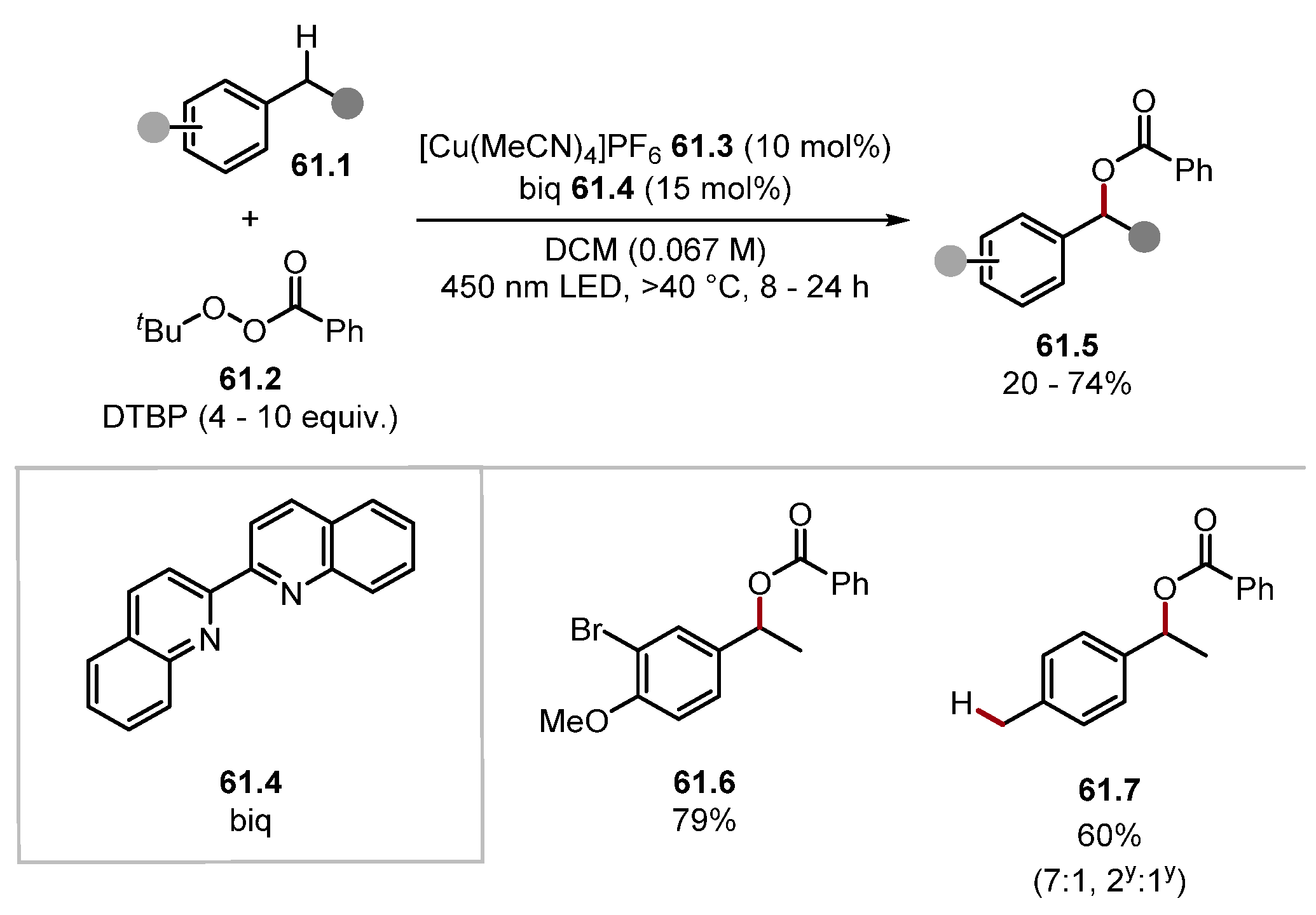
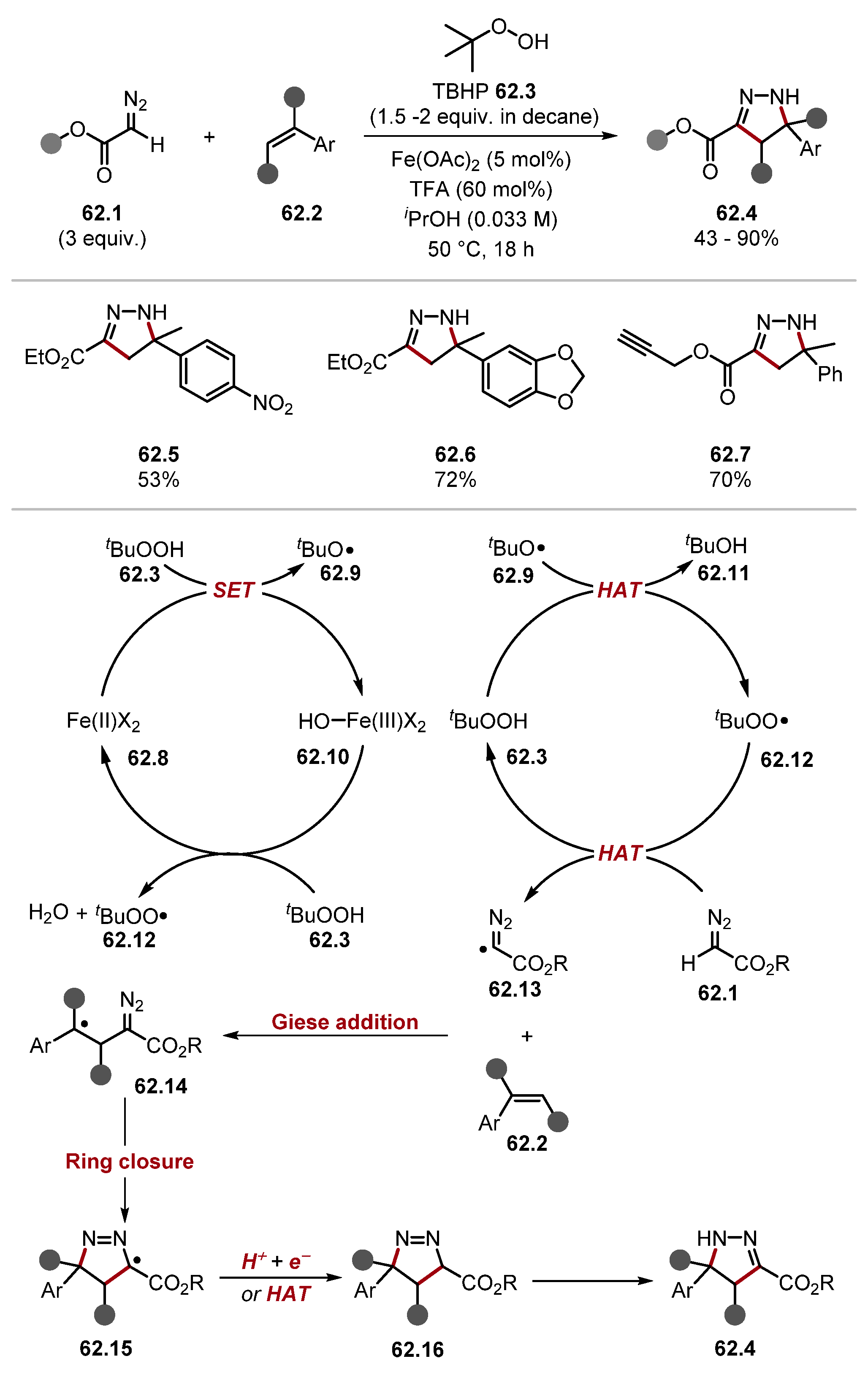


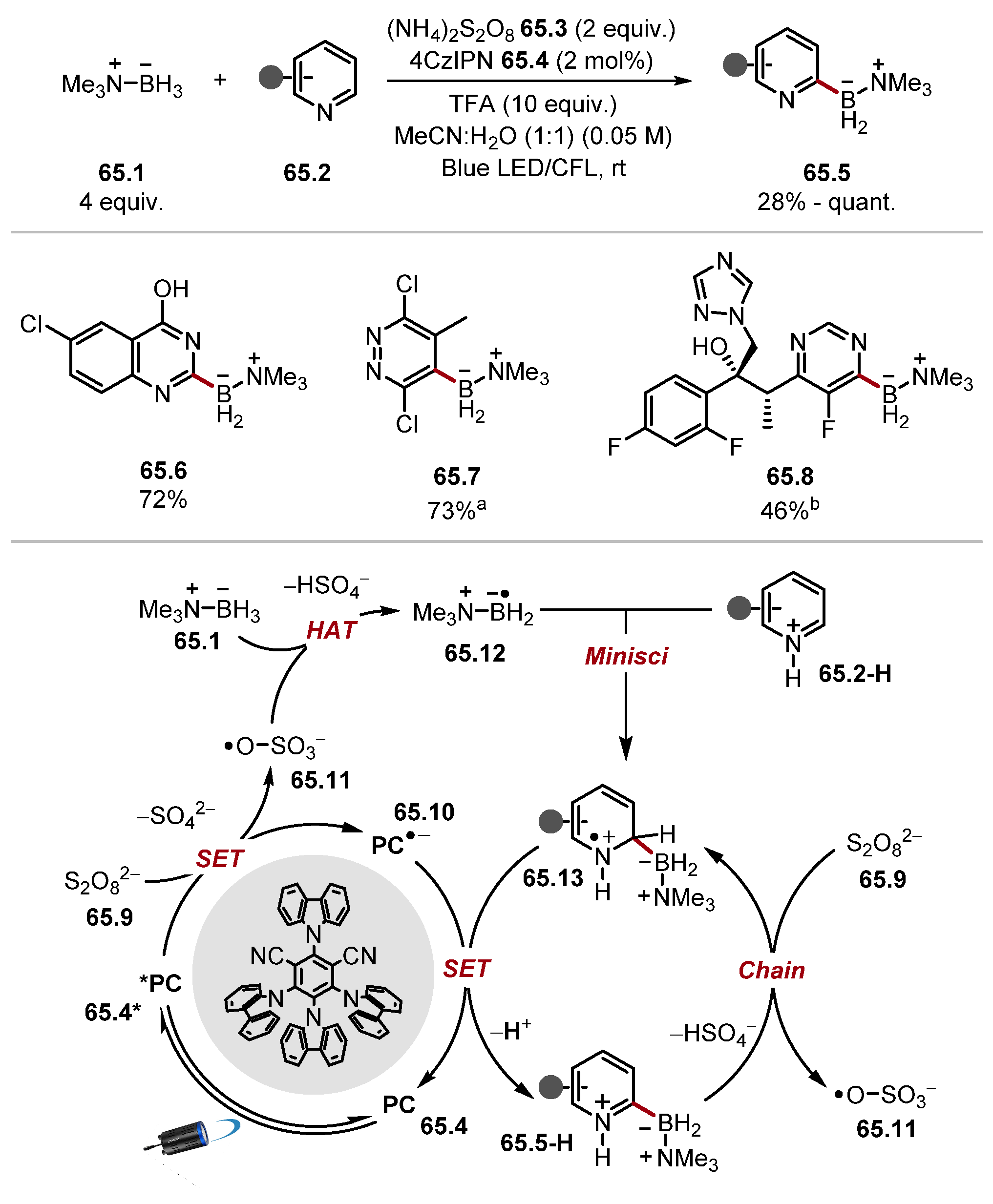
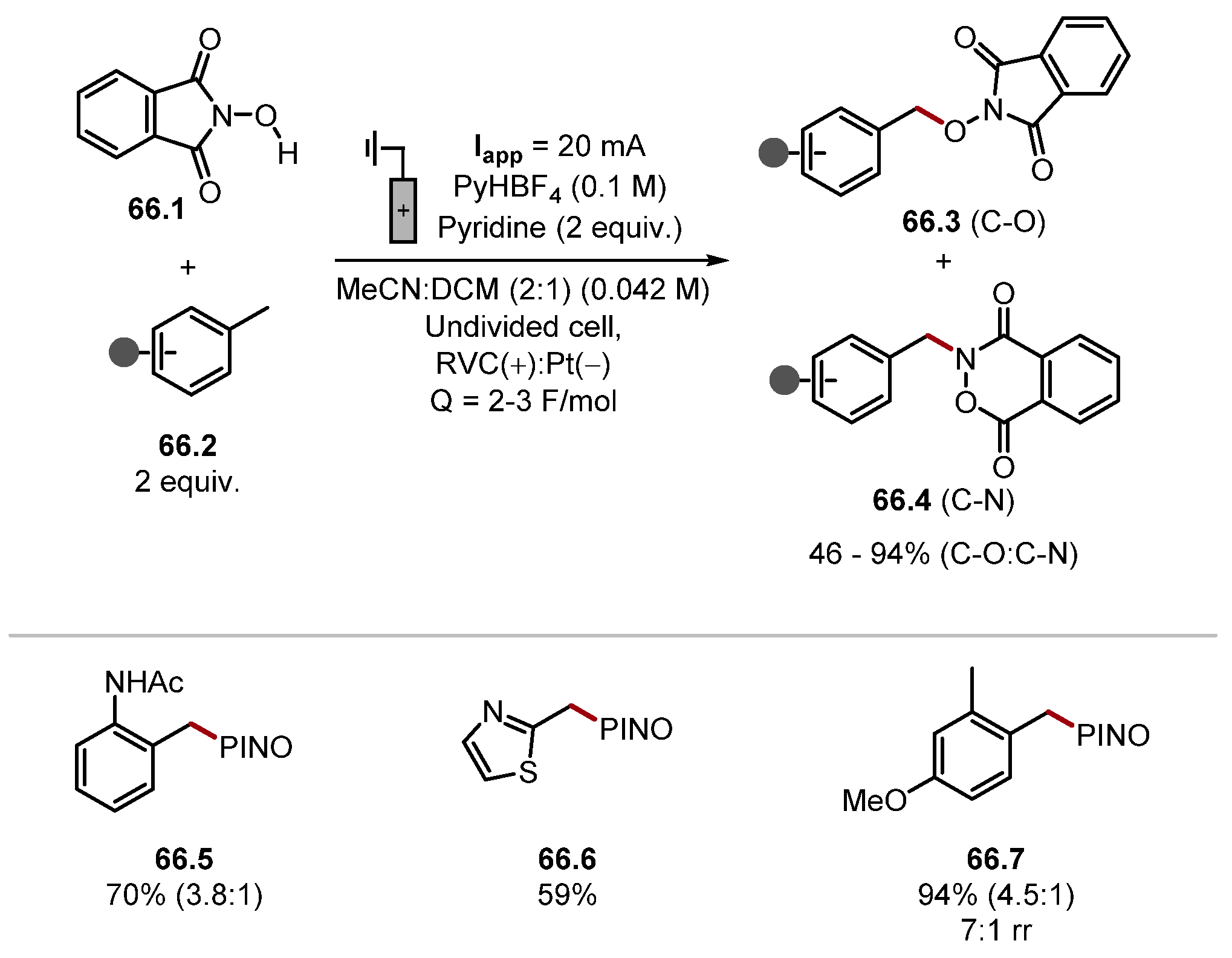
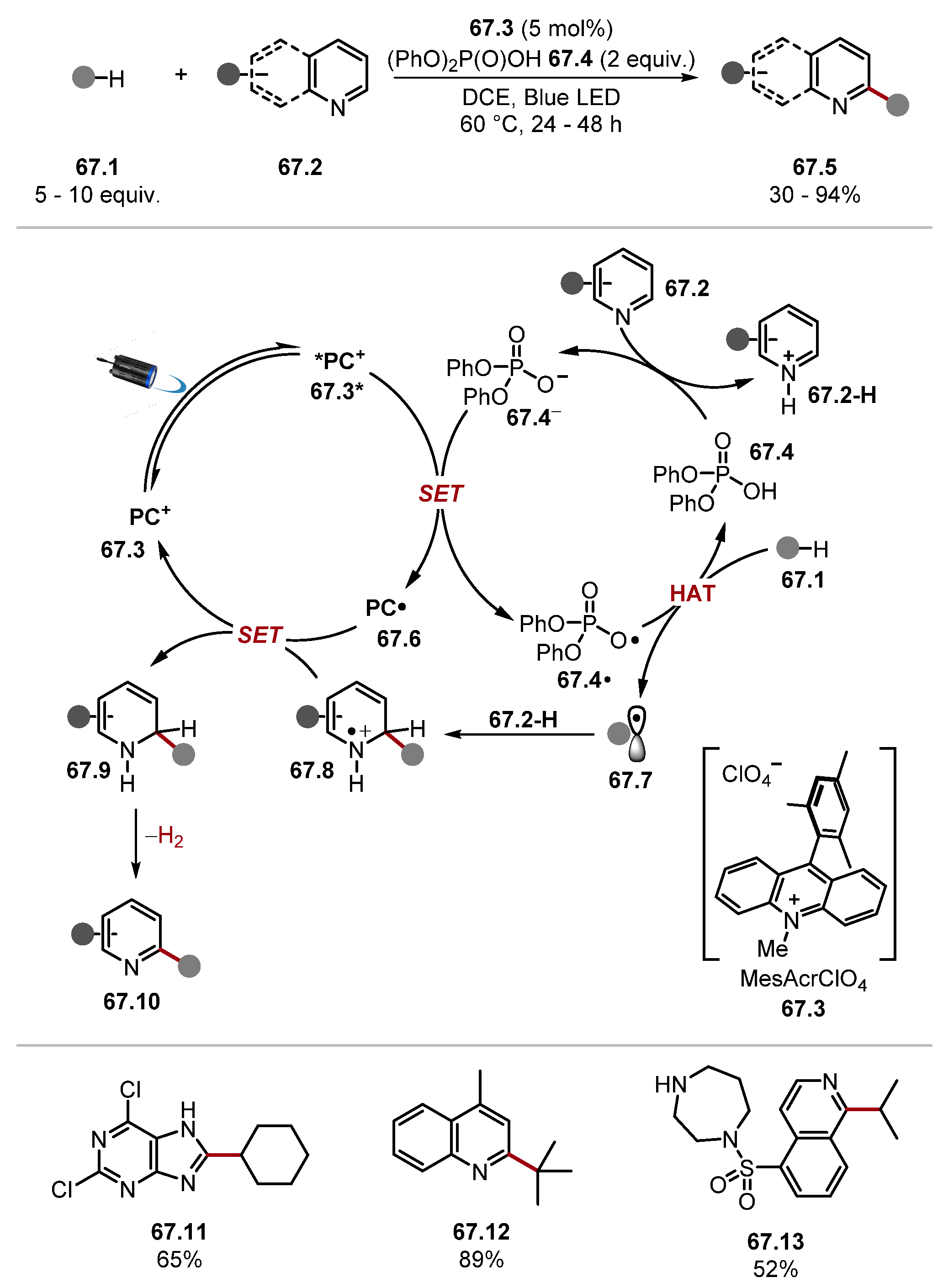
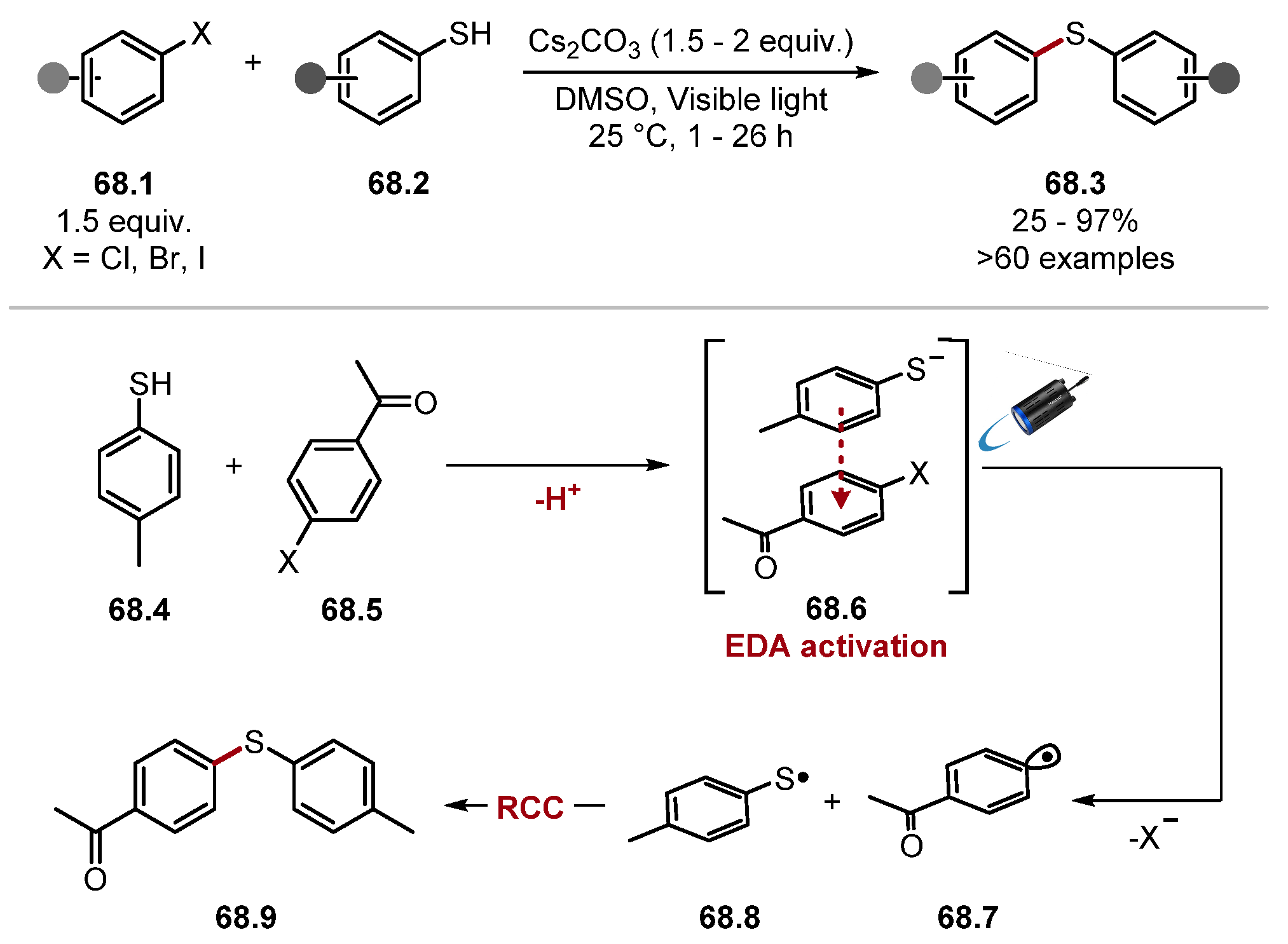
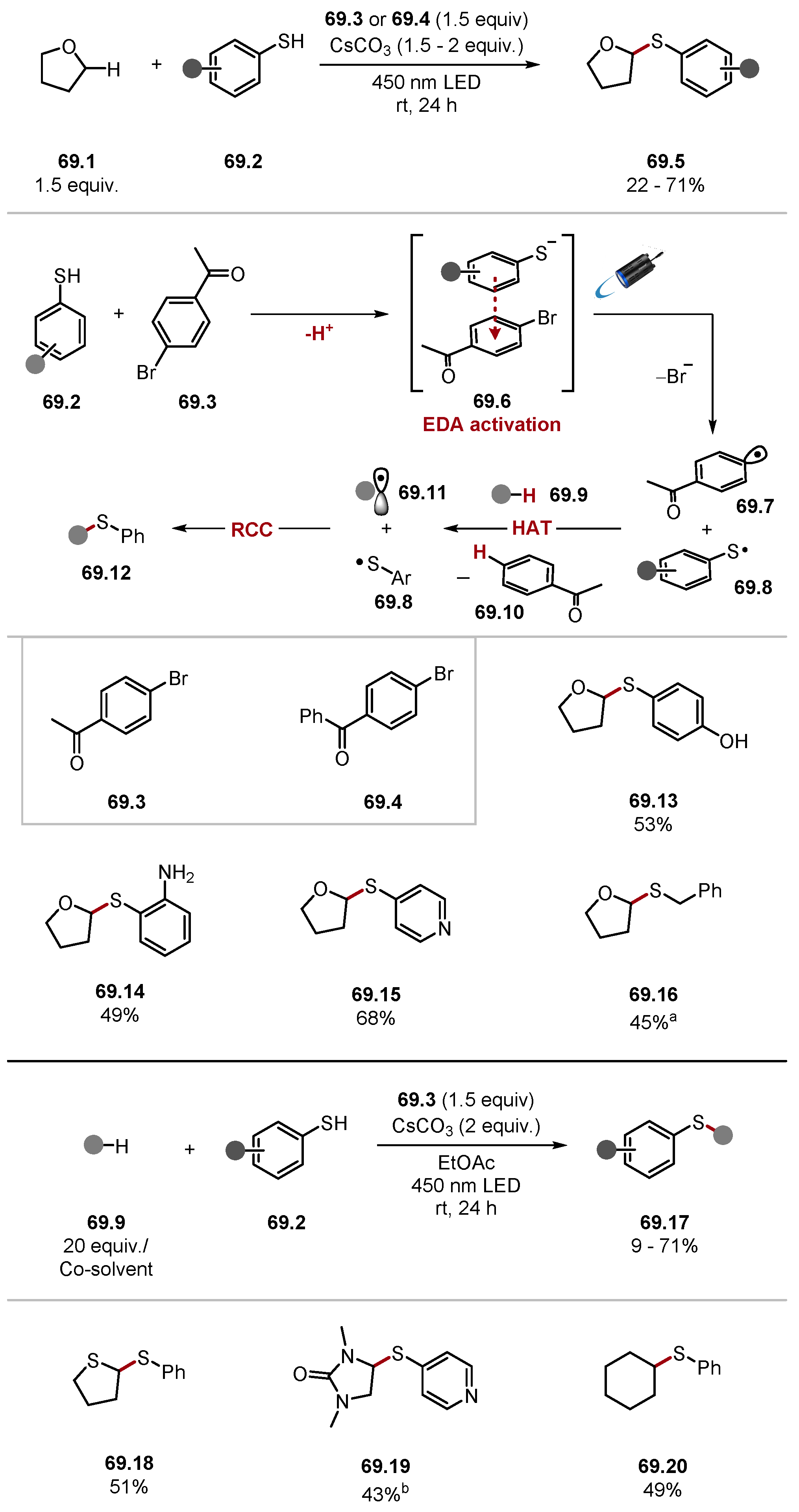
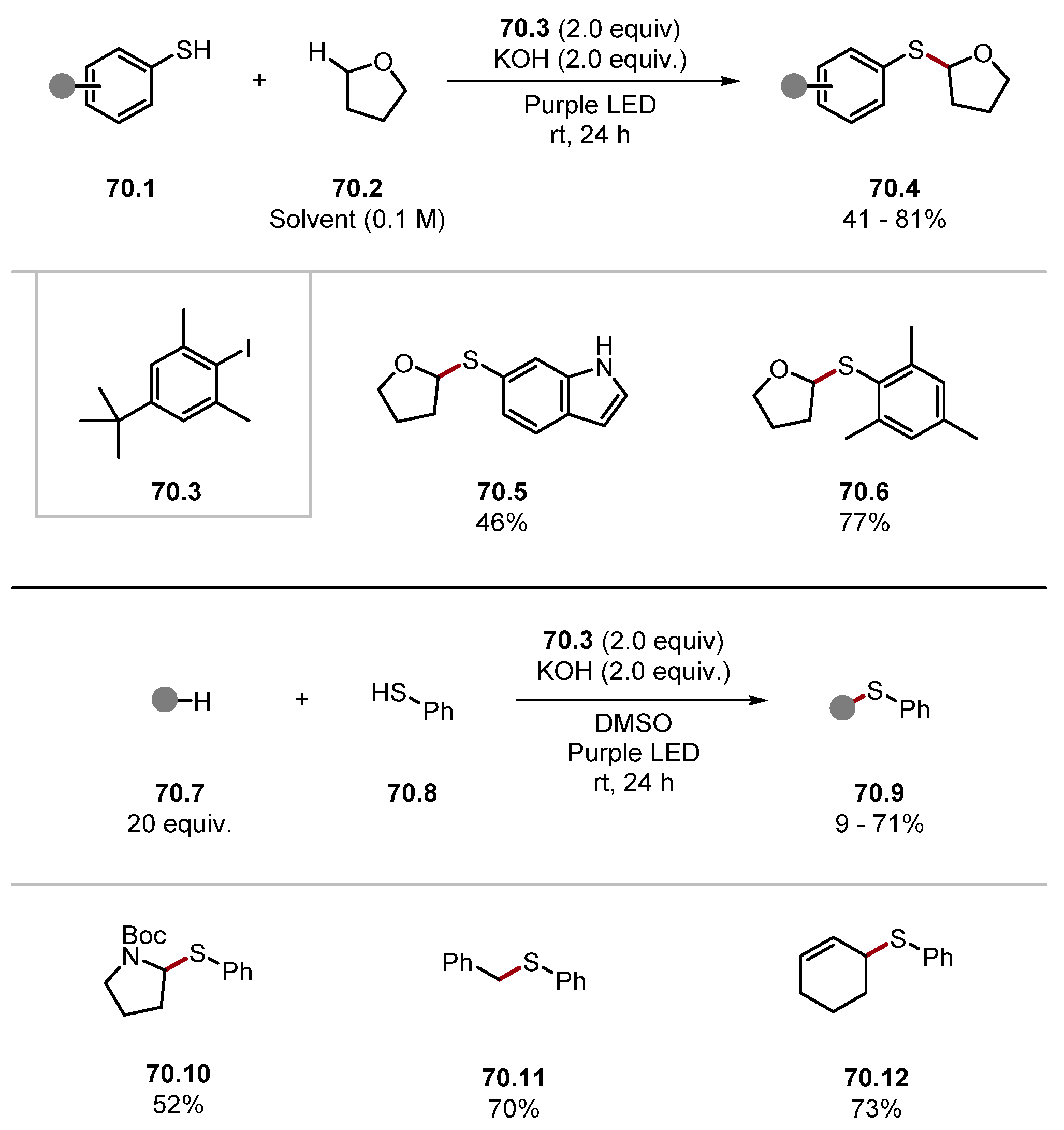
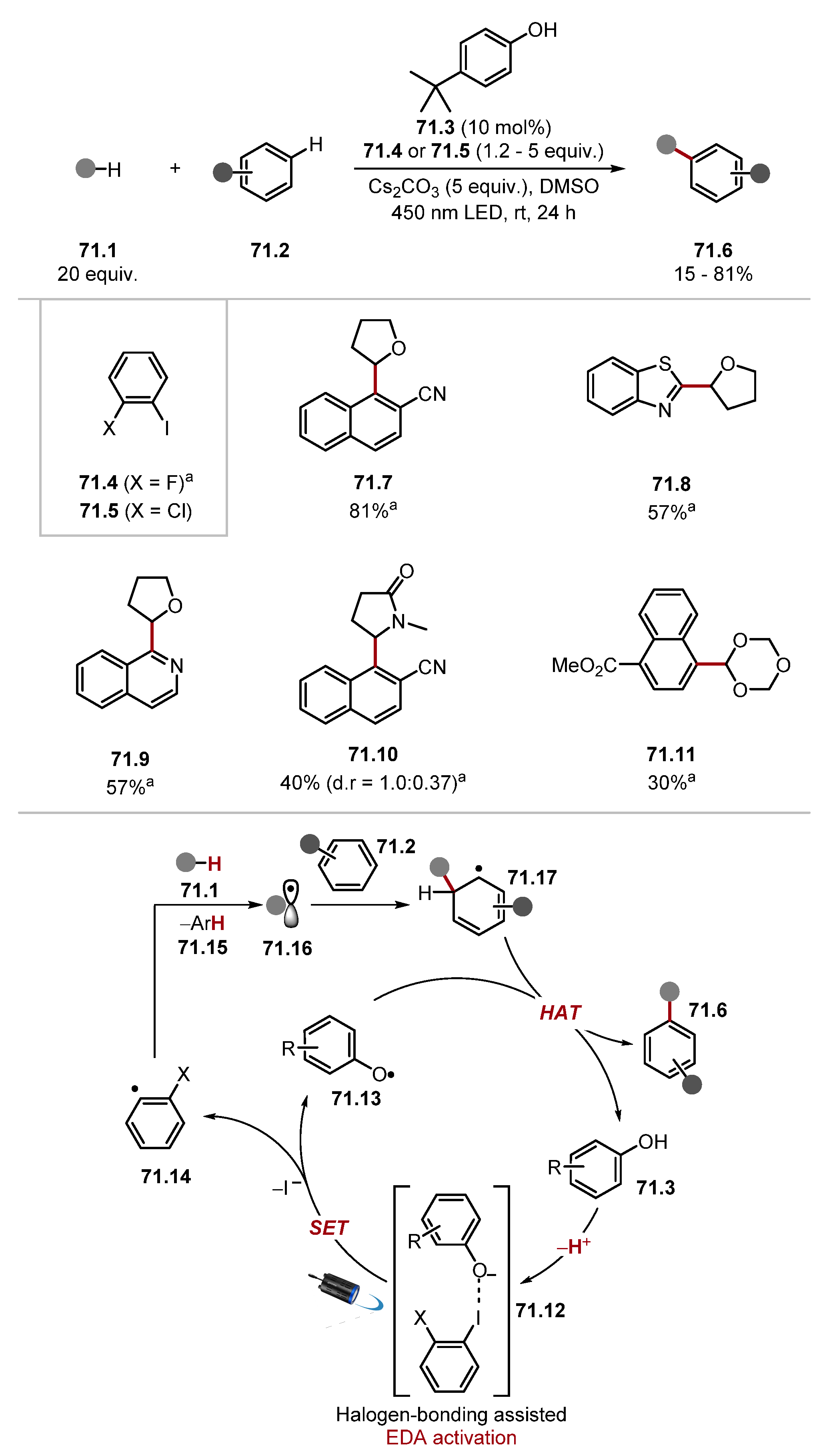
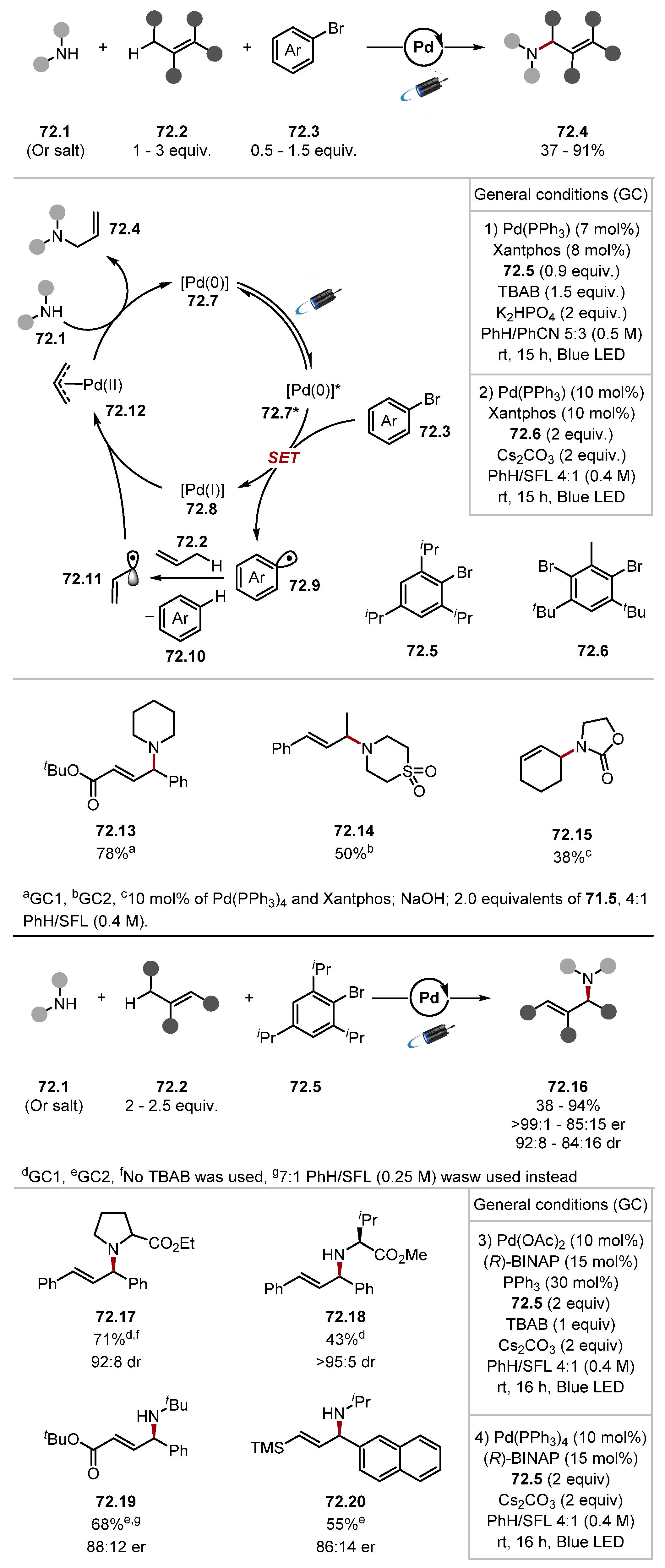
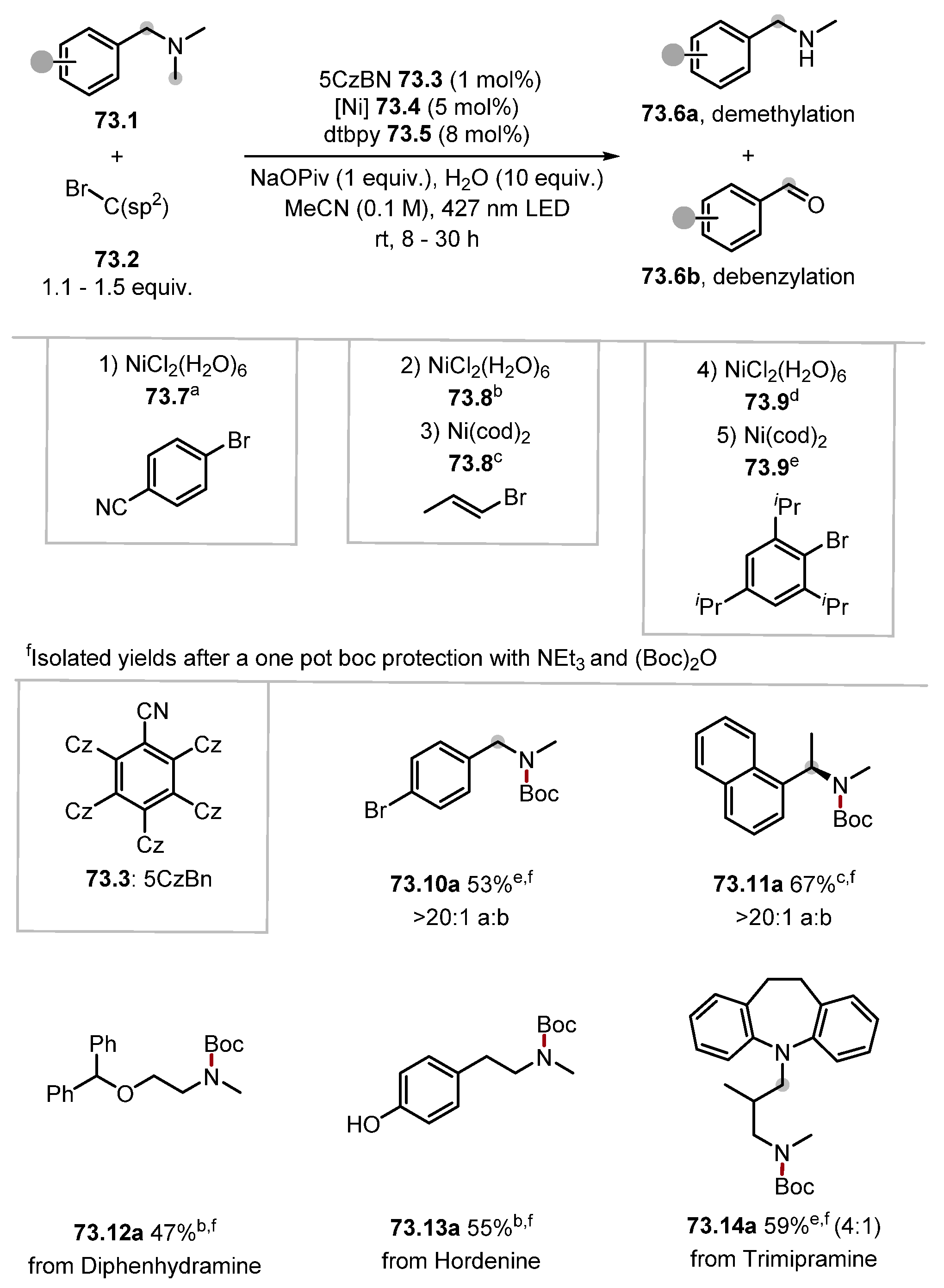
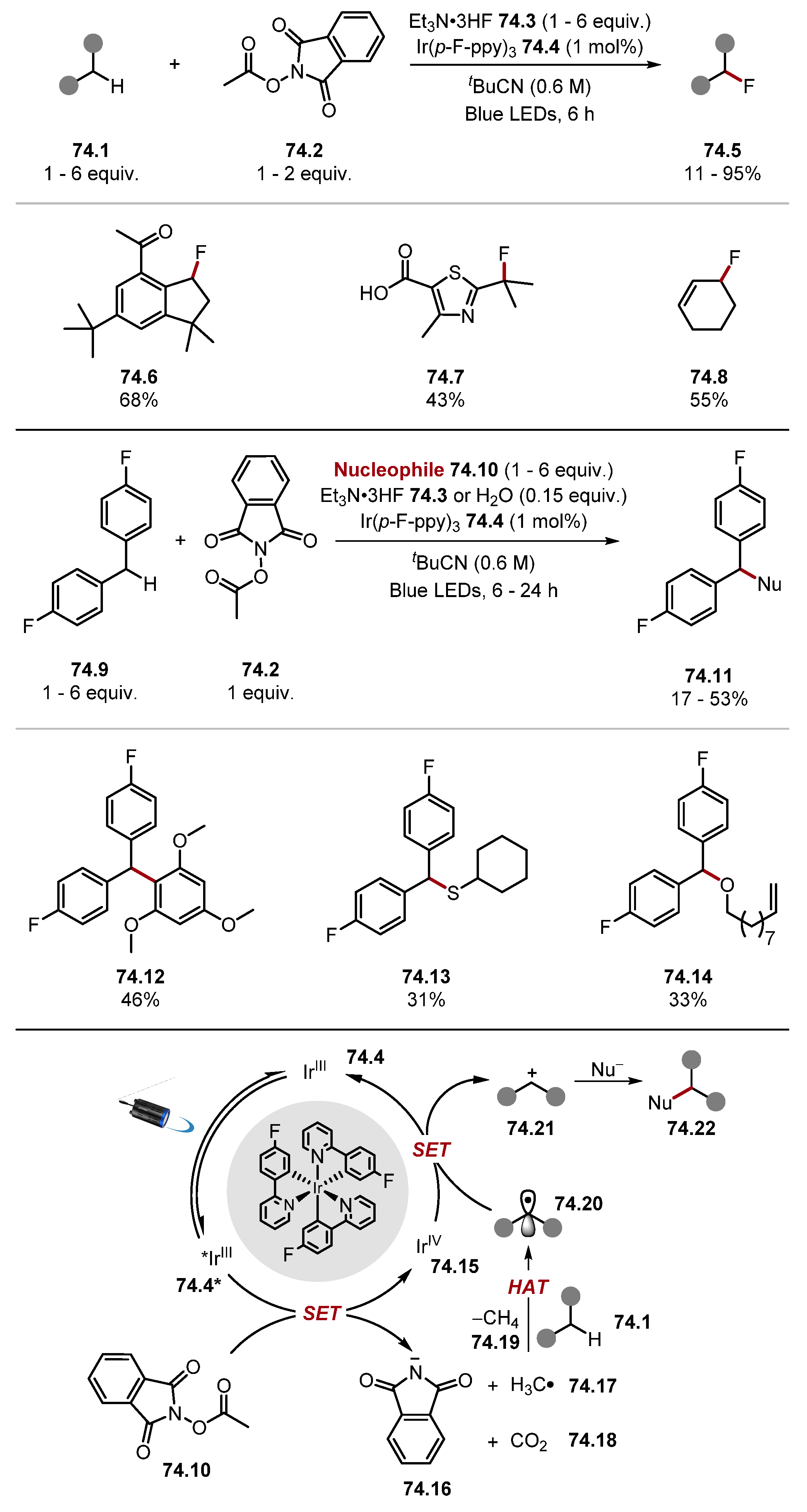
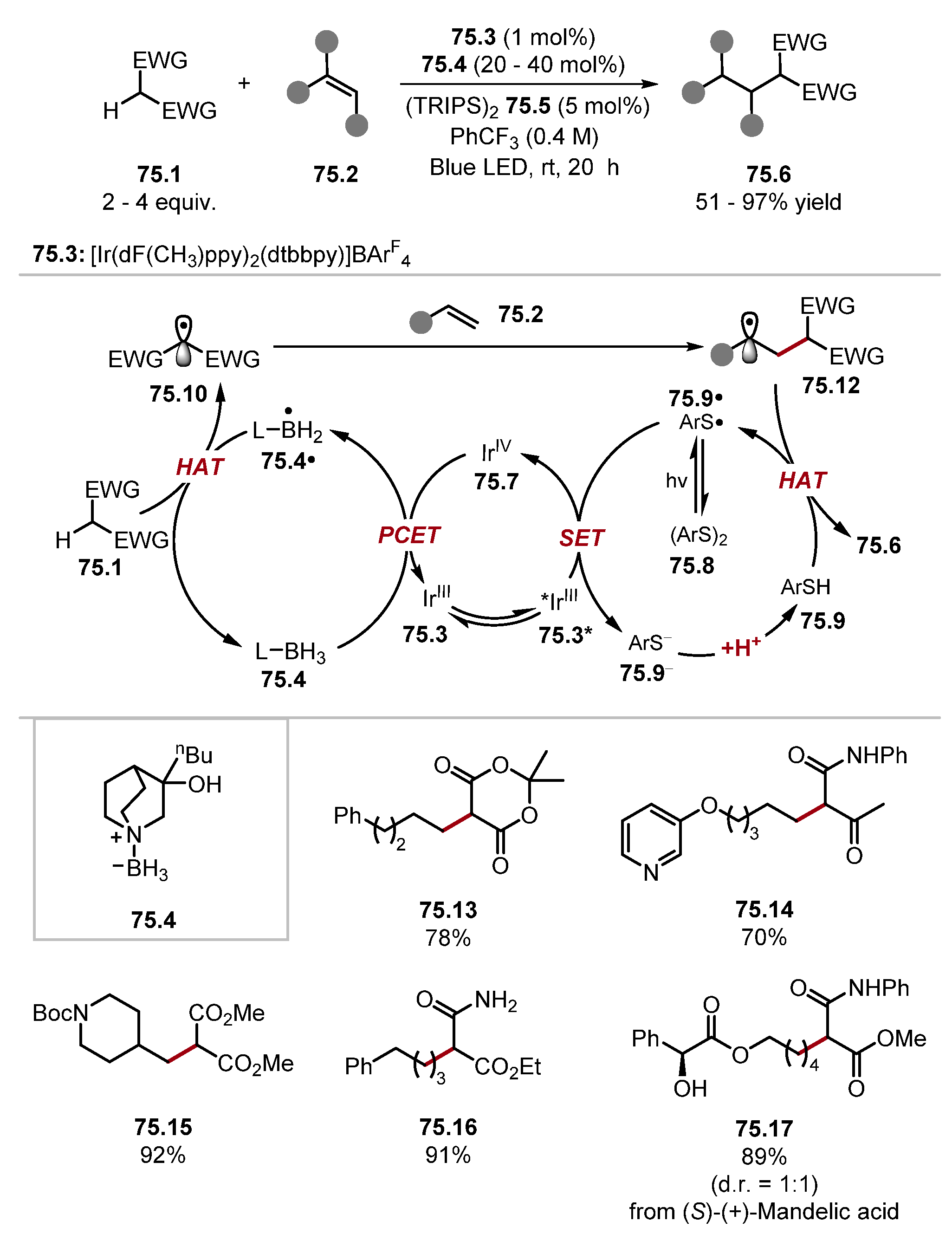
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meger, F.S.; Murphy, J.A. Recent Advances in C–H Functionalisation through Indirect Hydrogen Atom Transfer. Molecules 2023, 28, 6127. https://doi.org/10.3390/molecules28166127
Meger FS, Murphy JA. Recent Advances in C–H Functionalisation through Indirect Hydrogen Atom Transfer. Molecules. 2023; 28(16):6127. https://doi.org/10.3390/molecules28166127
Chicago/Turabian StyleMeger, Filip S., and John A. Murphy. 2023. "Recent Advances in C–H Functionalisation through Indirect Hydrogen Atom Transfer" Molecules 28, no. 16: 6127. https://doi.org/10.3390/molecules28166127
APA StyleMeger, F. S., & Murphy, J. A. (2023). Recent Advances in C–H Functionalisation through Indirect Hydrogen Atom Transfer. Molecules, 28(16), 6127. https://doi.org/10.3390/molecules28166127