

The Use of Extraction on C18-Silica-Modified Magnetic Nanoparticles for the Determination of Ciprofloxacin and Ofloxacin in Meat Tissues

Abstract
:1. Introduction
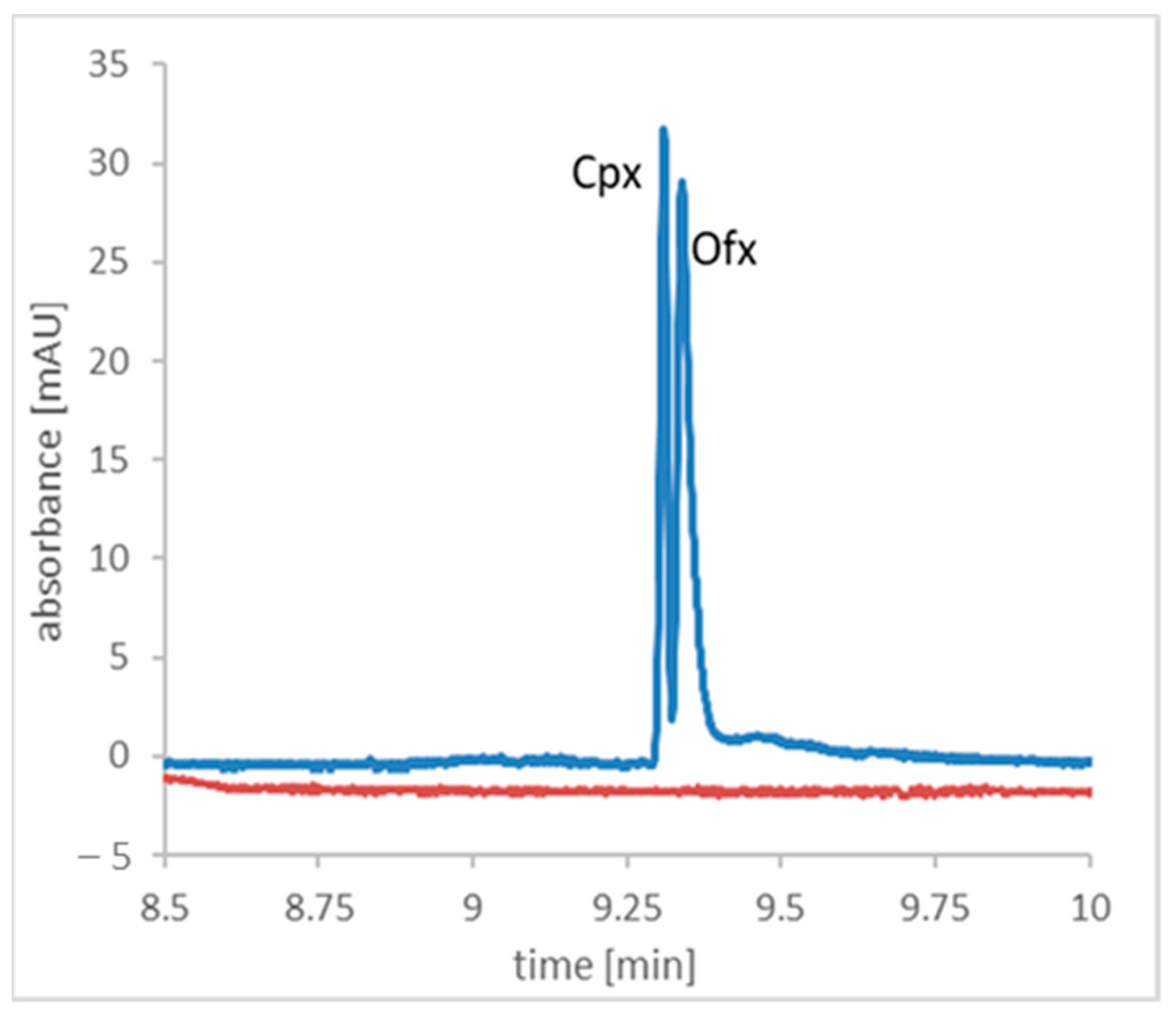
2. Results and Discussion
2.1. Sample Preparation
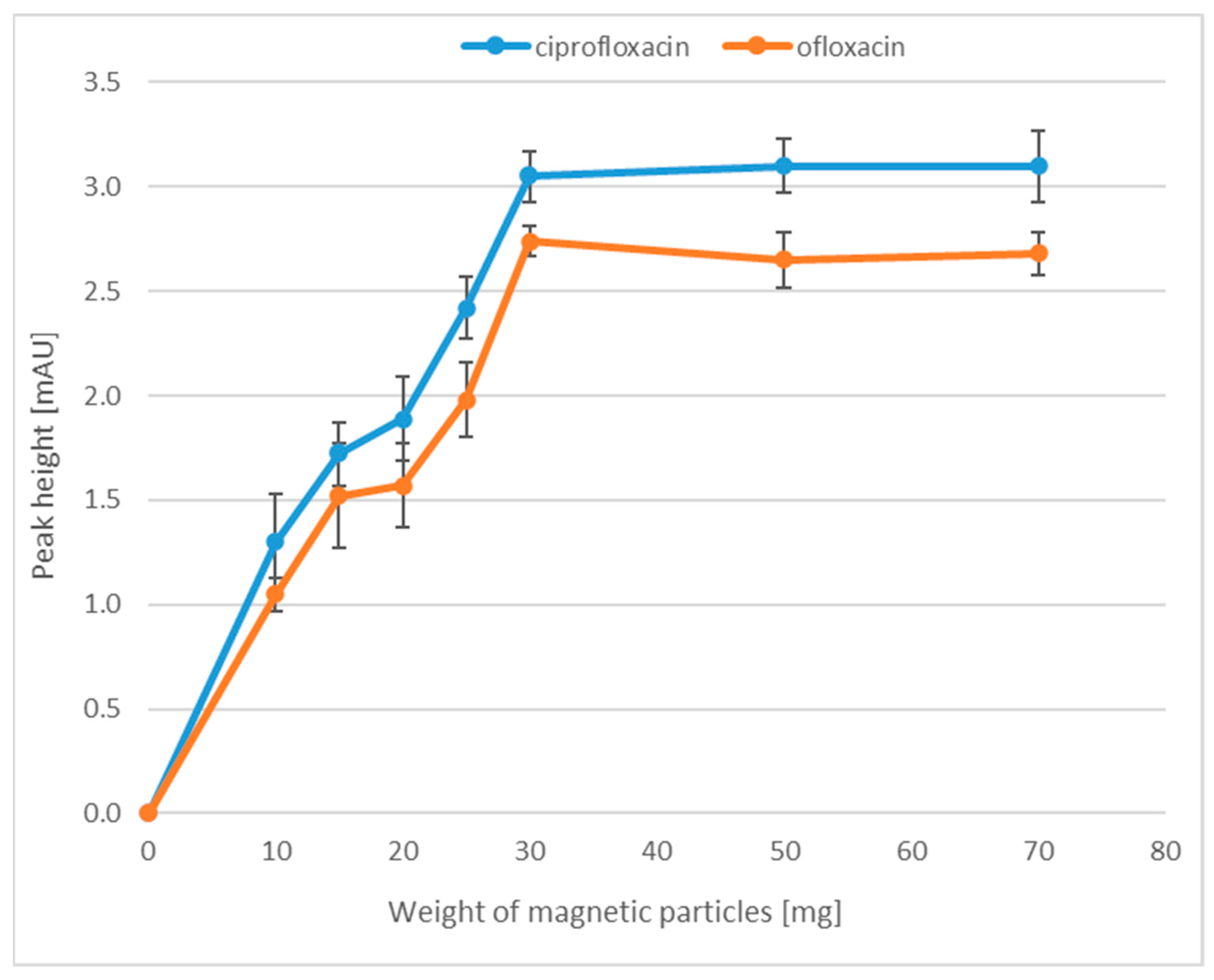
2.1.1. Weight of the Magnetic Nanoparticles
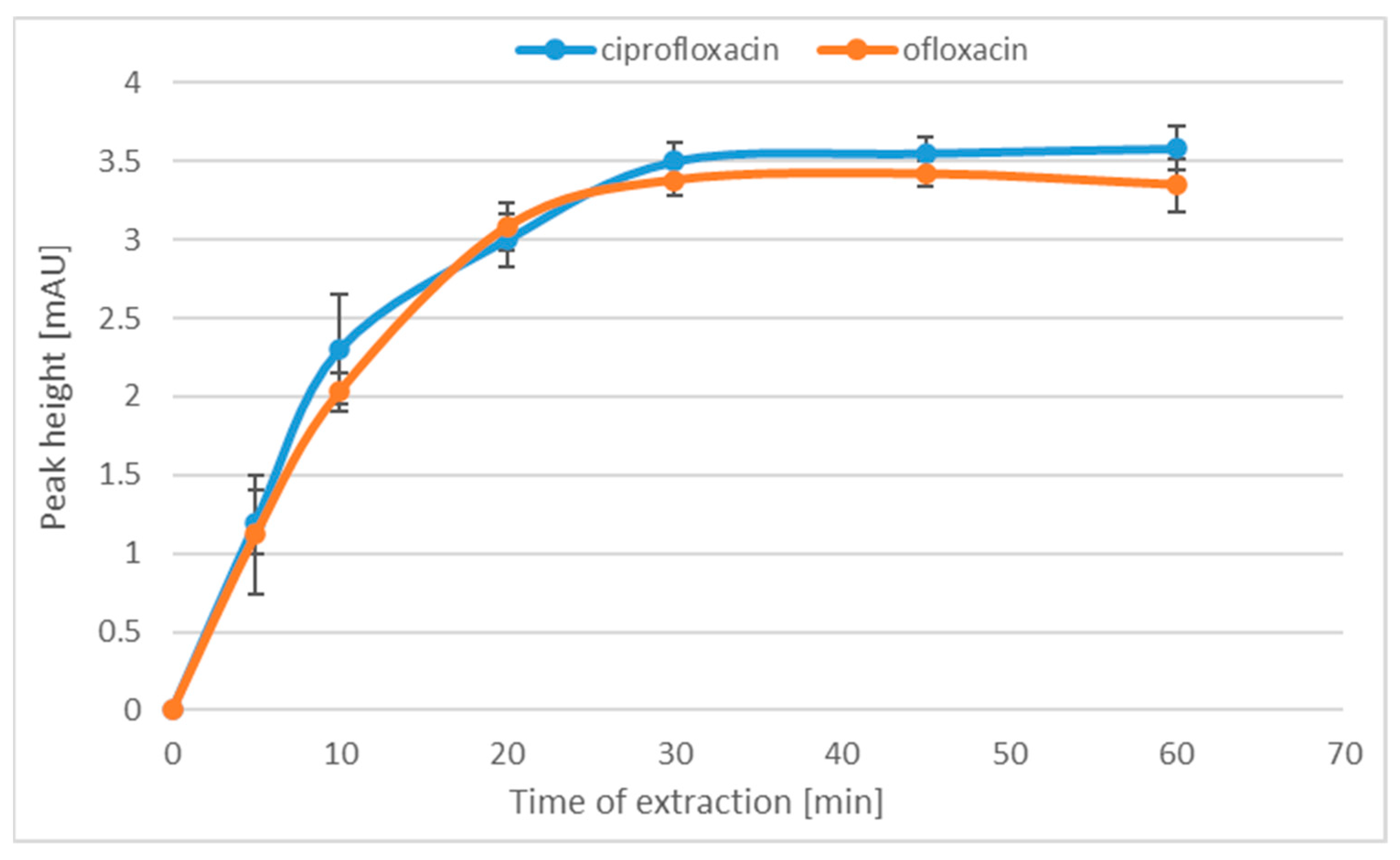
2.1.2. Time of Adsorption
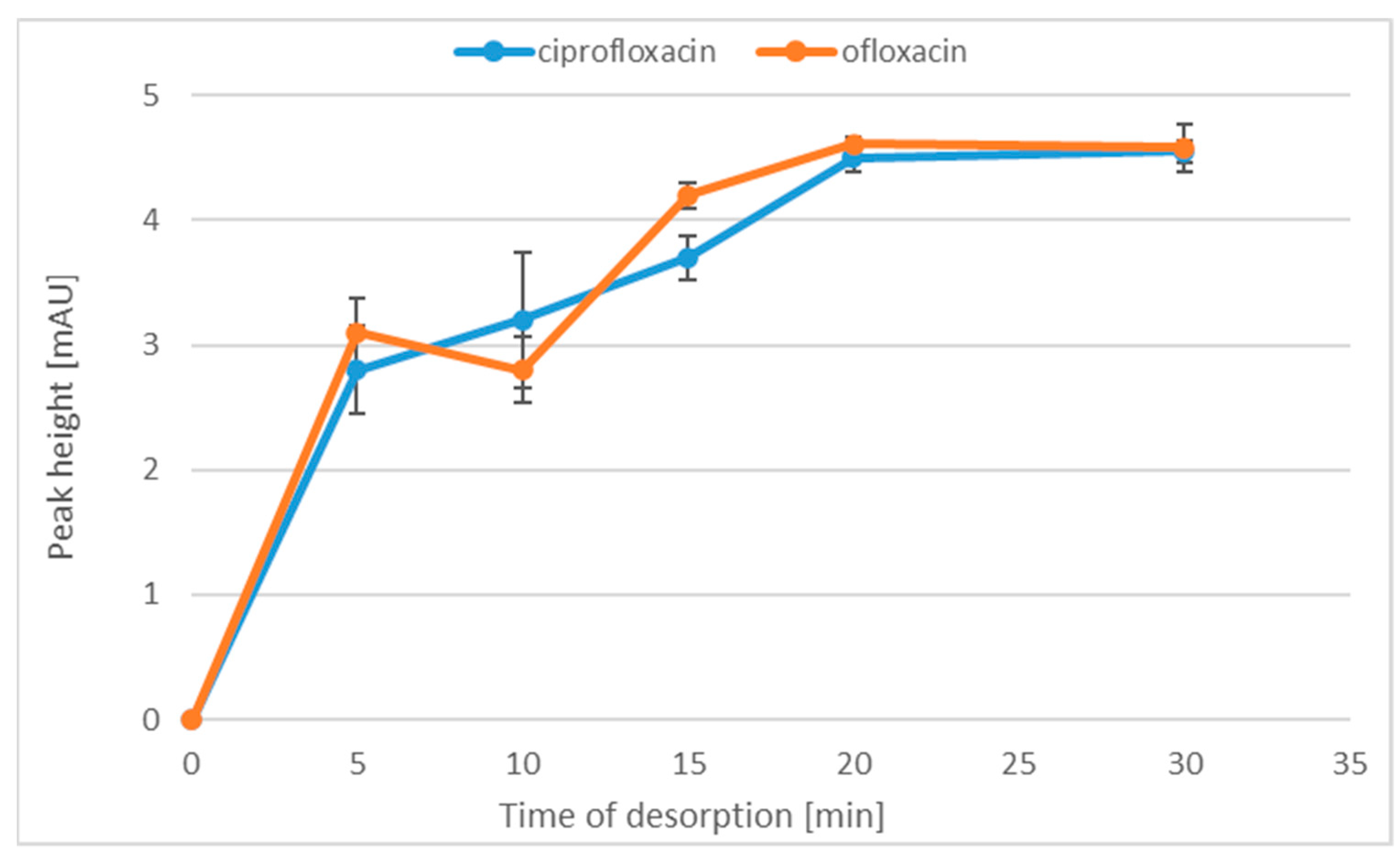
2.1.3. Time of Desorption
2.1.4. The Volume of the Desorbing Solvent
2.1.5. Sensitivity Enhancement Factor
2.1.6. Calibration and Other Validation Data
3. Materials and Methods
3.1. Instruments
3.2. Chemicals
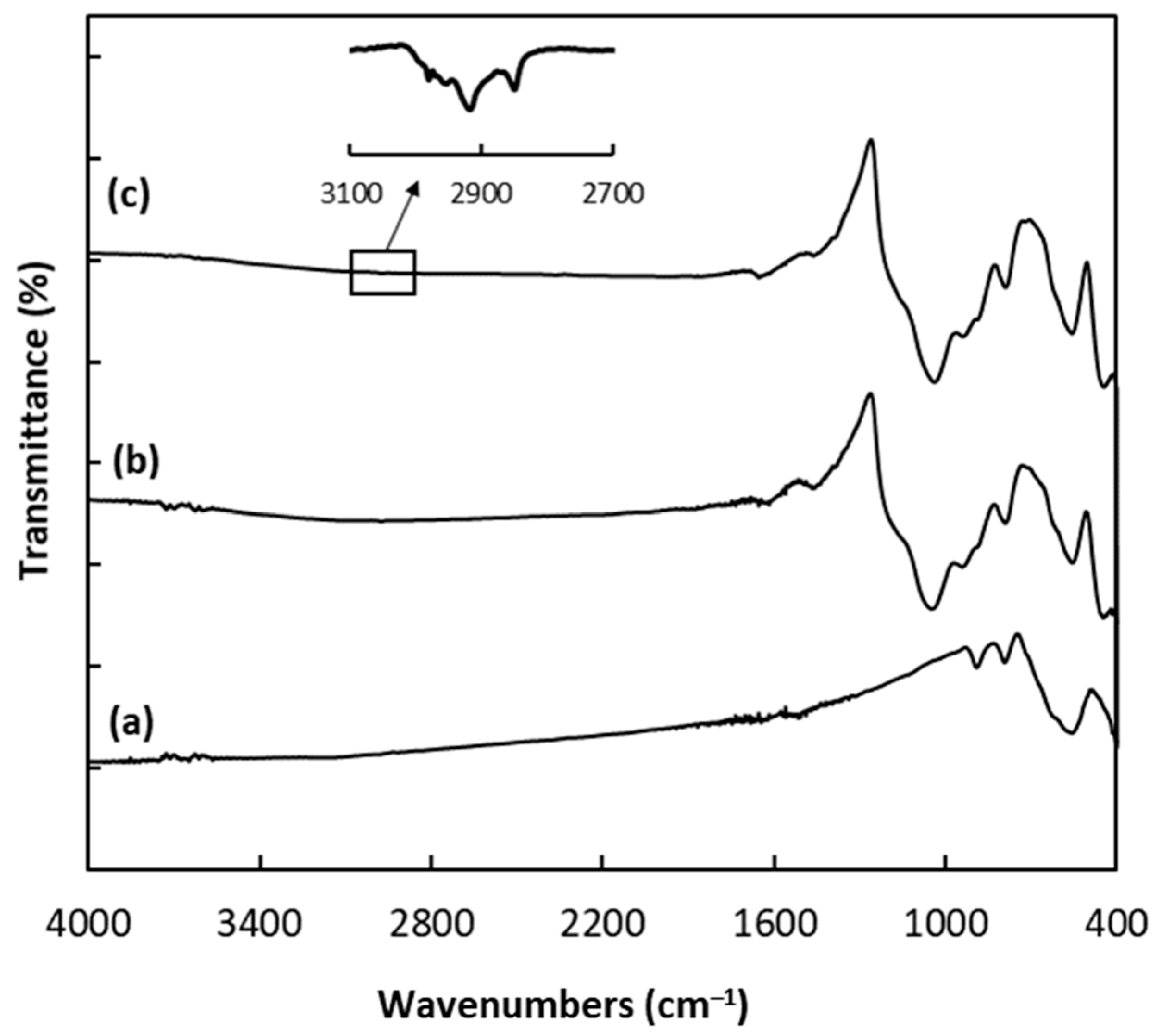
3.3. Preparation of the Magnetic Particles
3.4. Capillary Preconditioning
3.5. Electrophoretic Conditions
3.6. Sample Preparation
3.7. Method Validation
3.8. Calibration of the Method
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wang, Z.; Wang, X.Y.; Tian, H.; Wei, Q.H.; Liu, B.S.; Bao, G.M.; Liao, M.L.; Peng, J.L.; Huang, X.Q.; Wang, L.Q. High through-put determination of 28 veterinary antibiotic residues in swine wastewater by one-step dispersive solid phase extraction sample cleanup coupled with ultra-performance liquid chromatography-tandem mass spectrometry. Chemosphere 2019, 230, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Aresta, A.; Cotugno, P.; Zambonin, C. Determination of ciprofloxacin, enrofloxacin, and marbofloxacin in bovine urine, serum, and milk by microextraction by a packed sorbent coupled to ultra-high performance liquid chromatography. Food Anal. 2018, 52, 790–802. [Google Scholar] [CrossRef]
- Zheng, W.; El-Aty, A.M.; Kim, S.K.; Choi, J.M.; Park, D.H.; Yoo, K.H.; Kang, Y.S.; Jeon, J.S.; Hacımüftüoğlu, A.; Shim, J.H.; et al. Development and validation of a solid-phase extraction method coupled with LC-MS/MS for the simultaneous determination of 16 antibiotic residues in duck meat. Biomed. Chromatogr. 2019, 33, e4501. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.-Z.; Wu, H.-L.; Fang, H.; Wang, T.; Sun, X.-D.; Chang, Y.-Y.; Ding, Y.-J.; Yu, R.-Q. Rapid and simultaneous determination of three fluoroquinolones in animal-derived foods using excitation-emission matrix fluorescence coupled with second-order calibration method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 224, 117458. [Google Scholar] [CrossRef] [PubMed]
- Adjei, M.D.; Deck, J.; Heinze, T.M.; Freeman, J.P.; Williams, A.J.; Sutherland, J.B. Identification of metabolites produced from N-phenylpiperazine by Mycobacterium spp. J. Ind. Microbiol. Biotechnol. 2006, 34, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Plenis, A. Simultaneous determination of six quinolone antibiotics in poultry and porcine samples by capillary electrophoresis. Bull. Vet. Inst. Pulawy 2008, 52, 81–85. [Google Scholar]
- Medical Economics Staff. Physicians’ Desk Reference, 55th ed.; Medical Economics Co.: Montvale, NJ, USA, 2001; p. 852. [Google Scholar]
- Egunova, O.R.; Reshetnikova, I.S.; Kazimirova, K.O.; Shtykov, S.N. Magnetic solid-phase extraction and fluorimetric determination of some fluoroquinolones. J. Anal. Chem. 2020, 75, 24–33. [Google Scholar] [CrossRef]
- Wang, M.; Gao, M.; Zhang, K.; Wang, W.; Fu, Q.; Gao, D. Magnetic covalent organic frameworks with core-shell structure as sorbents for solid phase extraction of fluoroquinolones, and their quantitation by HPLC. Microchim. Acta 2019, 186, 827. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Wei, S. Selective extraction and determination of fluoroquinolones in bovine milk samples with montmorillonite magnetic molecularly imprinted polymers and capillary electrophoresis. Anal. Bioanal. Chem. 2016, 408, 589–598. [Google Scholar] [CrossRef]
- de Oliveira, L.G.; Barreto, F.; Hoff, R.; Rübensam, G.; Scherer Kurz, M.H.; Galle, G.; Gonçalves, F.F. Validation of a method for sedatives and β-blockers determination in swine, bovine and equine kidney using liquid chromatography coupled with tandem mass spectrometry. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2017, 34, 32–39. [Google Scholar] [CrossRef]
- Ghoufran, K.; Mohammad, M.; Oussama, M.; Alhaj, S.A. Analytical methods of ciprofloxacin and its combinations review. Res. J. Pharm. Technol. 2018, 11, 2139–2148. [Google Scholar]
- Czyrski, A. Analytical Methods for Determining Third and Fourth Generation Fluoroquinolones: A Review. Chromatographia 2017, 80, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.S.; Tóth, V.I.; Segundo, M.A.; Lima, H.L.F.C. Fluoroquinolones and sulfonamides: Features of their determination in water. A review. Int. J. Environ. Anal. Chem. 2016, 96, 185–202. [Google Scholar] [CrossRef]
- Cairoli, S.; Simeoli, R.; Tarchi, M.; Dionisi, M.; Vitale, A.; Perioli, L.; Dionisi-Vici, C.; Goffredo, B.M. A new HPLC–DAD method for contemporary quantification of 10 antibiotics for therapeutic drug monitoring of critically ill pediatric patients. Biomed. Chromatogr. 2020, 34, e4880. [Google Scholar] [CrossRef]
- Yıldırım, S.; Karakoç, H.N.; Ya¸sar, A.; Köksal, I. Determination of levofloxacin, ciprofloxacin, moxifloxacin and gemifloxacin in urine and plasma by HPLC–FLD–DAD using pentafluorophenyl core–shell column: Application to drug monitoring. Biomed. Chromatogr. 2020, 34, e4925. [Google Scholar] [CrossRef] [PubMed]
- Selahle, S.K.; Nomngongo, P.N. Determination of fluoroquinolones in the environmental samples using vortex assisted dispersive liquid–liquid microextraction coupled with high performance liquid chromatography. Int. J. Environ. Anal. Chem. 2020, 100, 282–294. [Google Scholar] [CrossRef]
- Magalhães, D.; Freitas, A.; Sofia Vila Pouca, A.; Barbosa, J.; Ramos, F. The use of ultra-high-pressure-liquid-chromatography tandem time-of-flight mass spectrometry as a confirmatory method in drug residue analysis: Application to the determination of antibiotics in piglet liver. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1153, 122264. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ren, X.; Diao, Y.; Chen, Y.; Wang, Q.; Jin, W.; Zhou, P.; Fan, Q.; Zhang, Y.; Liu, H. Multiclass analysis of 25 veterinary drugs in milk by ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem. 2018, 257, 259–264. [Google Scholar] [CrossRef]
- Maia, A.S.; Paíga, P.; Delerue-Matos, C.; Castro, P.M.L.; Tiritan, M.E. Quantification of fluoroquinolones in wastewaters by liquid chromatography–tandem mass spectrometry. Environ. Pollut. 2020, 259, 113927. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, Y.; Zhao, M.Z.; Zhou, Y.L.; Zhang, X.X. Highly-sensitive detection of eight typical fluoroquinolone antibiotics by capillary electrophoresis-mass spectroscopy coupled with immunoaffinity extraction. RSC Adv. 2018, 8, 4063–4071. [Google Scholar] [CrossRef]
- Martinez-Perez-Cejuela, H.; Benavente, F.; Simo-Alfonso, E.F.; Herrero-Martinez, J.M. A hybrid nano-MOF/polymer material for trace analysis of fluoroquinolones in complex matrices at microscale by on-line solid-phase extraction capillary electrophoresis. Talanta 2021, 233, 122529. [Google Scholar] [CrossRef]
- Gao, W.; Chen, G.; Chen, Y.; Zhang, X.; Yin, Y.; Hu, Z. Application of single drop liquid-liquid-liquid microextraction for the determination of fluoroquinolones in human urine by capillary electrophoresis. J. Chromatogr. B 2011, 879, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Purgat, K.; Olejarz, P.; Kośka, I.; Głowacki, R.; Kubalczyk, P. Determination of homocysteine thiolactone in human urine by capillary zone electrophoresis and single drop microextraction. Anal. Biochem. 2020, 596, 113640. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Lin, C.Y.; Luo, D.; Suo, L.L.; Chen, J.L.; Feng, Y.Q. Magnetic solid-phase extraction using magnetic hypercrosslinked polymer for rapid determination of illegal drugs in urine. J. Sep. Sci. 2011, 34, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, I.S.; Rodr´ıguez, J.A.; Paez-Hernandez, M.E.; Santos, E.M.; Miranda, J.M. Determination of quinolones in milk samples using a combination of magnetic solid-phase extraction and capillary electrophoresis. Electrophoresis 2012, 33, 2041–2048. [Google Scholar] [CrossRef]
- Tennico, Y.H.; Remcho, V.T. In-line extraction employing functionalized magnetic particles for capillary and microchip electrophoresis. Electrophoresis 2010, 31, 2548–2557. [Google Scholar] [CrossRef]
- Gasilova, N.; Gassner, A.L.; Girault, H.H. Analysis of major milk whey proteins by immunoaffinity capillary electrophoresis coupled with MALDI-MS. Electrophoresis 2012, 33, 2390–2398. [Google Scholar] [CrossRef]
- Chen, H.-X.; Busnel, J.-M.; Gassner, A.-L.; Peltre, G.; Zhang, X.-X.; Girault, H.H. Capillary electrophoresis immunoassay using magnetic beads. Electrophoresis 2008, 29, 3414–3421. [Google Scholar] [CrossRef]
- Morales-Cid, G.; Diez-Masa, J.C.; de Frutos, M. On-line immunoaffinity capillary electrophoresis based on magnetic beads for the determination of alpha-1 acid glycoprotein isoforms profile to facilitate its use as biomarker. Anal. Chim. Acta 2013, 773, 89–96. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Contains Nonbinding Recommendations. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 17 August 2023).
- Mejías, C.; Santos, J.L.; Martín, J.; Aparicio, I.; Alonso, E. Automatised on-line SPE-chiral LC-MS/MS method for the enantiomeric determination of main fluoroquinolones and their metabolites in environmental water samples. Microchem. J. 2023, 185, 108217. [Google Scholar] [CrossRef]
- Wei, D.; Guo, M. Facile preparation of magnetic graphene oxide/nanoscale zerovalent iron adsorbent for magnetic solid-phase extraction of ultra-trace quinolones in milk samples. J. Sep. Sci. 2020, 43, 3093–3102. [Google Scholar] [CrossRef]
- Horká, M.; Vykydalová, M.; Růžička, F.; Šalplachta, J.; Holá, V.; Dvořáčková, M.; Kubesová, A.; Šlais, K. CIEF separation, UV detection, and quantification of ampholytic antibiotics and bacteria from different matrices. Anal. Bioanal. Chem. 2014, 406, 6285–6296. [Google Scholar] [CrossRef]
- Paul, P.; Van Laeken, C.; Sanger-van de Griend, C.; Adams, E.; Van Schepdael, A. CE-C4D method development and validation for the assay of ciprofloxacin. J. Pharm. Biomed. Anal. 2016, 129, 1–8. [Google Scholar] [CrossRef]
- Sun, H.-W.; He, P.; Lv, Y.-K.; Liang, S.-X. Effective separation and simultaneous determination of seven fluoroquinolones by capillary electrophoresis with diode-array detector. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 852, 145–151. [Google Scholar] [CrossRef]
- Kośka, I.; Purgat, K.; Głowacki, R.; Kubalczyk, P. Simultaneous Determination of Ciprofloxacin and Ofloxacin in Animal Tissues with the Use of Capillary Electrophoresis with Transient Pseudo-Isotachophoresis. Molecules 2021, 26, 6931. [Google Scholar] [CrossRef] [PubMed]
- Baciu, T.; Borrull, F.; Neus¨uß, C.; Aguilar, C.; Calull, M. Capillary electrophoresis combined in-line with solid-phase extraction using magnetic particles as new adsorbents for the determination of drugs of abuse in human urine. Electrophoresis 2016, 37, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.T. Deposition kinetics of silicon dioxide from tetraethylorthosilicate by PECVD. Thin Solid Films 2000, 360, 60–68. [Google Scholar] [CrossRef]
- Nair, K.K.; Kaur, R.; Iqbal, N.; Hasan, A.; Alam, S.; Raza, S.K. High yield, facile aqueous synthesis and characterization of C18 functionalized iron oxide nanoparticles. Mater. Res. Express 2015, 2, 045014. [Google Scholar] [CrossRef]
- Hong, R.Y.; Li, J.H.; Zhang, S.Z.; Li, H.Z.; Zheng, Y.; Ding, J.; Wei, D.G. Preparation and characterization of silica-coated Fe3O4 nanoparticles used as precursor of ferrofluids. Appl. Surf. Sci. 2009, 255, 3485–3492. [Google Scholar] [CrossRef]
- EMEA. Committee for Veterinary Medicinal Products, EMEA/MRL/820/02-FINAL. 2002. Available online: https://www.ema.europa.eu/en/documents/mrl-report/enrofloxacin-extension-all-food-producing-species-summary-report-5-committee-veterinary-medicinal_en.pdf (accessed on 16 August 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LOD | LOQ | Calibration Concentration Range | Equations of the Calibration Curve | R2 |
|---|---|---|---|---|
| Ciprofloxacin | ||||
| 0.04 nmol/g tissue | 0.15 nmol/g tissue | 2 to 10 nmol/g tissue | y = (23.648 ± 0.310)x + (0.465 ± 0.026) | 0.9995 |
| Ofloxacin | ||||
| 0.04 nmol/g tissue | 0.15 nmol/g tissue | 2 to 10 nmol/g tissue | y = (16.021 ± 0.263)x + (0.715 ± 0.017) | 0.9992 |
| Added * (nmol/g Tissue) | Intra-Day | Inter-Day | ||||||
|---|---|---|---|---|---|---|---|---|
| Found ± SD (nmol/g Tissue) | Confidence Interval (nmol/g Tissue | RSD (%) | Accuracy (%) | Found ± SD (nmol/g Tissue) | Confidence Interval (nmol/g Tissue | RSD (%) | Accuracy (%) | |
| Ciprofloxacin | ||||||||
| 3.0 | 3.1 ± 0.2 | 3.1 ± 0.4 | 6.4 | 97.9 | 3.0 ± 0.2 | 3.0 ± 0.4 | 5.7 | 98.8 |
| 5.0 | 5.0 ± 0.2 | 5.0 ± 0.5 | 3.2 | 99.5 | 5.3 ± 0.4 | 5.3 ± 0.9 | 7.0 | 94.6 |
| 9.0 | 8.9 ± 1.0 | 8.9 ± 2.5 | 11.2 | 98.4 | 9.3 ± 0.3 | 9.3 ± 0.8 | 3.7 | 96.6 |
| Ofloxacin | ||||||||
| 3.0 | 3.0 ± 0.1 | 3.0 ± 0.3 | 3.7 | 95.7 | 2.8 ± 0.1 | 2.8 ± 0.1 | 2.1 | 94.6 |
| 5.0 | 5.3 ± 0.4 | 5.3 ± 0.9 | 7.8 | 93.0 | 4.7 ± 0.4 | 4.7 ± 0.9 | 7.7 | 93.0 |
| 9.0 | 9.3 ± 0.9 | 9.3 ± 2.2 | 9.3 | 94.0 | 9.9 ± 0.3 | 9.9 ± 0.7 | 3.0 | 90.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kośka, I.; Kubalczyk, P.; Cichomski, M.; Kisielewska, A. The Use of Extraction on C18-Silica-Modified Magnetic Nanoparticles for the Determination of Ciprofloxacin and Ofloxacin in Meat Tissues. Molecules 2023, 28, 6123. https://doi.org/10.3390/molecules28166123
Kośka I, Kubalczyk P, Cichomski M, Kisielewska A. The Use of Extraction on C18-Silica-Modified Magnetic Nanoparticles for the Determination of Ciprofloxacin and Ofloxacin in Meat Tissues. Molecules. 2023; 28(16):6123. https://doi.org/10.3390/molecules28166123
Chicago/Turabian StyleKośka, Izabella, Paweł Kubalczyk, Michał Cichomski, and Aneta Kisielewska. 2023. "The Use of Extraction on C18-Silica-Modified Magnetic Nanoparticles for the Determination of Ciprofloxacin and Ofloxacin in Meat Tissues" Molecules 28, no. 16: 6123. https://doi.org/10.3390/molecules28166123
APA StyleKośka, I., Kubalczyk, P., Cichomski, M., & Kisielewska, A. (2023). The Use of Extraction on C18-Silica-Modified Magnetic Nanoparticles for the Determination of Ciprofloxacin and Ofloxacin in Meat Tissues. Molecules, 28(16), 6123. https://doi.org/10.3390/molecules28166123