
Assessing the Performance of Non-Equilibrium Thermodynamic Integration in Flavodoxin Redox Potential Estimation

 , ,
, ,
Abstract
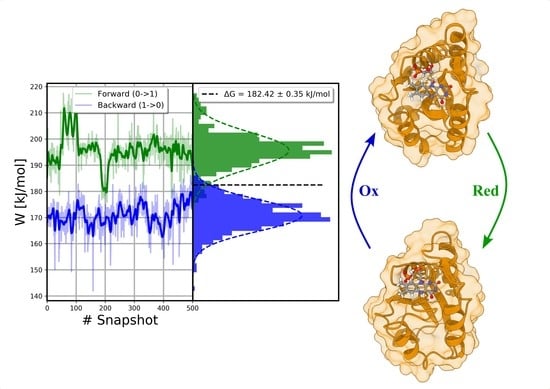
1. Introduction
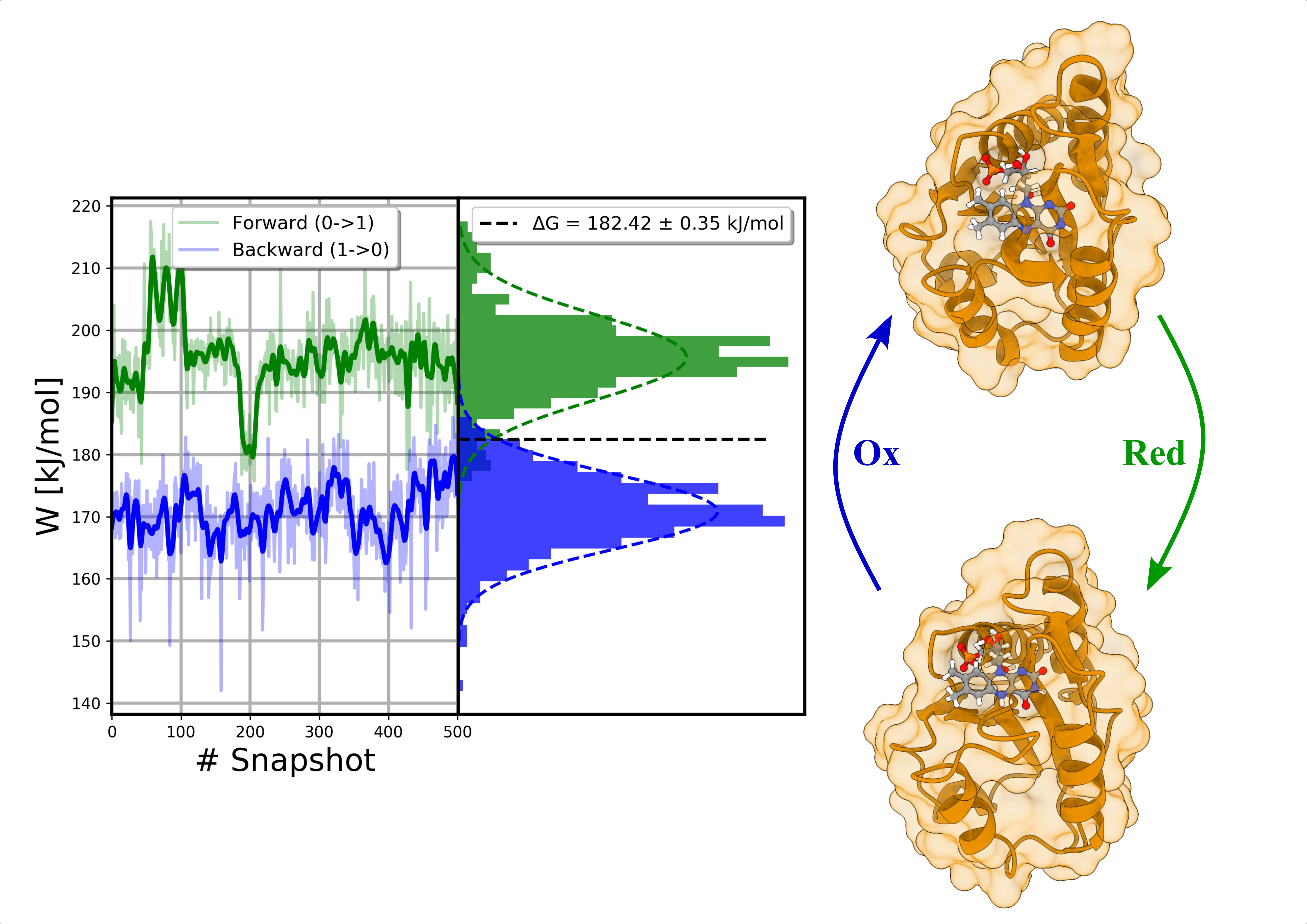
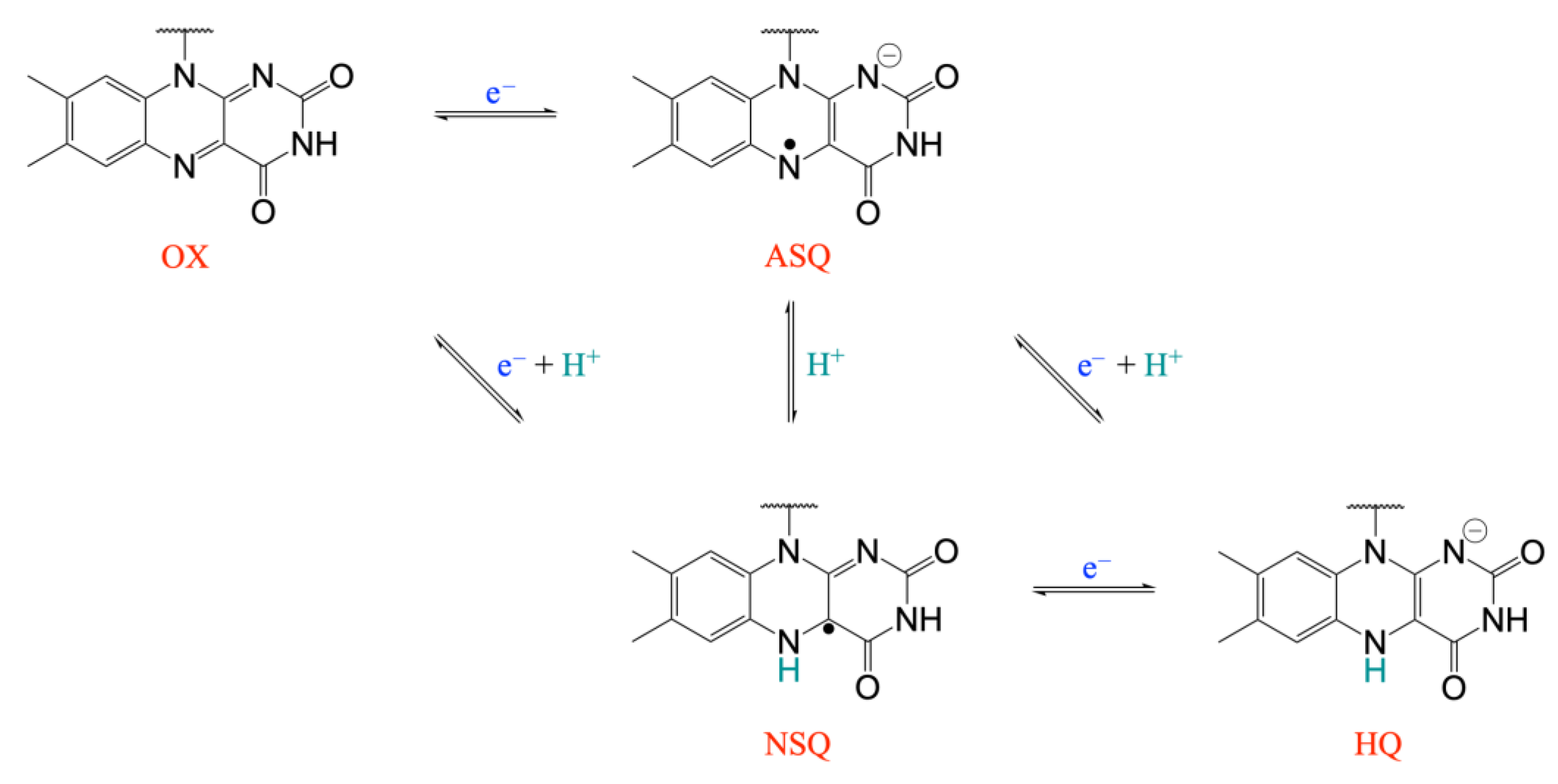
2. Results and Discussion
3. Materials and Methods
3.1. Simulated Systems
3.2. MD Simulations
3.3. TI Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Leys, D.; Scrutton, N.S. Sweating the Assets of Flavin Cofactors: New Insight of Chemical Versatility from Knowledge of Structure and Mechanism. Curr. Opin. Struct. Biol. 2016, 41, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Iyanagi, T. Molecular Mechanism of Metabolic NAD(P)H-Dependent Electron-Transfer Systems: The Role of Redox Cofactors. Biochim. Biophys. Acta BBA Bioenerg. 2019, 1860, 233–258. [Google Scholar] [CrossRef]
- Ishikita, H. Contributions of Protein Environment to the Reduction Potentials of Flavin-Containing Proteins; Hille, R., Miller, S., Palfey, B., Eds.; Walter De Gruyter Gmbh: Berlin, Germany, 2013; ISBN 978-3-11-029834-5. [Google Scholar]
- Galuzzi, B.G.; Mirarchi, A.; Viganò, E.L.; De, G.; Damiani, C.; Arrigoni, F. Machine Learning for Efficient Prediction of Protein Redox Potential: The Flavoproteins Case. J. Chem. Inf. Model. 2022, 62, 4748–4759. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Roberts, E.R.H.; Vujaskovic, Z.; Leong, K.W.; Spasojevic, I. SOD Therapeutics: Latest Insights into Their Structure-Activity Relationships and Impact on the Cellular Redox-Based Signaling Pathways. Antioxid. Redox Signal. 2014, 20, 2372–2415. [Google Scholar] [CrossRef] [PubMed]
- Legrand, Y.-M.; Gray, M.; Cooke, G.; Rotello, V.M. Model Systems for Flavoenzyme Activity: Relationships between Cofactor Structure, Binding and Redox Properties. J. Am. Chem. Soc. 2003, 125, 15789–15795. [Google Scholar] [CrossRef]
- Munro, A.W.; Taylor, P.; Walkinshaw, M.D. Structures of Redox Enzymes. Curr. Opin. Biotechnol. 2000, 11, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.; Medina, M. Anaerobic Stopped-Flow Spectrophotometry with Photodiode Array Detection in the Presteady State: An Application to Elucidate Oxidoreduction Mechanisms in Flavoproteins. In Flavins and Flavoproteins; Barile, M., Ed.; Springer: New York, NY, USA, 2021; Volume 2280, pp. 135–155. ISBN 978-1-07-161285-9. [Google Scholar]
- Dijkman, W.P.; de Gonzalo, G.; Mattevi, A.; Fraaije, M.W. Flavoprotein Oxidases: Classification and Applications. Appl. Microbiol. Biotechnol. 2013, 97, 5177–5188. [Google Scholar] [CrossRef]
- Van Berkel, W.J.H.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein Monooxygenases, a Diverse Class of Oxidative Biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, D.F.; Gonzalez, C.F.; Park, C.H.; Blake, R.; Keyhan, M.; Matin, A. Chromate-Reducing Properties of Soluble Flavoproteins from Pseudomonas Putida and Escherichia Coli. Appl Env. Microbiol 2004, 70, 873–882. [Google Scholar] [CrossRef]
- Zhou, Z.; Swenson, R.P. The Cumulative Electrostatic Effect of Aromatic Stacking Interactions and the Negative Electrostatic Environment of the Flavin Mononucleotide Binding Site Is a Major Determinant of the Reduction Potential for the Flavodoxin from Desulfovibrio Vulgaris [Hildenborough]. Biochemistry 1996, 35, 15980–15988. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Pattridge, K.A.; Metzger, A.L.; Dixon, M.M.; Eren, M.; Feng, Y.; Swenson, R.P. Control of Oxidation−Reduction Potentials in Flavodoxin from Clostridium Beijerinckii: The Role of Conformation Changes. Biochemistry 1997, 36, 1259–1280. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.-C.; Swenson, R.P. The Midpoint Potentials for the Oxidized−Semiquinone Couple for Gly57 Mutants of the Clostridium Beijerinckii Flavodoxin Correlate with Changes in the Hydrogen-Bonding Interaction with the Proton on N(5) of the Reduced Flavin Mononucleotide Cofactor As Measured by NMR Chemical Shift Temperature Dependencies. Biochemistry 1999, 38, 7168–7176. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Burnett, R.M.; Darling, G.D.; Ludwig, M.L. Structure of the Semiquinone Form of Flavodoxin from Clostridium MP. J. Mol. Biol. 1977, 117, 195–225. [Google Scholar] [CrossRef] [PubMed]
- Watt, W.; Tulinsky, A.; Swenson, R.P.; Watenpaugh, K.D. Comparison of the Crystal Structures of a Flavodoxin in Its Three Oxidation States at Cryogenic Temperatures. J. Mol. Biol. 1991, 218, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Franken, H.-D.; Ruterjans, H.; Muller, F. Nuclear-Magnetic-Resonance Investigation of 15N-Labeled Flavins, Free and Bound to Megasphaera Elsdenii Apoflavodoxin. Eur. J. Biochem. 1984, 138, 481–489. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Schopfer, L.M.; Metzger, A.L.; Pattridge, K.A.; Massey, V. Structure and Oxidation-Reduction Behavior of 1-Deaza-FMN Flavodoxins: Modulation of Redox Potentials in Flavodoxins. Biochemistry 1990, 29, 10364–10375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Swenson, R.P. Electrostatic Effects of Surface Acidic Amino Acid Residues on the Oxidation-Reduction Potentials of the Flavodoxin from Desulfovibrio Vulgaris (Hildenborough). Biochemistry 1995, 34, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Swenson, R.P.; Krey, G.D. Site-Directed Mutagenesis of Tyrosine-98 in the Flavodoxin from Desulfovibrio Vulgaris (Hildenborough): Regulation of Oxidation-Reduction Properties of the Bound FMN Cofactor by Aromatic, Solvent, and Electrostatic Interactions. Biochemistry 1994, 33, 8505–8514. [Google Scholar] [CrossRef] [PubMed]
- Stockman, B.J.; Richardson, T.E.; Swenson, R.P. Structural Changes Caused by Site-Directed Mutagenesis of Tyrosine-98 in Desulfovibrio Vulgaris Flavodoxin Delineated by 1H and 15N NMR Spectroscopy: Implications for Redox Potential Modulation. Biochemistry 1994, 33, 15298–15308. [Google Scholar] [CrossRef]
- Bradley, L.H.; Swenson, R.P. Role of Glutamate-59 Hydrogen Bonded to N(3)H of the Flavin Mononucleotide Cofactor in the Modulation of the Redox Potentials of the Clostridium Beijerinckii Flavodoxin. Glutamate-59 Is Not Responsible for the pH Dependency but Contributes to the Stabilization of the Flavin Semiquinone. Biochemistry 1999, 38, 12377–12386. [Google Scholar] [CrossRef]
- Ishikita, H. Influence of the Protein Environment on the Redox Potentials of Flavodoxins from Clostridium Beijerinckii. J. Biol. Chem. 2007, 282, 25240–25246. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.H.; Swenson, R.P. Role of Hydrogen Bonding Interactions to N(3)H of the Flavin Mononucleotide Cofactor in the Modulation of the Redox Potentials of the Clostridium Beijerinckii Flavodoxin. Biochemistry 2001, 40, 8686–8695. [Google Scholar] [CrossRef] [PubMed]
- Druhan, L.J.; Swenson, R.P. Role of Methionine 56 in the Control of the Oxidation−Reduction Potentials of the Clostridium Beijerinckii Flavodoxin: Effects of Substitutions by Aliphatic Amino Acids and Evidence for a Role of Sulfur−Flavin Interactions. Biochemistry 1998, 37, 9668–9678. [Google Scholar] [CrossRef]
- Gillet, N.; Lévy, B.; Moliner, V.; Demachy, I.; de la Lande, A. Theoretical Estimation of Redox Potential of Biological Quinone Cofactors. J. Comput. Chem. 2017, 38, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, G.M.; Dumit, V.I.; Bombarda, E. Methods Based on Continuum Electrostatics and Their Application to Flavoproteins—A Review; Hille, R., Miller, S., Palfey, B., Eds.; Walter De Gruyter Gmbh: Berlin, Germany, 2013; ISBN 978-3-11-029834-5. [Google Scholar]
- North, M.A.; Bhattacharyya, S.; Truhlar, D.G. Improved Density Functional Description of the Electrochemistry and Structure−Property Descriptors of Substituted Flavins. J. Phys. Chem. B 2010, 114, 14907–14915. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, M.; Ensing, B. First and Second One-Electron Reduction of Lumiflavin in Water—A First Principles Molecular Dynamics Study. J. Chem. Theory Comput. 2013, 9, 3889–3899. [Google Scholar] [CrossRef]
- Mueller, R.M.; North, M.A.; Yang, C.; Hati, S.; Bhattacharyya, S. Interplay of Flavin’s Redox States and Protein Dynamics: An Insight from QM/MM Simulations of Dihydronicotinamide Riboside Quinone Oxidoreductase 2. J. Phys. Chem. B 2011, 115, 3632–3641. [Google Scholar] [CrossRef]
- Sattelle, B.M.; Sutcliffe, M.J. Calculating Chemically Accurate Redox Potentials for Engineered Flavoproteins from Classical Molecular Dynamics Free Energy Simulations. J. Phys. Chem. A 2008, 112, 13053–13057. [Google Scholar] [CrossRef]
- Aldeghi, M.; Gapsys, V.; de Groot, B.L. Accurate Estimation of Ligand Binding Affinity Changes upon Protein Mutation. ACS Cent. Sci. 2018, 4, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Nilsson, I.; Ryde, U. Binding Affinities of Factor Xa Inhibitors Estimated by Thermodynamic Integration and MM/GBSA. J. Chem. Inf. Model. 2011, 51, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Madaj, R.; Gostynski, B.; Pawlowska, R.; Chworos, A. Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches. Biomolecules 2021, 11, 1104. [Google Scholar] [CrossRef] [PubMed]
- Lawrenz, M.; Wereszczynski, J.; Ortiz-Sánchez, J.M.; Nichols, S.E.; McCammon, J.A. Thermodynamic Integration to Predict Host-Guest Binding Affinities. J. Comput. Aided Mol. Des. 2012, 26, 569–576. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Den Bosch, M.; Swart, M.; Snijders†, J.G.; Berendsen, H.J.C.; Mark, A.E.; Oostenbrink, C.; Van Gunsteren, W.F.; Canters, G.W. Calculation of the Redox Potential of the Protein Azurin and Some Mutants. ChemBioChem 2005, 6, 738–746. [Google Scholar] [CrossRef]
- Steiner, D.; Oostenbrink, C.; Van Gunsteren, W.F. Calculation of the Relative Free Energy of Oxidation of Azurin at pH 5 and pH 9. J. Comput. Chem. 2012, 33, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.G.; Nardi, A.N.; Amadei, A.; D’Abramo, M. Theoretical Modeling of Redox Potentials of Biomolecules. Molecules 2022, 27, 1077. [Google Scholar] [CrossRef]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef]
- Hendrix, D.A.; Jarzynski, C. A “fast Growth” Method of Computing Free Energy Differences. J. Chem. Phys. 2001, 114, 5974–5981. [Google Scholar] [CrossRef]
- Crooks, G.E. Nonequilibrium Measurements of Free Energy Differences for Microscopically Reversible Markovian Systems. J. Stat. Phys. 1998, 90, 1481–1487. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Kasim, M.; Swenson, R.P. Alanine-Scanning of the 50’s Loop in the Clostridium Beijerinckii Flavodoxin: Evaluation of Additivity and the Importance of Interactions Provided by the Main Chain in the Modulation of the Oxidation−Reduction Potentials. Biochemistry 2001, 40, 13548–13555. [Google Scholar] [CrossRef]
- Li, W.; Baldus, I.B.; Gräter, F. Redox Potentials of Protein Disulfide Bonds from Free-Energy Calculations. J. Phys. Chem. B 2015, 119, 5386–5391. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; Pérez-Benito, L.; Aldeghi, M.; Seeliger, D.; Van Vlijmen, H.; Tresadern, G.; De Groot, B.L. Large Scale Relative Protein Ligand Binding Affinities Using Non-Equilibrium Alchemy. Chem. Sci. 2020, 11, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; Yildirim, A.; Aldeghi, M.; Khalak, Y.; Van Der Spoel, D.; De Groot, B.L. Accurate Absolute Free Energies for Ligand–protein Binding Based on Non-Equilibrium Approaches. Commun. Chem. 2021, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, A. A Molecular Mechanics Model for Flavins. J. Comput. Chem. 2019, 40, 2834–2842. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-Range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Protein Thermostability Calculations Using Alchemical Free Energy Simulations. Biophys. J. 2010, 98, 2309–2316. [Google Scholar] [CrossRef]
- Shirts, M.R.; Bair, E.; Hooker, G.; Pande, V.S. Equilibrium Free Energies from Nonequilibrium Measurements Using Maximum-Likelihood Methods. Phys. Rev. Lett. 2003, 91, 140601. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
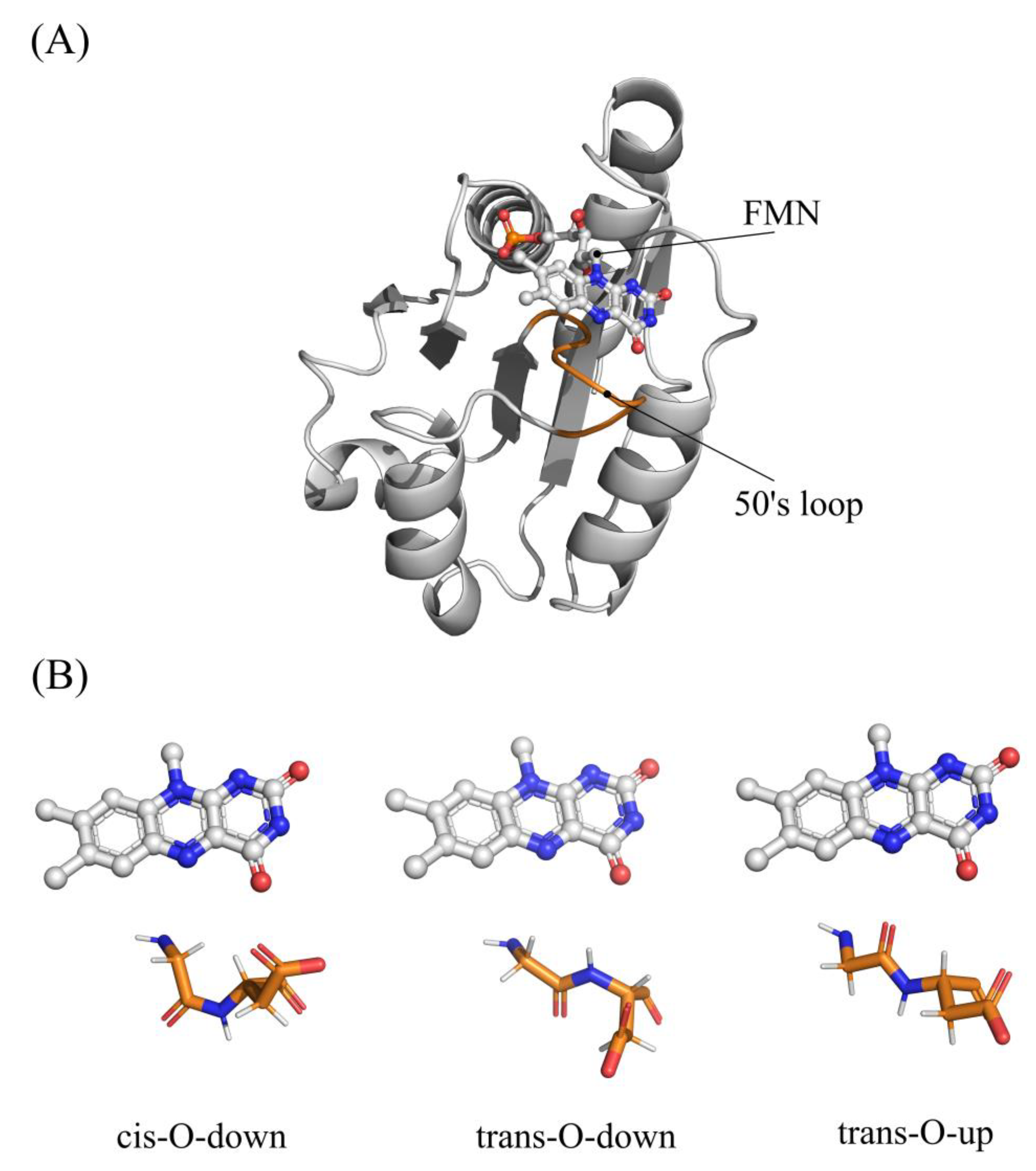
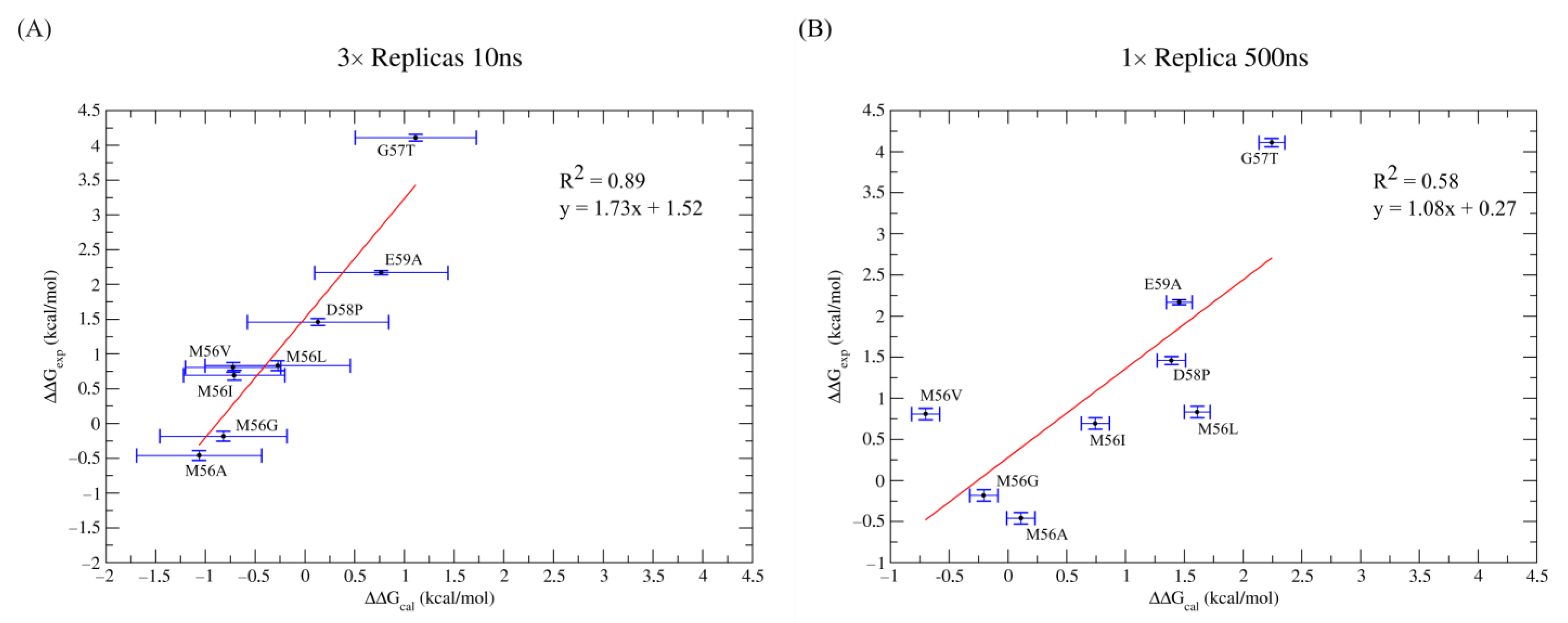
| WT | Exp. Proportion | Replica a | b | b,c | Standard Error b | MAE b | RMSE b |
|---|---|---|---|---|---|---|---|
| Cis-O-down | 50% | 1 | −43.31 ± 0.05 | −42.92 | 0.31 | 1.37 | 1.54 |
| 2 | −43.44 ± 0.04 | ||||||
| 3 | −42.48 ± 0.04 | ||||||
| Trans-O-down | 20% | 1 | −42.92 ± 0.05 | −42.91 | 0.19 | 0.64 | 0.87 |
| 2 | −43.97 ± 0.04 | ||||||
| 3 | −43.73 ± 0.05 | ||||||
| Trans-O-up | 30% | 1 | −43.17 ± 0.05 | −43.76 | 0.32 | 1.37 | 1.54 |
| 2 | −42.70 ± 0.04 | ||||||
| 3 | −43.33 ± 0.04 | ||||||
| Overall | −43.17 d | 0.29 d | 1.06 e | 1.25 e |
| 3 × 10 ns Simulations | ||||||||
|---|---|---|---|---|---|---|---|---|
| a | a,b | a | a | a | a,c,d | a,b | Absolute Error a | |
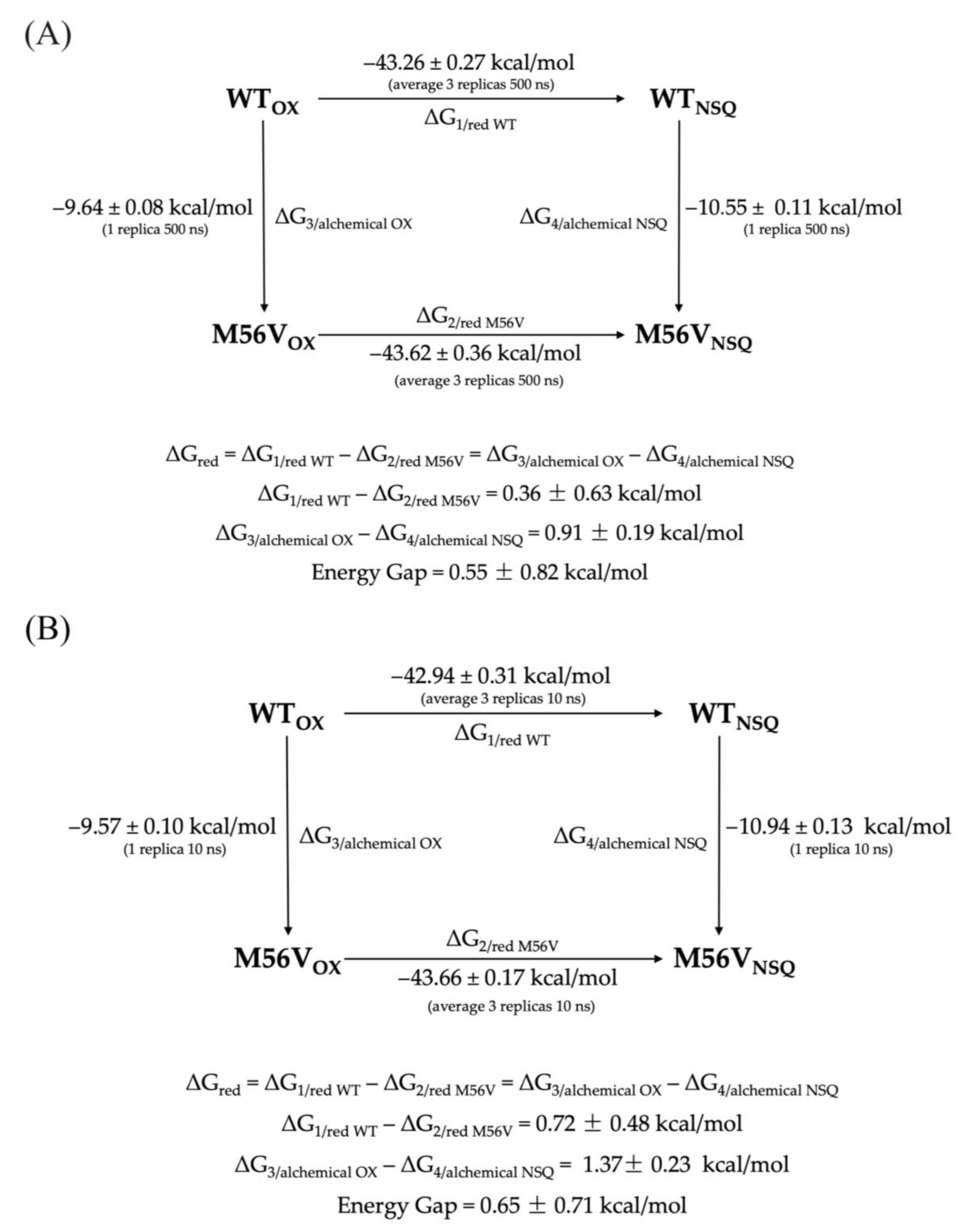
| WT | 2.12 ± 0.11 | - | −43.33 ± 0.05 | −43.46 ± 0.04 | −42.50 ± 0.04 | −42.94 ± 0.31 | - | - |
| G57T | 6.23 ± 0.11 | 4.11 ± 0.23 | −41.17 ± 0.04 | −42.12 ± 0.05 | −42.05 ± 0.04 | −41.83 ± 0.30 | 1.11 ± 0.61 | 3.00 |
| D58P | 3.58 ± 0.11 | 1.46 ± 0.23 | −41.98 ± 0.05 | −42.81 ± 0.06 | −43.31 ± 0.03 | −42.81 ± 0.40 | 0.13 ± 0.71 | 1.33 |
| E59A | 4.29± 0.12 | 2.17 ± 0.23 | −41.64 ± 0.04 | −42.53 ± 0.05 | −43.22 ± 0.05 | −42.18 ± 0.36 | 0.76 ± 0.67 | 1.41 |
| M56A | 1.66 ± 0.18 | −0.46 ± 0.30 | −43.98 ± 0.04 | −43.66 ± 0.06 | −44.56 ± 0.05 | −44.01 ± 0.32 | −1.06 ± 0.63 | 0.60 |
| M56G | 1.94 ± 0.18 | −0.18 ± 0.30 | −44.14 ± 0.06 | −43.31 ± 0.05 | −44.45 ± 0.07 | −43.76 ± 0.33 | −0.82 ± 0.64 | 1.24 |
| M56L | 2.95 ± 0.18 | 0.83 ± 0.30 | −42.93 ± 0.06 | −43.14 ± 0.05 | −44.20 ± 0.05 | −43.21 ± 0.42 | −0.27 ± 0.73 | 1.10 |
| M56I | 2.81± 0.18 | 0.69 ± 0.30 | −43.18 ± 0.07 | −43.93 ± 0.06 | −43.68 ± 0.08 | −43.65 ± 0.20 | −0.71 ± 0.51 | 1.40 |
| M56V | 2.93 ± 0.18 | 0.81 ± 0.30 | −43.95 ± 0.05 | −43.61 ± 0.05 | −43.49 ± 0.05 | −43.66 ± 0.17 | −0.72 ± 0.48 | 1.53 |
| MAE | 1.37 | |||||||
| RMSσ | 0.06 | |||||||
| RMSE | 1.54 | |||||||
| 1 × 500 ns Simulations | ||||||||
| a | a,b | a | a,b | Absolute Error a | ||||
| WT | 2.12 ± 0.11 | - | −43.60 ± 0.08 | - | - | |||
| G57T | 6.23 ± 0.11 | 4.11 ± 0.23 | −41.45 ± 0.04 | 2.15 ± 0.12 | 1.96 | |||
| D58P | 3.58 ± 0.11 | 1.46 ± 0.23 | −42.21 ± 0.04 | 1.39 ± 0.12 | 0.07 | |||
| E59A | 4.29± 0.12 | 2.17 ± 0.23 | −42.14 ± 0.04 | 1.46 ± 0.12 | 0.71 | |||
| M56A | 1.66 ± 0.18 | −0.46 ± 0.30 | −43.49 ± 0.04 | 0.11 ± 0.12 | 0.57 | |||
| M56G | 1.94 ± 0.18 | −0.18 ± 0.30 | −43.80 ± 0.05 | −0.20 ± 0.13 | 0.02 | |||
| M56L | 2.95 ± 0.18 | 0.83 ± 0.30 | −41.99 ± 0.03 | 1.61 ± 0.11 | 0.78 | |||
| M56I | 2.81± 0.18 | 0.69 ± 0.30 | −42.86 ± 0.05 | 0.74 ± 0.13 | 0.05 | |||
| M56V | 2.93 ± 0.18 | 0.81 ± 0.30 | −44.30 ± 0.04 | −0.70 ± 0.12 | 1.51 | |||
| MAE | 0.70 | |||||||
| RMS | 0.95 | |||||||
| Sistem | OX | NSQ | ||
|---|---|---|---|---|
| Structure a | 50’s Loop b | Structure a | 50’s loop b | |
| WT | 5NLL | cis-O-down | 2FOX | trans-O-up |
| G57T | 1FLD | trans-O-down | 5NUL | trans-O-down |
| D58P | 4NUL | cis-O-down | 5FLN | trans-O-down |
| E59A | 5NLL in silico mutagenesis | cis-O-down | 2FOX in silico mutagenesis | trans-O-up |
| M56A | 5NLL in silico mutagenesis | cis-O-down | 2FOX in silico mutagenesis | trans-O-up |
| M56G | 5NLL in silico mutagenesis | cis-O-down | 2FOX in silico mutagenesis | trans-O-up |
| M56L | 5NLL in silico mutagenesis | cis-O-down | 2FOX in silico mutagenesis | trans-O-up |
| M56I | 5NLL in silico mutagenesis | cis-O-down | 2FOX in silico mutagenesis | trans-O-up |
| M56V | 5NLL in silico mutagenesis | cis-O-down | 2FOX in silico mutagenesis | trans-O-up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silvestri, G.; Arrigoni, F.; Persico, F.; Bertini, L.; Zampella, G.; De Gioia, L.; Vertemara, J. Assessing the Performance of Non-Equilibrium Thermodynamic Integration in Flavodoxin Redox Potential Estimation. Molecules 2023, 28, 6016. https://doi.org/10.3390/molecules28166016
Silvestri G, Arrigoni F, Persico F, Bertini L, Zampella G, De Gioia L, Vertemara J. Assessing the Performance of Non-Equilibrium Thermodynamic Integration in Flavodoxin Redox Potential Estimation. Molecules. 2023; 28(16):6016. https://doi.org/10.3390/molecules28166016
Chicago/Turabian StyleSilvestri, Giuseppe, Federica Arrigoni, Francesca Persico, Luca Bertini, Giuseppe Zampella, Luca De Gioia, and Jacopo Vertemara. 2023. "Assessing the Performance of Non-Equilibrium Thermodynamic Integration in Flavodoxin Redox Potential Estimation" Molecules 28, no. 16: 6016. https://doi.org/10.3390/molecules28166016
APA StyleSilvestri, G., Arrigoni, F., Persico, F., Bertini, L., Zampella, G., De Gioia, L., & Vertemara, J. (2023). Assessing the Performance of Non-Equilibrium Thermodynamic Integration in Flavodoxin Redox Potential Estimation. Molecules, 28(16), 6016. https://doi.org/10.3390/molecules28166016