
Evolutionary Algorithm-Based Crystal Structure Prediction of CuxZnyOz Ternary Oxides

Abstract
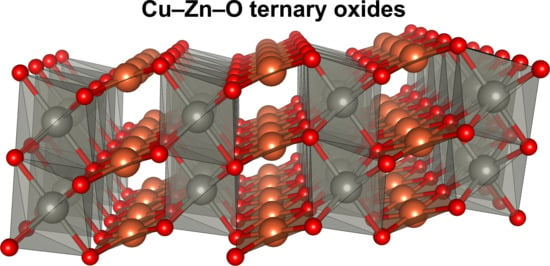
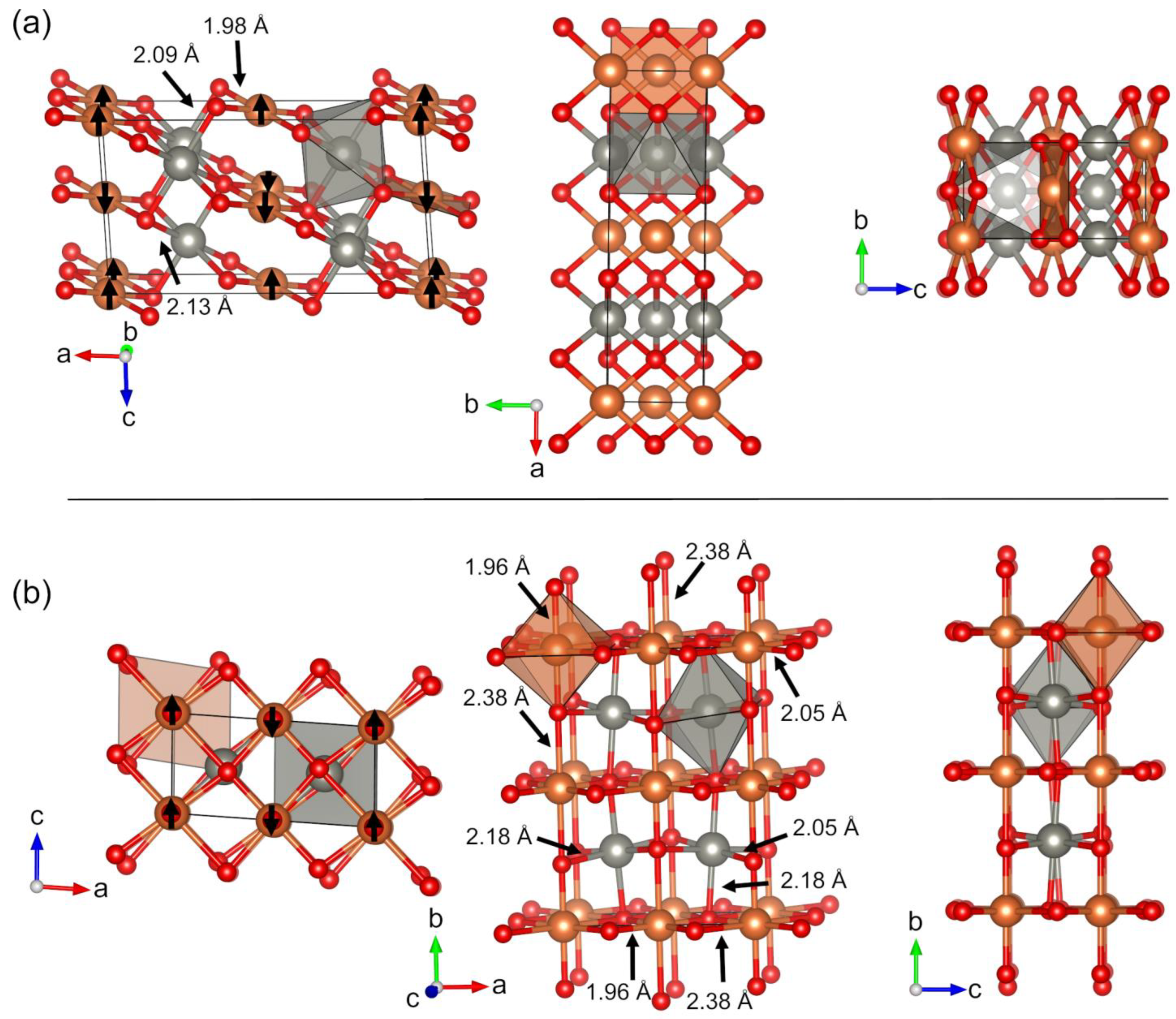
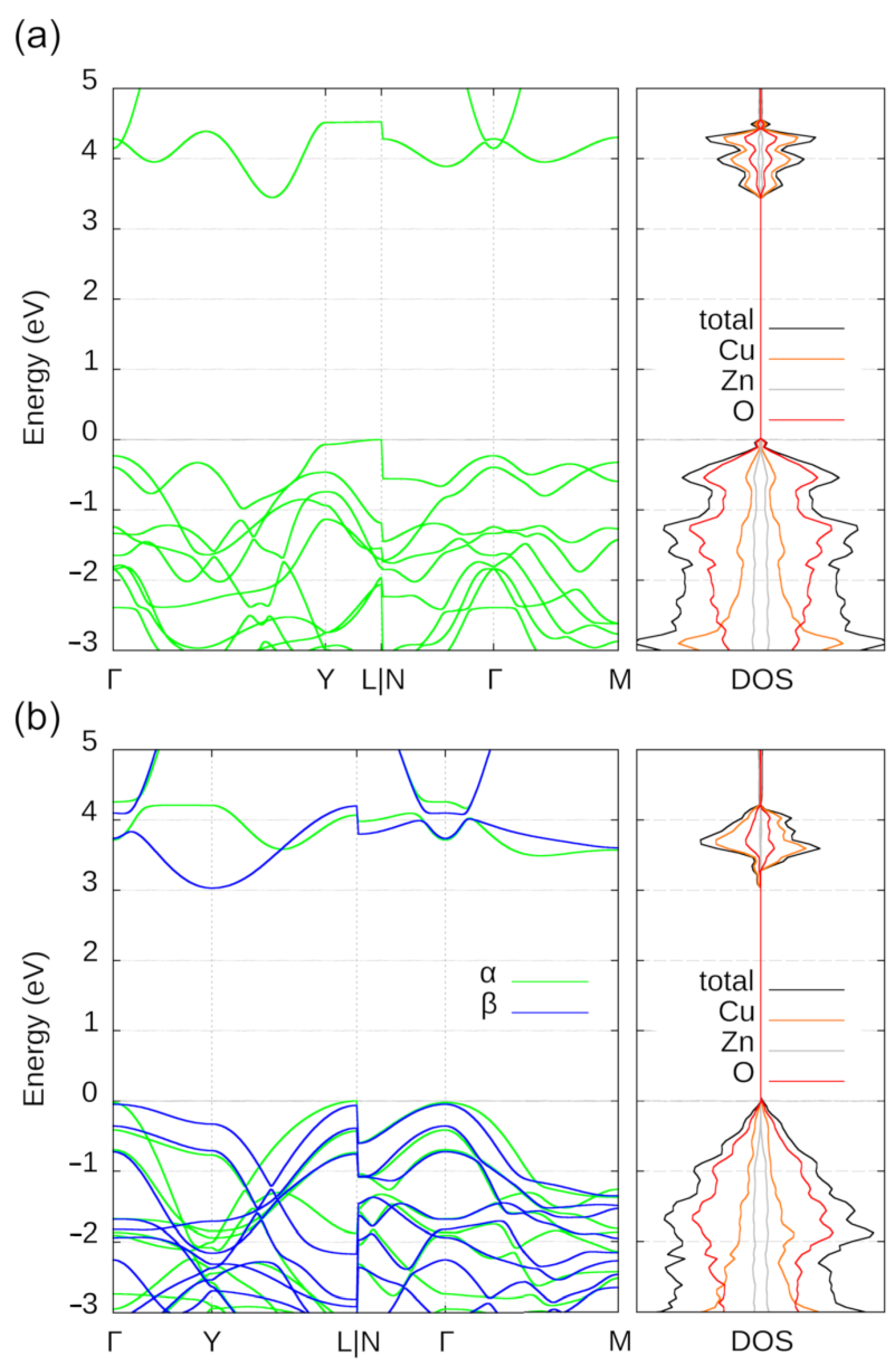
1. Introduction
2. Results
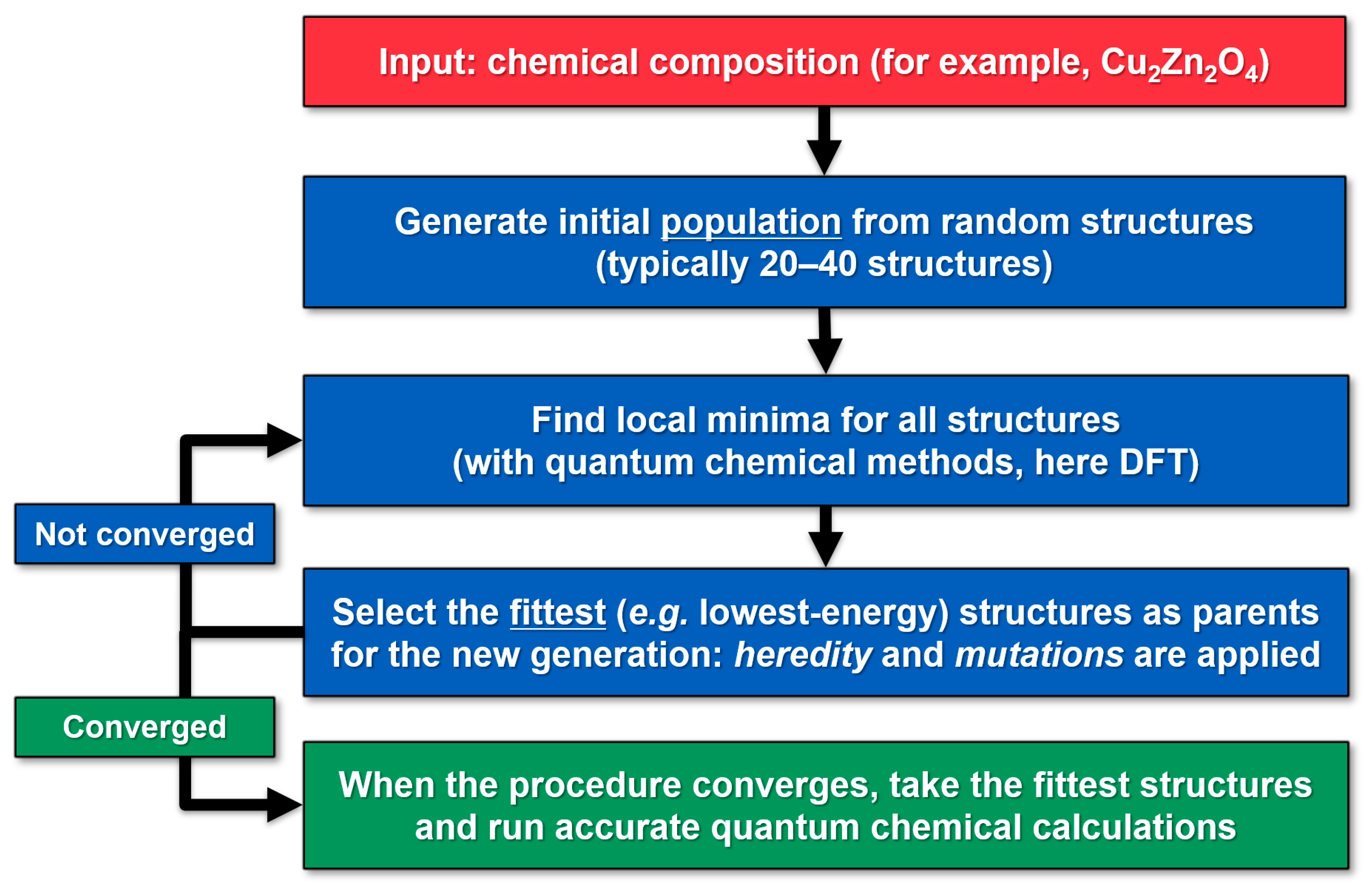
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Védrine, J.C. Metal Oxides in Heterogeneous Oxidation Catalysis: State of the Art and Challenges for a More Sustainable World. ChemSusChem 2019, 12, 577–588. [Google Scholar] [CrossRef]
- Dixon, S.C.; Scanlon, D.O.; Carmalt, C.J.; Parkin, I.P. N-Type Doped Transparent Conducting Binary Oxides: An Overview. J. Mater. Chem. C 2016, 4, 6946–6961. [Google Scholar] [CrossRef]
- Spencer, J.A.; Mock, A.L.; Jacobs, A.G.; Schubert, M.; Zhang, Y.; Tadjer, M.J. A Review of Band Structure and Material Properties of Transparent Conducting and Semiconducting Oxides: Ga2O3, Al2O3, In2O3, ZnO, SnO2, CdO, NiO, CuO, and Sc2O3. Appl. Phys. Rev. 2022, 9, 011315. [Google Scholar] [CrossRef]
- Walia, S.; Balendhran, S.; Nili, H.; Zhuiykov, S.; Rosengarten, G.; Wang, Q.H.; Bhaskaran, M.; Sriram, S.; Strano, M.S.; Kalantar-zadeh, K. Transition Metal Oxides—Thermoelectric Properties. Prog. Mater. Sci. 2013, 58, 1443–1489. [Google Scholar] [CrossRef]
- Díez-García, M.I.; Gómez, R. Progress in Ternary Metal Oxides as Photocathodes for Water Splitting Cells: Optimization Strategies. Sol. RRL 2022, 6, 2100871. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, J.; Zhu, P.; Gao, W.; Cui, Q. The Phase Transition of Zn0.854Cu0.146O under High Pressure. Phys. Status Solidi B 2011, 248, 1128–1131. [Google Scholar] [CrossRef]
- Prabhakaran, D.; Boothroyd, A.T. Single Crystal Growth of Zn-Doped CuO by the Floating-Zone Method. J. Cryst. Growth 2003, 250, 77–82. [Google Scholar] [CrossRef]
- Bououdina, M.; Mamouni, N.; Lemine, O.M.; Al-Saie, A.; Jaafar, A.; Ouladdiaf, B.; El Kenz, A.; Benyoussef, A.; Hlil, E.K. Neutron Diffraction Study and Ab-Initio Calculations of Nanostructured Doped ZnO. J. Alloys Compd. 2012, 536, 66–72. [Google Scholar] [CrossRef]
- Volnianska, O.; Bogusławski, P. Green Luminescence and Calculated Optical Properties of Cu Ions in ZnO. J. Alloys Compd. 2019, 782, 1024–1030. [Google Scholar] [CrossRef]
- Wattoo, A.G.; Song, Z.; Iqbal, M.Z.; Rizwan, M.; Saeed, A.; Ahmad, S.; Ali, A.; Naz, N.A. Effect of Zinc Concentration on Physical Properties of Copper Oxide (Cu1−xZnxO). J. Mater. Sci. Mater. Electron. 2015, 26, 9795–9800. [Google Scholar] [CrossRef]
- Amaral, J.B.; Araujo, R.M.; Pedra, P.P.; Meneses, C.T.; Duque, J.G.S.; Rezende, M.D.S. Doping Effect on the Structural Properties of Cu1−x(Ni, Zn, Al and Fe)xO Samples (0 < x < 0.10): An Experimental and Computational Study. J. Solid State Chem. 2016, 241, 26–29. [Google Scholar] [CrossRef]
- Oganov, A.R.; Pickard, C.J.; Zhu, Q.; Needs, R.J. Structure Prediction Drives Materials Discovery. Nat. Rev. Mater. 2019, 4, 331–348. [Google Scholar] [CrossRef]
- Kuklin, M.S.; Eklund, K.; Linnera, J.; Ropponen, A.; Tolvanen, N.; Karttunen, A.J. Structural Properties and Magnetic Ground States of 100 Binary D-Metal Oxides Studied by Hybrid Density Functional Methods. Molecules 2022, 27, 874. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.V.; Görne, A.L.; Tchougréeff, A.L.; Dronskowski, R. Relative Stability of Diamond and Graphite as Seen through Bonds and Hybridizations. Phys. Chem. Chem. Phys. 2019, 21, 10961–10969. [Google Scholar] [CrossRef] [PubMed]
- Civalleri, B.; Presti, D.; Dovesi, R.; Savin, A. On Choosing the Best Density Functional Approximation. In Chemical Modelling; Springborg, M., Ed.; Royal Society of Chemistry: Cambridge, UK, 2012; Volume 9, pp. 168–185. ISBN 978-1-84973-412-7. [Google Scholar]
- Linnera, J.; Karttunen, A.J. Ab Initio Study of the Lattice Thermal Conductivity of Cu2O Using the Generalized Gradient Approximation and Hybrid Density Functional Methods. Phys. Rev. B 2017, 96, 014304. [Google Scholar] [CrossRef]
- Glass, C.W.; Oganov, A.R.; Hansen, N. USPEX—Evolutionary Crystal Structure Prediction. Comput. Phys. Commun. 2006, 175, 713–720. [Google Scholar] [CrossRef]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How Evolutionary Crystal Structure Prediction Works—And Why. Acc. Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef]
- Lyakhov, A.O.; Oganov, A.R.; Stokes, H.T.; Zhu, Q. New Developments in Evolutionary Structure Prediction Algorithm USPEX. Comput. Phys. Commun. 2013, 184, 1172–1182. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for High-Throughput DFT Calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation Energies of Transition Metal Oxides within the GGA+U Framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Schrön, A.; Rödl, C.; Bechstedt, F. Crystalline and Magnetic Anisotropy of the 3d-Transition Metal Monoxides MnO, FeO, CoO, and NiO. Phys. Rev. B 2012, 86, 115134. [Google Scholar] [CrossRef]
- Noh, J.; Osman, O.I.; Aziz, S.G.; Winget, P.; Brédas, J.L. A Density Functional Theory Investigation of the Electronic Structure and Spin Moments of Magnetite. Sci. Technol. Adv. Mater. 2014, 15, 044202. [Google Scholar] [CrossRef]
- Lima, A.F. Density Functional Theory Study on the Magnetic Properties of Co3O4with Normal Spinel Structure. J. Phys. Chem. Solids 2016, 91, 86–89. [Google Scholar] [CrossRef]
- Singh, V.; Kosa, M.; Majhi, K.; Major, D.T. Putting DFT to the Test: A First-Principles Study of Electronic, Magnetic, and Optical Properties of Co3O4. J. Chem. Theory Comput. 2015, 11, 64–72. [Google Scholar] [CrossRef]
- Deng, H.X.; Li, J.; Li, S.S.; Xia, J.B.; Walsh, A.; Wei, S.H. Origin of Antiferromagnetism in CoO: A Density Functional Theory Study. Appl. Phys. Lett. 2010, 96, 162508. [Google Scholar] [CrossRef]
- Bredow, T.; Gerson, A.R. Effect of Exchange and Correlation on Bulk Properties of MgO, NiO, and CoO. Phys. Rev. B 2000, 61, 5194–5201. [Google Scholar] [CrossRef]
- Rollmann, G.; Rohrbach, A.; Entel, P.; Hafner, J. First-Principles Calculation of the Structure and Magnetic Phases of Hematite. Phys. Rev. B 2004, 69, 165107. [Google Scholar] [CrossRef]
- Kuklin, M.S.; Karttunen, A.J. Crystal Structure Prediction of Magnetic Transition Metal Oxides by Using Evolutionary Algorithm and Hybrid DFT Methods. J. Phys. Chem. C 2018, 122, 24949–24957. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Rödl, C.; Fuchs, F.; Furthmüller, J.; Bechstedt, F. Quasiparticle Band Structures of the Antiferromagnetic Transition-Metal Oxides MnO, FeO, CoO, and NiO. Phys. Rev. B 2009, 79, 235114. [Google Scholar] [CrossRef]
- Kulik, H.J.; Marzari, N. Transition-Metal Dioxides: A Case for the Intersite Term in Hubbard-Model Functionals. J. Chem. Phys. 2011, 134, 094103. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Parker, D.; Du, M.H.; Singh, D.J. Potential Thermoelectric Performance of Hole-Doped Cu2O. New J. Phys. 2013, 15, 043029. [Google Scholar] [CrossRef]
- Seo, D.H.; Urban, A.; Ceder, G. Calibrating Transition-Metal Energy Levels and Oxygen Bands in First-Principles Calculations: Accurate Prediction of Redox Potentials and Charge Transfer in Lithium Transition-Metal Oxides. Phys. Rev. B 2015, 92, 115118. [Google Scholar] [CrossRef]
- Gillen, R.; Robertson, J. Accurate Screened Exchange Band Structures for the Transition Metal Monoxides MnO, FeO, CoO and NiO. J. Phys. Condens. Matter 2013, 25, 165502. [Google Scholar] [CrossRef]
- Kuklin, M.S.; Maschio, L.; Usvyat, D.; Kraus, F.; Karttunen, A.J. Evolutionary Algorithm-Based Crystal Structure Prediction for Copper(I) Fluoride. Chem. Eur. J. 2019, 25, 11528–11537. [Google Scholar] [CrossRef]
- Eklund, K.; Kuklin, M.S.; Kraus, F.; Karttunen, A.J. Evolutionary Algorithm-based Crystal Structure Prediction for Gold(I) Fluoride. ChemPhysChem 2020, 21, 802–808. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. FINDSYM: Program for Identifying the Space-Group Symmetry of a Crystal. J. Appl. Crystallogr. 2005, 38, 237–238. [Google Scholar] [CrossRef]
- Pyykko, P.; Runeberg, N.; Mendizabal, F. Theory of the D10-D10 Closed-Shell Attraction: 1. Dimers Near Equilibrium. Chem. Soc. Rev. 1997, 3, 1451–1457. [Google Scholar] [CrossRef]
- Hermann, H.L.; Boche, G.; Schwerdtfeger, P. Metallophilic Interactions in Closed-Shell Copper (I) Compounds—A Theoretical Study. Chem Eur J 2001, 7, 5333–5342. [Google Scholar] [CrossRef] [PubMed]
- Grady, E.O.; Kaltsoyannis, N. Does Metallophilicity Increase or Decrease down Group 11? Computational Investigations of [Cl–M–PH3]2 (M = Cu, Ag, Au, [111]). Phys Chem Chem Phys 2004, 6, 680–687. [Google Scholar] [CrossRef]
- Angels Carvajal, M.; Alvarez, S.; Novoa, J.J. The Nature of Intermolecular CuI...CuI Interactions: A Combined Theoretical and Structural Database Analysis. Chem Eur J 2004, 10, 2117–2132. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [PubMed]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the Vibration Frequencies of A-Quartz: The Effect of Hamiltonian and Basis Set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; Lopez Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of Crystalline Compounds and Its Implementation in the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Åsbrink, S.; Norrby, L.J. A Refinement of the Crystal Structure of Copper(II) Oxide with a Discussion of Some Exceptional e.s.d.’s. Acta Crystallogr. B 1970, 26, 8–15. [Google Scholar] [CrossRef]
- Forsyth, J.B.; Brown, P.J.; Wanklyn, B.M. Magnetism in Cupric Oxide. J. Phys. C Solid State Phys. 1988, 21, 2917–2929. [Google Scholar] [CrossRef]
- Yang, B.; Tranquada, J.; Shirane, G. Neutron Scattering Studies of the Magnetic Structure of Cupric Oxide. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 38, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.X.; Thurston, T.R.; Tranquada, J.M.; Shirane, G. Magnetic Neutron Scattering Study of Single-Crystal Cupric Oxide. Phys. Rev. B 1989, 39, 4343–4349. [Google Scholar] [CrossRef] [PubMed]
- Rödl, C.; Sottile, F.; Reining, L. Quasiparticle Excitations in the Photoemission Spectrum of CuO from First Principles: A GW Study. Phys. Rev. B Condens. Matter Mater. Phys. 2015, 91, 045102. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
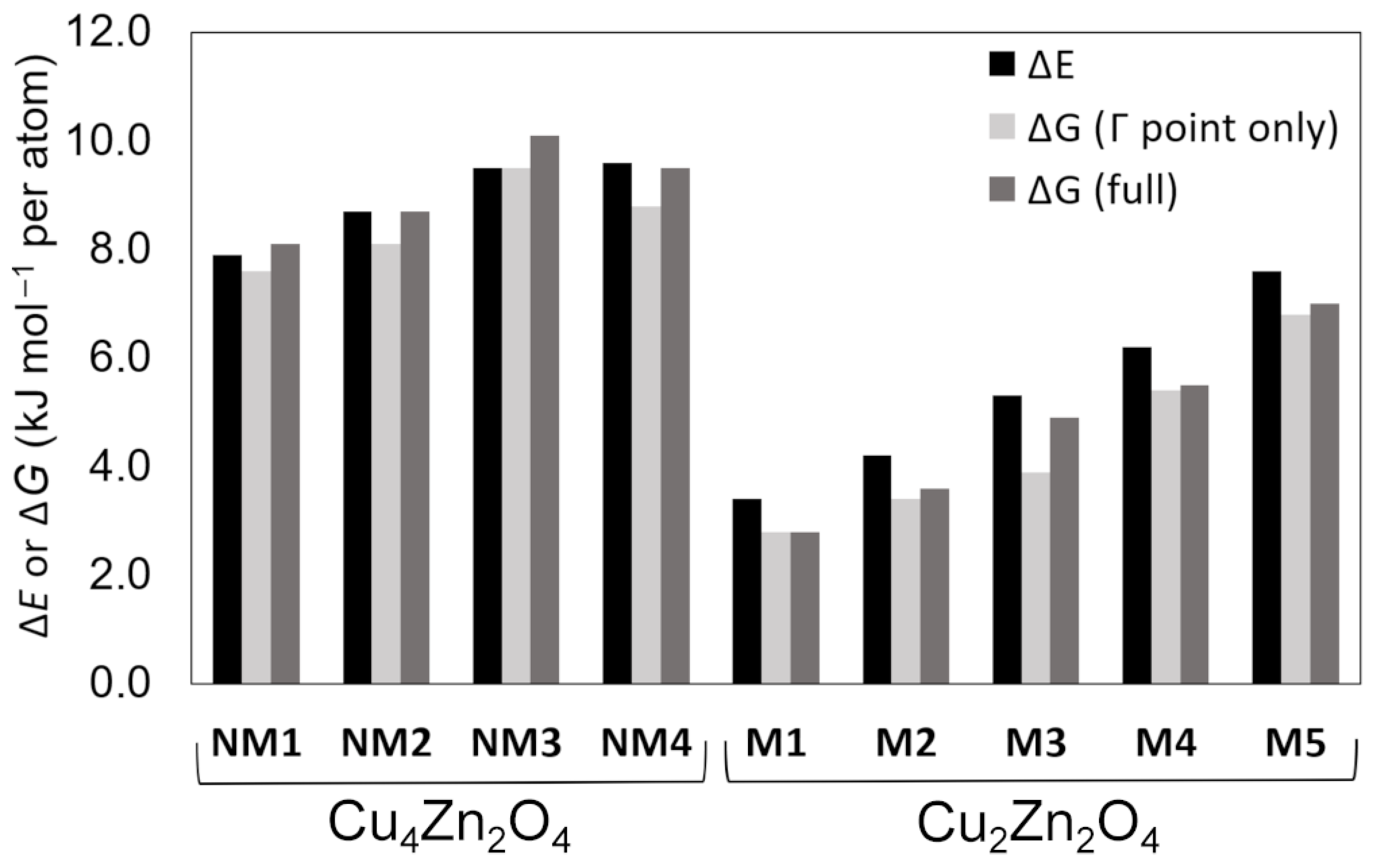
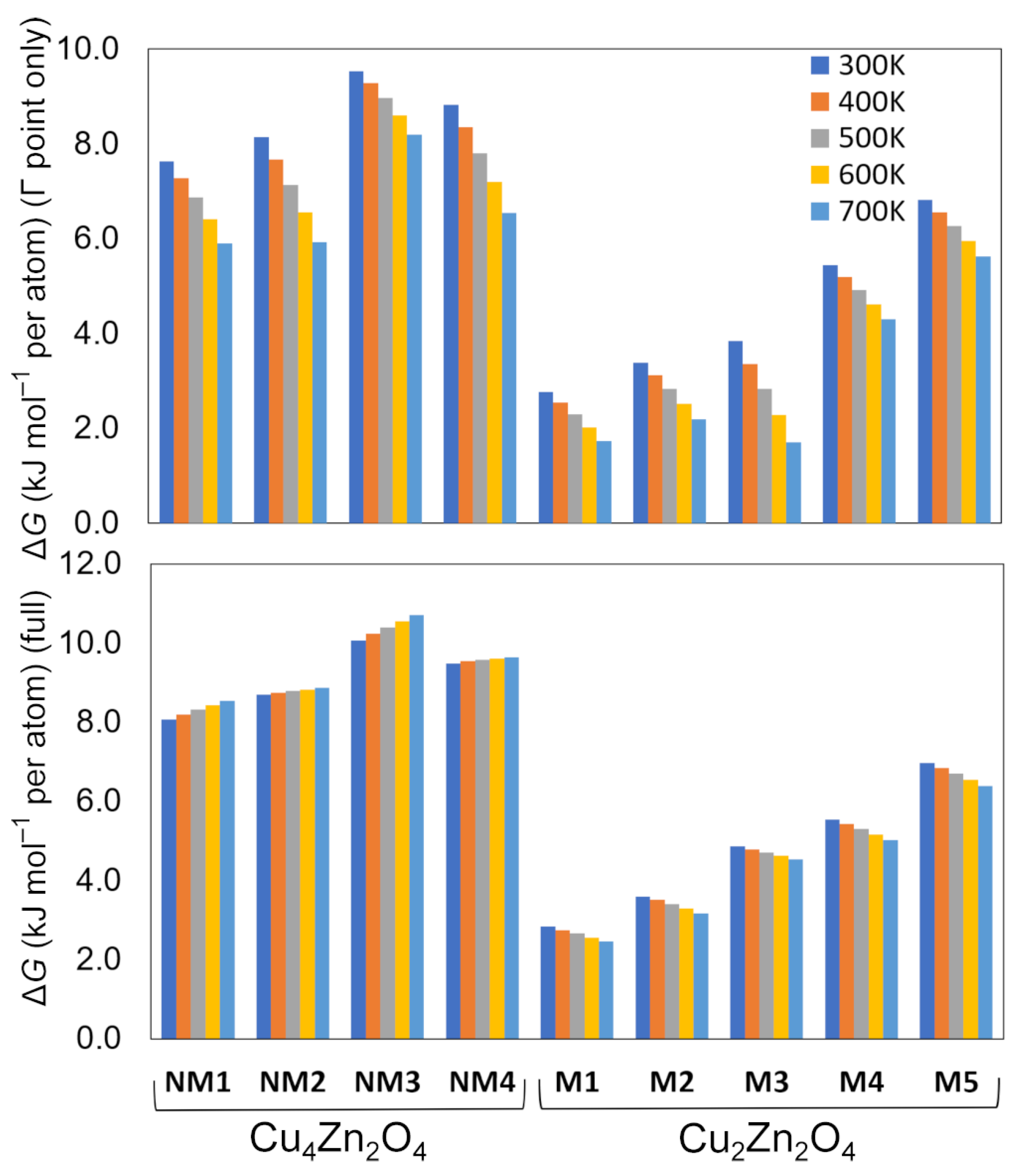
| Crystal Structure | Space Group | ΔE (kJ mol−1 per atom) | ΔGΓ (kJ mol−1 per atom) b | ΔG (kJ mol−1 per atom) c | Band Gap (eV) | CuII Magnetic Moment (μB) | Coordination Number of Cu | Coordination Number of Zn | |
|---|---|---|---|---|---|---|---|---|---|
| Cu4Zn2O4 | NM1 | Pc (7) | 7.9 | 7.6 | 8.1 | 2.4 | – | 2 | 4 |
| NM2 | I212121 (24) | 8.7 | 8.1 | 8.7 | 2.2 | – | 2 | 4 | |
| NM3 | Fdd2(43) | 9.5 | 9.5 | 10.1 | 2.4 | – | 2 | 4 | |
| NM4 | P-1 (2) | 9.6 | 8.8 | 9.5 | 2.2 | – | 2 | 5 | |
| Cu2Zn2O4 | M1 | C2/m (12) | 3.4 | 2.8 | 2.8 | 3.4 | 0.7 | 4 | 6 |
| M2 | C2/m (12) | 4.2 | 3.4 | 3.6 | 3.1 | 0.7 | 6 | 6 | |
| M3 | P-1 (2)/C2/c (15) a | 5.3 | 3.9 | 4.9 | 3.4 | 0.7 | 6 | 6 | |
| M4 | P1 (1) | 6.2 | 5.4 | 5.5 | 3.3 | 0.7 | 6 | 6 | |
| M5 | Pm (6)/Pmn21 (31) a | 7.6 | 6.8 | 7.0 | 3.1 | 0.7 | 5 | 5 | |
| Crystal Structure | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | |
|---|---|---|---|---|---|---|---|
| Cu4Zn2O4 | NM1 | 3.34 | 7.73 | 5.74 | 90 | 125.1 | 90 |
| NM2 | 3.14 | 9.88 | 8.06 | 90 | 90 | 90 | |
| NM3 | 7.94 | 7.64 | 8.08 | 90 | 90 | 90 | |
| NM4 | 3.05 | 6.26 | 6.49 | 73.6 | 79.1 | 82.2 | |
| Cu2Zn2O4 | M1 | 9.91 | 2.87 | 5.46 | 90 | 93.1 | 90 |
| M2 | 5.98 | 8.67 | 2.95 | 90 | 93.6 | 90 | |
| M3 | 2.95 | 2.95 | 10.01 | 98.0 | 96.1 | 117.3 | |
| M4 | 2.95 | 5.13 | 5.28 | 97.6 | 92.7 | 103.7 | |
| M5 | 4.98 | 2.84 | 5.67 | 90 | 90.0 | 90 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuklin, M.S.; Karttunen, A.J. Evolutionary Algorithm-Based Crystal Structure Prediction of CuxZnyOz Ternary Oxides. Molecules 2023, 28, 5986. https://doi.org/10.3390/molecules28165986
Kuklin MS, Karttunen AJ. Evolutionary Algorithm-Based Crystal Structure Prediction of CuxZnyOz Ternary Oxides. Molecules. 2023; 28(16):5986. https://doi.org/10.3390/molecules28165986
Chicago/Turabian StyleKuklin, Mikhail S., and Antti J. Karttunen. 2023. "Evolutionary Algorithm-Based Crystal Structure Prediction of CuxZnyOz Ternary Oxides" Molecules 28, no. 16: 5986. https://doi.org/10.3390/molecules28165986
APA StyleKuklin, M. S., & Karttunen, A. J. (2023). Evolutionary Algorithm-Based Crystal Structure Prediction of CuxZnyOz Ternary Oxides. Molecules, 28(16), 5986. https://doi.org/10.3390/molecules28165986