
Employing Molecular Conformations for Ligand-Based Virtual Screening with Equivariant Graph Neural Network and Deep Multiple Instance Learning


Abstract

1. Introduction
2. Results
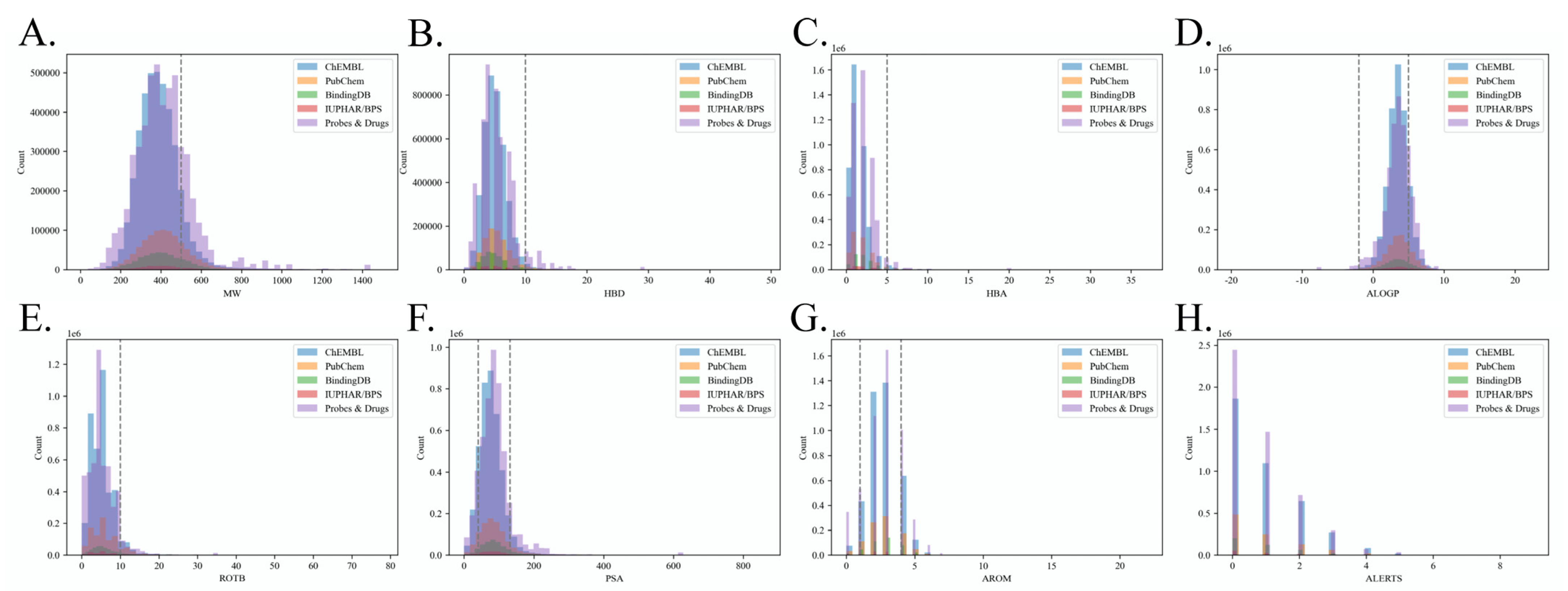
2.1. Benchmark Dataset Quality Analysis
2.2. Model Performance Comparison
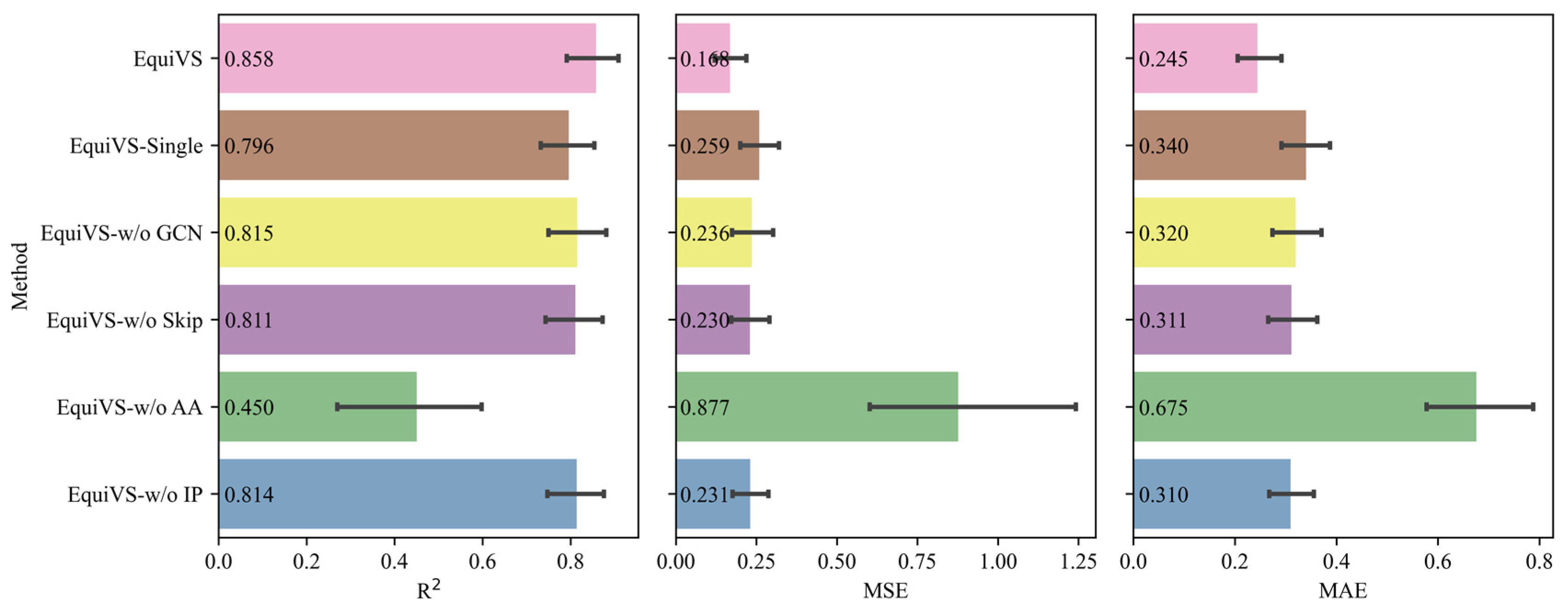
2.3. Ablation Study
- EquiVS-Single: EquiVS architecture which adopts one conformer for each molecule for structural representation learning.
- EquiVS-w/o GCN: EquiVS architecture which replaces the GCN layer with a simple linear layer.
- EquiVS-w/o Skip: EquiVS architecture which deletes the skip connection between different EGNN layers.
- EquiVS-w/o AA: EquiVS architecture which replaces the attention-based conformer representation aggregation process with a simple sum calculation.
- EquiVS-w/o IP: EquiVS architecture which deletes the conformer-level predictor and the corresponding conformer-level prediction loss.
2.4. Case Study: Optimal Molecular Conformer Discovery
3. Materials and Methods
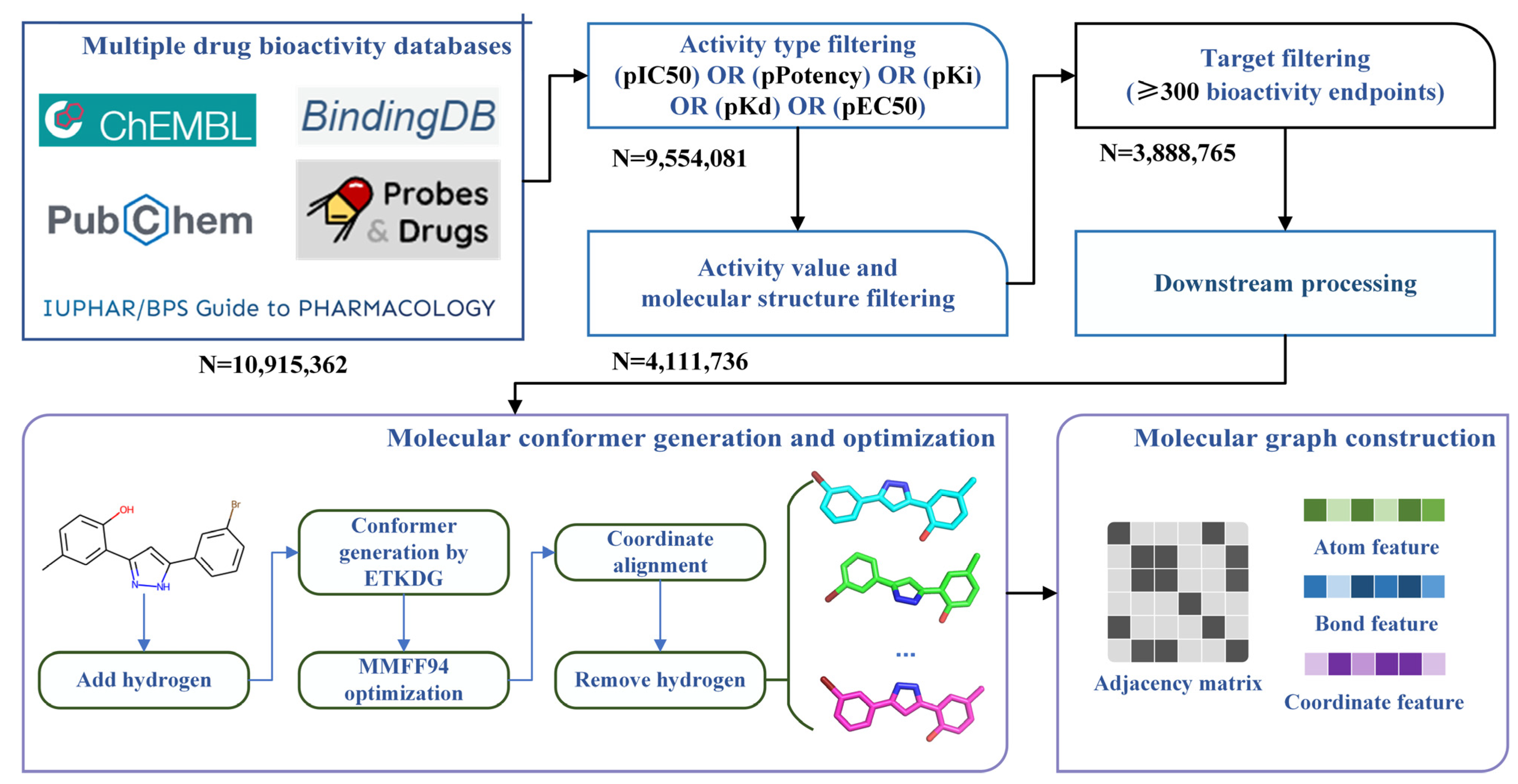
3.1. Bioactivity Data Collecting, Integrating and Filtering
3.2. Molecular Conformer and Graph Generating
3.3. Bioactivity Prediction Model Construction
3.4. Training Optimization
| Algorithm 1: The computing flow of EquiVS |
| Input: A molecule graph , the set of atoms , the set of bonds . Its node feature , edge feature , and coordinate feature . A set of molecular conformation for . |
| Output: Predicted bioactivity for on a specific target. |
| 1: Initialize the trainable parameters in EquiVS. 2: Acquire 2D topological graph representation by Equation (3). 3: for Molecular conformers do 4: for EGNN layer do 5: Acquire 3D structural graph representation by Equation (8). 6: end for 7: Concatenate the conformer representation by Equation (13). 8: end for 9: Acuiqre aggregated graph representation with attention mechanism by Equations (14)–(16). 10: Generate conformer-level prediction by Equation (17). 11: Generate molecule-level prediction by Equation (18). 12: Calculate conformer-lebel loss and molecule-level loss by Equations (19) and (20). 13: Update parameters by optimizing Equation (21). 14: Ouput . |
3.5. Model Interpretation
3.6. Settings
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Bajorath, J. Selected concepts and investigations in compound classification, molecular descriptor analysis, and virtual screening. J. Chem. Inf. Comput. Sci. 2001, 41, 233–245. [Google Scholar] [CrossRef]
- Garcia-Hernandez, C.; Fernández, A.; Serratosa, F. Ligand-Based Virtual Screening Using Graph Edit Distance as Molecular Similarity Measure. J. Chem. Inf. Model. 2019, 59, 1410–1421. [Google Scholar] [CrossRef]
- Sun, H. Pharmacophore-based virtual screening. Curr. Med. Chem. 2008, 15, 1018–1024. [Google Scholar] [CrossRef]
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G.M.; Liedl, K.R.; Wolber, G. How to optimize shape-based virtual screening: Choosing the right query and including chemical information. J. Chem. Inf. Model. 2009, 49, 678–692. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Kong, W.; Wang, W.; An, J. Prediction of 5-hydroxytryptamine transporter inhibitors based on machine learning. Comput. Biol. Chem. 2020, 87, 107303. [Google Scholar] [CrossRef]
- Kong, W.; Tu, X.; Huang, W.; Yang, Y.; Xie, Z.; Huang, Z. Prediction and optimization of NaV1. 7 sodium channel inhibitors based on machine learning and simulated annealing. J. Chem. Inf. Model. 2020, 60, 2739–2753. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Huang, W.; Peng, C.; Zhang, B.; Duan, G.; Ma, W.; Huang, Z. Multiple machine learning methods aided virtual screening of NaV1. 5 inhibitors. J. Cell. Mol. Med. 2023, 27, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: Hoboken, NJ, USA, 1990. [Google Scholar]
- Wang, M.; Wang, Z.; Sun, H.; Wang, J.; Shen, C.; Weng, G.; Chai, X.; Li, H.; Cao, D.; Hou, T. Deep learning approaches for de novo drug design: An overview. Curr. Opin. Struct. Biol. 2022, 72, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, J.; Wang, Y.; Zhou, J.; Zhang, L.; Liu, Z. DeepScaffold: A comprehensive tool for scaffold-based de novo drug discovery using deep learning. J. Chem. Inf. Model. 2019, 60, 77–91. [Google Scholar] [CrossRef]
- Gu, Y.; Zhang, B.; Zheng, S.; Yang, F.; Li, J. Predicting Drug ADMET Properties Based on Graph Attention Network. Data Anal. Knowl. Discov. 2021, 5, 76–85. [Google Scholar] [CrossRef]
- Yang, L.; Jin, C.; Yang, G.; Bing, Z.; Huang, L.; Niu, Y.; Yang, L. Transformer-based deep learning method for optimizing ADMET properties of lead compounds. Phys. Chem. Chem. Phys. 2023, 25, 2377–2385. [Google Scholar] [CrossRef]
- Gu, Y.; Zheng, S.; Yin, Q.; Jiang, R.; Li, J. REDDA: Integrating multiple biological relations to heterogeneous graph neural network for drug-disease association prediction. Comput. Biol. Med. 2022, 150, 106127. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zheng, S.; Zhang, B.; Kang, H.; Li, J. MilGNet: A Multi-instance Learning-based Heterogeneous Graph Network for Drug repositioning. In Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Las Vegas, NV, USA, 6–8 December 2022; pp. 430–437. [Google Scholar]
- Kimber, T.B.; Chen, Y.; Volkamer, A. Deep Learning in Virtual Screening: Recent Applications and Developments. Int. J. Mol. Sci. 2021, 22, 4435. [Google Scholar] [CrossRef] [PubMed]
- Yaowen, G.; Si, Z.; Fengchun, Y.; Jiao, L. GNN-MTB: An Anti-Mycobacterium Drug Virtual Screening Model Based on Graph Neural Network. Data Anal. Knowl. Discov. 2023, 6, 93–102. [Google Scholar]
- Liu, Z.; Du, J.; Fang, J.; Yin, Y.; Xu, G.; Xie, L. DeepScreening: A deep learning-based screening web server for accelerating drug discovery. Database 2019, 2019, baz104. [Google Scholar] [CrossRef]
- Stojanovic, L.; Popovic, M.; Tijanic, N.; Rakocevic, G.; Kalinic, M. Improved scaffold hopping in ligand-based virtual screening using neural representation learning. J. Chem. Inf. Model. 2020, 60, 4629–4639. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Hu, H.; Yang, Z.; Xu, H.; Wu, J. Realvs: Toward enhancing the precision of top hits in ligand-based virtual screening of drug leads from large compound databases. J. Chem. Inf. Model. 2021, 61, 4924–4939. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Q.; Wu, W.; Pang, T.; Hu, H.; Chan, W.K.B.; Ke, X.; Zhang, Y. WDL-RF: Predicting bioactivities of ligand molecules acting with G protein-coupled receptors by combining weighted deep learning and random forest. Bioinformatics 2018, 34, 2271–2282. [Google Scholar] [CrossRef]
- Wu, J.; Liu, B.; Chan, W.K.B.; Wu, W.; Pang, T.; Hu, H.; Yan, S.; Ke, X.; Zhang, Y. Precise modelling and interpretation of bioactivities of ligands targeting G protein-coupled receptors. Bioinformatics 2019, 35, i324–i332. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Altalib, M.K.; Salim, N. Hybrid-Enhanced Siamese Similarity Models in Ligand-Based Virtual Screen. Biomolecules 2022, 12, 1719. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Méndez-Lucio, O.; Ahmad, M.; del Rio-Chanona, E.A.; Wegner, J.K. A geometric deep learning approach to predict binding conformations of bioactive molecules. Nat. Mach. Intell. 2021, 3, 1033–1039. [Google Scholar] [CrossRef]
- Sauer, W.H.; Schwarz, M.K. Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. [Google Scholar] [CrossRef]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef]
- Shang, J.; Dai, X.; Li, Y.; Pistolozzi, M.; Wang, L. HybridSim-VS: A web server for large-scale ligand-based virtual screening using hybrid similarity recognition techniques. Bioinformatics 2017, 33, 3480–3481. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminformatics 2013, 5, 26. [Google Scholar] [CrossRef]
- Zankov, D.V.; Matveieva, M.; Nikonenko, A.V.; Nugmanov, R.I.; Baskin, I.I.; Varnek, A.; Polishchuk, P.; Madzhidov, T.I. QSAR modeling based on conformation ensembles using a multi-instance learning approach. J. Chem. Inf. Model. 2021, 61, 4913–4923. [Google Scholar] [CrossRef] [PubMed]
- Isigkeit, L.; Chaikuad, A.; Merk, D. A Consensus Compound/Bioactivity Dataset for Data-Driven Drug Design and Chemogenomics. Molecules 2022, 27, 2513. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- Yusof, I.; Segall, M.D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today 2013, 18, 659–666. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Škuta, C.; Southan, C.; Bartůněk, P. Will the chemical probes please stand up? RSC Med. Chem. 2021, 12, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.D.; Armstrong, J.F.; Faccenda, E.; Southan, C.; Alexander, S.P.H.; Davenport, A.P.; Pawson, A.J.; Spedding, M.; Davies, J.A. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: Curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res. 2022, 50, D1282–D1294. [Google Scholar] [CrossRef]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef]
- Morgan, H.L. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Swain, M. MolVS: Molecule Validation and Standardization. 2018. Available online: https://molvs.readthedocs.io/en/latest/ (accessed on 3 June 2023).
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8, 31. [Google Scholar]
- Riniker, S.; Landrum, G.A. Better informed distance geometry: Using what we know to improve conformation generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Shi, X.; Xing, F.; Xie, Y.; Zhang, Z.; Cui, L.; Yang, L. Loss-based attention for deep multiple instance learning. Proc. AAAI Conf. Artif. Intell. 2020, 34, 5742–5749. [Google Scholar] [CrossRef]
- Polton, D. Installation and operational experiences with MACCS (Molecular Access System). Online Rev. 1982, 6, 235–242. [Google Scholar] [CrossRef]
- Drucker, H.; Cortes, C. Boosting decision trees. Adv. Neural Inf. Process. Syst. 1995, 8. [Google Scholar]
- Chen, T.; He, T.; Benesty, M.; Khotilovich, V.; Tang, Y.; Cho, H.; Chen, K.; Mitchell, R.; Cano, I.; Zhou, T. Xgboost: Extreme Gradient Boosting. R Package Version 0.4-2. 2023; Volume 1, 1–4. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2020, 63, 8749–8760. [Google Scholar] [CrossRef]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, H.; Liu, W.; Lasenby, J.; Guo, H.; Tang, J. Pre-training molecular graph representation with 3d geometry. arXiv 2021, arXiv:2110.07728. [Google Scholar]
- Stärk, H.; Beaini, D.; Corso, G.; Tossou, P.; Dallago, C.; Günnemann, S.; Liò, P. 3D infomax improves gnns for molecular property prediction. In Proceedings of the 39th International Conference on Machine Learning, Baltimore, MD, USA, 17–23 July 2022; pp. 20479–20502. [Google Scholar]
- Jiao, R.; Han, J.; Huang, W.; Rong, Y.; Liu, Y. 3D equivariant molecular graph pretraining. arXiv 2022, arXiv:2207.08824. [Google Scholar]
- Gu, Y.; Zheng, S.; Li, J. CurrMG: A Curriculum Learning Approach for Graph Based Molecular Property Prediction. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 2686–2693. [Google Scholar]
- Gu, Y.; Zheng, S.; Xu, Z.; Yin, Q.; Li, L.; Li, J. An efficient curriculum learning-based strategy for molecular graph learning. Brief. Bioinform. 2022, 23, bbac099. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.-J.; Tkatchenko, A.; Müller, K.-R. Schnet–a deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef]
- Gasteiger, J.; Becker, F.; Günnemann, S. Gemnet: Universal directional graph neural networks for molecules. Adv. Neural Inf. Process. Syst. 2021, 34, 6790–6802. [Google Scholar]
- Schütt, K.; Unke, O.; Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra. In Proceedings of the International Conference on Machine Learning, Virtual, 18–24 July 2021; pp. 9377–9388. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Method | Feature | Compared LBVS Methods |
|---|---|---|---|
| WDL-RF [22] | Convolutional neural network and random forest | Molecular atomic properties | Random forest-based LBVS methods with NGFP, MACCS, ECFP2, ECFP4, ECFP8, ECFP10, FCFP2, FCFP4, FCFP8, and FCFP10 fingerprints. |
| DeepScreening [19] | Deep neural network | Multiple molecular fingerprints extracted from 2D molecular structures | - |
| SED [23] | Deep neural network | Molecular ECFP fingerprints extracted from 2D molecular structures | Different length of ECFP fingerprints and multiple machine learning-based LBVS methods (GBDT, SVR, and RF) |
| Winter et al. [24] | Recurrent neural network and convolutional neural network | Tokenized molecular representations from 1D molecular sequences | Similarity-based LBVS methods with lAval, TT, lECFP4, lECFP6, ECFP4, RDK5, Avalon, ECFP6, FCFP4, MACCS, FCFC4, and ECFC0 fingerprints. |
| GATNN [20] | Graph attention network | 2D molecular graphs | Similarity-based LBVS methods with ECFP4, TT, MHFP6, and ECFC0 fingerprints. |
| RealVS [21] | Graph attention network | 2D molecular graphs | Multiple graph neural network-based LBVS methods (GCN, GAT, GIN, NeuralFP, Weave, MPNN, WDL-RF, and AttentiveFP). |
| Altalib et. al. [25] | Hybrid Siamese convolutional neural network | Molecular ECFC fingerprints extracted from 2D molecular structures | Similarity-, Bayesian inference-, deep belief network-, and single Siamese convolutional neural network-based LBVS with ECFC4 and ECFP4 fingerprints. |
| Bioactivity | Statistics | ChEMBL | PubChem | BindingDB | IUPHAR/BPS | Probes&Drugs |
|---|---|---|---|---|---|---|
| Value | Avg. | 5.33 | 6.29 | 6.17 | 7.31 | 5.52 |
| Std. | 1.25 | 1.52 | 1.58 | 1.48 | 1.48 | |
| Min. | 0.10 | 0.10 | 0.10 | 0.80 | 0.10 | |
| Max. | 15.90 | 13.00 | 12.20 | 18.00 | 21.80 | |
| Category | Num. Active | 939,036 | 540,384 | 228,907 | 78,483 | 1,656,557 |
| PCT. Active | 23.56% | 56.72% | 54.9% | 81.42% | 32.93% | |
| Num. Inactive | 3,047,145 | 412,288 | 188,067 | 17,915 | 3,373,950 | |
| PCT. Inactive | 76.44% | 43.28% | 45.1% | 18.58% | 67.07% |
| Type | Method | R2 | MSE | MAE |
|---|---|---|---|---|
| Machine Learning | LR_ECFP | −4.543 ± 5.443 | 24.844 ± 24.904 | 4.960 ± 4.956 |
| LR_MACCS | −3.438 ± 5.302 | 19.853 ± 24.339 | 4.063 ± 4.798 | |
| GBDT_ECFP | 0.831 ± 0.189 | 0.240 ± 0.256 | 0.325 ± 0.176 | |
| GBDT_MACCS | 0.810 ± 0.202 | 0.273 ± 0.286 | 0.328 ± 0.190 | |
| XGB_ECFP | 0.830 ± 0.188 | 0.243 ± 0.254 | 0.329 ± 0.175 | |
| XGB_MACCS | 0.808 ± 0.201 | 0.276 ± 0.285 | 0.331 ± 0.189 | |
| Deep Learning | GCN | 0.768 ± 0.960 | 0.509 ± 3.868 | 0.280 ± 0.412 |
| GAT | 0.760 ± 0.838 | 0.413 ± 2.962 | 0.259 ± 0.282 | |
| Weave | 0.705 ± 1.056 | 0.601 ± 4.051 | 0.286 ± 0.297 | |
| AttentiveFP | 0.746 ± 0.390 | 0.359 ± 0.587 | 0.330 ± 0.300 | |
| EquiVS | 0.833 ± 0.243 | 0.208 ± 0.282 | 0.257 ± 0.189 |
| Database | Description | Version | Num. Molecule |
|---|---|---|---|
| ChEMBL | Contains the bioactivity data of more than 2.1 million experimentally determined drug-like molecules | 28 | 1,131,947 |
| PubChem | Contains the bioactivity data and physiochemical properties of more than 1.1 million molecules | 11.01.21 | 444,152 |
| BindingDB | Contains binding affinity data of approximately 26,000 drug-like molecules regarding specific biological targets | 25.02.21 | 26,856 |
| Probes&Drugs | Contains manually collected biological target and bioactivity data of pharmacologically active compounds | 2021.1 | 34,211 |
| IUPHAR/BPS | Contains bioactivity data, target, and signaling pathway information of approximately 29,000 compounds derived from 30 public and commercial libraries | 02b_2021 | 7371 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Li, J.; Kang, H.; Zhang, B.; Zheng, S. Employing Molecular Conformations for Ligand-Based Virtual Screening with Equivariant Graph Neural Network and Deep Multiple Instance Learning. Molecules 2023, 28, 5982. https://doi.org/10.3390/molecules28165982
Gu Y, Li J, Kang H, Zhang B, Zheng S. Employing Molecular Conformations for Ligand-Based Virtual Screening with Equivariant Graph Neural Network and Deep Multiple Instance Learning. Molecules. 2023; 28(16):5982. https://doi.org/10.3390/molecules28165982
Chicago/Turabian StyleGu, Yaowen, Jiao Li, Hongyu Kang, Bowen Zhang, and Si Zheng. 2023. "Employing Molecular Conformations for Ligand-Based Virtual Screening with Equivariant Graph Neural Network and Deep Multiple Instance Learning" Molecules 28, no. 16: 5982. https://doi.org/10.3390/molecules28165982
APA StyleGu, Y., Li, J., Kang, H., Zhang, B., & Zheng, S. (2023). Employing Molecular Conformations for Ligand-Based Virtual Screening with Equivariant Graph Neural Network and Deep Multiple Instance Learning. Molecules, 28(16), 5982. https://doi.org/10.3390/molecules28165982