
Computer-Aided Designing Peptide Inhibitors of Human Hematopoietic Prostaglandin D2 Synthase Combined Molecular Docking and Molecular Dynamics Simulation

Abstract
1. Introduction
2. Results and Discussion
2.1. Generating Peptide Conformation
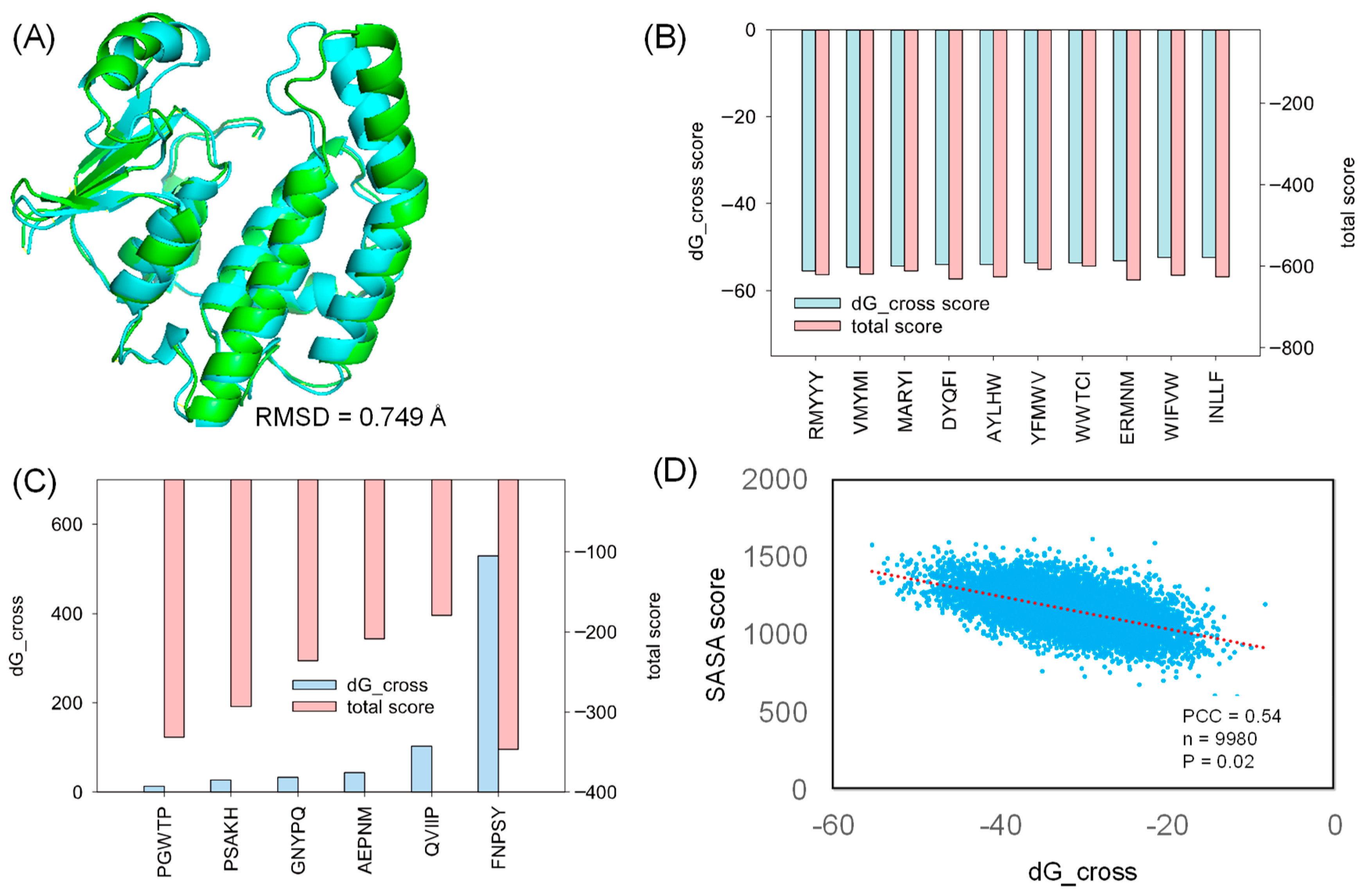
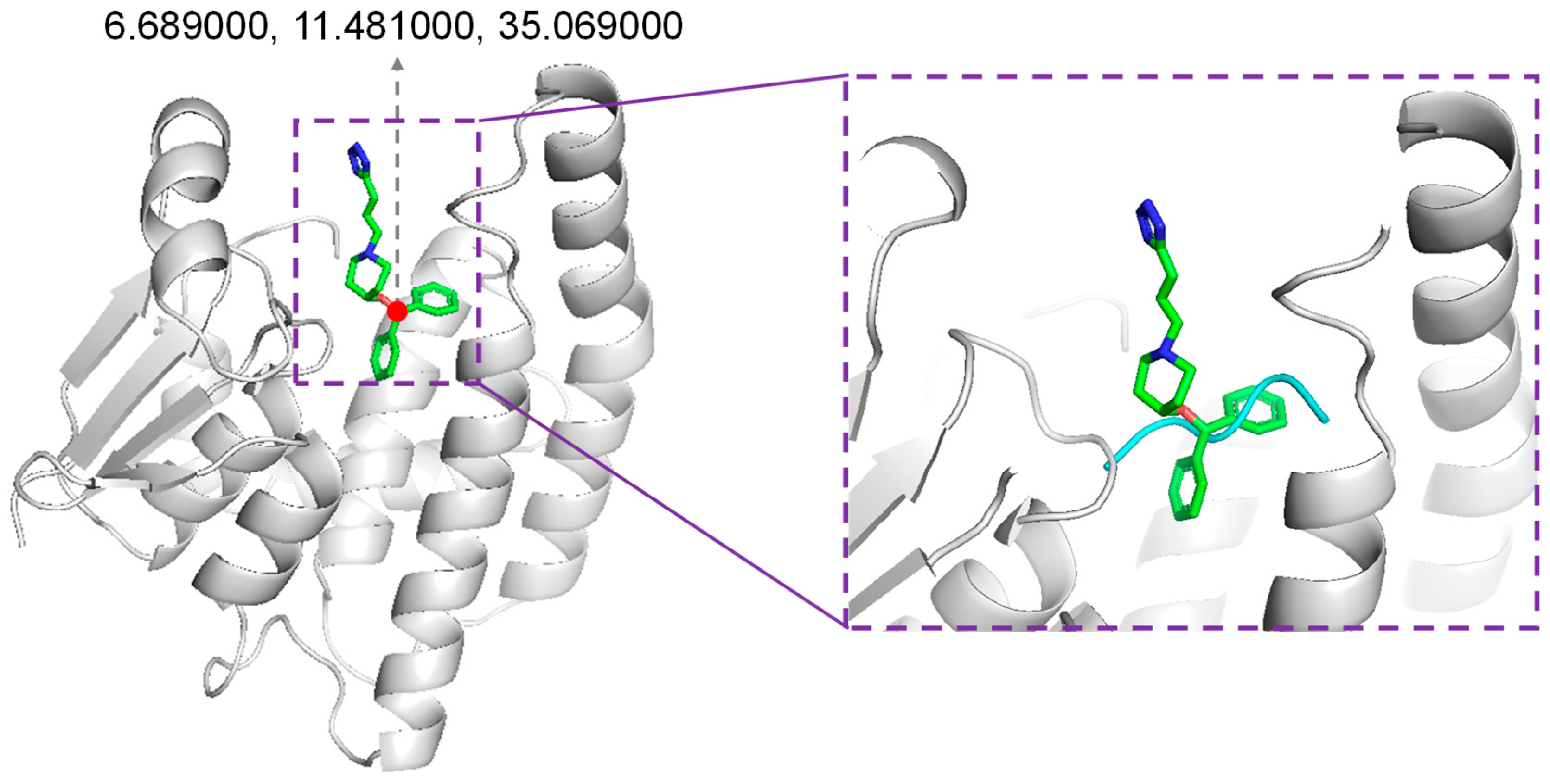
2.2. Molecular Docking
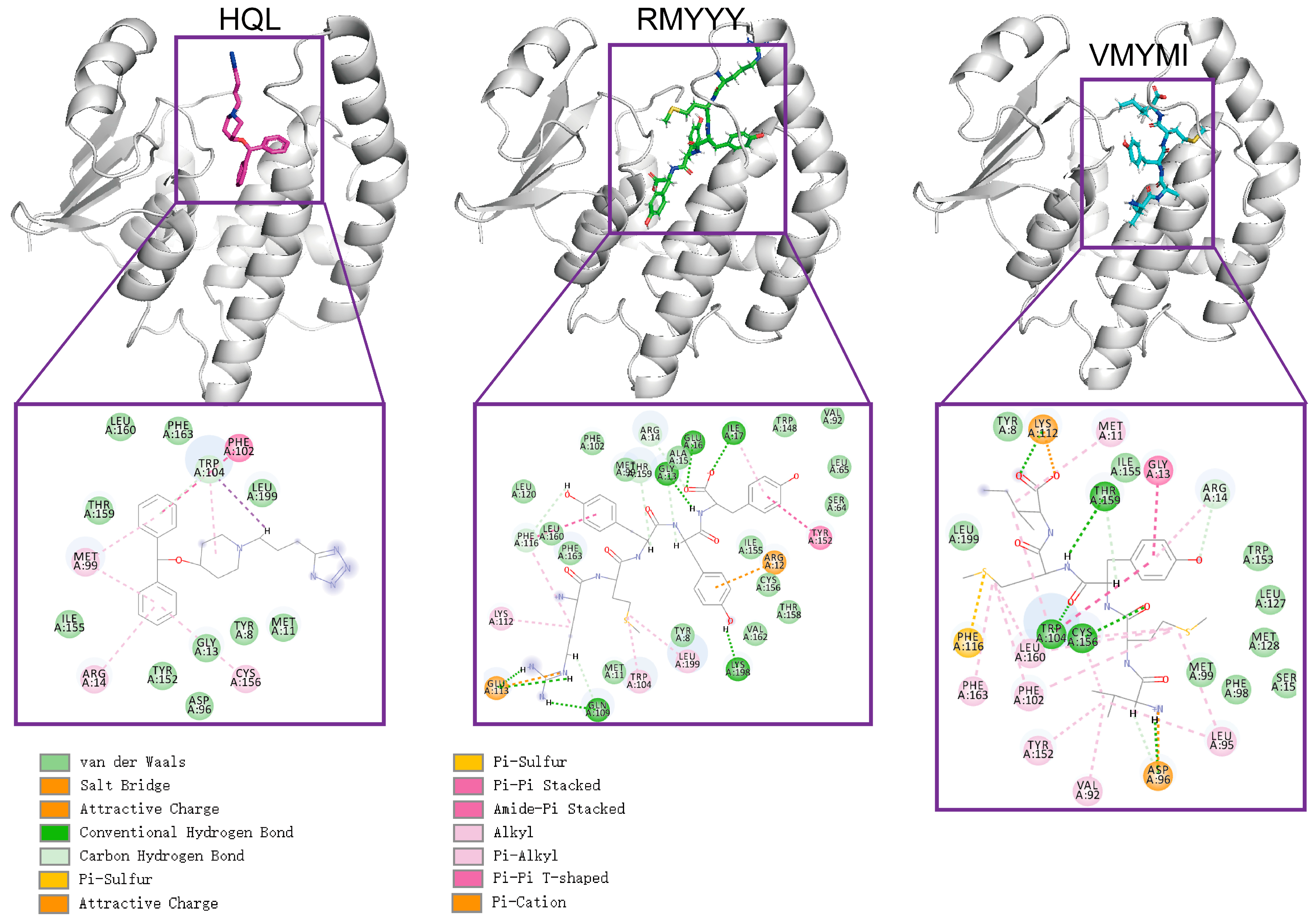
2.3. Comparative Study
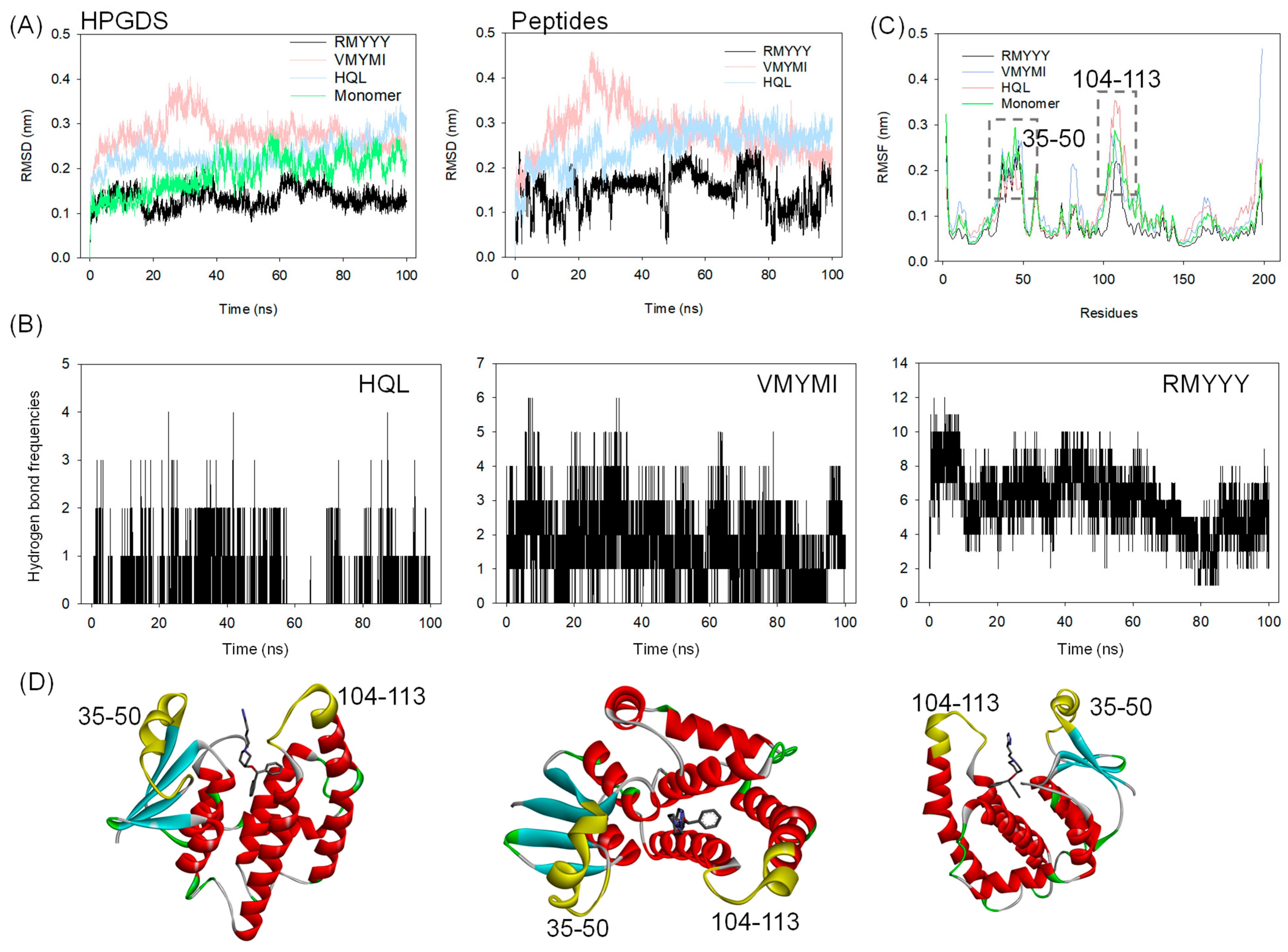
2.4. Molecular Dynamics Simulation
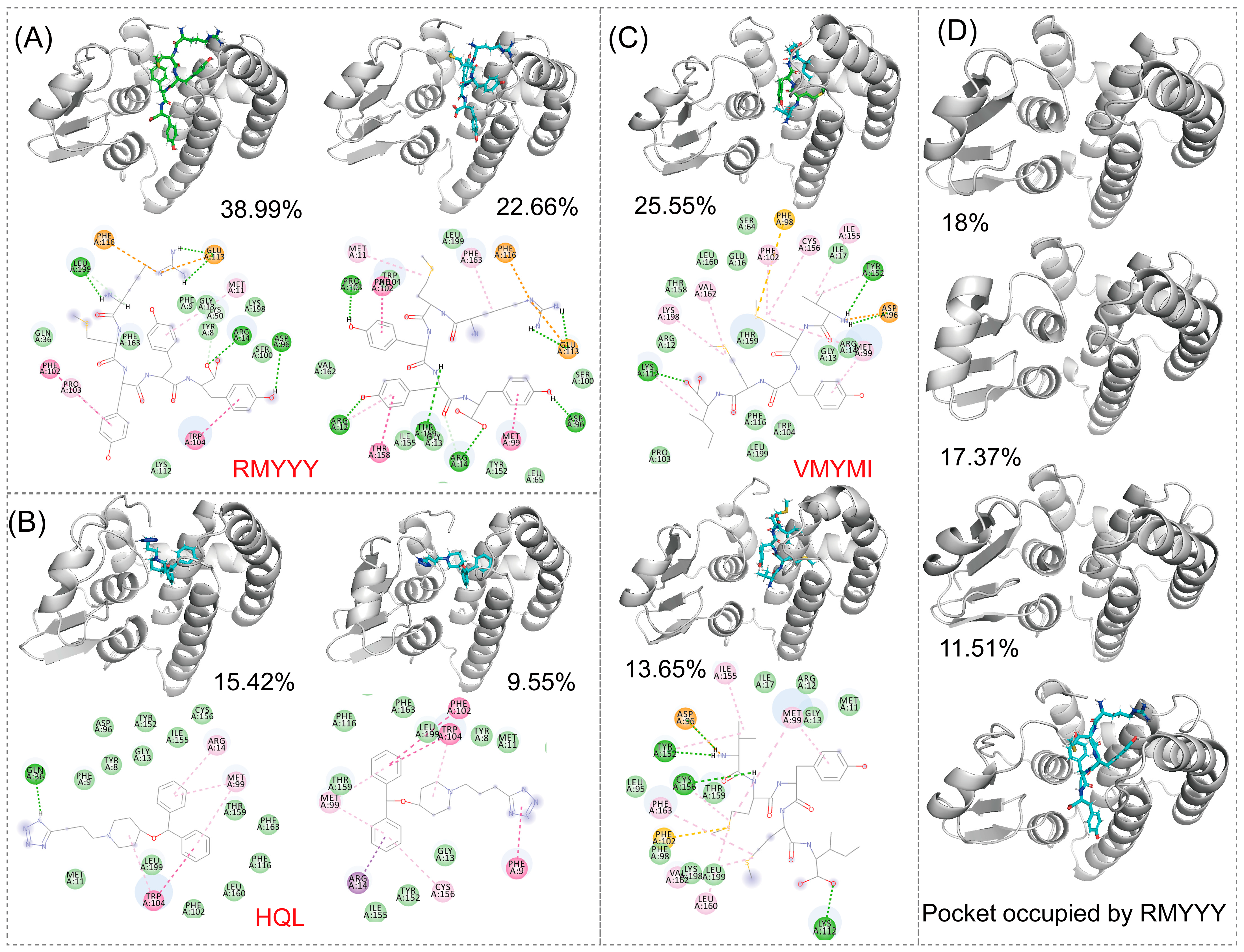
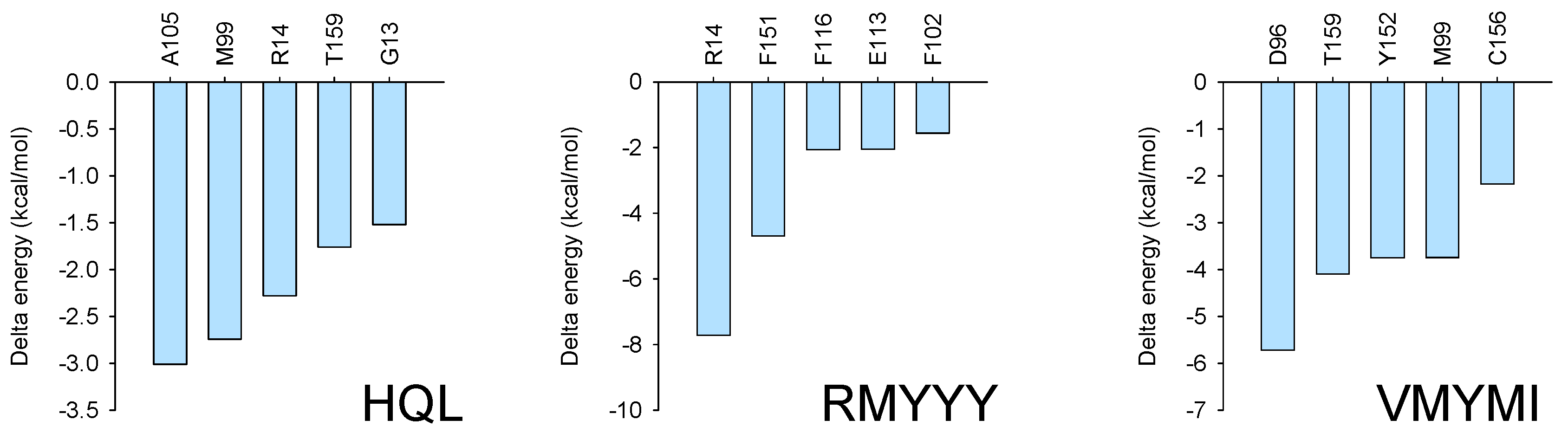
2.5. Identification of Key Binding Residues
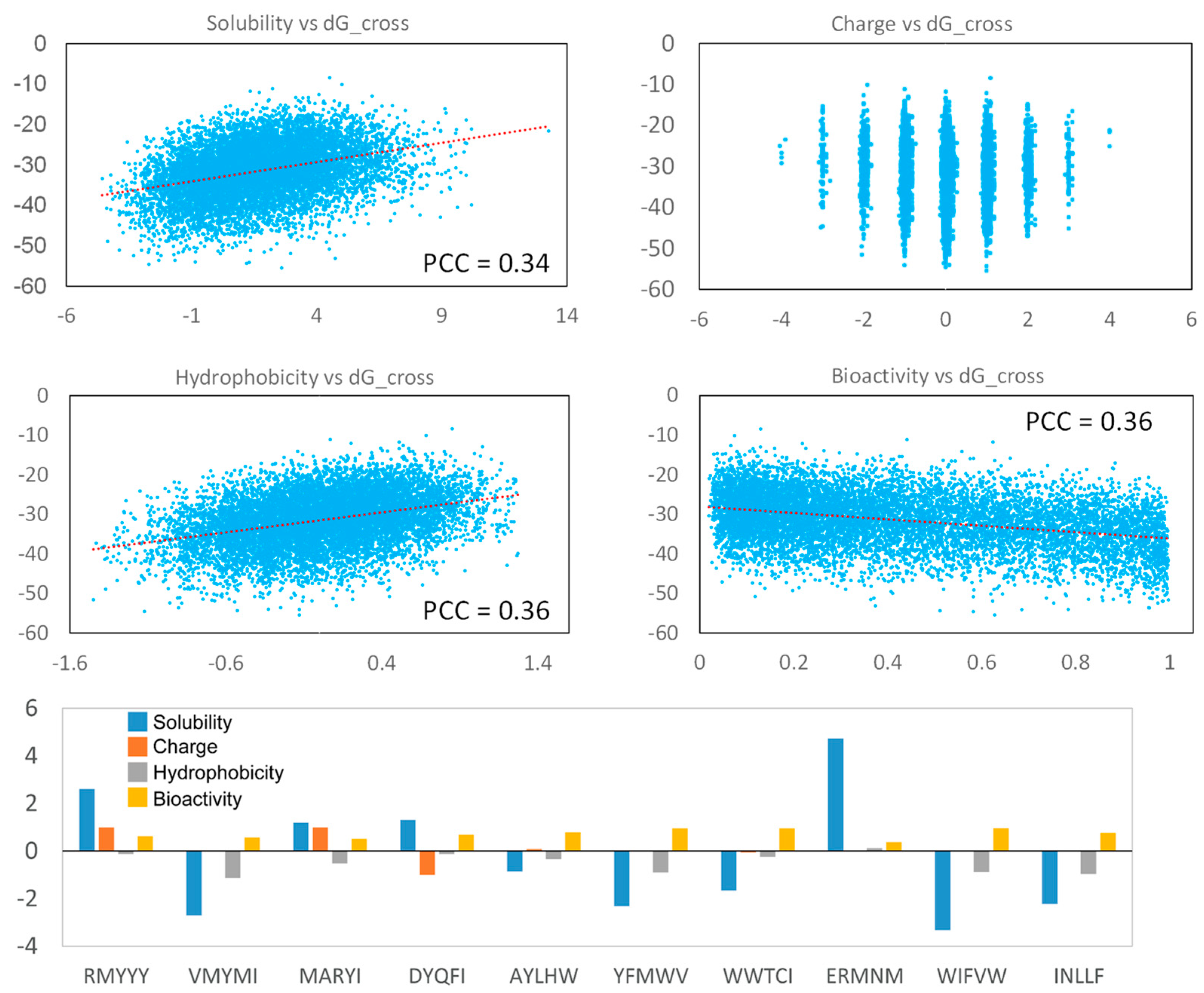
2.6. Analyzing Peptide Properties
3. Materials and Methods
3.1. Docking Receptor Preparation
3.2. Building a Random Peptide Library
3.3. Integrating Rosetta Script for Protein-Peptide Docking
3.4. Molecular Dynamics Simulation
3.5. MMPBSA Analysis
3.6. Peptide Properties Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Suzuki, K.; Suzuki, S.; Ishii, Y.; Okamura, M.; Matsubara, T.; Fujita, H.; Nozawa, N.; Kobayashi, S.; Hirata, K. Plasma prostaglandin D2 synthase levels in sleep and neurological diseases. J. Neurol. Sci. 2020, 411, 116692. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yu, Y. Prostaglandin D2 signaling and cardiovascular homeostasis. J. Mol. Cell. Cardiol. 2022, 167, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.R.; Bak, S.-S.; Kim, M.K.; Joo, H.W.; Mali, N.M.; Shin, M.J.; Kim, M.K.; Kim, J.C.; Oh, J.W.; Sung, Y.K. Expression Level of Prostaglandin D2 Receptor 2 Regulates Hair Regression. J. Invest. Dermatol. 2019, 139, 1824–1828.e2. [Google Scholar] [CrossRef] [PubMed]
- Napora, P.; Kobrzycka, A.; Pierzchała-Koziec, K.; Wieczorek, M. Effect of selective cyclooxygenase inhibitors on animal behaviour and monoaminergic systems of the rat brain. Behav. Brain Res. 2023, 438, 114143. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.K.; Mori, S. Density functional studies on isomerization of prostaglandin H2 to prostacyclin catalyzed by cytochrome P450. Chemistry 2009, 15, 4464–4473. [Google Scholar] [CrossRef]
- Rajakariar, R.; Hilliard, M.; Lawrence, T.; Trivedi, S.; Colville-Nash, P.; Bellingan, G.; Fitzgerald, D.; Yaqoob, M.M.; Gilroy, D.W. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyΔ12–14 PGJ2. Proc. Natl. Acad. Sci. USA 2007, 104, 20979–20984. [Google Scholar] [CrossRef]
- Meleza, C.; Thomasson, B.; Ramachandran, C.; O’Neill, J.W.; Michelsen, K.; Lo, M.-C. Development of a scintillation proximity binding assay for high-throughput screening of hematopoietic prostaglandin D2 synthase. Anal. Biochem. 2016, 511, 17–23. [Google Scholar] [CrossRef]
- Mary, R.; Chalmin, F.; Accogli, T.; Bruchard, M.; Hibos, C.; Melin, J.; Truntzer, C.; Limagne, E.; Derangère, V.; Thibaudin, M.; et al. Hematopoietic Prostaglandin D2 Synthase Controls Tfh/Th2 Communication and Limits Tfh Antitumor Effects. Cancer. Immunol. Res. 2022, 10, 900–916. [Google Scholar] [CrossRef]
- Joo, M.; Sadikot, R.T. PGD Synthase and PGD2 in Immune Resposne. Mediat. Inflamm. 2012, 2012, 503128. [Google Scholar] [CrossRef]
- Urade, Y.; Hayaishi, O. Prostaglandin D2 and sleep/wake regulation. Sleep Med. Rev. 2011, 15, 411–418. [Google Scholar] [CrossRef]
- Cadilla, R.; Deaton, D.N.; Do, Y.; Elkins, P.A.; Ennulat, D.; Guss, J.H.; Holt, J.; Jeune, M.R.; King, A.G.; Klapwijk, J.C.; et al. The exploration of aza-quinolines as hematopoietic prostaglandin D synthase (H-PGDS) inhibitors with low brain exposure. Bioorg. Med. Chem. 2020, 28, 115791. [Google Scholar] [CrossRef]
- Trivedi, S.G.; Newson, J.; Rajakariar, R.; Jacques, T.S.; Hannon, R.; Kanaoka, Y.; Eguchi, N.; Colville-Nash, P.; Gilroy, D.W. Essential role for hematopoietic prostaglandin D2 synthase in the control of delayed type hypersensitivity. PNAS 2006, 103, 5179–5184. [Google Scholar] [CrossRef]
- Olson, K.L.; Holt, M.C.; Ciske, F.L.; Kramer, J.B.; Heiple, P.E.; Collins, M.L.; Johnson, C.M.; Ho, C.S.; Morano, M.I.; Barrett, S.D. Novel amide and imidazole compounds as potent hematopoietic prostaglandin D2 synthase inhibitors. Bioorganic Med. Chem. Lett. 2021, 34, 127759. [Google Scholar] [CrossRef]
- Beura, S.; Chetti, P. Identification of potential inhibitors for Hematopoietic Prostaglandin D2 synthase: Computational modeling and molecular dynamics simulations. J. Mol. Struct. 2022, 1259, 132704. [Google Scholar] [CrossRef]
- Garza, L.A.; Liu, Y.; Yang, Z.; Alagesan, B.; Lawson, J.A.; Norberg, S.M.; Loy, D.E.; Zhao, T.; Blatt, H.B.; Stanton, D.C.; et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Sci. Transl. Med. 2012, 4, 126ra134. [Google Scholar] [CrossRef]
- Butterfield, J.H.; Singh, R.J. Divergent PGD2 and leukotriene C4 metabolite excretion following aspirin therapy: Ten patients with systemic mastocytosis. Prostaglandins Other Lipid Mediat. 2021, 155, 106563. [Google Scholar] [CrossRef]
- Khanna, S.; Chichester, K.; Devine, K.; Gao, P.; Saini, S.; Oliver, E. Increased local production of PGD2 in skin lesions of patients with chronic spontaneous urticaria. J. Allergy Clin. 2022, 149, AB221. [Google Scholar] [CrossRef]
- Rossi, S.P.; Windschüttl, S.; Matzkin, M.E.; Rey-Ares, V.; Terradas, C.; Ponzio, R.; Puigdomenech, E.; Levalle, O.; Calandra, R.S.; Mayerhofer, A.; et al. Reactive oxygen species (ROS) production triggered by prostaglandin D2 (PGD2) regulates lactate dehydrogenase (LDH) expression/activity in TM4 Sertoli cells. Mol. Cell. Endocrinol. 2016, 434, 154–165. [Google Scholar] [CrossRef]
- Zhao, G.; Yu, R.; Deng, J.; Zhao, Q.; Li, Y.; Joo, M.; van Breemen, R.B.; Christman, J.W.; Xiao, L. Pivotal role of reactive oxygen species in differential regulation of lipopolysaccharide-induced prostaglandins production in macrophages. Mol. Pharmacol. 2013, 83, 167–178. [Google Scholar] [CrossRef]
- Nabe, T.; Kuriyama, Y.; Mizutani, N.; Shibayama, S.; Hiromoto, A.; Fujii, M.; Tanaka, K.; Kohno, S. Inhibition of hematopoietic prostaglandin D synthase improves allergic nasal blockage in guinea pigs. Prostaglandins Other Lipid Mediat. 2011, 95, 27–34. [Google Scholar] [CrossRef]
- Takeshita, E.; Komaki, H.; Shimizu-Motohashi, Y.; Ishiyama, A.; Sasaki, M.; Takeda, S. A phase I study of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2018, 5, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, P.; Zhao, B.; Liu, S. AI-assisted food enzymes design and engineering: A critical review. Syst. Microbiol. Biomanufacturing. 2023, 3, 75–87. [Google Scholar] [CrossRef]
- Wang, X.; Xu, K.; Tan, Y.; Liu, S.; Zhou, J. Possibilities of Using De Novo Design for Generating Diverse Functional Food Enzymes. Int. J. Mol. Sci. 2023, 24, 3827. [Google Scholar] [CrossRef] [PubMed]
- Moussa, N.; Hassan, A.; Gharaghani, S. Pharmacophore model, docking, QSAR, and molecular dynamics simulation studies of substituted cyclic imides and herbal medicines as COX-2 inhibitors. Heliyon 2021, 7, e06605. [Google Scholar] [CrossRef]
- Moshawih, S.; Lim, A.F.; Ardianto, C.; Goh, K.W.; Kifli, N.; Goh, H.P.; Jarrar, Q.; Ming, L.C. Target-Based Small Molecule Drug Discovery for Colorectal Cancer: A Review of Molecular Pathways and In Silico Studies. Biomolecules 2022, 12, 878. [Google Scholar] [CrossRef]
- Halder, D.; Das, S.; Aiswarya, R.; Jeyaprakash, R.S. Molecular docking and dynamics based approach for the identification of kinase inhibitors targeting PI3Kα against non-small cell lung cancer: A computational study. RSC Adv. 2022, 12, 21452–21467. [Google Scholar] [CrossRef]
- Wang, X.; Du, J.; Zhao, B.; Wang, H.; Rao, S.; Du, G.; Zhou, J.; Chen, J.; Liu, S. Significantly Improving the Thermostability and Catalytic Efficiency of Streptomyces mobaraenesis Transglutaminase through Combined Rational Design. J. Agric. Food Chem. 2021, 69, 15268–15278. [Google Scholar] [CrossRef]
- Nardo, A.E.; Añón, M.C.; Quiroga, A.V. Identification of renin inhibitors peptides from amaranth proteins by docking protocols. J. Funct. Foods. 2020, 64, 103683. [Google Scholar] [CrossRef]
- Alam, N.; Goldstein, O.; Xia, B.; Porter, K.A.; Kozakov, D.; Schueler-Furman, O. High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. PLoS Comput. Biol. 2017, 13, e1005905. [Google Scholar] [CrossRef]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.-C.; Brylinski, M. DeepDrug3D: Classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 2019, 15, e1006718. [Google Scholar] [CrossRef]
- Gupta, S.; Azadvari, N.; Hosseinzadeh, P. Design of Protein Segments and Peptides for Binding to Protein Targets. BioDesign Res. 2022, 2022, 9783197. [Google Scholar] [CrossRef]
- Vadevoo, S.M.P.; Gurung, S.; Lee, H.-S.; Gunassekaran, G.R.; Lee, S.-M.; Yoon, J.-W.; Lee, Y.-K.; Lee, B. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 2023, 55, 1099–1109. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Li, T.; Lu, X.-M.; Zhang, M.-R.; Hu, K.; Li, Z. Peptide-based nanomaterials: Self-assembly, properties and applications. Bioact. Mater. 2022, 11, 268–282. [Google Scholar] [CrossRef]
- Aritake, K.; Kado, Y.; Inoue, T.; Miyano, M.; Urade, Y. Structural and Functional Characterization of HQL-79, an Orally Selective Inhibitor of Human Hematopoietic Prostaglandin D Synthase. J. Biol. Chem. 2006, 281, 15277–15286. [Google Scholar] [CrossRef]
- Park, H.; Bradley, P.; Greisen, P., Jr.; Liu, Y.; Mulligan, V.K.; Kim, D.E.; Baker, D.; DiMaio, F. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J. Chem. Theory Comput. 2016, 12, 6201–6212. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Leaver-Fay, A.; Corn, J.E.; Strauch, E.-M.; Khare, S.D.; Koga, N.; Ashworth, J.; Murphy, P.; Richter, F.; Lemmon, G.; et al. RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLoS ONE 2011, 6, e20161. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Barley, M.H.; Turner, N.J.; Goodacre, R. Improved Descriptors for the Quantitative Structure–Activity Relationship Modeling of Peptides and Proteins. J. Chem. Theory. Comput. 2018, 58, 234–243. [Google Scholar] [CrossRef]
- Mooney, C.; Haslam, N.J.; Pollastri, G.; Shields, D.C. Towards the Improved Discovery and Design of Functional Peptides: Common Features of Diverse Classes Permit Generalized Prediction of Bioactivity. PLoS ONE 2012, 7, e45012. [Google Scholar] [CrossRef]
- Nivón, L.G.; Moretti, R.; Baker, D. A Pareto-Optimal Refinement Method for Protein Design Scaffolds. PLoS ONE 2013, 8, e59004. [Google Scholar] [CrossRef] [PubMed]
- Bazzoli, A.; Kelow, S.P.; Karanicolas, J. Enhancements to the Rosetta Energy Function Enable Improved Identification of Small Molecules that Inhibit Protein-Protein Interactions. PLoS ONE 2015, 10, e0140359. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory. Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1st Round | 2nd Round | |
|---|---|---|
| nstruct_1 | −423.413 | −657.422 |
| nstruct_2 | −423.324 | −660.113 |
| nstruct_3 | −423.27 | −666.091 |
| nstruct_4 | −423.159 | −657.826 |
| nstruct_5 | −423.096 | −665.231 |
| nstruct_6 | −423.486 | −664.487 |
| nstruct_7 | −424.494 | −662.3 |
| nstruct_8 | −424.38 | −664.144 |
| nstruct_9 | −424.675 | −662.181 |
| nstruct_10 | −424.429 | −659.845 |
| nstruct_11 | −424.675 | −662.543 |
| nstruct_12 | −424.704 | −659.206 |
| nstruct_13 | −423.319 | −662.416 |
| nstruct_14 | −424.675 | −661.017 |
| nstruct_15 | −424.543 | −665.381 |
| nstruct_16 | −424.733 | −661.849 |
| nstruct_17 | −424.197 | −661.809 |
| nstruct_18 | −424.163 | −664.934 |
| nstruct_19 | −424.733 | −661.004 |
| nstruct_20 | −424.675 | −664.397 |
| nstruct_21 | −423.413 | −660.583 |
| nstruct_22 | −424.675 | −668.228 |
| nstruct_23 | −424.143 | −660.682 |
| nstruct_24 | −424.145 | −662.245 |
| nstruct_25 | −424.068 | −665.338 |
| nstruct_26 | −423.234 | −664.834 |
| nstruct_27 | −424.149 | −661.667 |
| nstruct_28 | −424.335 | −663.85 |
| nstruct_29 | −424.775 | −658.93 |
| nstruct_30 | −424.675 | −658.555 |
| nstruct_31 | −424.647 | −664.356 |
| nstruct_32 | −424.675 | −665.182 |
| nstruct_33 | −424.068 | −664.331 |
| nstruct_34 | −424.773 | −664.757 |
| nstruct_35 | −424.675 | −663.109 |
| nstruct_36 | −424.814 | −662.415 |
| nstruct_37 | −424.733 | −663.211 |
| nstruct_38 | −424.733 | −665.031 |
| nstruct_39 | −424.659 | −665.633 |
| nstruct_40 | −423.159 | −665.943 |
| nstruct_41 | −423.128 | −663.531 |
| nstruct_42 | −423.413 | −662.742 |
| nstruct_43 | −424.675 | −665.409 |
| nstruct_44 | −423.138 | −664.335 |
| nstruct_45 | −423.267 | −658.936 |
| nstruct_46 | −423.267 | −660.002 |
| nstruct_47 | −425.471 | −658.008 |
| nstruct_48 | −424.675 | −664.131 |
| nstruct_49 | −424.742 | −658.943 |
| nstruct_50 | −424.741 | −665.313 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Feng, Y.; Yang, T.; Wang, X.; Tang, H. Computer-Aided Designing Peptide Inhibitors of Human Hematopoietic Prostaglandin D2 Synthase Combined Molecular Docking and Molecular Dynamics Simulation. Molecules 2023, 28, 5933. https://doi.org/10.3390/molecules28155933
Cui J, Feng Y, Yang T, Wang X, Tang H. Computer-Aided Designing Peptide Inhibitors of Human Hematopoietic Prostaglandin D2 Synthase Combined Molecular Docking and Molecular Dynamics Simulation. Molecules. 2023; 28(15):5933. https://doi.org/10.3390/molecules28155933
Chicago/Turabian StyleCui, Jing, Yongwei Feng, Ting Yang, Xinglong Wang, and Heng Tang. 2023. "Computer-Aided Designing Peptide Inhibitors of Human Hematopoietic Prostaglandin D2 Synthase Combined Molecular Docking and Molecular Dynamics Simulation" Molecules 28, no. 15: 5933. https://doi.org/10.3390/molecules28155933
APA StyleCui, J., Feng, Y., Yang, T., Wang, X., & Tang, H. (2023). Computer-Aided Designing Peptide Inhibitors of Human Hematopoietic Prostaglandin D2 Synthase Combined Molecular Docking and Molecular Dynamics Simulation. Molecules, 28(15), 5933. https://doi.org/10.3390/molecules28155933