
Computer-Aided Drug Design of Novel Derivatives of 2-Amino-7,9-dihydro-8H-purin-8-one as Potent Pan-Janus JAK3 Inhibitors

 ,
,  ,
,  and
and
Abstract
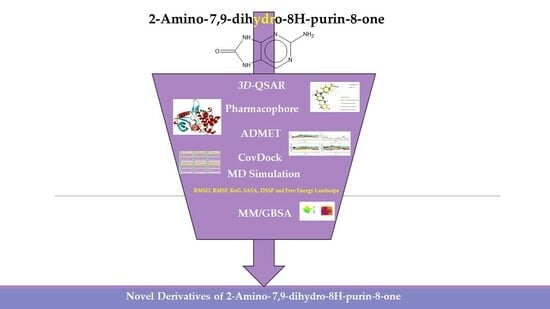
1. Introduction
2. Results and Discussion
2.1. Three-Dimensional-QSAR Models
2.1.1. Analysis Statistics (Field-Based and Atom-Based)
2.1.2. Contours Maps Analysis (Field-Based)
2.1.3. Contours Maps Analysis (Atom-Based)
2.2. Pharmacophore Model
Comparing Field-Based and Atom-Based Models with the DHHHR Pharmacophore Model
2.3. Three-dimensional-QSAR Models Insights for Designing Novel JAK3 Ligands
2.4. Pharmacophore Validation
2.5. Predicted Activity Using 3D-QSAR Models of New Ligands’ Design
2.6. ADMET and Screening Using Covalent Docking
2.7. Physicochemical Property
2.8. Covalent Docking (CovDock)
2.9. Molecular Dynamics Simulation Analysis
2.9.1. DSSP Analysis
2.9.2. Free Energy Landscape Analysis (FEL)
2.10. MM/GBSA Analysis
3. Conclusions
4. Methods and Materials
4.1. Data Set
Software
4.2. Three-Dimensional-QSAR
4.3. QSAR Methodology
4.4. Pharmacophore Hypothesis
4.5. ADMET
4.6. Molecular Docking (MD)
4.6.1. CovDock and Molecular Docking-Based Virtual Screening
4.6.2. Molecular Docking Standard (MDS)
4.7. MD Simulation
4.8. Free Binding Energy (MM/GBSA)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The Role of JAK/STAT Signaling Pathway and Its Inhibitors in Diseases. Int. Immunopharm. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Akahane, K.; McNally, R.; Reyskens, K.M.; Ficarro, S.B.; Liu, S.; Herter-Sprie, G.S.; Koyama, S.; Pattison, M.J.; Labella, K. Development of Selective Covalent Janus Kinase 3 Inhibitors. J. Med. Chem. 2015, 58, 6589–6606. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.C.; Rajapaksa, N.S. Kinase Inhibitors for the Treatment of Immunological Disorders: Recent Advances. J. Med. Chem. 2018, 61, 9030–9058. [Google Scholar] [CrossRef]
- Alunno, A.; Padjen, I.; Fanouriakis, A.; Boumpas, D.T. Pathogenic and Therapeutic Relevance of JAK/STAT Signaling in Systemic Lupus Erythematosus: Integration of Distinct Inflammatory Pathways and the Prospect of Their Inhibition with an Oral Agent. Cells 2019, 8, 898. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Javadian, M.; Babaloo, Z.; Baradaran, B. Janus Kinase Inhibitors: A Therapeutic Strategy for Cancer and Autoimmune Diseases. J. Cell. Phys. 2020, 235, 5903–5924. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus Kinase Inhibitors in Autoimmune Diseases. Ann. Rheum. Dis. 2013, 72, ii111–ii115. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus Kinases in Immune Cell Signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef]
- Malemud, C.J.; Pearlman, E. Targeting JAK/STAT Signaling Pathway in Inflammatory Diseases. Curr. Signal Transduct. Ther. 2009, 4, 201–221. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Vande Casteele, N. JAK–STAT Pathway Targeting for the Treatment of Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Yin, Y.; Chen, C.-J.; Yu, R.-N.; Shu, L.; Wang, Z.-J.; Zhang, T.-T.; Zhang, D.-Y. Novel 1H-Pyrazolo[3,4-d]Pyrimidin-6-Amino Derivatives as Potent Selective Janus Kinase 3 (JAK3) Inhibitors. Evaluation of Their Improved Effect for the Treatment of Rheumatoid Arthritis. Bioorg. Chem. 2020, 98, 103720. [Google Scholar] [CrossRef]
- Dürr, R.; Keppler, O.; Christ, F.; Crespan, E.; Garbelli, A.; Maga, G.; Dietrich, U. Targeting Cellular Cofactors in HIV Therapy. In Therapy of Viral Infections; Diederich, W.E., Steuber, H., Eds.; Topics in Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2015; pp. 183–222. ISBN 978-3-662-46759-6. [Google Scholar]
- Expression Levels of Jak/Stat Signaling Genes in Newly Diagnosed, Drug Sensitive and Resistant Chronic Myeloid Leukemia Patients—ProQuest. Available online: https://www.proquest.com/openview/cecbb9fcb2e75cead444e247a2026151/1?pq-origsite=gscholar&cbl=2026366&diss=y (accessed on 1 June 2023).
- Selective JAK3 Inhibitors with a Covalent Reversible Binding Mode Targeting a New Induced Fit Binding Pocket—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S2451945616303865 (accessed on 1 June 2023).
- Hallenbeck, K.K.; Turner, D.M.; Renslo, A.R.; Arkin, M.R. Targeting Non-Catalytic Cysteine Residues Through Structure-Guided Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.; Eastwood, P.; González, J.; Gómez, E.; Alonso, J.A.; Fonquerna, S.; Lozoya, E.; Orellana, A.; Maldonado, M.; Calaf, E. Identification of 2-Imidazopyridine and 2-Aminopyridone Purinones as Potent Pan-Janus Kinase (JAK) Inhibitors for the Inhaled Treatment of Respiratory Diseases. J. Med. Chem. 2019, 62, 9045–9060. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Q.; Yan, L.; Zhong, G.; Zhang, L.; Tan, X.; Wang, Y. Approaching the Active Conformation of 1,3-Diaminopyrimidine Based Covalent Inhibitors of Bruton’s Tyrosine Kinase for Treatment of Rheumatoid Arthritis. Bioorg. Med. Chem. Lett. 2016, 26, 1954–1957. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhu, C.; Ze, S.; Wang, H.; Yang, X.; Liu, M.; Xie, Q.; Lu, W.; Wang, Y. Design, Synthesis, and Bioevaluation of 2-Aminopteridin-7(8H)-One Derivatives as Novel Potent Adenosine A2A Receptor Antagonists for Cancer Immunotherapy. J. Med. Chem. 2022, 65, 4367–4386. [Google Scholar] [CrossRef]
- Wang, F.; Sun, L.; Wang, S.; Davis, J.M.; Matteson, E.L.; Murad, M.H.; Luo, F.; Vassallo, R. Efficacy and Safety of Tofacitinib, Baricitinib, and Upadacitinib for Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Mayo Clin. Proc. 2020, 95, 1404–1419. [Google Scholar] [CrossRef]
- McInnes, I.B.; Byers, N.L.; Higgs, R.E.; Lee, J.; Macias, W.L.; Na, S.; Ortmann, R.A.; Rocha, G.; Rooney, T.P.; Wehrman, T.; et al. Comparison of Baricitinib, Upadacitinib, and Tofacitinib Mediated Regulation of Cytokine Signaling in Human Leukocyte Subpopulations. Arthritis Res. Ther. 2019, 21, 183. [Google Scholar] [CrossRef]
- Traves, P.G.; Murray, B.; Campigotto, F.; Galien, R.; Meng, A.; Paolo, J.A.D. JAK Selectivity and the Implications for Clinical Inhibition of Pharmacodynamic Cytokine Signalling by Filgotinib, Upadacitinib, Tofacitinib and Baricitinib. Ann. Rheum. Dis. 2021, 80, 865–875. [Google Scholar] [CrossRef]
- Hu, L.; Liu, R.; Zhang, L. Advance in Bone Destruction Participated by JAK/STAT in Rheumatoid Arthritis and Therapeutic Effect of JAK/STAT Inhibitors. Int. Immunopharm. 2022, 111, 109095. [Google Scholar] [CrossRef] [PubMed]
- The New Era of Drug Discovery: The Power of Computer-Aided Drug Design (CADD)|Bentham Science. Available online: https://www.eurekaselect.com/article/122306 (accessed on 28 July 2023).
- Dos Santos Nascimento, I.J.; de Aquino, T.M.; da Silva-Júnior, E.F. Drug Repurposing: A Strategy for Discovering Inhibitors against Emerging Viral Infections. Curr. Med. Chem. 2021, 28, 2887–2942. [Google Scholar]
- Vora, J.; Patel, S.; Athar, M.; Sinha, S.; Chhabria, M.T.; Jha, P.C.; Shrivastava, N. Pharmacophore Modeling, Molecular Docking and Molecular Dynamics Simulation for Screening and Identifying Anti-Dengue Phytocompounds. J. Biomol. Struct. Dyn. 2020, 38, 1726–1740. [Google Scholar] [CrossRef]
- Alqahtani, S. In Silico ADME-Tox Modeling: Progress and Prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Sainath, S.B.; Shrikanya, K.V.L. In Silico Validation and ADMET Analysis for the Best Lead Molecules. In Brucella Melitensis; Elsevier: Amsterdam, The Netherlands, 2021; pp. 133–176. [Google Scholar]
- Erdman, V.V.; Nasibullin, T.R.; Tuktarova, I.A.; Somova, R.S.; Mustafina, O.E. Association Analysis of Polymorphic Gene Variants in the JAK/STAT Signaling Pathway with Aging and Longevity. Russ. J. Genet. 2019, 55, 728–737. [Google Scholar] [CrossRef]
- Wahnschaffe, L.; Braun, T.; Timonen, S.; Giri, A.K.; Schrader, A.; Wagle, P.; Almusa, H.; Johansson, P.; Bellanger, D.; López, C. JAK/STAT-Activating Genomic Alterations Are a Hallmark of T-PLL. Cancers 2019, 11, 1833. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Sanachai, K.; Mahalapbutr, P.; Hengphasatporn, K.; Shigeta, Y.; Seetaha, S.; Tabtimmai, L.; Langer, T.; Wolschann, P.; Kittikool, T.; Yotphan, S. Pharmacophore-Based Virtual Screening and Experimental Validation of Pyrazolone-Derived Inhibitors toward Janus Kinases. ACS Omega 2022, 7, 33548–33559. [Google Scholar] [CrossRef]
- Voiculescu, D.I.; Roman, D.L.; Ostafe, V.; Isvoran, A. A Cheminformatics Study Regarding the Human Health Risks Assessment of the Stereoisomers of Difenoconazole. Molecules 2022, 27, 4682. [Google Scholar] [CrossRef]
- Mendie, L.E.; Hemalatha, S. Bioactive Compounds from Nyctanthes Arbor Tristis Linn as Potential Inhibitors of Janus Kinases (JAKs) Involved in Rheumatoid Arthritis. Appl. Biochem. Biotechnol. 2023, 195, 314–330. [Google Scholar] [CrossRef]
- Faris, A.; Hadni, H.; Ibrahim, I.M.; Elhallaoui, M. In Silico Discovery of Potent and Selective Janus Kinase 3 (JAK3) Inhibitors through 3D-QSAR, Covalent Docking, ADMET Analysis, Molecular Dynamics Simulations, and Binding Free Energy of Pyrazolopyrimidine Derivatives. J. Biomol. Struct. Dyn. 2023. [Google Scholar] [CrossRef]
- Tian, C.; Wang, M.; Han, Z.; Fang, F.; Zhang, Z.; Wang, X.; Liu, J. Design, Synthesis and Biological Evaluation of Novel 6-Substituted Pyrrolo [3,2-d] Pyrimidine Analogues as Antifolate Antitumor Agents. Eur. J. Med. Chem. 2017, 138, 630–643. [Google Scholar] [CrossRef]
- Chan, G.; Changelian, P.S. Inhibitors Targeting JAK3. In Immunotherapy in Transplantation; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2010; pp. 273–289. ISBN 978-1-4443-5562-8. [Google Scholar]
- Yamaoka, K. Tofacitinib for the Treatment of Rheumatoid Arthritis: An Update. Expert Rev. Clin. Immunol. 2019, 15, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, W.; Li, Q.; Chen, Y.; Zhao, G.; Peng, Y.; Zheng, J. Tofacitinib Is a Mechanism-Based Inactivator of Cytochrome P450 3A4. Chem. Res. Toxicol. 2019, 32, 1791–1800. [Google Scholar] [CrossRef]
- Walton, A.; Paik, J.; Quebe, A.; Kannowski, C.L.; Choong, C.; Anderson, S.; Owensby, J.K. Frequency of Prescription Claims for Drugs That May Interact with Janus Kinase Inhibitors Among Patients with Rheumatoid Arthritis in the US. Rheumatol. Ther. 2021, 8, 599–607. [Google Scholar] [CrossRef]
- Coleman, M.D. Human Drug Metabolism; John Wiley & Sons: Hoboken, NJ, USA, 2020; ISBN 978-1-119-45856-2. [Google Scholar]
- Rationalization of Stereoselectivity in Enzyme Reactions—Chan—2019—WIREs Computational Molecular Science—Wiley Online Library. Available online: https://wires.onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1403 (accessed on 26 July 2023).
- Targeting JAK/STAT Signaling to Prevent Rejection after Kidney Transplantation. Available online: https://journals.lww.com/transplantjournal/Fulltext/2016/09000/Targeting_JAK_STAT_Signaling_to_Prevent_Rejection.14.aspx (accessed on 26 July 2023).
- Faquetti, M.L.; Grisoni, F.; Schneider, P.; Schneider, G.; Burden, A.M. Identification of Novel off Targets of Baricitinib and Tofacitinib by Machine Learning with a Focus on Thrombosis and Viral Infection. Sci. Rep. 2022, 12, 7843. [Google Scholar] [CrossRef]
- Zhong, H.A.; Almahmoud, S. Docking and Selectivity Studies of Covalently Bound Janus Kinase 3 Inhibitors. Int. J. Mol. Sci. 2023, 24, 6023. [Google Scholar] [CrossRef] [PubMed]
- En-nahli, F.; Baammi, S.; Hajji, H.; Alaqarbeh, M.; Lakhlifi, T.; Bouachrine, M. High-Throughput Virtual Screening Approach of Natural Compounds as Target Inhibitors of Plasmepsin-II. J. Biomol. Struct. Dyn. 2022. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, S.; Silva, A.C.; Borba, J.V.; Ramos, P.I.; Paveley, R.A.; Muratov, E.N.; Andrade, C.H.; Furnham, N. Computationally-Guided Drug Repurposing Enables the Discovery of Kinase Targets and Inhibitors as New Schistosomicidal Agents. PLoS Comput. Biol. 2018, 14, e1006515. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-1; Maestro, Schrödinger, LLC: New York, NY, USA, 2021.
- Abraham, M.; Alekseenko, A.; Bergh, C.; Blau, C.; Briand, E.; Doijade, M.; Fleischmann, S.; Gapsys, V.; Garg, G.; Gorelov, S.; et al. GROMACS 2023.1 Manual; GROMACS: Groningen, The Netherlands, 2023. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/49/W1/W5/6249611?login=false (accessed on 20 July 2023).
- Free Download: BIOVIA Discovery Studio Visualizer—Dassault Systèmes. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 5 February 2023).
- Free Chemical Drawing Software for Students|ChemSketch|ACD/Labs. Available online: https://www.acdlabs.com/resources/free-chemistry-software-apps/chemsketch-freeware/ (accessed on 18 March 2023).
- Verma, H.; Narendra, G.; Raju, B.; Kumar, M.; Jain, S.K.; Tung, G.K.; Singh, P.K.; Silakari, O. 3D-QSAR and Scaffold Hopping Based Designing of Benzo[d]Ox-Azol-2(3H)-One and 2-Oxazolo[4,5-b]Pyridin-2(3H)-One Derivatives as Selective Aldehyde Dehydrogenase 1A1 Inhibitors: Synthesis and Biological Evaluation. Arch. Der Pharm. 2022, 355, 2200108. [Google Scholar] [CrossRef]
- Ahmad, S.; Hassan, M.I.; Gupta, D.; Dwivedi, N.; Islam, A. Design and Evaluation of Pyrimidine Derivatives as Potent Inhibitors of ABCG2, a Breast Cancer Resistance Protein. 3 Biotech 2022, 12, 182. [Google Scholar] [CrossRef]
- Lankala, V.R.; Joginipally, V.R.; Gunda, S.K. 3D-QSAR and molecular docking studies of natural flavonoids as A431 cell line inhibitors. J. Pharm. Negat. Results 2022, 13, 6955–6969. [Google Scholar]
- Stortz, C.A.; Johnson, G.P.; French, A.D.; Csonka, G.I. Comparison of Different Force Fields for the Study of Disaccharides. Carbohydr. Res. 2009, 344, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Dror, O.; Shulman-Peleg, A.; Nussinov, R.; Wolfson, H.J. Predicting Molecular Interactions in Silico: I. an Updated Guide to Pharmacophore Identification and Its Applications to Drug Design. Curr. Med. Chem. 2006, 11, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Molecular Similarity Methods and QSAR Models as Tools for Virtual Screening. In Drug Discovery Handbook; Wiley: Hoboken, NJ, USA, 2005; pp. 87–122. [Google Scholar]
- Muegge, I.; Martin, Y.C. A General and Fast Scoring Function for Protein–Ligand Interactions: A Simplified Potential Approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in Drug Design—A Review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. 3D QSAR in Drug Design: Volume 1: Theory Methods and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1993; Volume 1. [Google Scholar]
- Kubinyi, H.; Folkers, G.; Martin, Y.C. 3D QSAR in Drug Design: Recent Advances; Springer Science & Business Media: Berlin, Germany, 2006; ISBN 978-0-306-46858-2. [Google Scholar]
- Tenenhaus, M. La Régression PLS: Théorie et Pratique; Editions Technip: Paris, France, 1998. [Google Scholar]
- Yang, S.-Y. Pharmacophore Modeling and Applications in Drug Discovery: Challenges and Recent Advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Pharmaceuticals|Free Full-Text|Drug Design by Pharmacophore and Virtual Screening Approach. Available online: https://www.mdpi.com/1424-8247/15/5/646 (accessed on 30 May 2023).
- Mali, S.N.; Pandey, A.; Bhandare, R.R.; Shaik, A.B. Identification of Hydantoin Based Decaprenylphosphoryl-β-D-Ribose Oxidase (DprE1) Inhibitors as Antimycobacterial Agents Using Computational Tools. Sci. Rep. 2022, 12, 16368. [Google Scholar] [CrossRef]
- Dearden, J.C. In Silico Prediction of ADMET Properties: How Far Have We Come? Expert Opin. Drug Metab. Toxicol. 2007, 3, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Stanzione, F.; Giangreco, I.; Cole, J.C. Chapter Four—Use of Molecular Docking Computational Tools in Drug Discovery. In Progress in Medicinal Chemistry; Witty, D.R., Cox, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 60, pp. 273–343. [Google Scholar]
- Scarpino, A.; Ferenczy, G.G.; Keserű, G.M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model. 2018, 58, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Bianco, G.; Forli, S.; Goodsell, D.S.; Olson, A.J. Covalent Docking Using Autodock: Two-Point Attractor and Flexible Side Chain Methods. Protein Sci. 2016, 25, 295–301. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields—Vanommeslaeghe—2010—Journal of Computational Chemistry—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.21367 (accessed on 31 May 2023).
- Monte Carlo Simulation of Ions in a Magnetron Plasma|IEEE Journals & Magazine|IEEE Xplore. Available online: https://ieeexplore.ieee.org/abstract/document/106828 (accessed on 28 May 2023).
- VMD—Visual Molecular Dynamics. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 14 March 2023).
- Faris, A.; Hadni, H.; Saleh, B.A.; Khelfaoui, H.; Harkati, D.; Ait Ahsaine, H.; Elhallaoui, M.; El-Hiti, G.A. In Silico Screening of a Series of 1,6-Disubstituted 1H-Pyrazolo[3,4-d]Pyrimidines as Potential Selective Inhibitors of the Janus Kinase 3. J. Biomol. Struct. Dyn. 2023. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Aqvist, J. Ion-Water Interaction Potentials Derived from Free Energy Perturbation Simulations. J. Phys. Chem. 1990, 94, 8021–8024. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03, Revision B. 03 and Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Calculations Were Performed Using Gaussian, D.F.T. Program: Gaussian 09 (Revision A. 02); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kim, J.; Kim, S.; Schaumann, G.E. Reliable Predictive Computational Toxicology Methods for Mixture Toxicity: Toward the Development of Innovative Integrated Models for Environmental Risk Assessment. Rev. Environ. Sci. Bio/Technol. 2013, 12, 235–256. [Google Scholar] [CrossRef]
- Izadyar, M.; Housaindokht, M.R.; Zavvar, N.; Khavani, M.; Reisi-vanani, A. Secondary Structure Effects on the Acidity of Histidine and Lysine-Based Peptides Model; A Theoretical Study. Phys. Chem. Res. 2015, 3, 67–77. [Google Scholar]
- Palacios-Prado, N.; Soto, P.A.; López, X.; Choi, E.J.; Marquez-Miranda, V.; Rojas, M.; Duarte, Y.; Lee, J.; González-Nilo, F.D.; Sáez, J.C. Endogenous Pannexin1 Channels Form Functional Intercellular Cell–Cell Channels with Characteristic Voltage-Dependent Properties. Proc. Natl. Acad. Sci. USA 2022, 119, e2202104119. [Google Scholar] [CrossRef]
- Jaffar, S. Optimizing Selectivity in Heterocycle CH Functionalization through Computational Design. Ph.D. Thesis, University of Oxford, Oxford, UK, 2015. [Google Scholar]
- Dong, Z.; Liu, C.-H.; Wang, Y.; Lin, M.; Yu, Z.-X. Gold (I)-Catalyzed Endo-Selective Intramolecular a-Alkenylation of b-Yne-Furans: Synthesis of Seven-Membered-Ring-Fused Furans and DFT Calculations. Angew. Chem. Int. Ed. Engl. 2013, 52, 14157–14161. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.; Atkins, P.W.; de Paula, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein- Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Mondal, S.; Masse, C.; Greenwood, J.; Harriman, G.; Ashwell, M.A.; Bhat, S.; Wester, R.; Frye, L.; Kapeller, R. Accelerating Drug Discovery through Tight Integration of Expert Molecular Design and Predictive Scoring. Curr. Opin. Struct. Biol. 2017, 43, 38–44. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | SD | R2 | R2CV | R2 Scramble | F | p-Value | RMSE | Q2 | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.52 | 0.62 | 0.49 | 0.30 | 42.50 | 0.00 | 0.39 | 0.69 | 0.91 |
| 2 | 0.38 | 0.81 | 0.40 | 0.49 | 53.00 | 0.00 | 0.35 | 0.75 | 0.87 |
| 3 | 0.30 | 0.88 | 0.48 | 0.63 | 59.30 | 0.00 | 0.30 | 0.81 | 0.91 |
| 4 | 0.24 | 0.93 | 0.51 | 0.73 | 78.50 | 0.00 | 0.25 | 0.87 | 0.94 |
| Factors | Steric | Electrostatic | Hydrophobic | H-Bond Acceptor | H-Bond Donor |
|---|---|---|---|---|---|
| 1 | 0.579 | 0.071 | 0.161 | 0.131 | 0.059 |
| 2 | 0.514 | 0.078 | 0.22 | 0.155 | 0.034 |
| 3 | 0.491 | 0.087 | 0.213 | 0.182 | 0.028 |
| 4 | 0.47 | 0.092 | 0.206 | 0.203 | 0.029 |
| Factors | SD | R2 | R2CV | R2 Scramble | Stability | F | p-Value | RMSE | Q2 | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.50 | 0.65 | 0.51 | 0.38 | 0.95 | 48.70 | 0.00 | 0.34 | 0.77 | 0.94 |
| 2 | 0.33 | 0.86 | 0.51 | 0.53 | 0.79 | 74.30 | 0.00 | 0.40 | 0.67 | 0.86 |
| 3 | 0.28 | 0.90 | 0.51 | 0.68 | 0.74 | 71.10 | 0.00 | 0.31 | 0.80 | 0.91 |
| 4 | 0.23 | 0.94 | 0.47 | 0.78 | 0.61 | 85.30 | 0.00 | 0.26 | 0.86 | 0.93 |
| Factors | H-Bond Donor | Hydrophobic/Non-Polar | Electron Withdrawal |
|---|---|---|---|
| 1 | 0.042 | 0.753 | 0.205 |
| 2 | 0.018 | 0.813 | 0.169 |
| 3 | 0.02 | 0.818 | 0.162 |
| 4 | 0.029 | 0.804 | 0.167 |
| Model | Survival | Site | Vector | Volume | Selectivity | Num-Matched | Inactive | Adjusted | Sites | PhaseHypo |
|---|---|---|---|---|---|---|---|---|---|---|
| DHRRR1 | 5.88 | 0.83 | 0.99 | 0.76 | 2.02 | 19 | 2.27 | 3.61 | 8.85 | 8.55 |
| DHRRR2 | 5.86 | 0.83 | 0.99 | 0.75 | 2.02 | 19 | 2.27 | 3.60 | 8.85 | 8.55 |
| DHRRR3 | 5.86 | 0.82 | 0.99 | 0.76 | 2.02 | 19 | 2.28 | 3.58 | 8.85 | 8.55 |
| DHRRR4 | 5.85 | 0.83 | 0.98 | 0.75 | 2.01 | 19 | 2.28 | 3.57 | 8.85 | 8.55 |
| DHRRR5 | 5.83 | 0.80 | 0.98 | 0.74 | 2.04 | 19 | 2.27 | 3.56 | 8.85 | 8.55 |
| DHRRR6 | 5.83 | 0.80 | 0.98 | 0.74 | 2.03 | 19 | 2.24 | 3.59 | 8.85 | 8.55 |
| DHRRR7 | 5.81 | 0.82 | 0.99 | 0.72 | 2.01 | 19 | 2.21 | 3.60 | 8.85 | 8.55 |
| DHRRR8 | 5.80 | 0.80 | 0.97 | 0.72 | 2.02 | 19 | 2.14 | 3.66 | 8.85 | 8.55 |
| DHRRR9 | 5.76 | 0.82 | 0.97 | 0.69 | 2.01 | 19 | 2.09 | 3.67 | 8.85 | 8.55 |
| DHRR10 | 5.37 | 0.83 | 0.99 | 0.76 | 1.51 | 19 | 2.22 | 3.14 | 8.85 | 8.55 |
| DRRR11 | 5.34 | 0.93 | 0.99 | 0.80 | 1.34 | 19 | 2.23 | 3.11 | 8.85 | 8.55 |
| DHRR1 | 5.33 | 0.81 | 0.99 | 0.75 | 1.50 | 19 | 2.26 | 3.07 | 8.85 | 8.55 |
| DHRR2 | 5.34 | 0.83 | 0.98 | 0.75 | 1.50 | 19 | 2.23 | 3.11 | 8.85 | 8.55 |
| DHRR3 | 5.35 | 0.83 | 0.98 | 0.75 | 1.51 | 19 | 2.25 | 3.10 | 8.85 | 8.55 |
| DHRR4 | 5.34 | 0.88 | 1.00 | 0.72 | 1.47 | 19 | 2.16 | 3.18 | 8.85 | 8.55 |
| DHRR5 | 5.35 | 0.87 | 1.00 | 0.74 | 1.46 | 19 | 2.22 | 3.13 | 8.85 | 8.55 |
| HRRR1 | 5.33 | 0.83 | 0.99 | 0.76 | 1.48 | 19 | 2.70 | 2.63 | 8.85 | 8.55 |
| HRRR2 | 5.32 | 0.83 | 0.98 | 0.76 | 1.48 | 19 | 2.76 | 2.57 | 8.85 | 8.55 |
| DHRR5 | 5.34 | 0.81 | 0.99 | 0.76 | 1.49 | 19 | 2.24 | 3.10 | 8.85 | 8.55 |
| Hypothesis | DHRRR_1 |
|---|---|
| PhaseHypo Score | 1.35 |
| EF1% | 2.3 |
| BEDROC160.9 | 0.96 |
| ROC | 0.87 |
| AUAC | 0.74 |
| Ave Outranking Decoys | 4.29 |
| Total Actives | 17 |
| Ranked Actives | 17 |
| Matches | 4 of 5 |
| Excluded Volumes | Yes |
| Reference pIC50 = 9.15 |  | ||
|---|---|---|---|
| ID | Two-Bimensional Compound Structure | Field-Based pIC50 (Pred) | Atom-Based pIC50 (Pred) |
| D1 |  | 7.97 | 8.00 |
| D2 |  | 8.00 | 7.93 |
| D3 |  | 7.92 | 7.91 |
| D4 |  | 8.011 | 7.91 |
| D5 |  | 8.40 | 8.04 |
| D6 |  | 8.01 | 8.07 |
| D7 |  | 8.41 | 8.07 |
| D8 |  | 8.05 | 8.01 |
| D9 |  | 7.94 | 7.94 |
| D10 |  | 7.84 | 7.97 |
| D11 |  | 8.27 | 8.06 |
| D12 |  | 8.03 | 8.22 |
| D13 |  | 8.39 | 8.23 |
| ADMET | Rule | D1 | D2 | D3 | D4 | D5 | D6 | D7 | D8 | D9 | D10 | D11 | D12 | D13 | Tofacitinib |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LogS | −4–0.5 | −3.582 | −3.387 | −3.928 | −3.464 | −3.861 | −3.469 | −3.941 | −3.944 | −3.463 | −3.463 | −3.388 | −3.388 | −3.872 | −2.176 |
| LogD | 1–3 | 3.341 | 2.983 | 2.875 | 1.636 | 2.88 | 1.635 | 2.88 | 3.122 | 1.315 | 1.315 | 1.329 | 1.329 | 3.159 | 1.426 |
| LogP | 0–3 | 2.929 | 2.256 | 2.297 | 1.197 | 2.251 | 1.253 | 2.195 | 2.887 | 0.939 | 0.939 | 0.861 | 0.861 | 2.711 | 1.174 |
| HIA | >30 | 0.849 | 0.64 | 0.938 | 0.42 | 0.853 | 0.797 | 0.848 | 0.155 | 0.252 | 0.252 | 0.462 | 0.462 | 0.438 | 0.934 |
| Caco-2 | >−5.15 | −5.084 | −5.132 | −5.284 | −5.829 | −5.186 | −5.824 | −5.294 | −5.17 | −5.751 | −5.751 | −5.744 | −5.744 | −4.964 | −4.655 |
| MDCK | >20 × 10−6 | 1.33 × 10−5 | 1.07 × 10−5 | 4.10 × 10−6 | 7.24 × 10−6 | 3.84 × 10−6 | 4.97 × 10−6 | 4.26 × 10−6 | 8.33 × 10−6 | 4.48 × 10−6 | 4.48 × 10−6 | 5.18 × 10−6 | 5.18 × 10−6 | 5.59 × 10−6 | 6.3 × 10−6 |
| BBB | 0–0.3 | 0.041 | 0.027 | 0.012 | 0.107 | 0.011 | 0.073 | 0.01 | 0.009 | 0.35 | 0.35 | 0.073 | 0.073 | 0.037 | |
| VDss | 0.04–20 | 0.561 | 0.589 | 0.484 | 1.51 | 0.433 | 1.363 | 0.481 | 0.38 | 1.012 | 1.012 | 1.226 | 1.226 | 0.655 | |
| 1A2-inh | Yes | No | No | No | No | No | No | Yes | No | No | No | No | No | Yes | |
| 1A2-sub | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | |
| 2C19-inh | Yes | Yes | Yes | No | No | No | No | Yes | No | No | No | No | Yes | No | |
| 2C19-sub | No | No | No | No | No | No | No | No | No | No | No | No | No | No | |
| 2C9-inh | Yes | Yes | Yes | Yes | Yes | No | No | Yes | No | No | No | No | No | No | |
| 2C9-sub | No | No | No | No | No | No | No | No | No | No | No | No | No | No | |
| 2D6-inh | No | No | No | No | No | No | No | No | No | No | No | No | No | No | |
| 2D6-sub | No | No | No | No | No | No | No | No | No | No | No | No | No | No | |
| 3A4-inh | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | |
| 3A4-sub | No | No | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | No | No | |
| CL | ≥5 | 7.891 | 7.891 | 8.039 | 5.676 | 5.94 | 7.485 | 7.953 | 7.953 | 5.648 | 6.664 | 7.141 | 6.372 | 6.753 | 8.737 |
| Ames | No | No | No | No | No | No | No | Yes | No | No | No | No | No | No |
| P. P | nHA | nHD | TPSA | nRot | nRing | MaxRing | nHet | fChar | nStereo | MW | Lipinski |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rule | 0~12 | 0~7 | 0~14 | 0~11 | 0~6 | 0~6 | 1~15 | −4~4 | ≤2 | 100~600 | Accepted |
| D1 | 8 | 3 | 111.69 | 3 | 4 | 9 | 10 | 0 | 0 | 402.040 | |
| D2 | 8 | 3 | 111.69 | 3 | 4 | 9 | 9 | 0 | 0 | 368.080 | |
| D3 | 9 | 4 | 127.48 | 2 | 5 | 9 | 10 | 0 | 0 | 393.070 | |
| D4 | 11 | 3 | 151.61 | 4 | 5 | 9 | 12 | 0 | 0 | 477.140 | |
| D5 | 9 | 4 | 127.48 | 2 | 5 | 9 | 10 | 0 | 0 | 393.070 | |
| D6 | 11 | 4 | 154.51 | 4 | 5 | 9 | 12 | 0 | 0 | 465.140 | |
| D7 | 9 | 4 | 127.48 | 2 | 5 | 9 | 10 | 0 | 0 | 393.070 | |
| D8 | 9 | 3 | 116.62 | 2 | 5 | 9 | 10 | 0 | 0 | 393.070 | |
| D9 | 12 | 3 | 156.54 | 4 | 5 | 9 | 13 | 0 | 0 | 466.140 | |
| D10 | 12 | 3 | 156.54 | 4 | 5 | 9 | 13 | 0 | 0 | 466.140 | |
| D11 | 12 | 4 | 167.4 | 4 | 5 | 9 | 13 | 0 | 0 | 466.140 | |
| D12 | 12 | 4 | 167.4 | 4 | 5 | 9 | 13 | 0 | 0 | 466.140 | |
| D13 | 8 | 3 | 111.69 | 2 | 4 | 9 | 9 | 0 | 0 | 360.110 | |
| Tofacitinib | 7 | 1 | 88.910 | 4 | 18 | 9 | 7 | 0 | 2 | 312.170 |
| Compound | D1 | D2 | D3 | D4 | D5 | D6 | D7 | D8 | D9 | D10 | D11 | D12 | D13 | Tofacitinib |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Affinity (Kcal/mol) | −9.10 | −9.55 | −9.37 | −9.53 | −9.5 | −5.63 | −6.71 | −7–24 | −6.36 | −4.15 | −7.42 | −6.66 | −8.10 | −7.50 |
| Delta Energy (Kcal/mol) | D1 | D2 | D3 | D4 | D5 | Tofacitinib |
|---|---|---|---|---|---|---|
| ΔVDWAALS | −33.95 | −36.93 | −40.29 | −32.60 | −37.09 | −22.82 |
| ΔEEL | −30.27 | −29.89 | −33.86 | −32.77 | −19.55 | −32.93 |
| ΔEGB | 42.36 | 46.61 | 48.34 | 45.61 | 34.48 | 55.89 |
| ΔESURF | −5.00 | −4.78 | −5.55 | −4.78 | −4.64 | −3.34 |
| ΔGGAS | −64.22 | −66.82 | −74.15 | −65.37 | −56.64 | −55.75 |
| ΔGSOLV | 37.35 | 41.83 | 42.79 | 40.84 | 29.84 | 52.55 |
| ΔTOTAL | −26.87 | −24.99 | −31.37 | −24.54 | −26.80 | −3.20 |
| Model 3D-QSAR | Field-Based | Atom-Based | |||
|---|---|---|---|---|---|
| No. | Compound | pIC50 (Exp) | QSAR | pIC50 (Pred) | |
| 1 |  | 8.77 | training | 9.18 | 9.25 |
| 2 |  | 8.59 | training | 8.58 | 8.53 |
| 3 |  | 7.47 | training | 9.12 | 9.00 |
| 4 |  | 7.41 | test | 8.68 | 8.46 |
| 5 |  | 7.28 | test | 8.84 | 9.02 |
| 6 |  | 7.23 | training | 8.53 | 8.52 |
| 7 |  | 7.31 | training | 8.66 | 8.45 |
| 8 |  | 7.33 | training | 8.51 | 8.88 |
| 9 |  | 7.10 | test | 8.55 | 8.60 |
| 10 |  | 7.02 | training | 8.42 | 8.59 |
| 11 |  | 7.10 | training | 8.68 | 8.39 |
| 12 |  | 6.31 | training | 8.18 | 8.24 |
| 13 |  | 6.59 | training | 8.49 | 8.25 |
| 14 |  | 8.31 | training | 8.73 | 8.63 |
| 15 |  | 8.08 | test | 8.39 | 8.43 |
| 16 |  | 7.19 | training | 8.46 | 8.60 |
| 17 |  | 9.00 | training | 8.13 | 8.33 |
| 18 |  | 6.38 | training | 8.31 | 8.05 |
| 19 |  | 8.57 | test | 8.14 | 8.14 |
| 20 |  | 8.52 | training | 7.81 | 8.24 |
| 21 |  | 8.41 | training | 7.39 | 7.55 |
| 22 |  | 8.55 | training | 7.42 | 7.39 |
| 23 |  | 8.96 | training | 7.72 | 7.51 |
| 24 |  | 8.96 | training | 8.09 | 7.91 |
| 25 |  | 8.62 | test | 7.23 | 7.34 |
| 26 |  | 7.80 | training | 7.71 | 7.46 |
| 27 |  | 8.47 | training | 7.25 | 7.29 |
| 28 |  | 7.40 | training | 7.39 | 7.29 |
| 29 |  | 8.22 | training | 7.42 | 7.41 |
| 30 |  | 8.85 | training | 7.15 | 6.98 |
| 31 |  | 8.44 | test | 7.30 | 7.06 |
| 32 |  | 8.85 | training | 7.11 | 6.92 |
| 33 |  | 7.74 | training | 6.38 | 6.56 |
| 34 |  | 8.46 | training | 6.38 | 6.34 |
| 35 |  | 9.15 | training | 6.30 | 6.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faris, A.; Ibrahim, I.M.; Al kamaly, O.; Saleh, A.; Elhallaoui, M. Computer-Aided Drug Design of Novel Derivatives of 2-Amino-7,9-dihydro-8H-purin-8-one as Potent Pan-Janus JAK3 Inhibitors. Molecules 2023, 28, 5914. https://doi.org/10.3390/molecules28155914
Faris A, Ibrahim IM, Al kamaly O, Saleh A, Elhallaoui M. Computer-Aided Drug Design of Novel Derivatives of 2-Amino-7,9-dihydro-8H-purin-8-one as Potent Pan-Janus JAK3 Inhibitors. Molecules. 2023; 28(15):5914. https://doi.org/10.3390/molecules28155914
Chicago/Turabian StyleFaris, Abdelmoujoud, Ibrahim M. Ibrahim, Omkulthom Al kamaly, Asmaa Saleh, and Menana Elhallaoui. 2023. "Computer-Aided Drug Design of Novel Derivatives of 2-Amino-7,9-dihydro-8H-purin-8-one as Potent Pan-Janus JAK3 Inhibitors" Molecules 28, no. 15: 5914. https://doi.org/10.3390/molecules28155914
APA StyleFaris, A., Ibrahim, I. M., Al kamaly, O., Saleh, A., & Elhallaoui, M. (2023). Computer-Aided Drug Design of Novel Derivatives of 2-Amino-7,9-dihydro-8H-purin-8-one as Potent Pan-Janus JAK3 Inhibitors. Molecules, 28(15), 5914. https://doi.org/10.3390/molecules28155914