

Pyronaridine as a Bromodomain-Containing Protein 4-N-Terminal Bromodomain (BRD4-BD1) Inhibitor: In Silico Database Mining, Molecular Docking, and Molecular Dynamics Simulation

 ,
,  ,
,  , , , ,
, , , ,  ,
,  and
and
Abstract
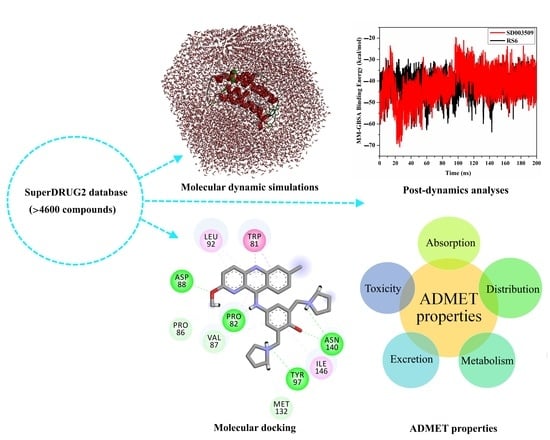
1. Introduction
2. Results and Discussion
2.1. Docking Protocol Validation
2.2. Virtual Screening of the SuperDRUG2 Database
2.3. Molecular Dynamics (MD)
2.4. Post-MD Analyses
2.4.1. Binding Energy Per Trajectory
2.4.2. H-Bond Analysis
2.4.3. Center-of-Mass Distance (CoM)
2.4.4. Root-Mean-Square Deviation (RMSD)
2.4.5. Root-Mean-Square Fluctuation (RMSF)
2.5. ADMET Characteristics
2.6. Drug-Likeness Characteristics
3. Computational Methods
3.1. BRD4-BD1 Preparation
3.2. Database Preparation
3.3. Docking Calculations
3.4. Molecular Dynamics (MD)
3.5. Binding Energy Evaluation
3.6. ADMET Characteristics
3.7. Drug-Likeness Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Owen, D.J.; Ornaghi, P.; Yang, J.C.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS J. 2012, 586, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS J. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Chen, R.; Yik, J.H.; Lew, Q.J.; Chao, S.H. Brd4 and HEXIM1: Multiple roles in P-TEFb regulation and cancer. Biomed Res. Int. 2014, 2014, 232870. [Google Scholar] [CrossRef]
- Cheng, Z.; Gong, Y.; Ma, Y.; Lu, K.; Lu, X.; Pierce, L.A.; Thompson, R.C.; Muller, S.; Knapp, S.; Wang, J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 2013, 19, 1748–1759. [Google Scholar] [CrossRef]
- Gacias, M.; Gerona-Navarro, G.; Plotnikov, A.N.; Zhang, G.; Zeng, L.; Kaur, J.; Moy, G.; Rusinova, E.; Rodriguez, Y.; Matikainen, B.; et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014, 21, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, J.; Ye, F.; Xiong, H.; Peng, L.; Zheng, Z.; Xu, F.; Cui, M.; Wei, C.; Wang, X.; et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int. J. Mol. Sci. 2015, 16, 1928–1948. [Google Scholar] [CrossRef]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef]
- Bid, H.K.; Kerk, S. BET bromodomain inhibitor (JQ1) and tumor angiogenesis. Oncoscience 2016, 3, 316–317. [Google Scholar] [CrossRef]
- Bid, H.K.; Phelps, D.A.; Xaio, L.; Guttridge, D.C.; Lin, J.; London, C.; Baker, L.H.; Mo, X.; Houghton, P.J. The Bromodomain BET Inhibitor JQ1 Suppresses Tumor Angiogenesis in Models of Childhood Sarcoma. Mol. Cancer Ther. 2016, 15, 1018–1028. [Google Scholar] [CrossRef]
- Hao, J.; Yang, Z.; Wang, L.; Zhang, Y.; Shu, Y.; Jiang, L.; Hu, Y.; Lv, W.; Dong, P.; Liu, Y. Downregulation of BRD4 inhibits gallbladder cancer proliferation and metastasis and induces apoptosis via PI3K/AKT pathway. Int. J. Oncol. 2017, 51, 823–831. [Google Scholar] [CrossRef][Green Version]
- Chiang, C.M. Nonequivalent response to bromodomain-targeting BET inhibitors in oligodendrocyte cell fate decision. Chem. Biol. 2014, 21, 804–806. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Liu, Z.; Tian, B.; Chen, H.; Wang, P.; Brasier, A.R.; Zhou, J. Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur. J. Med. Chem. 2018, 151, 450–461. [Google Scholar] [CrossRef]
- Lin, S.; Du, L. The therapeutic potential of BRD4 in cardiovascular disease. Hypertens. Res. 2020, 43, 1006–1014. [Google Scholar] [CrossRef]
- Ding, D.; Zheng, R.; Tian, Y.; Jimenez, R.; Hou, X.; Weroha, S.J.; Wang, L.; Shi, L.; Huang, H. Retinoblastoma protein as an intrinsic BRD4 inhibitor modulates small molecule BET inhibitor sensitivity in cancer. Nat. Commun. 2022, 13, 6311. [Google Scholar] [CrossRef]
- Lu, T.; Lu, W.; Luo, C. A patent review of BRD4 inhibitors (2013–2019). Expert Opin. Ther. Pat. 2020, 30, 57–81. [Google Scholar] [CrossRef]
- Karim, M.R.; Schonbrunn, E. Crystal structure of the first bromodomain (BD1) of human BRD4 in complex with dual BRD4-JAK2 inhibitor MA9-086. 2021. Available online: https://www.wwpdb.org/pdb?id=pdb_00007rek (accessed on 9 June 2023).
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Croft, S.L.; Duparc, S.; Arbe-Barnes, S.J.; Craft, J.C.; Shin, C.S.; Fleckenstein, L.; Borghini-Fuhrer, I.; Rim, H.J. Review of pyronaridine anti-malarial properties and product characteristics. Malar. J. 2012, 11, 270. [Google Scholar] [CrossRef]
- Garbuzenko, O.B.; Kbah, N.; Kuzmov, A.; Pogrebnyak, N.; Pozharov, V.; Minko, T. Inhalation treatment of cystic fibrosis with lumacaftor and ivacaftor co-delivered by nanostructured lipid carriers. J. Control. Release 2019, 296, 225–231. [Google Scholar] [CrossRef]
- Cholon, D.M.; Esther, C.R., Jr.; Gentzsch, M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the F508del-CFTR mutation. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 235–243. [Google Scholar] [CrossRef]
- Goekjian, P.G.; Jirousek, M.R. Protein kinase C inhibitors as novel anticancer drugs. Expert Opin. Investig. Drugs 2001, 10, 2117–2140. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. In In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 95–113. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Lennernas, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019, 10, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Siramshetty, V.B.; Eckert, O.A.; Gohlke, B.O.; Goede, A.; Chen, Q.; Devarakonda, P.; Preissner, S.; Preissner, R. SuperDRUG2: A one stop resource for approved/marketed drugs. Nucleic Acids Res. 2018, 46, D1137–D1143. [Google Scholar] [CrossRef]
- OMEGA, version 2.5.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013.
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- SZYBKI, version 1.9.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Jaragh-Alhadad, L.A.; Atia, M.A.M.; Alzahrani, O.R.; Ahmed, M.N.; Moustafa, M.S.; Soliman, M.E.S.; Shawky, A.M.; Pare, P.W.; et al. Exploring Toxins for Hunting SARS-CoV-2 Main Protease Inhibitors: Molecular Docking, Molecular Dynamics, Pharmacokinetic Properties, and Reactome Study. Pharmaceuticals 2022, 15, 153. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Shawky, A.M.; Mekhemer, G.A.H.; Alrumaihi, F.; Moustafa, M.F.; Atia, M.A.M. Prospective drug candidates as human multidrug transporter ABCG2 inhibitors: An in silico drug discovery study. Cell Biochem. Biophys. 2021, 79, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Alzahrani, O.R.; Alshabrmi, F.M.; Khalaf, E.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; Soliman, M.E.S.; et al. Non-beta-lactam allosteric inhibitors target methicillin-resistant staphylococcus aureus: An in silico drug discovery study. Antibiotics 2021, 10, 934–956. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Mishra, B.K.; Karthikeyan, S.; Ramanathan, V. Tuning the C-H...Pi interaction by different substitutions in benzene-acetylene complexes. J. Chem. Theory Comput. 2012, 8, 1935–1942. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA, Discovery Studio Visualizer, version 2019; Dassault Systèmes: San Diego, CA, USA, 2019.
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Shen, M.; Tian, S.; Li, Y.; Li, Q.; Xu, X.; Wang, J.; Hou, T. Drug-likeness analysis of traditional Chinese medicines: 1. property distributions of drug-like compounds, non-drug-like compounds and natural compounds from traditional Chinese medicines. J. Cheminformatics 2012, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Inhibitor Name/Code | Two-Dimensional Chemical Structure | Docking Score (kcal/mol) | Binding Features | |
|---|---|---|---|---|---|
| Standard | Expensive | ||||
| R6S |  | −9.9 | −10.0 | ASN140 (H-bond, 2.71, 1.92 Å), PRO82 (H-bond, 3.04 Å; π-Alkyl, 5.07 Å), LYS91 (H- bond, 2.45 Å), ASP88 (H- bond, 1.93 Å), LEU92 (π-Alkyl, 4.75, 4.85, 5.49 Å), VAL87 (π-Alkyl, 5.22, 4.92 Å), LEU94 (π-Alkyl, 5.10 Å), PHE83 (π-Alkyl, 4.17 Å), CYS136 (π-Alkyl, 5.36 Å), ILE146 (π-Alkyl, 4.19 Å), TRP81 (π-Alkyl, 5.03 Å; π-π T-shaped, 5.25 Å) | |
| 1 | Pyronaridine (SD003509) |  | −10.1 | −10.2 | ASN140 (H-bond, 2.32, 2.06 Å), TYR97 (H-bond, 2.67 Å), ASP88 (H-bond, 1.95 Å), PRO82 (H-bond, 2.23 Å; π-Alkyl, 5.13, 5.47 Å), LEU92 (π-Alkyl, 4.39, 4.68 Å), VAL87 (π-Alkyl, 4.44 Å), ILE146 (π-Alkyl, 4.90 Å), TRRP81 (π-π T-shaped, 4.89, 4.92 Å) |
| 2 | Lumacaftor (SD003873) |  | −10.1 | −10.1 | GLN85 (H-bond, 2.45, 3.08 Å), LEU92 (π-Alkyl, 4.42 Å), VAL87 (π-Alkyl, 4.87 Å), ILE146 (π-Alkyl, 4.21, 4.68 Å), TRP81 (π-Alkyl, 4.87 Å; π-π T-shaped, 5.15 Å) |
| 3 | N-benzoylstaurosporine (SD006001) |  | −9.9 | −10.0 | LEU92 (π-Alkyl, 4.50, 4.99 Å), VAL87 (π-Alkyl, 5.22, 4.86 Å), CYS136 (π-Alkyl, 5.13 Å), TRRP81 (π-π T-shaped, 5.29, 5.21 Å) |
| Inhibitor Name/SuperDRUG2 Code | MM-GBSA Binding Energy (kcal/mol) | |
|---|---|---|
| 50 ns | 200 ns | |
| R6S | −43.9 | −41.5 |
| Pyronaridine (SD003509) | −46.2 | −42.7 |
| Lumacaftor (SD003873) | −27.8 | --- a |
| N-benzoylstaurosporine (SD006001) | −20.0 | --- a |
| Inhibitor Code | Absorption (A) | Distribution (D) | Metabolism (M) | Excretion (E) | Toxicity (T) | |
|---|---|---|---|---|---|---|
| Caco2 Permeability (cm/s) | Human Intestinal Absorption (HIA) | VDss (Human) | CYP3A4 Inhibitor/Substrate | Total Clearance | AMES Toxicity | |
| Pyronaridine (SD003509) | 0.62 | 93.60 | 1.41 | Yes | 1.21 | No |
| R6S | 1.12 | 90.84 | 1.15 | Yes | 0.57 | No |
| Compound Name | MLogP | TPSA | nON | nOHNH | Nrotb | MWt | %ABS |
|---|---|---|---|---|---|---|---|
| R6S | 6.0 | 76.1 | 7 | 2 | 6 | 552.7 | 82.7 |
| Pyronaridine (SD003509) | 2.8 | 90.5 | 5 | 4 | 7 | 520.1 | 77.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Abdelhamid, M.M.H.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Mekhemer, G.A.H.; Sidhom, P.A.; Sayed, S.R.M.; Paré, P.W.; Hegazy, M.-E.F.; Shoeib, T. Pyronaridine as a Bromodomain-Containing Protein 4-N-Terminal Bromodomain (BRD4-BD1) Inhibitor: In Silico Database Mining, Molecular Docking, and Molecular Dynamics Simulation. Molecules 2023, 28, 5713. https://doi.org/10.3390/molecules28155713
Ibrahim MAA, Abdelhamid MMH, Abdeljawaad KAA, Abdelrahman AHM, Mekhemer GAH, Sidhom PA, Sayed SRM, Paré PW, Hegazy M-EF, Shoeib T. Pyronaridine as a Bromodomain-Containing Protein 4-N-Terminal Bromodomain (BRD4-BD1) Inhibitor: In Silico Database Mining, Molecular Docking, and Molecular Dynamics Simulation. Molecules. 2023; 28(15):5713. https://doi.org/10.3390/molecules28155713
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Mahmoud M. H. Abdelhamid, Khlood A. A. Abdeljawaad, Alaa H. M. Abdelrahman, Gamal A. H. Mekhemer, Peter A. Sidhom, Shaban R. M. Sayed, Paul W. Paré, Mohamed-Elamir F. Hegazy, and Tamer Shoeib. 2023. "Pyronaridine as a Bromodomain-Containing Protein 4-N-Terminal Bromodomain (BRD4-BD1) Inhibitor: In Silico Database Mining, Molecular Docking, and Molecular Dynamics Simulation" Molecules 28, no. 15: 5713. https://doi.org/10.3390/molecules28155713
APA StyleIbrahim, M. A. A., Abdelhamid, M. M. H., Abdeljawaad, K. A. A., Abdelrahman, A. H. M., Mekhemer, G. A. H., Sidhom, P. A., Sayed, S. R. M., Paré, P. W., Hegazy, M.-E. F., & Shoeib, T. (2023). Pyronaridine as a Bromodomain-Containing Protein 4-N-Terminal Bromodomain (BRD4-BD1) Inhibitor: In Silico Database Mining, Molecular Docking, and Molecular Dynamics Simulation. Molecules, 28(15), 5713. https://doi.org/10.3390/molecules28155713