
A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination
Abstract
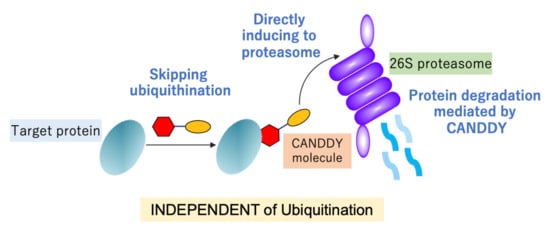
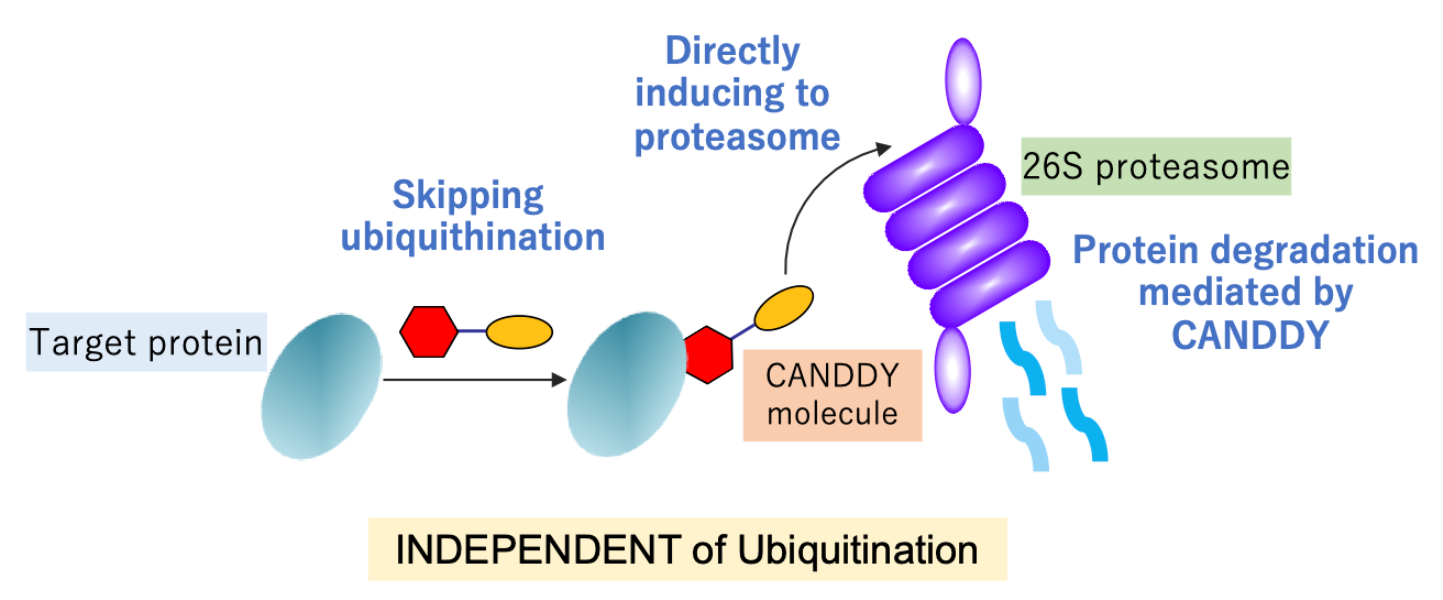{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
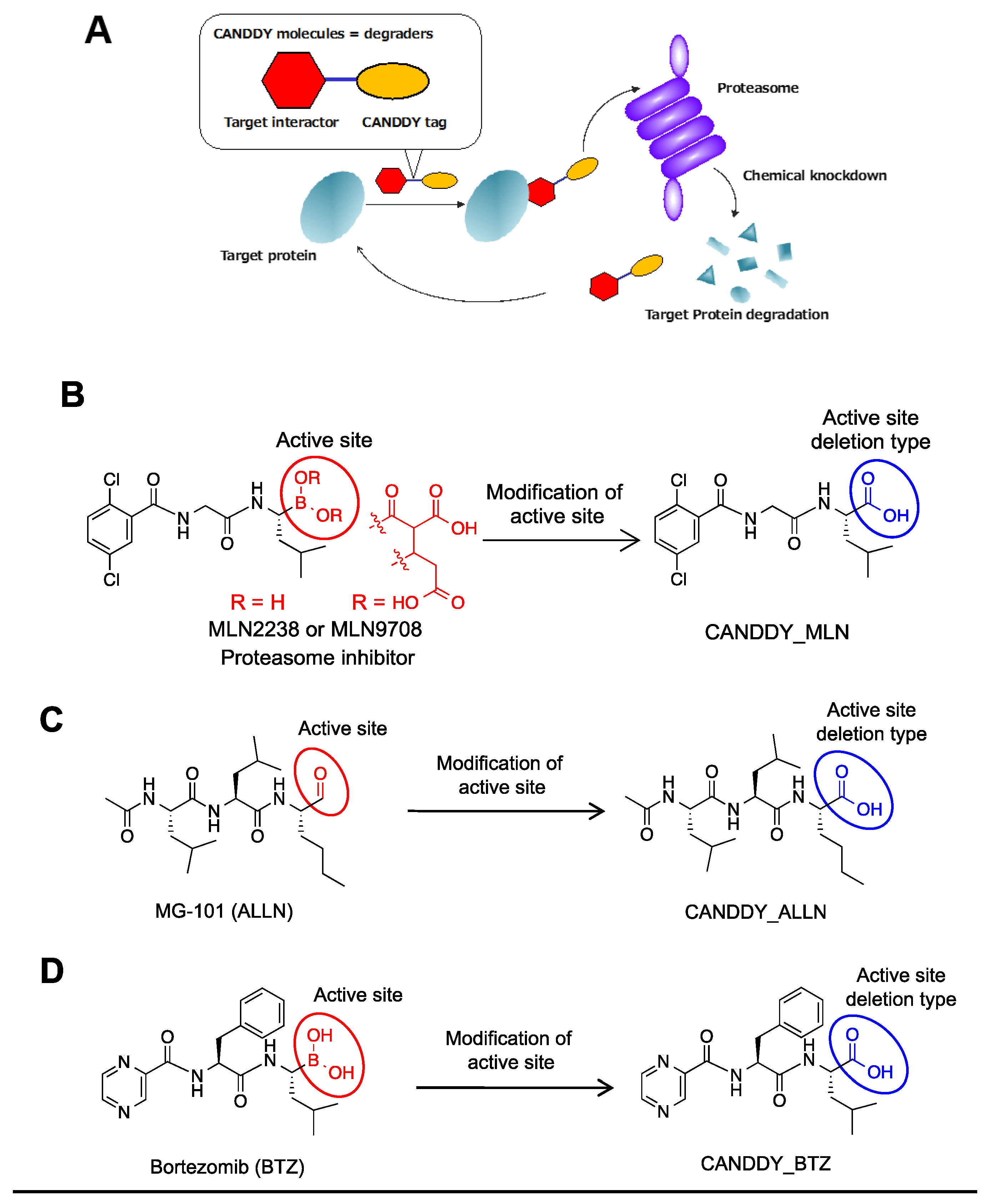
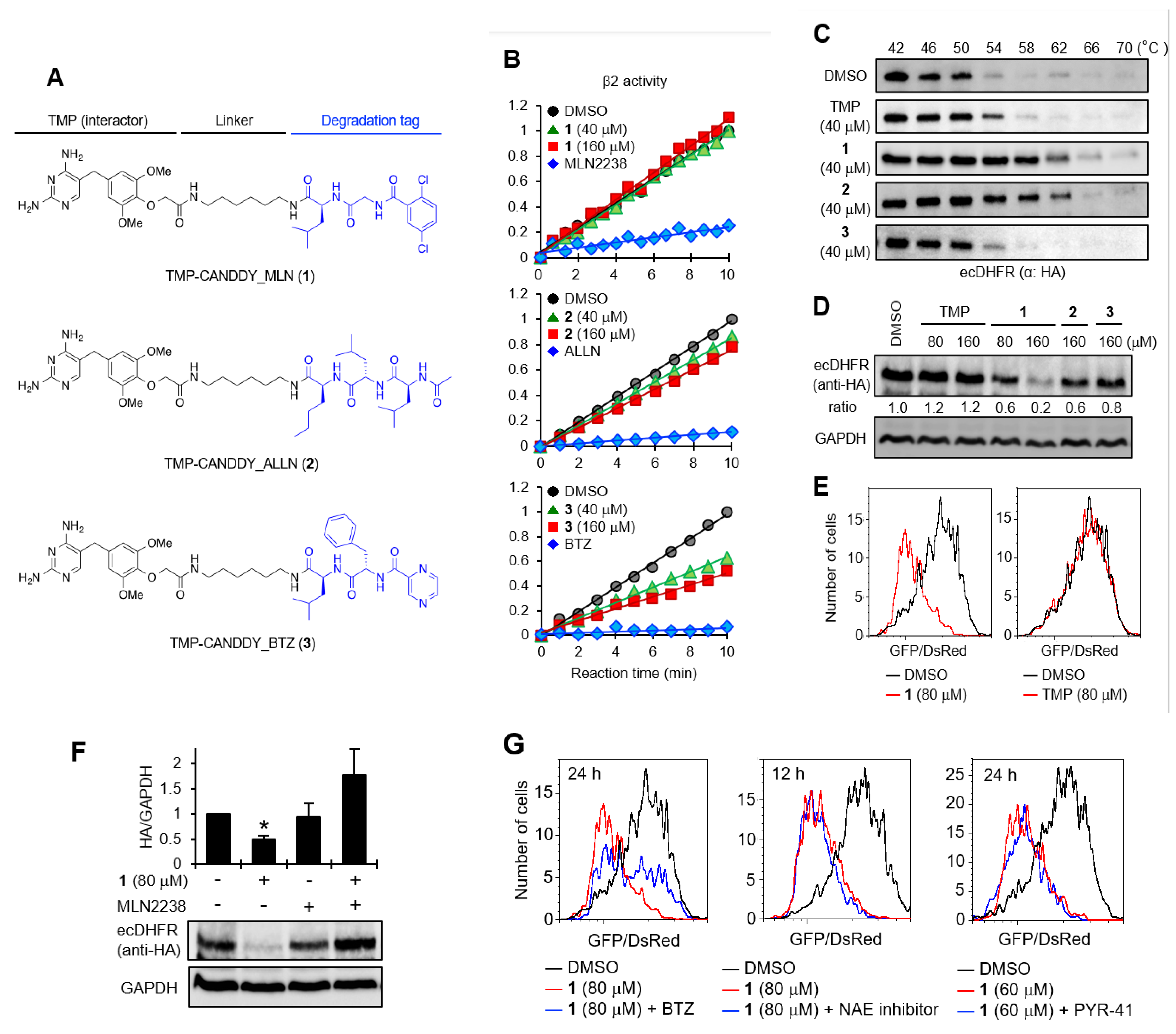
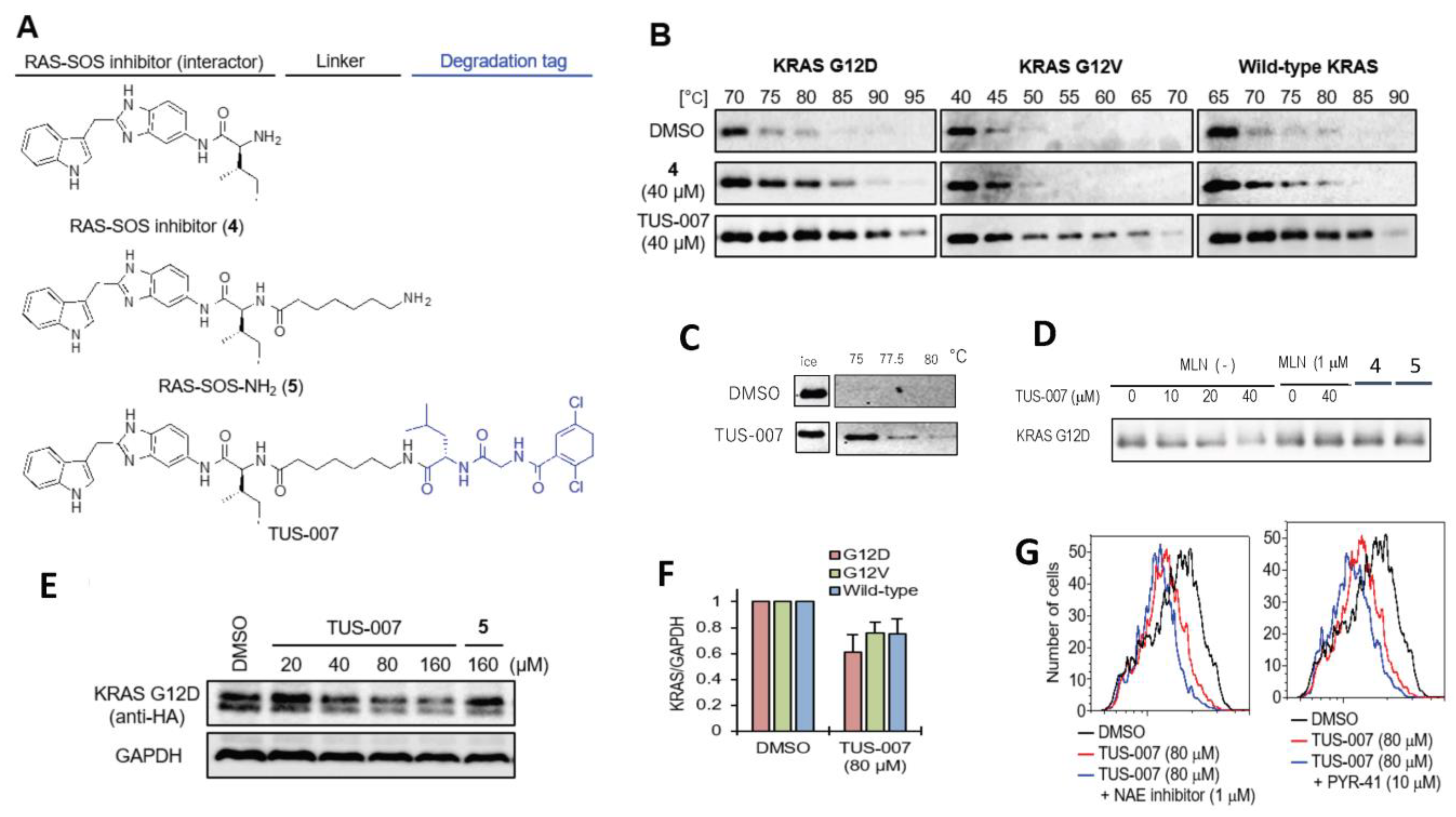
2.1. Proof of Concept for the CANDDY Strategy Mediated by Degradation Tags Derived from Proteasome Inhibitors
2.2. Chemical Knockdown of KRAS Mutants In Vitro by TUS-007
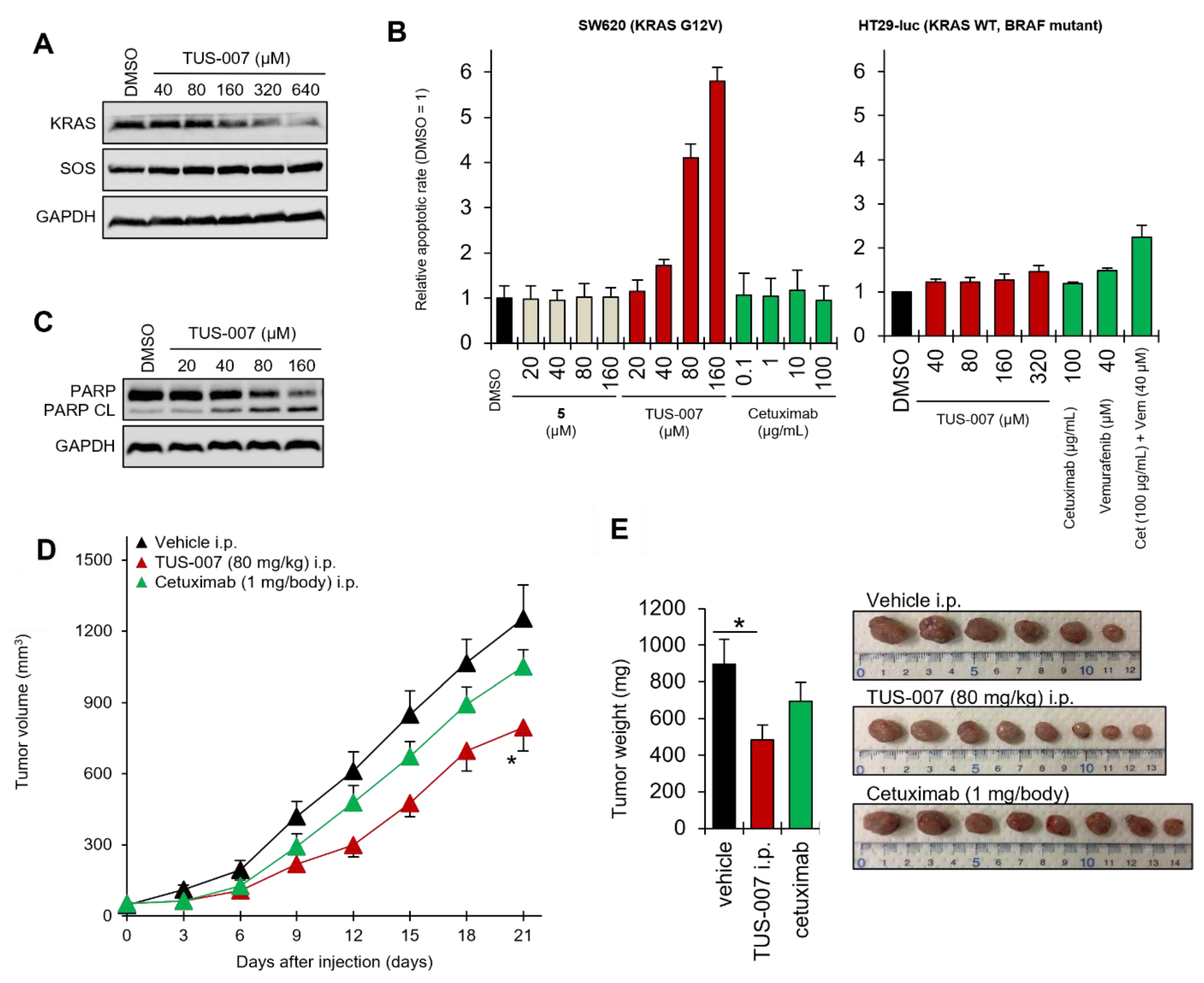
2.3. Apoptosis Induction and Tumor Suppression in Cetuximab-Resistant Human Colon Carcinoma Cells by TUS-007
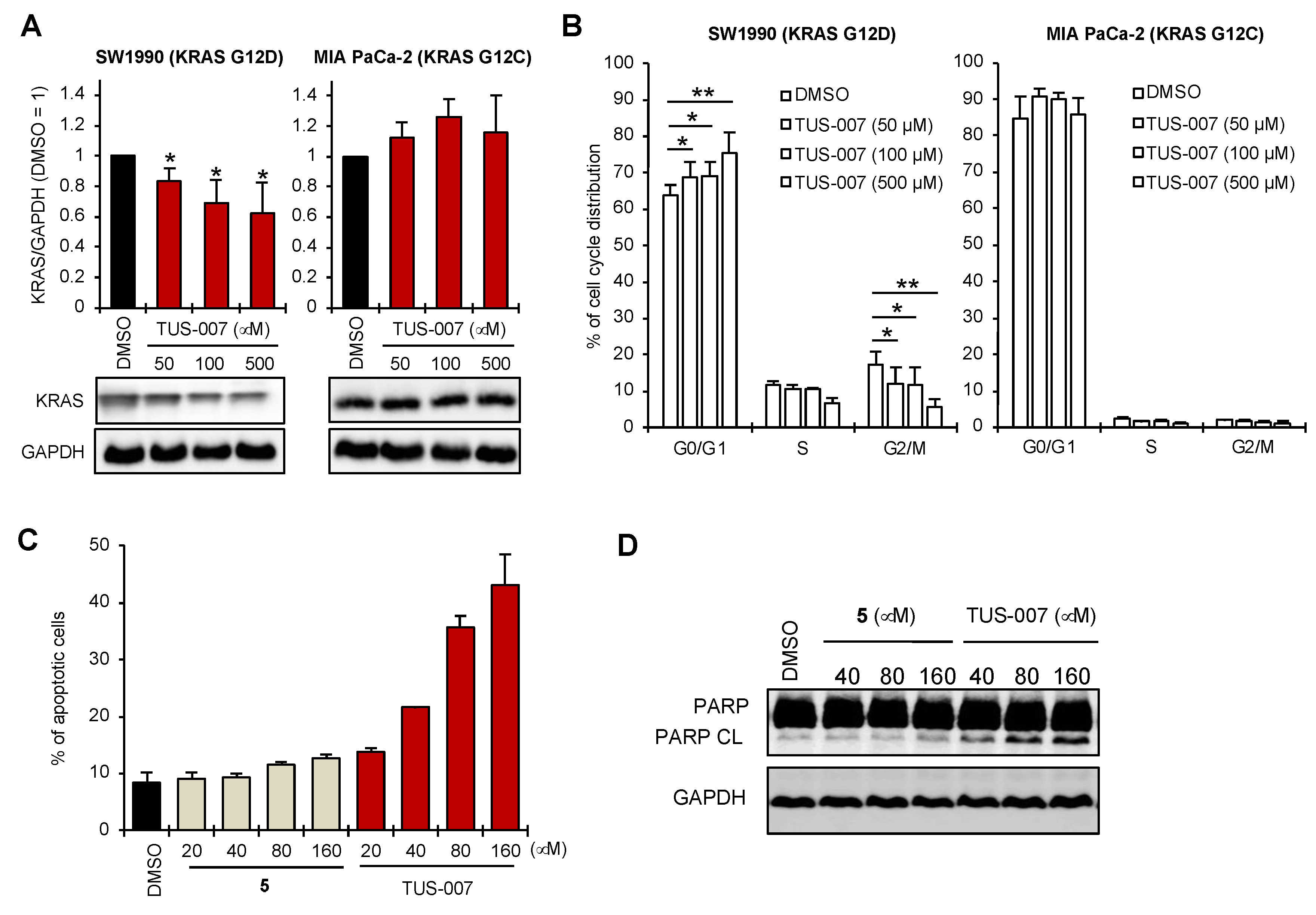
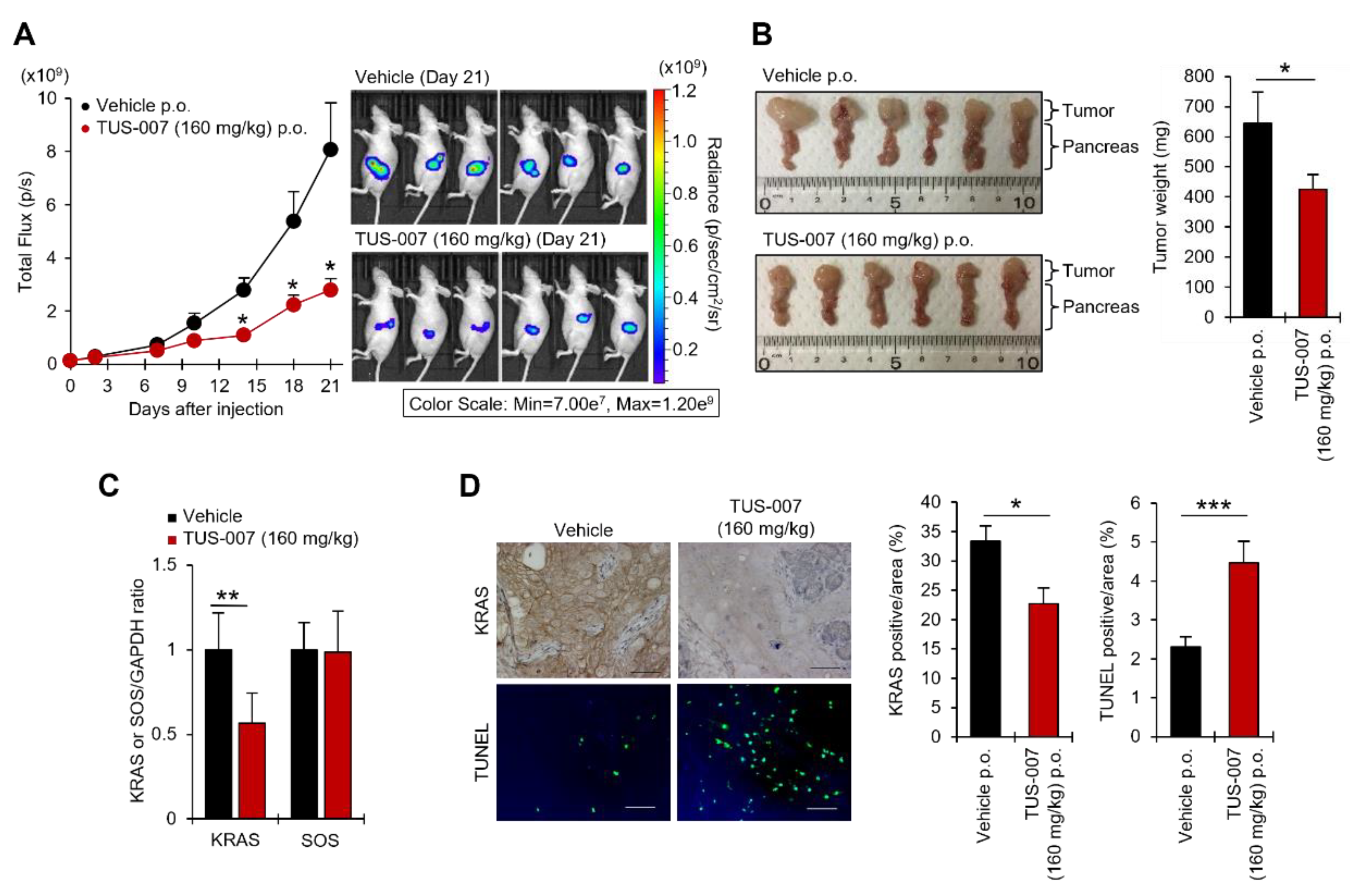
2.4. Apoptosis Induction and Tumor Suppression in Human Pancreatic Carcinoma Cells by TUS-007
3. Discussion
4. Materials and Methods
4.1. Synthesis of CANDDY Molecules
4.2. Evaluation of Affinity of TUS-007 for KRAS and Proteasome in a Thermal Shift Assay
4.3. Evaluation of KRAS Degradation Induced by TUS-007 in a Cell-Free System
4.4. Cell Proliferation Assay by WST-8
4.5. KRAS Degradation Assays of TUS-007 by Western Blotting
4.6. Analysis of Cell Cycle Distribution
4.7. KRAS Degradation Assays by Enzyme Linked Immunosorbent Assay (ELISA)
4.8. Analysis of TUS-007-Induced Apoptosis by Western Blotting
4.9. Analysis of TUS-007-Induced Apoptotic Cells by Flow Cytometry
4.10. Antitumor Evaluation of TUS-007 in a Subcutaneous Xenograft Model
4.11. Antitumor Evaluation of TUS-007 in an Orthotopic Xenograft Model
4.12. KRAS Degradation Analysis of TUS-007 in Xenograft Tumors
4.13. RAS Family Degradation Assay of TUS-007 in Normal Tissue of Mice
4.14. TUNEL Assay of Xenograft Tumors Treated with TUS-007
4.15. Pharmacokinetic Analysis
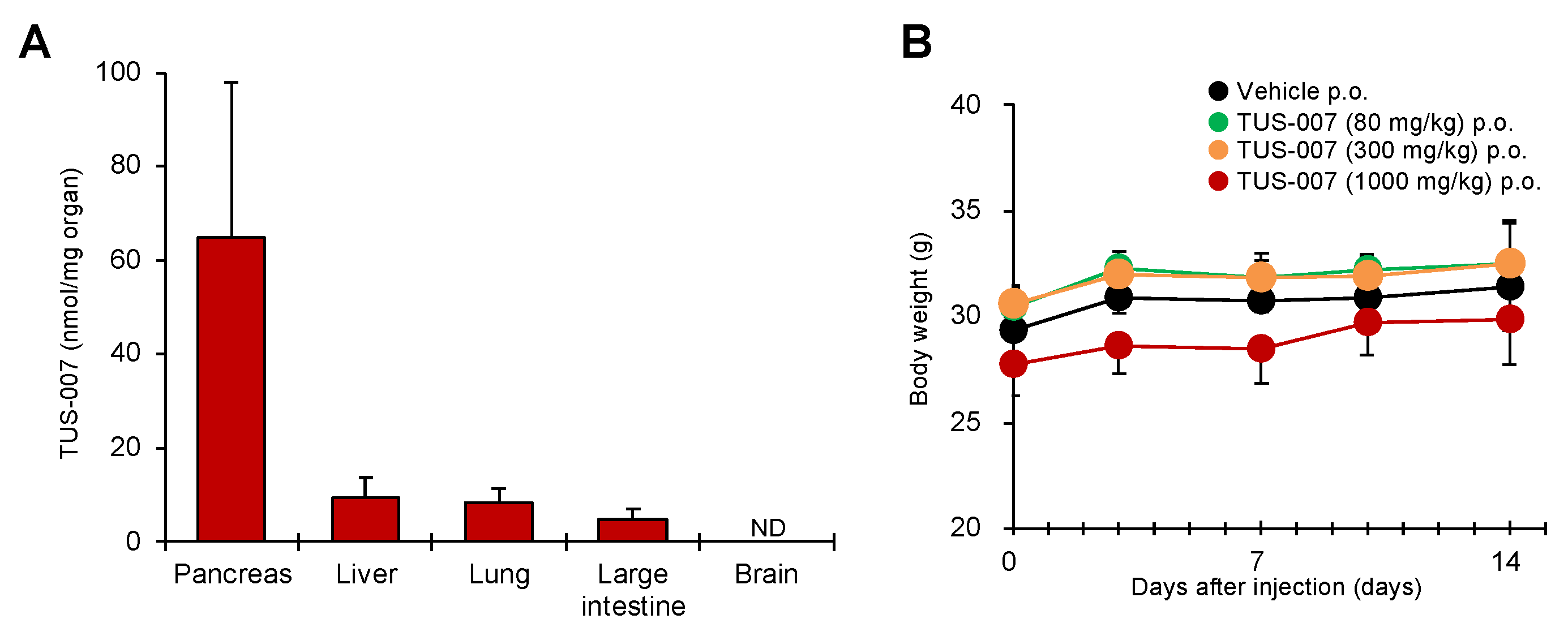
4.16. Tissue Distribution of TUS-007
4.17. In Vivo Toxicity Test of TUS-007
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fujimori, S.; Hirai, N.; Ohashi, H.; Masuoka, K.; Nishikimi, A.; Fukui, Y.; Washio, T.; Oshikubo, T.; Yamashita, T.; Miyamoto-Sato, E. Next-generation sequencing coupled with a cell-free display technology for high-throughput production of reliable interactome data. Sci. Rep. 2012, 2, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New Hope for drugging the “undruggable”. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118570–118586. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging Ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Burke, J.P.; Phan, J.; Burns, M.C.; Olejniczak, E.T.; Waterson, A.G.; Lee, T.; Rossanese, O.W.; Fesik, S.W. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew. Chem. Int. Ed. 2012, 51, 6140–6143. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS alleles: The devil is in the detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Sheridan, C. Grail of RAS cancer drugs within reach. Nat. Biotechnol. 2020, 38, 6–8. [Google Scholar] [CrossRef]
- Shin, S.M.; Choi, D.-K.; Jung, W.K.; Bae, J.; Kim, J.S.; Park, S.-W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090–15104. [Google Scholar] [CrossRef]
- Yin, W.; Rogge, M. Targeting RNA: A transformative therapeutic strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253–eaal5266. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Scudellari, M. Protein-slaying drugs could be the next blockbuster therapies. Nature 2019, 567, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Crews, C.M. Targeted protein degradation: From chemical biology to drug discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras–(PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Churcher, I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones? J. Med. Chem. 2018, 61, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Dohlman, H.G.; Campbell, S.L. Regulation of large and small G proteins by ubiquitination. J. Biol. Chem. 2019, 294, 18613–18623. [Google Scholar] [CrossRef]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted degradation of oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Miyamoto, E.; Ozawa, M. Protein Degradation Inducing Tag and Usage Thereof. U.S. Patent No. 10,976,306 B2, 13 April 2021. [Google Scholar]
- Miyamoto, E. Ras Protein Degradation Inducing Molecule and Pharmaceutical Composition. U.S. Patent No. 11052154B2, 6 July 2021. [Google Scholar]
- Shi, Y.; Long, M.J.C.; Rosenberg, M.M.; Li, S.; Kobiack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc3Arg-Linked Ligands Induce Degradation by Localizing Target Proteins to the 20S Proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor mediated protein degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef]
- Baccanari, D.P.; Daluge, S.; King, R.W. Inhibition of dihydrofolate reductase: Effect of reduced nicotinamide adenine dinucleotide phosphate on the selectivity and affinity of diaminobenzylpyrimidines. Biochemistry 1982, 21, 5068–5075. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Ross, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent small-molecule pan-RAS inhibitors. Cell 2017, 168, 878–889. [Google Scholar] [CrossRef]
- Pant, S.; Hubbard, J.; Martinelli, E.; Bekaii-Saab, T. Clinical update on K-Ras targeted therapy in gastrointestinal cancers. Crit. Rev. Oncol. Hematol. 2018, 130, 78–91. [Google Scholar] [CrossRef]
- Bashore, C.; Prakash, S.; Johnson, M.C.; Conrad, R.J.; Kekessie, I.A.; Scales, S.J.; Ishisoko, N.; Kleinheinz, T.; Liu, P.S.; Popovych, N.; et al. Targeted degradation via direct 26S proteasome recruitment. Nat. Chem. Bio. 2023, 19, 55–63. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyamoto-Sato, E.; Imanishi, S.; Huang, L.; Itakura, S.; Iwasaki, Y.; Ishizaka, M. A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination. Molecules 2023, 28, 5600. https://doi.org/10.3390/molecules28145600
Miyamoto-Sato E, Imanishi S, Huang L, Itakura S, Iwasaki Y, Ishizaka M. A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination. Molecules. 2023; 28(14):5600. https://doi.org/10.3390/molecules28145600
Chicago/Turabian StyleMiyamoto-Sato, Etsuko, Satoshi Imanishi, Lijuan Huang, Shoko Itakura, Yoichi Iwasaki, and Masamichi Ishizaka. 2023. "A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination" Molecules 28, no. 14: 5600. https://doi.org/10.3390/molecules28145600
APA StyleMiyamoto-Sato, E., Imanishi, S., Huang, L., Itakura, S., Iwasaki, Y., & Ishizaka, M. (2023). A First-Class Degrader Candidate Targeting Both KRAS G12D and G12V Mediated by CANDDY Technology Independent of Ubiquitination. Molecules, 28(14), 5600. https://doi.org/10.3390/molecules28145600