

Different Structures—Similar Effect: Do Substituted 5-(4-Methoxyphenyl)-1H-indoles and 5-(4-Methoxyphenyl)-1H-imidazoles Represent a Common Pharmacophore for Substrate Selective Inhibition of Linoleate Oxygenase Activity of ALOX15?

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
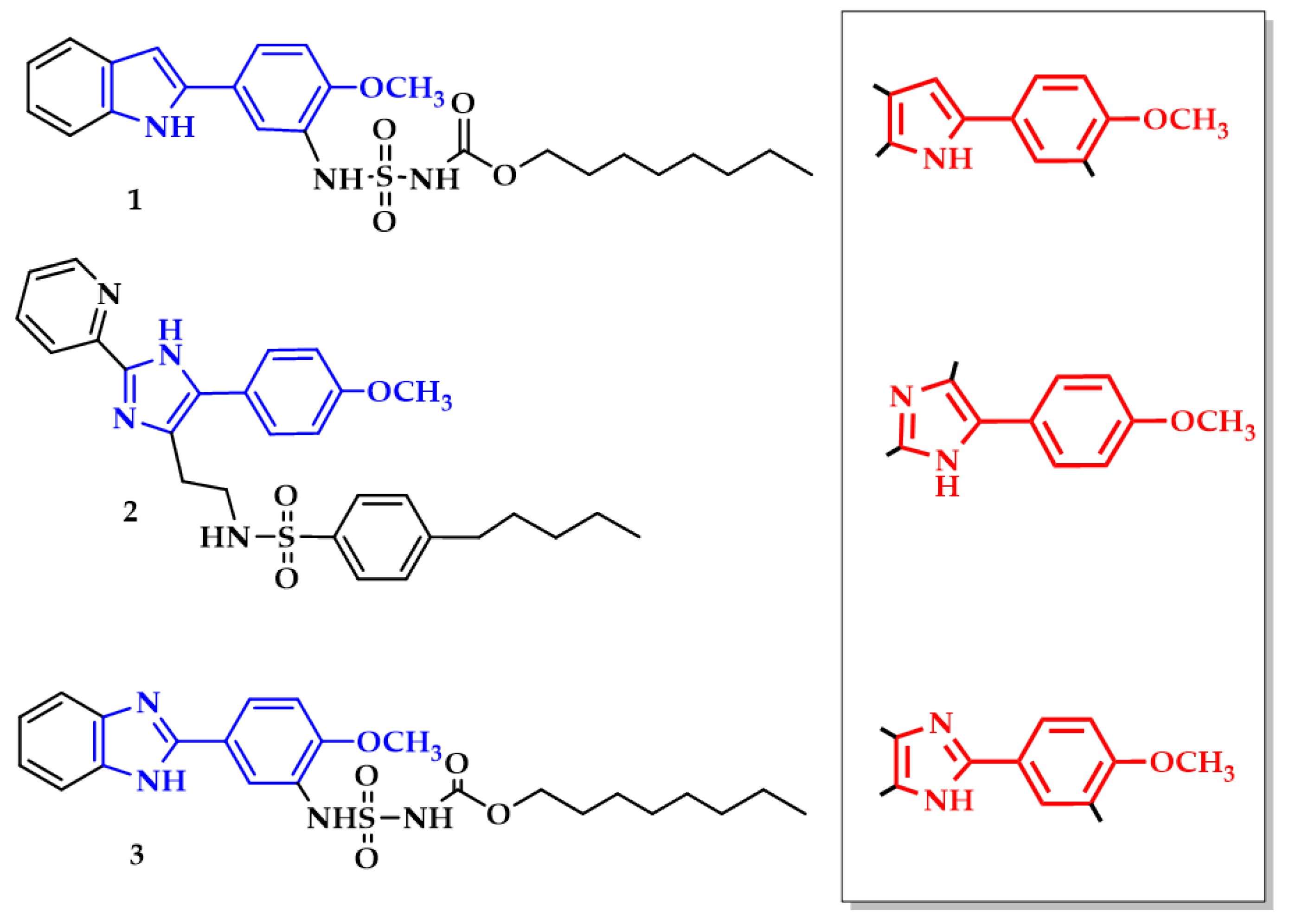
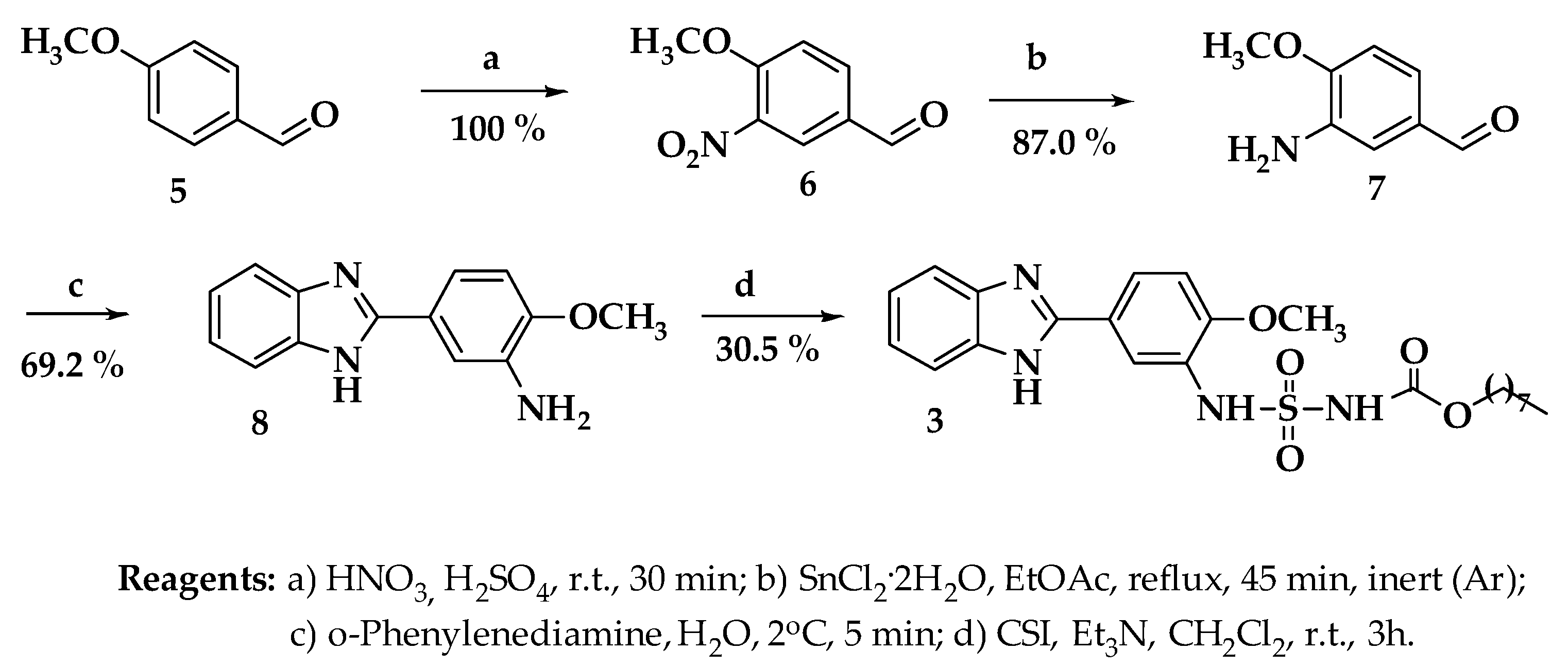
2.1. Chemical Synthesis of Target Compounds
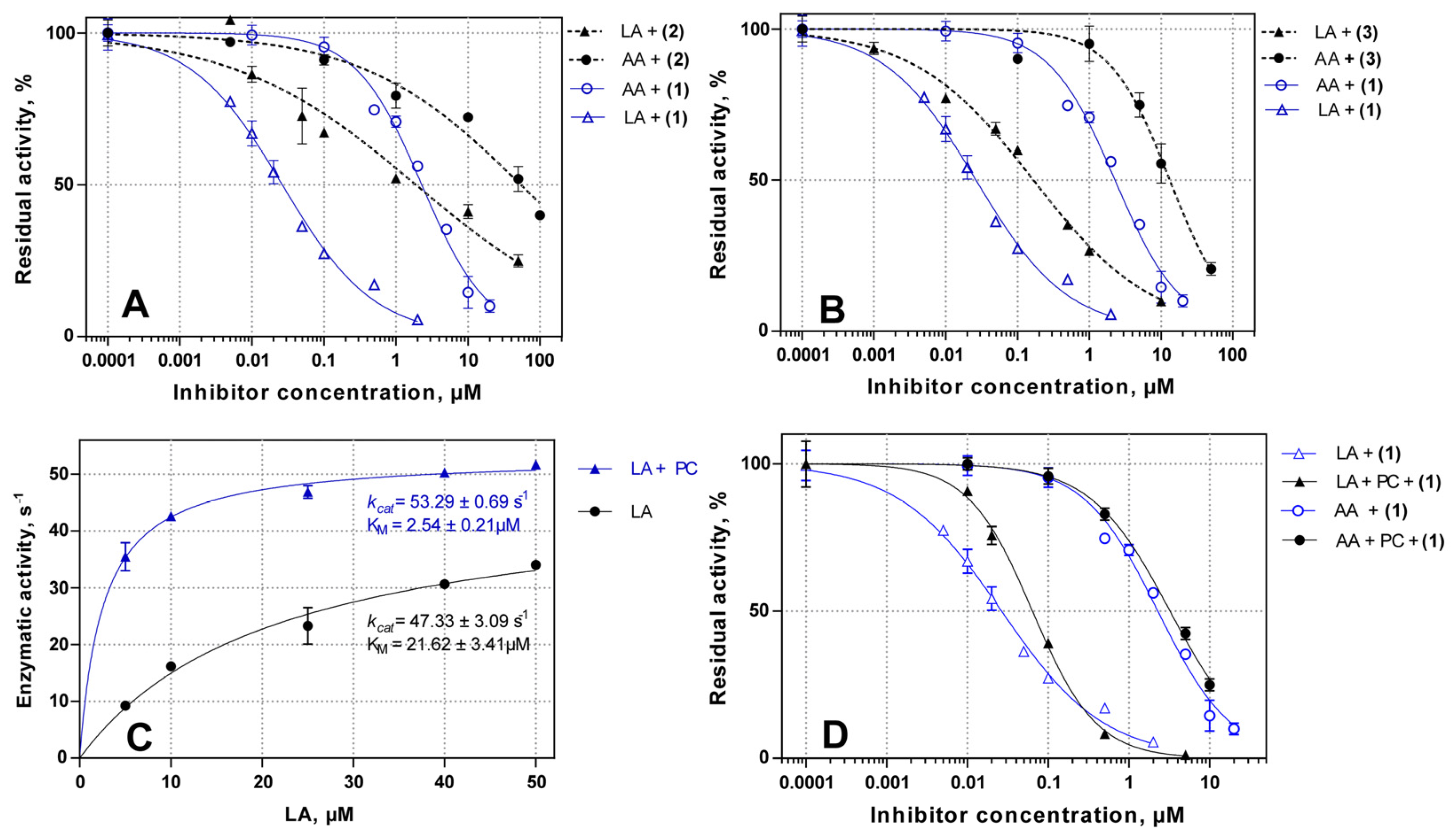
2.2. Inhibitory Potency of Substituted 4-Methoxyphenyl-1H-indole- and 4-Methoxyphenyl-1H-imidazol-Based Inhibitors 1–3
2.3. Substrate Selective Inhibition of LA-Oxygenase Activity of ALOX15 in the Presence of Liposomes
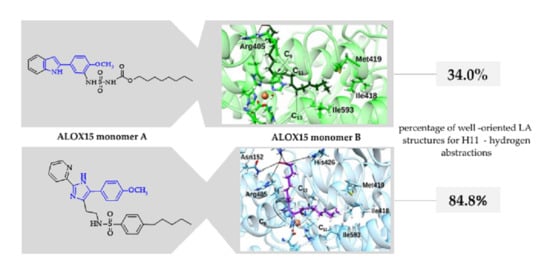
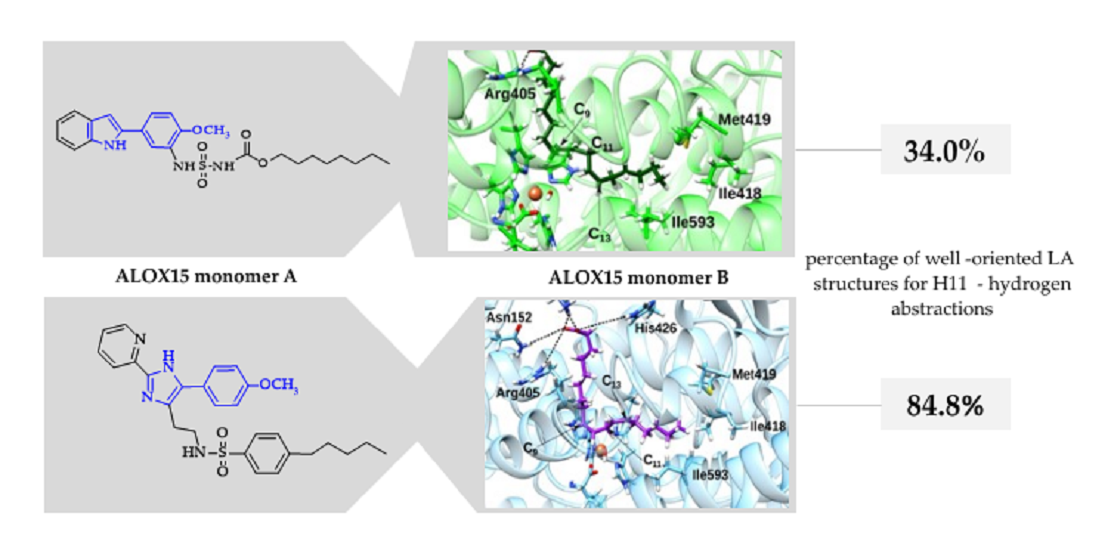
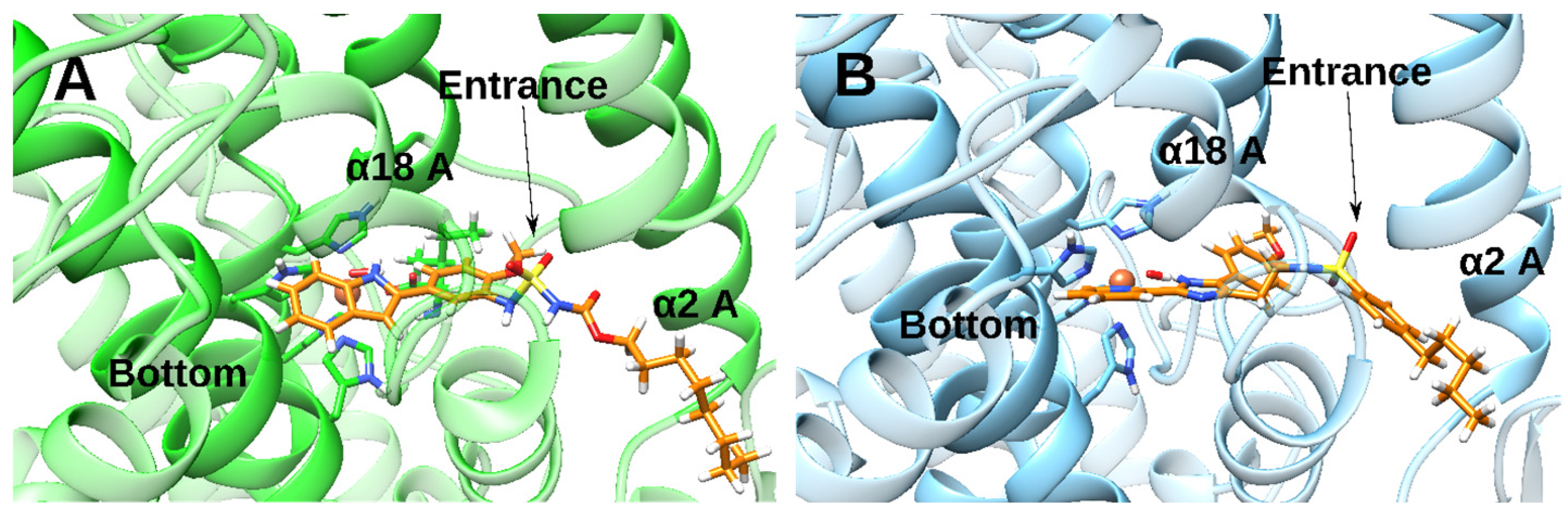
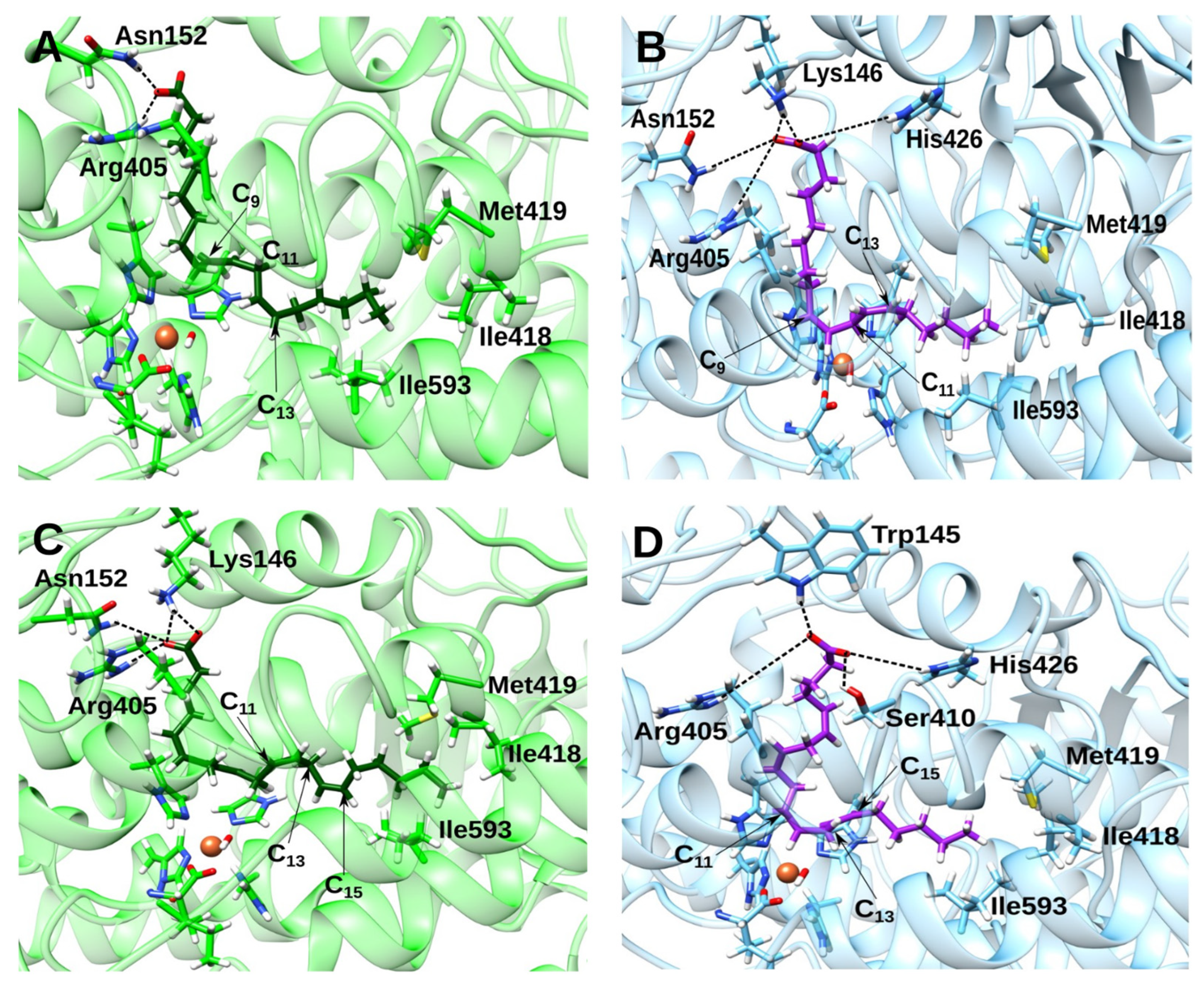
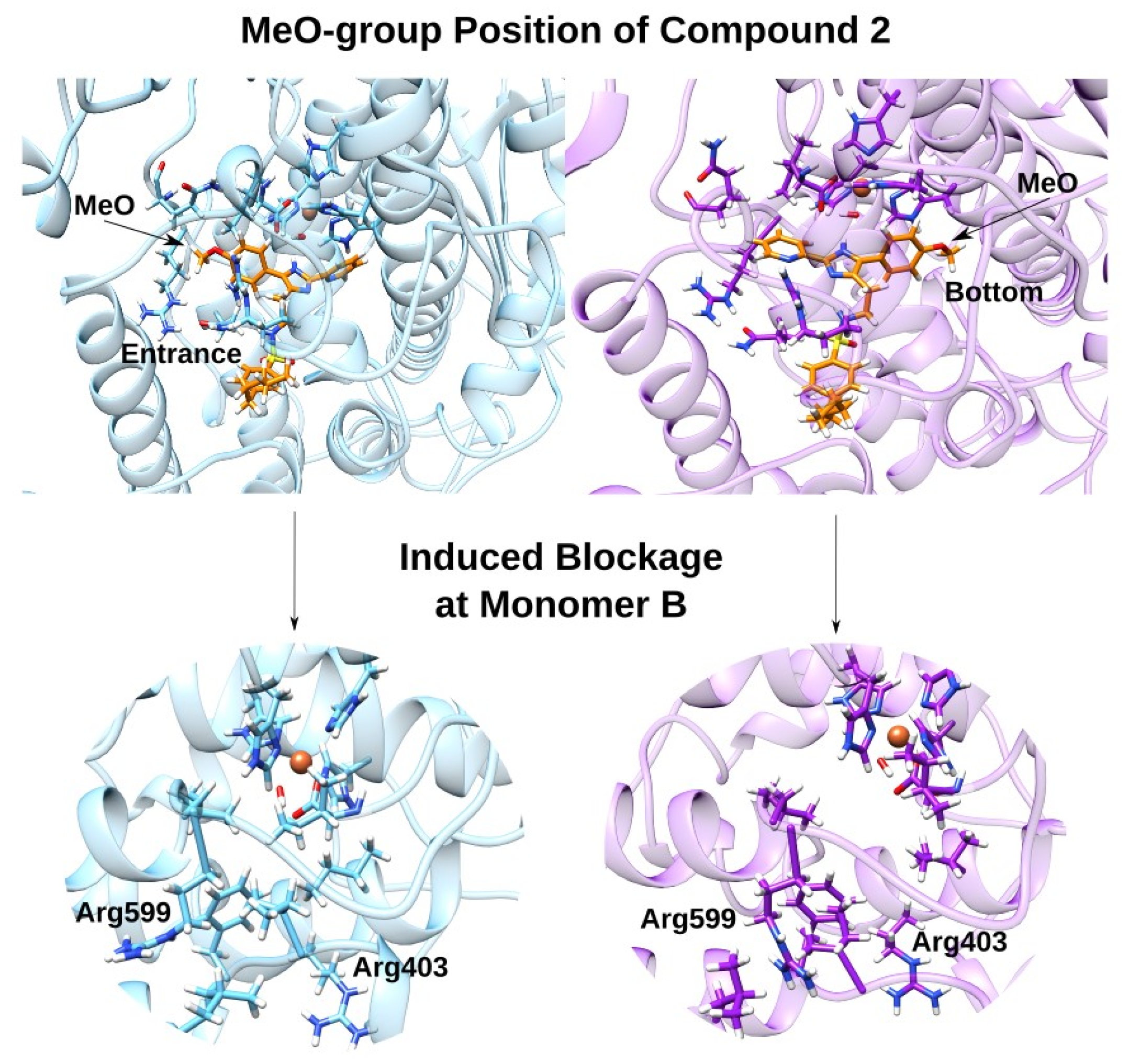
2.4. Docking and MD Simulations of Enzyme–Inhibitor Complexes
2.5. Impact of the Allosteric Inhibitors on Binding of LA and AA along Their MD Simulations
3. Discussion
3.1. Liposomes Do Not Affect the Substrate Selective Inhibition of LA-Oxygenase Activity of ALOX15
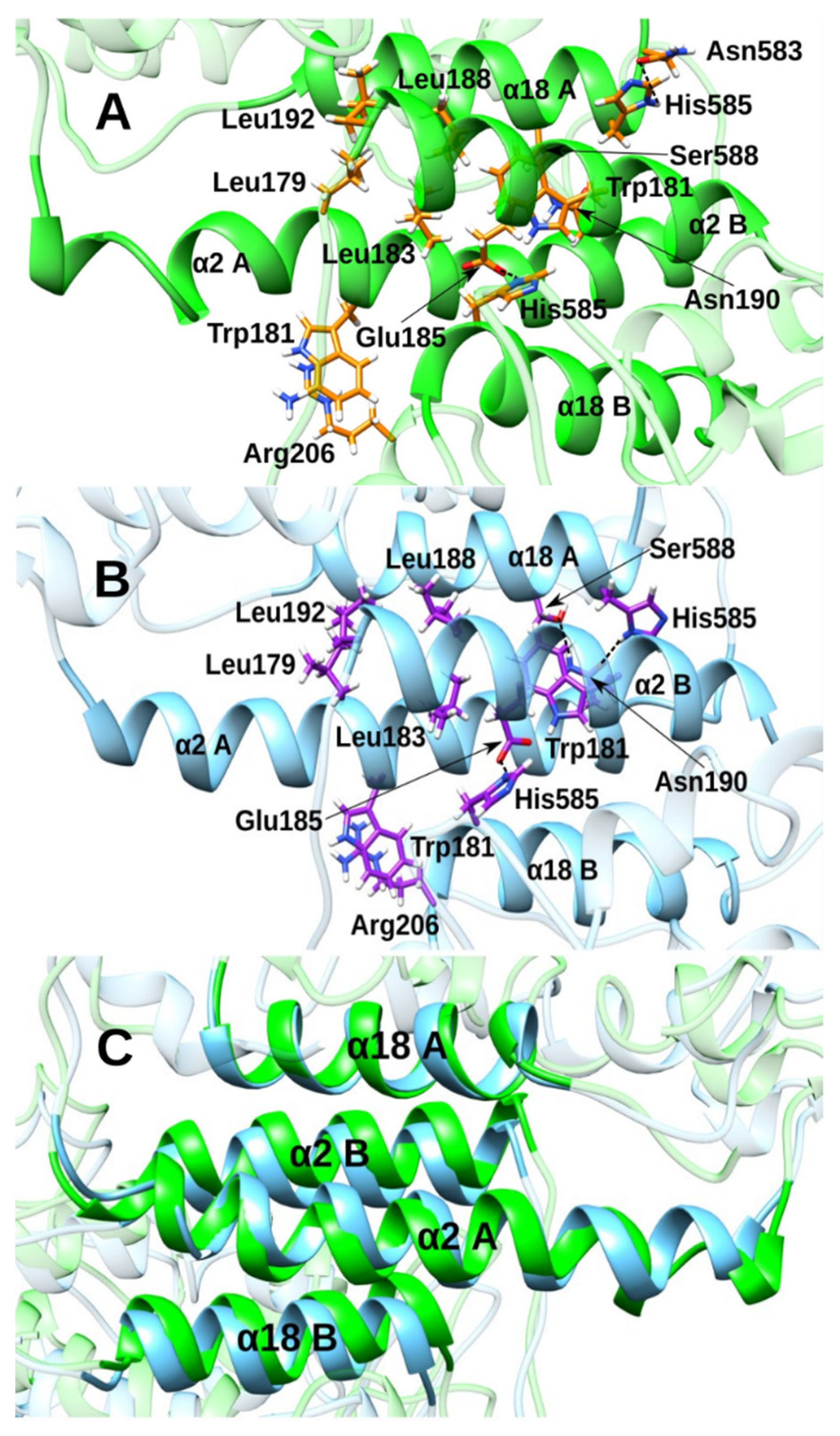
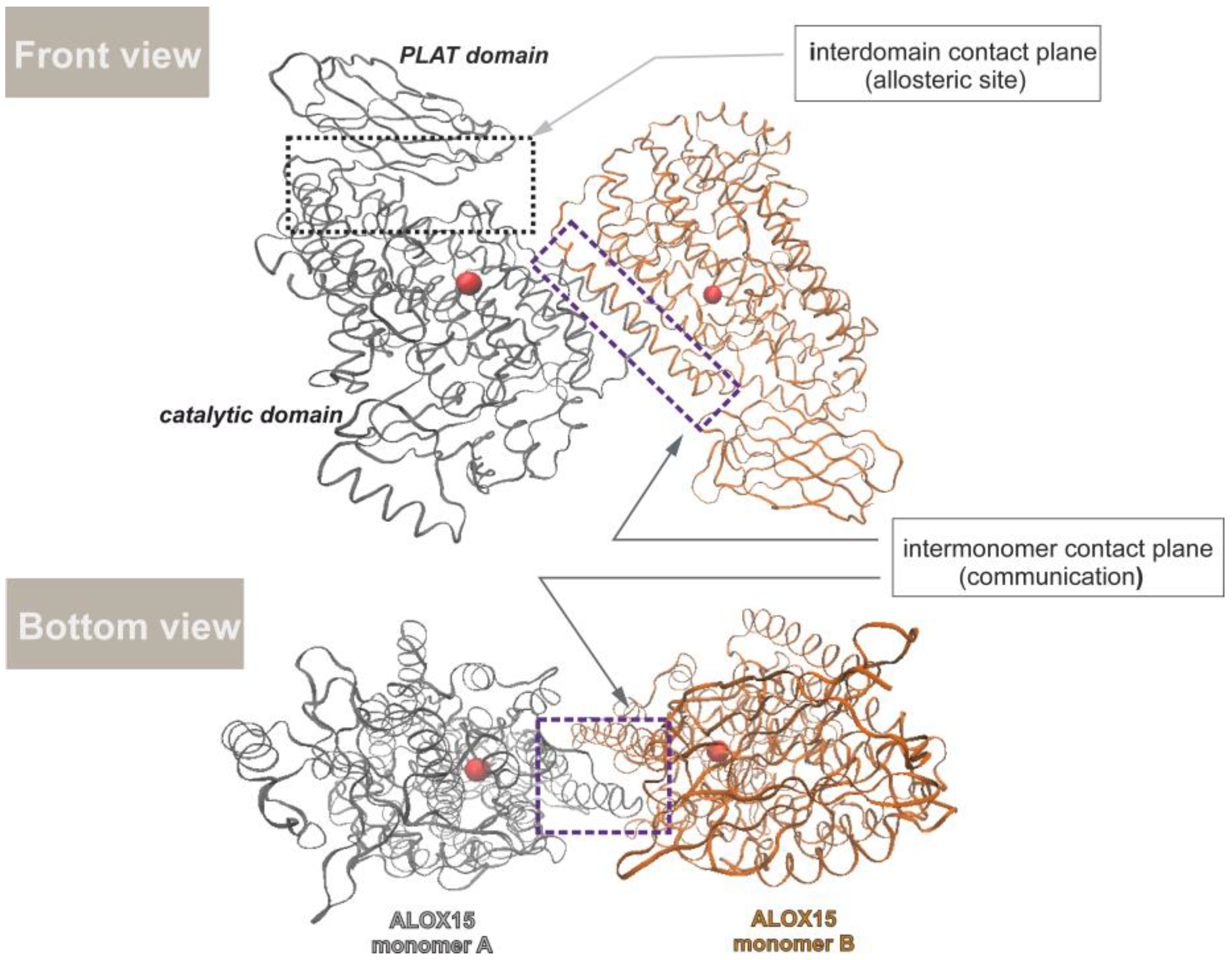
3.2. Effect of the Inhibitor Structure on Cooperativity of Rabbit ALOX15 Monomers
4. Materials and Methods
4.1. Chemistry
4.2. Biochemistry and Biotechnology
4.2.1. Expression of Rabbit ALOX15 in Bioreactor
4.2.2. Enzyme Purification
4.2.3. Liposome Preparations
4.2.4. Rabbit ALOX15 Kinetic Assay
4.2.5. Inhibitor Potency Assay
4.2.6. Molecular Docking Studies
4.2.7. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cyrus, T.; Witztum, J.L.; Rader, D.J.; Tangirala, R.; Fazio, S.; Linton, M.F.; Funk, C.D. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Investig. 1999, 103, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Pidgeon, G.P.; Lysaght, J.; Krishnamoorthy, S.; Reynolds, J.V.; O’Byrne, K.; Nie, D.; Honn, K.V. Lipoxygenase metabolism: Roles in tumor progression and survival. Cancer Metastasis Rev. 2007, 26, 503–524. [Google Scholar]
- Cimen, I.; Tuncay, S.; Banerjee, S. 15-Lipoxygenase-1 expression suppresses the invasive properties of colorectal carcinoma cell lines HCT-116 and HT-29. Cancer Sci. 2009, 100, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, N.; Kwon, Y.G. 15-lipoxygenase-1 in the vasculature: Expanding roles in angiogenesis. Circ. Res. 2008, 102, 143–145. [Google Scholar] [CrossRef]
- Zhao, L.; Grosser, T.; Fries, S.; Kadakia, L.; Wang, H.; Zhao, J.; Falotico, R. Lipoxygenase and prostaglandin G/H synthase cascades in cardiovascular disease. Expert Rev. Clin. Immunol. 2006, 2, 649–658. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli JP, F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar]
- Sears, D.D.; Miles, P.D.; Chapman, J.; Ofrecio, J.M.; Almazan, F.; Thapar, D.; Miller, Y.I. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS ONE 2009, 4, e7250. [Google Scholar] [CrossRef]
- Lieb, D.C.; Brotman, J.J.; Hatcher, M.A.; Aye, M.S.; Cole, B.K.; Haynes, B.A.; Wohlgemuth, S.D.; Fontana, M.A.; Beydoun, H.; Nadler, J.L.; et al. Adipose tissue 12/15 lipoxygenase pathway in human obesity and diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E1713–E1720. [Google Scholar] [CrossRef]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kühn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Vangaveti, V.; Shashidhar, V.; Collier, F.; Hodge, J.; Rush, C.; Malabu, U.; Baune, B.; Kennedy, R.L. 9- and 13-HODE regulate fatty acid binding protein-4 in human macrophages, but does not involve HODE/GPR132 axis in PPAR-γ regulation of FABP4. Ther. Adv. Endocrinol. Metab. 2018, 9, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef]
- Souza, F.d.C.; Ferreira, M.T.; Colquhoun, A. Influence of Lipoxygenase Inhibition on Glioblastoma Cell Biology. Int. J. Mol. Sci. 2020, 21, 8395. [Google Scholar] [CrossRef]
- Hsi, L.C.; Wilson, L.C.; Eling, T.E. Opposing effects of 15-lipoxygenase-1 and -2 metabolites on MAPK signaling in prostate. Alteration in peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2002, 277, 40549–40556. [Google Scholar] [CrossRef]
- Spite, M.; Serhan, C.N. Novel lipid mediators promote resolution of acute inflammation: Impact of aspirin and statins. Circ. Res. 2010, 107, 1170–1184. [Google Scholar] [CrossRef]
- Sekheri, M.; El Kebir, D.; Edner, N.; Filep, J.G. 15-Epi-LXA(4) and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 7971–7980. [Google Scholar] [CrossRef]
- Leghmar, K.; Cenac, N.; Rolland, M.; Martin, H.; Rauwel, B.; Bertrand-Michel, J.; Le Faouder, P.; Bénard, M.; Casper, C.; Davrinche, C.; et al. Cytomegalovirus Infection Triggers the Secretion of the PPARγ Agonists 15-Hydroxyeicosatetraenoic Acid (15-HETE) and 13-Hydroxyoctadecadienoic Acid (13-HODE) in Human Cytotrophoblasts and Placental Cultures. PLoS ONE 2015, 10, e0132627. [Google Scholar] [CrossRef]
- Rai, G.; Joshi, N.; Jung, J.E.; Liu, Y.; Schultz, L.; Yasgar, A.; Perry, S.; Diaz, G.; Zhang, Q.; Kenyon, V.; et al. Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J. Med. Chem. 2014, 57, 4035–4048. [Google Scholar] [CrossRef]
- Ngu, K.; Weinstein, D.S.; Liu, W.; Langevine, C.; Combs, D.W.; Zhuang, S.; Chen, X.; Madsen, C.S.; Harper, T.W.; Ahmad, S.; et al. Pyrazole-based sulfonamide and sulfamides as potent inhibitors of mammalian 15-lipoxygenase. Bioorg. Med. Chem. Lett. 2011, 21, 4141–4145. [Google Scholar] [CrossRef]
- Weinstein, D.S.; Liu, W.; Ngu, K.; Langevine, C.; Combs, D.W.; Zhuang, S.; Chen, C.; Madsen, C.S.; Harper, T.W.; Robl, J.A. Discovery of selective imidazole-based inhibitors of mammalian 15-lipoxygenase: Highly potent against human enzyme within a cellular environment. Bioorg. Med. Chem. Lett. 2007, 17, 5115–5120. [Google Scholar] [CrossRef] [PubMed]
- Sendobry, S.M.; Cornicelli, J.A.; Welch, K.; Bocan, T.; Tait, B.; Trivedi, B.K.; Colbry, N.; Dyer, R.D.; Feinmark, S.J.; Daugherty, A. Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br. J. Pharmacol. 1997, 120, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Barvian, N.C.; O’brian, P.M.; Patt, W.C.; Picard, J.A.; Sliskovic, D.R. 1,2,4-Trisubstituted Benzenes as Inhibitors of 15-Lipoxygenase. Patent WO 096298 A2, 20 December 2001. [Google Scholar]
- Connor, D.; Roark, W.; Sorenson, R. Indole and Benzimidazole 15-Lipoxygenase Inhibitors. U.S. Patent 0038943 A1, 26 February 2004. [Google Scholar]
- Eleftheriadis, N.; Neochoritis, C.G.; Leus, N.G.; van der Wouden, P.E.; Dömling, A.; Dekker, F.J. Rational Development of a Potent 15-Lipoxygenase-1 Inhibitor with in Vitro and ex Vivo Anti-inflammatory Properties. J. Med. Chem. 2015, 58, 7850–7862. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.S.; Liu, W.; Gu, Z.; Langevine, C.; Ngu, K.; Fadnis, L.; Combs, D.W.; Sitkoff, D.; Ahmad, S.; Zhuang, S.; et al. Tryptamine and homotryptamine-based sulfonamides as potent and selective inhibitors of 15-lipoxygenase. Bioorg. Med. Chem. Lett. 2005, 15, 1435–1440. [Google Scholar] [CrossRef]
- Golovanov, A.; Zhuravlev, A.; Cruz, A.; Aksenov, V.; Shafiullina, R.; Kakularam, K.R.; Lluch, J.M.; Kuhn, H.; Gonzalez-Lafont, A.; Ivanov, I. N-Substituted 5-(1H-Indol-2-yl)-2-methoxyanilines Are Allosteric Inhibitors of the Linoleate Oxygenase Activity of Selected Mammalian ALOX15 Orthologs: Mechanism of Action. J. Med. Chem. 2022, 65, 1979–1995. [Google Scholar] [CrossRef]
- Choi, J.; Chon, J.K.; Kim, S.; Shin, W. Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins 2008, 70, 1023–1032. [Google Scholar] [CrossRef]
- Shang, W.; Ivanov, I.; Svergun, D.I.; Borbulevych, O.Y.; Aleem, A.M.; Stehling, S.; Jankun, J.; Kuhn, H.; Skrzypczak-Jankun, E. Probing dimerization and structural flexibility of mammalian lipoxygenases by small-angle X-ray scattering. J. Mol. Biol. 2011, 409, 654–668. [Google Scholar] [CrossRef]
- Ivanov, I.; Cruz, A.; Zhuravlev, A.; Di Venere, A.; Nicolai, E.; Stehling, S.; Lluch, J.M.; González-Lafont, À.; Kuhn, H. Conformational Heterogeneity and Cooperative Effects of Mammalian ALOX15. Int. J. Mol. Sci. 2021, 22, 3285. [Google Scholar] [CrossRef]
- Mobbs, J.I.; Black, K.A.; Tran, M.; Venugopal, H.; Holman, T.R.; Holinstat, M.; Thal, D.M.; Glukhova, A. Cryo-EM structures of human arachidonate 12S-Lipoxygenase (12-LOX) bound to endogenous and exogenous inhibitors. bioRxiv 2023. [Google Scholar] [CrossRef]
- Sokhraneva, V.A.; Yusupova, D.A.; Boriskin, V.S.; Groza, N.V. Obtaining substituted phenol derivatives with potential antimicrobial activity. Fine Chem. Technol. 2022, 17, 210–230. [Google Scholar] [CrossRef]
- Siwach, A.; Verma, P.K. Synthesis and therapeutic potential of imidazole containing compounds. BMC Chem. 2021, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Ermolenko, L.; Al-Mourabit, A. Selective autoxidation of benzylamines: Application to the synthesis of some nitrogen heterocycles. Green Chem. 2013, 15, 2713–2717. [Google Scholar] [CrossRef]
- Bahrami, K.; Khodaei, M.M.; Kavianinia, I. A Simple and Efficient One-Pot Synthesis of 2-Substituted Benzimidazoles. Synthesis 2007, 2007, 547–550. [Google Scholar] [CrossRef]
- Xiao, T.; Xiong, S.; Xie, Y.; Dong, X.; Zhou, L. Copper-catalyzed synthesis of benzazoles via aerobic oxidative condensation of o-amino/mercaptan/hydroxyanilines with benzylamines. RSC Adv. 2013, 3, 15592–15595. [Google Scholar] [CrossRef]
- Chen, G.-F.; Dong, X.-Y.; Meng, F.-Z.; Chen, B.-H.; Li, J.-T.; Wang, S.-X.; Bai, G.-Y. Synthesis of 2-Substituted Benzimidazoles Catalyzed by FeCl3/Al2O3 Under Ultrasonic Irradiation. Lett. Org. Chem. 2011, 8, 464–469. [Google Scholar] [CrossRef]
- Di Venere, A.; Horn, T.; Stehling, S.; Mei, G.; Masgrau, L.; González-Lafont, À.; Kuhn, H.; Ivanov, I. Role of Arg403 for thermostability and catalytic activity of rabbit 12/15-lipoxygenase. Biochim. Biophys. Acta 2013, 1831, 1079–1088. [Google Scholar] [CrossRef]
- Andersson, E.; Schain, F.; Svedling, M.; Claesson, H.E.; Forsell, P.K. Interaction of human 15-lipoxygenase-1 with phosphatidylinositol bisphosphates results in increased enzyme activity. Biochim. Biophys. Acta 2006, 1761, 1498–1505. [Google Scholar] [CrossRef]
- Ivanov, I.; Shang, W.; Toledo, L.; Masgrau, L.; Svergun, D.I.; Stehling, S.; Gomez, H.; Di Venere, A.; Mei, G.; Lluch, J.M.; et al. Ligand-induced formation of transient dimers of mammalian 12/15-lipoxygenase: A key to allosteric behavior of this class of enzymes? Proteins 2012, 80, 703–712. [Google Scholar] [CrossRef]
- Saura, P.; Suardíaz, R.; Masgrau, L.; Lluch, J.M.; González-Lafont, À. Unraveling How Enzymes Can Use Bulky Residues to Drive Site-Selective C–H Activation: The Case of Mammalian Lipoxygenases Catalyzing Arachidonic Acid Oxidation. ACS Catal. 2014, 4, 4351–4363. [Google Scholar] [CrossRef]
- Adel, S.; Karst, F.; González-Lafont, À.; Pekarova, M.; Saura, P.; Masgrau, L.; Lluch, J.M.; Stehling, S.; Horn, T.; Kuhn, H.; et al. Evolutionary alteration of ALOX15 specificity optimizes the biosynthesis of antiinflammatory and proresolving lipoxins. Proc. Natl. Acad. Sci. USA 2016, 113, E4266–E4275. [Google Scholar] [CrossRef]
- Ivanov, I.; Di Venere, A.; Horn, T.; Scheerer, P.; Nicolai, E.; Stehling, S.; Richter, C.; Skrzypczak-Jankun, E.; Mei, G.; Maccarrone, M.; et al. Tight association of N-terminal and catalytic subunits of rabbit 12/15-lipoxygenase is important for protein stability and catalytic activity. Biochim. Biophys. Acta 2011, 1811, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.; Wiesner, R.; Kuhn, H. Investigations into calcium-dependent membrane association of 15-lipoxygenase-1. Mechanistic roles of surface-exposed hydrophobic amino acids and calcium. J. Biol. Chem. 2004, 279, 3717–3725. [Google Scholar] [CrossRef] [PubMed]
- Dainese, E.; Sabatucci, A.; van Zadelhoff, G.; Angelucci, C.B.; Vachette, P.; Veldink, G.A.; Agro, A.F.; Maccarrone, M. Structural stability of soybean lipoxygenase-1 in solution as probed by small angle X-ray scattering. J. Mol. Biol. 2005, 349, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Börner, F.; Pace, S.; Jordan, P.M.; Gerstmeier, J.; Gomez, M.; Rossi, A.; Gilbert, N.C.; Newcomer, M.E.; Werz, O. Allosteric Activation of 15-Lipoxygenase-1 by Boswellic Acid Induces the Lipid Mediator Class Switch to Promote Resolution of Inflammation. Adv. Sci. 2022, 10, e2205604. [Google Scholar] [CrossRef]
- Kozlov, N.; Humeniuk, L.; Ufer, C.; Ivanov, I.; Golovanov, A.; Stehling, S.; Heydeck, D.; Kuhn, H. Functional characterization of novel ALOX15 orthologs representing key steps in mammalian evolution supports the Evolutionary Hypothesis of reaction specificity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 372–385. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pK(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Tosco, P. A mechanistic hypothesis for the aspirin-induced switch in lipid mediator production by cyclooxygenase-2. J. Am. Chem. Soc. 2013, 135, 10404–10410. [Google Scholar] [CrossRef]
- Ivanov, I.; Golovanov, A.B.; Ferretti, C.; Canyelles-Niño, M.; Heydeck, D.; Stehling, S.; Lluch, J.M.; González-Lafont, À.; Kühn, H. Mutations of Triad Determinants Changes the Substrate Alignment at the Catalytic Center of Human ALOX5. ACS Chem. Biol. 2019, 14, 2768–2782. [Google Scholar] [CrossRef]
- Cruz, A.; Di Venere, A.; Mei, G.; Zhuravlev, A.; Golovanov, A.; Stehling, S.; Heydeck, D.; Lluch, J.M.; González-Lafont, À.; Kuhn, H.; et al. A role of Gln596 in fine-tuning mammalian ALOX15 specificity, protein stability and allosteric properties. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158680. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; MacKerell, A.D., Jr. Automated conformational energy fitting for force-field development. J. Mol. Model. 2008, 14, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges—The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, S.; Gotz, A.W.; Walker, R.C. SPFP: Speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Leach, A.R. Molecular Modeling: Principles and Applications; Eddison Wesley Longman Limited: Essex, UK, 1996; p. 595. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H. Numerical-integration of cartesian equations of motion of a system with contraints—Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50(LA), µM | IC50(AA), µM | IC50(LA)/IC50(AA) Ratio |
|---|---|---|---|
| 1 | 0.03 ± 0.02 | 2.79 ± 0.18 | 0.010 |
| 1 + PC | 0.06 ± 0.04 | 3.45 ± 0.10 | 0.018 |
| 2 | 1.8 ± 1.2 | 54.02 ± 2.50 | 0.032 |
| 3 | 0.16 ± 0.02 | 13.21 ± 1.59 | 0.011 |
 | |||
|---|---|---|---|
| Group | Type of Interaction | Interaction Partner | Distance |
| A | π-π | His361 (sidechain) His366 (sidechain) | |
| B, NH | hydrogen bond | Fe(III)-OH− | d(H1-OH) = 1.982 Å |
| C, CH3O | hydrogen bonds | Arg403 (sidechain NH2) Gln596 (sidechain NH2) Gln601 (sidechain NH2 and backbone NH) | d(O1-HH12-Arg403) = 3.793 Å d(O1-HH22-Arg403) = 4.301 Å d(O1-HE21-Gln596) = 5.451 Å d(O1-HE22-Gln596) = 5.229 Å d(O1-HE22-Gln601) = 4.341 Å d(O1-H-Gln601) = 5.237 Å |
| D, NH | hydrogen bond | Arg403 | d(N4-HH11-Arg403) = 3.254 Å |
| E, SO2 | hydrogen bond | Leu408 (backbone NH) Gln596 (sidechain NH2) | d(O3-H-Leu408) = 3.421 Å d(O2-HE21-Gln596) = 3.731 Å |
 | |||
|---|---|---|---|
| Group | Type of Interaction | Interaction Partner | Distance |
| A, NH | hydrogen bond | Fe(III)-OH− | d(H1-OH) = 2.116 Å |
| B, CH3O | hydrogen bonds | Gln596 (sidechain NH2) Arg403 (sidechain NH2) | d(O1-HE22-Gln596) = 3.534 Å d(O1-HH11-Arg403) = 4.465 Å |
| B, CH3O | electrostatic | Arg599 | ** |
| C, SO2 | hydrogen bond | Gln596 (side chain NH2) | d(O2-HE21-Gln596) = 2.059 Å |
| D, CO | hydrogen bond | Leu408 (backbone NH) | (O4-H-Leu408) = 2.345 Å |
| Substrate | No Inhibitor | Compound 2 | Compound 1 |
|---|---|---|---|
| AA | 97.78 | 93.38 | 87.90 |
| LA | 90.09 | 84.80 | 34.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuravlev, A.; Cruz, A.; Aksenov, V.; Golovanov, A.; Lluch, J.M.; Kuhn, H.; González-Lafont, À.; Ivanov, I. Different Structures—Similar Effect: Do Substituted 5-(4-Methoxyphenyl)-1H-indoles and 5-(4-Methoxyphenyl)-1H-imidazoles Represent a Common Pharmacophore for Substrate Selective Inhibition of Linoleate Oxygenase Activity of ALOX15? Molecules 2023, 28, 5418. https://doi.org/10.3390/molecules28145418
Zhuravlev A, Cruz A, Aksenov V, Golovanov A, Lluch JM, Kuhn H, González-Lafont À, Ivanov I. Different Structures—Similar Effect: Do Substituted 5-(4-Methoxyphenyl)-1H-indoles and 5-(4-Methoxyphenyl)-1H-imidazoles Represent a Common Pharmacophore for Substrate Selective Inhibition of Linoleate Oxygenase Activity of ALOX15? Molecules. 2023; 28(14):5418. https://doi.org/10.3390/molecules28145418
Chicago/Turabian StyleZhuravlev, Alexander, Alejandro Cruz, Vladislav Aksenov, Alexey Golovanov, José M. Lluch, Hartmut Kuhn, Àngels González-Lafont, and Igor Ivanov. 2023. "Different Structures—Similar Effect: Do Substituted 5-(4-Methoxyphenyl)-1H-indoles and 5-(4-Methoxyphenyl)-1H-imidazoles Represent a Common Pharmacophore for Substrate Selective Inhibition of Linoleate Oxygenase Activity of ALOX15?" Molecules 28, no. 14: 5418. https://doi.org/10.3390/molecules28145418
APA StyleZhuravlev, A., Cruz, A., Aksenov, V., Golovanov, A., Lluch, J. M., Kuhn, H., González-Lafont, À., & Ivanov, I. (2023). Different Structures—Similar Effect: Do Substituted 5-(4-Methoxyphenyl)-1H-indoles and 5-(4-Methoxyphenyl)-1H-imidazoles Represent a Common Pharmacophore for Substrate Selective Inhibition of Linoleate Oxygenase Activity of ALOX15? Molecules, 28(14), 5418. https://doi.org/10.3390/molecules28145418