1. Introduction
Communities of microorganisms reside within humans, with the gut being the most densely colonised site [
1]. Microbial communities that cohabit the intestinal tract, known as gut microbiota, can be beneficial, neutral, or detrimental to the host. Gut microbiota can impact human health by mediating physiological homeostasis through immune function, digestion, vitamin synthesis, and pathogen colonisation [
2]. The effects are exerted through interfaces between gut microbiota and intestinal epithelial cells, the immune system, and dietary intake [
2,
3,
4]. Gut microbiota diversity and abundance is associated with health through processes such as reduced inflammation and disorders linked to inflammatory events [
3].
Microbial diversity is required for healthy gut function, with a loss of diversity associated with an increased risk of disease [
5]. In healthy adults, the gut microbiota comprises eight dominant phyla, including Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Fusobacteria, and Verrucomicrobia. The Firmicutes and Bacteroidetes together represent 90% of gut microbiota [
6,
7]. Bacteria in these phyla perform diverse roles in regulating host health [
7].
Bifidobacterium species comprise only 2% of the total gut microbiota but play a significant role in the breakdown of complex carbohydrates [
8,
9], protecting the host against pathogens through competitive exclusion, modulating the immune system, and providing vitamins and other nutrients for the host [
10]. The genus
Lactobacillus includes species that produce short-chain fatty acids (SCFAs) and regulate intestinal transit [
11,
12]. The production of SCFAs by these bacteria can improve insulin sensitivity and protect against diet-induced obesity [
9]. Other bacteria of importance to human health include
Roseburia,
Faecalibacterium,
Ruminococcus, and
Bacteroides [
13,
14,
15].
The presence of microbes and their interaction with the host and other gut microbes in the gastrointestinal tract can lead to adverse health outcomes [
1]. Metabolic products from gut microbiota have been linked to an increased risk of major adverse cardiovascular events [
16] and type 2 diabetes [
17]. Supporting an optimal composition of gut microbiota is vital for human health [
18]. The gut microbiota in healthy individuals includes shared common microbiota, with variations across age, ethnicity, and socioeconomic status [
19,
20,
21,
22,
23]. Although relatively stable throughout adulthood, variations in the common microbial genera are observed, with major shifts identified in older adults [
23,
24,
25,
26]. The composition of the gut microbiome is also affected by lifestyle [
27,
28]. Cigarette smoking and alcohol consumption impact the gut microbiome with changes identified with the cessation of smoking, and heavy alcohol consumption [
27,
28].
Diet has also been shown to be a significant factor affecting gut microbial composition [
18,
29]. Functional foods aim to improve health and wellbeing and have been shown to interact with gut microbiota [
30,
31,
32]. Prebiotics are non-digestible dietary ingredients that stimulate the growth of microbes in the gut after fermentation [
33]. Recent research identifies prebiotics that enhance microbial diversity to promote health and defend against the dysbiosis of gut microbiota [
4,
5]. Arabinoxylans (AXs) are the non-digestible fibre of cereal grains, including wheat, rice, rye, maize, and sorghum [
9]. The structures of AXs have been shown to influence substrate fermentation and degradation by gut microbiota [
34]. Francios et al. [
35] and Kjolbaek et al. [
36] showed that supplementation with wheat AX/AX oligosaccharides in humans led to changes in the microbiota profile, including increased beneficial bacteria, such as
Bifidobacterium. Lachnospiraceae have also been shown to be increased in AX-fed mice [
37]. The extent and mechanism of the effects of AXs on gut microbiota differs due to variations in the chemical structure and molecular weight [
38]. Recent in vivo research has focused on linear AXs due to their bioavailability and simple structures [
9,
39].
Rice bran arabinoxylan compound (RBAC), known as BioBran, MGN-3, and Ribraxx, is a modified AX compound [
40]. RBAC consists of a water-soluble hemicellulose-
fraction (degree of polymerisation approximately 200), partially decomposed by enzymes extracted from a cultured medium of
Lentinula edodes (shiitake mushrooms) [
40]. The production of RBAC typically involves the preparation of rice bran in a growth medium with sterilisation, followed by bioconversion or fermentation with
L. edodes enzyme for a set time before the extraction, purification, and drying of the compounds into powder form [
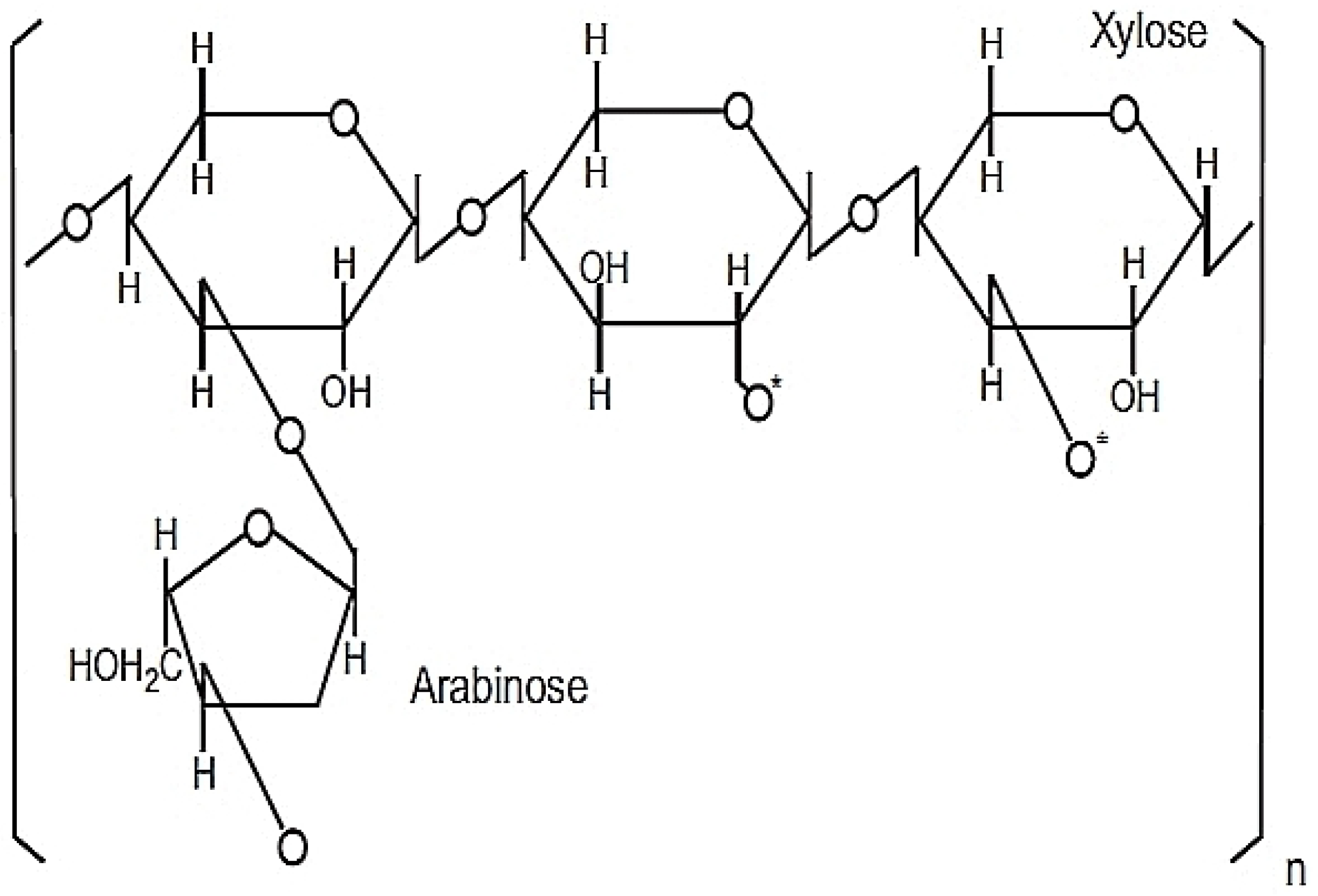
31]. The active ingredients of RBAC comprise heteropolysaccharides with a primary structure of arabinoxylan with a xylose in its main chain and an arabinose polymer side chain as shown in
Figure 1 [
41,
42].
RBAC is safe to consume with a median lethal dose (LD
50) above 36 g/kg body weight, and the no-observed-adverse-effect level is above 200 mg/kg/day [
43]. The compound also tested negative in a reverse mutagenicity array and did not elicit any allergic response in a controlled antigenicity test [
43]. A safety study conducted with 24 healthy volunteers given RBAC at different concentrations of 15, 30, and 45 mg/kg body weight daily for one month also reported no abnormalities detected in blood chemistry analysis, including liver enzymes, as compared to the baseline [
44]. RBAC also has an excellent safety record. In the systematic review of RBAC, no adverse events at the typical dosage of 1–3 g/day were reported in any of the included clinical trials (total 11) or clinical case reports (total 14) [
45]. Hence, RBAC is considered safe to consume with no known side effect at the typical dosages used in research and clinical settings.
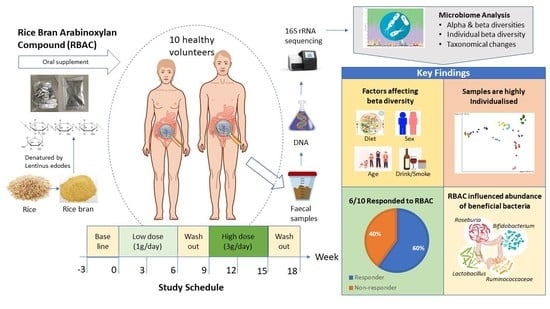
RBAC has been widely used as a dietary supplement, most notable for its immunomodulating property [
45]. However, knowledge of the effect of RBAC on gut microbiota, particularly in healthy adults, is lacking. Therefore, the purpose of this basic science study was to explore possible changes in gut microbiota in response to dietary RBAC supplementation in healthy adults. Additional observational analysis was also performed to assess the abundance of the beneficial bacteria
Bifidobacterium,
Lactobacillus,
Roseburia,
Ruminococcaceae, and
Faecalibacterium.
2. Results
2.1. Participant Characteristics and Dietary Intake
Characteristics of the study participants and their dietary intake from the screening survey and Australian Eating Survey® (AES) are summarised in
Table 1. The participant group consisted of balanced numbers of males and females (sex ratio 1:1). Ages ranged from 22 to 56 years (n = 10, average age of 30.6 years), with most participants (n = 8) being 30 years or younger (average age of 26.5 years), and two participants above 30 years (average age of 46.5 years). Nine participants did not smoke cigarettes, and six participants consumed alcohol. The dietary analysis showed that most participants did not follow a particular dietary type and were considered omnivores; however, two participants were either vegan or pescatarian. The Australian Recommended Food Score (ARFS) was calculated as an average from AES results obtained before intervention commencement and at the completion of the study. For the sampling, 80 faecal samples were collected, with 3 faecal samples lost in transit (P05 timepoints 3 and 4, and P09 timepoint 4). A total of 77 samples were analysed for this study.
2.2. DNA Quality
There were 14,883,521 high-quality 16S rRNA sequences obtained with a mean of 173,579 forward and reverse reads per sample from 77 samples.
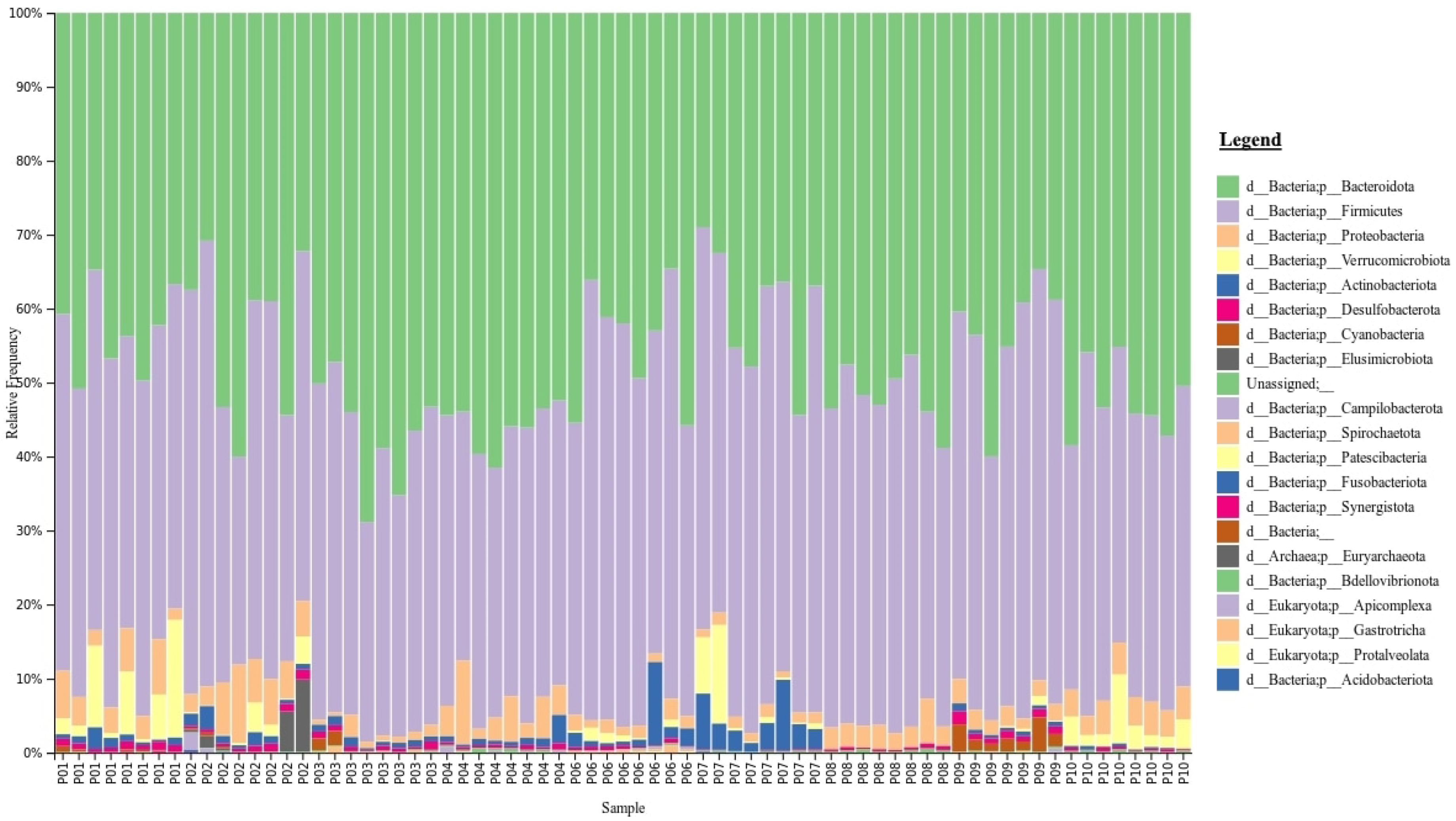
2.3. Phylogenetic Taxonomy of the Gut Microbiota
The gut microbiota taxonomy for all samples from each participant are visualised at the phylum level using taxa bar plots (
Figure 2). This graph shows that Bacteroidetes (green) and Firmicutes (purple) are the two dominant phyla across all samples. Furthermore, the plot shows that the taxa of gut microbiota in participants vary from each other. However, the ratio of Firmicutes/Bacteroidetes fluctuates over the time points and was not statistically significant.
2.4. Associations of Alpha Diversity of Gut Microbiota with Explanatory Factors
The rarefaction plot generated using Shannon’s index [
46] shows a maximum depth of 47,647 based on the median frequency value from the frequency per sample results. This is sufficient for analysis, as a levelling out on the
y-axis was observed at this depth, indicating that additional sequences beyond this depth would unlikely result in additional observed features.
The association of alpha diversity metrics with explanatory factors using Kruskal–Wallis analysis was performed on sequences pooled according to explanatory factors (
Table 2). These factors are participant, sex, age group, cigarette smoking, alcohol consumption, and ARFS group. The association of alpha diversity was also determined for the time point (1–8), experimental phase (baseline, low dose, washout, high dose, and post-intervention), RBAC dosage (0 g/day, 1 g/day, and 3 g/day), and interventional period (baseline to experimental period including washout and post-intervention). Participant was the only factor that showed a statistically significant effect on alpha diversity in Shannon’s evenness (
p = 9.66 × 10
) and Faith’s PD (
p = 0.002). Statistically significant changes in Shannon’s evenness were shown for alcohol consumption (
p = 7.92 × 10
, cigarette smoking (
p = 0.008), sex (
p = 0.003), and ARFS group (
p = 0.032). There were no significant changes in alpha diversity across age group, time point, experimental phase, RBAC dosage, and interventional period.
2.5. Associations of Beta Diversity of Gut Microbiota with Explanatory Factors
Associations of beta diversity with explanatory factors were determined using PERMANOVA (
Table 3). Participant sequence data were grouped according to the explanatory factor being tested using the same technique as alpha diversity analysis. Statistically significant differences in beta diversities were found across participant (
p = 0.001), sex (
p = 0.001), alcohol consumption (
p = 0.001), ARFS group (
p = 0.001), and age group (
p ≤ 0.013). Cigarette smoking was significantly associated with all matrices (
p = 0.001), except weighted unifrac (
p = 0.1). The analysis for time point, experimental phase, RBAC dosage, and interventional period did not show statistically significant associations with any beta diversity measurements.
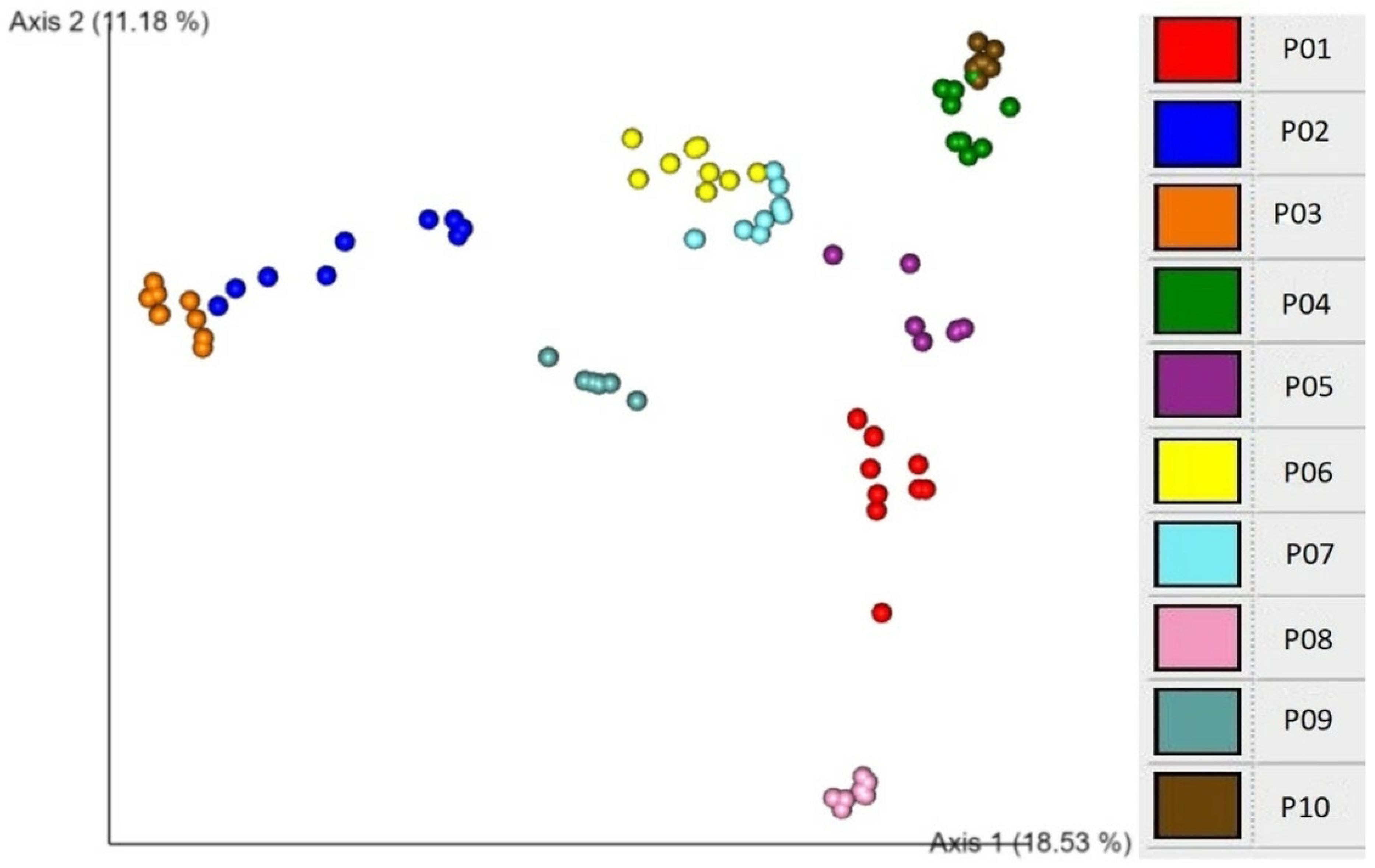
An EMPeror plot of Bray–Curtis beta diversity showing clustering according to individual participants is demonstrated in
Figure 3. These association analyses show that participant is the major explanatory factor associated with both alpha and beta diversity patterns. This finding suggests that individuality is a critical determinant of microbial diversity.
These findings suggest the requirement to analyse beta diversity for each participant to determine whether there are statistically significant longitudinal changes from the baseline across the experimental period. Individual beta diversity for each participant across samples (1–8) was calculated using weighted unifrac and Bray–Curtis methods (
Table 4).
The explanatory factors used for this analysis were RBAC dosage, experimental phase, and interventional period. In P02, the interventional period was significantly associated with weighted unifrac (p = 0.01) and Bray–Curtis (p = 0.012) beta diversity methods. In P03, the experimental phase and interventional period were significantly associated with both methods (p ≤ 0.052). P04 observed associated changes in RBAC dosage for both methods (p = 0.035 and p = 0.018, respectively), as well as the experimental phase with Bray–Curtis (p = 0.044). In P07, the experimental phase (p = 0.025) and interventional period (p = 0.025) were significantly associated with Bray–Curtis. In P08, the RBAC dosage was significantly associated with both methods (p < 0.05), and the experimental phase and interventional period were associated with Bray–Curtis only. P09 observed that the dosage and experimental phase are significantly associated (p < 0.05) with both methods. It is further noted that P05 and P10 showed marginally significant values in the Bray–Curtis method for the experimental phase.
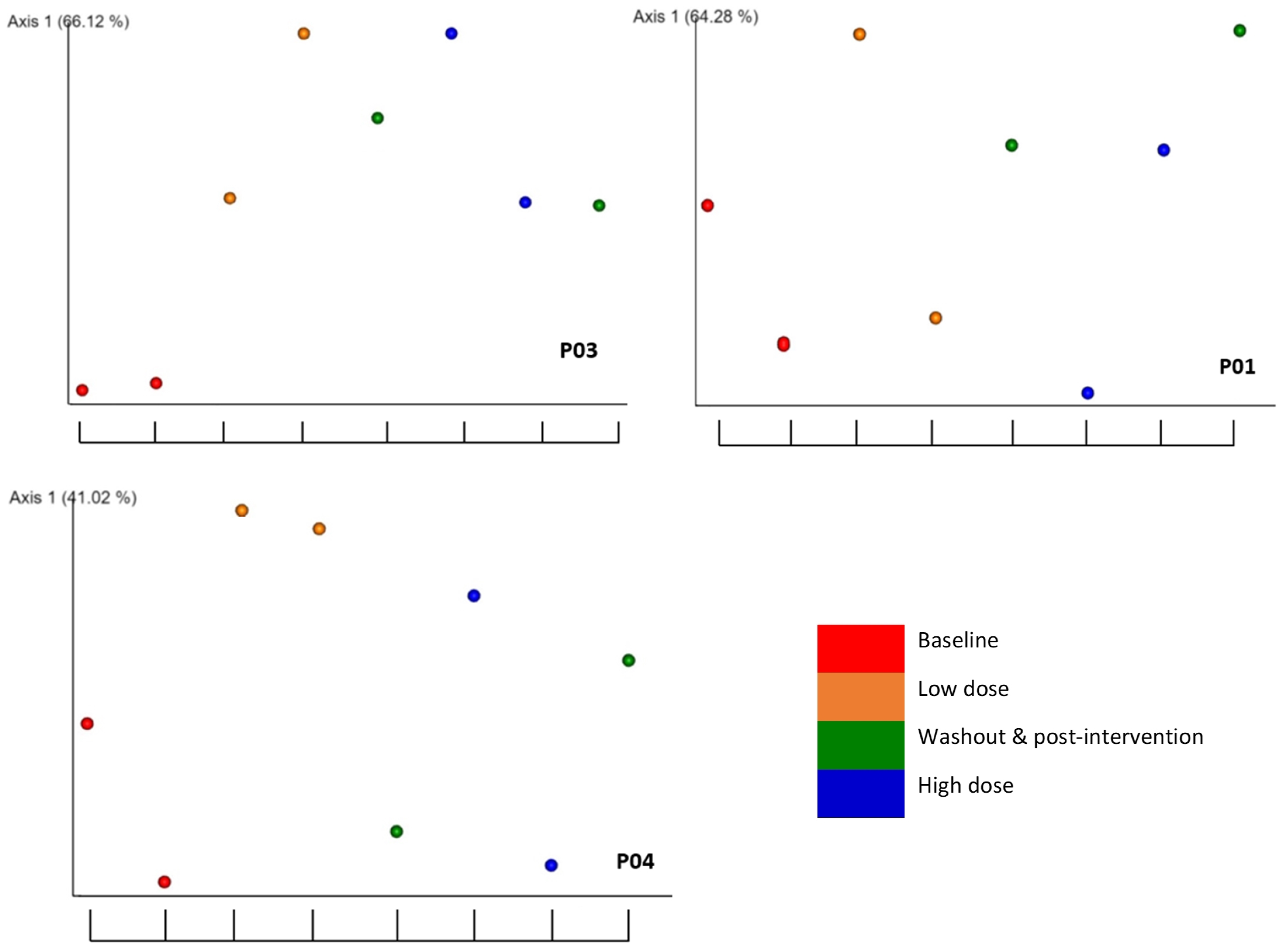
The longitudinal findings are illustrated using the weighted unifrac beta diversity distance matrices across the eight time points in the EMPeror plots for the selected participants (
Figure 4). For P03, all time points after baseline (red points) show a significant change. The graph for P04 illustrates an increase from the baseline in the low dose interventional period (orange points) but no change in the high dose (blue points) or washout (green points) periods. P01 shows no change across any time points from the baseline. Please refer to
Figure S1 of the
supplementary materials for the plots for all participants.
2.6. Detection of the Association of Microbial Diversity with Interactions of Explanatory Factors
Further investigation was carried out to determine whether multiple variables could explain the variation in participants’ gut microbiota and reduce false positives in the beta diversity analysis. The calculated unifrac distance matrix was used to perform an Adonis PERMANOVA test on participant and other explanatory factors, including RBAC dosage, experimental phase, time point, and interventional period. The results showed that the variable participant (R2 = 0.796, p = 0.001) explains approximately 80% of variance in the beta diversity. Regression analysis with linear models showed a statistical significance for the experimental phase (R2 = 0.019, p = 0.026) and intervention (R2 = 0.012, p = 0.011). Hence, after the participants’ variation is accounted for, the experimental and interventional periods explain approximately 3.1% of the variance observed in beta diversity with the remaining 18% of variance being unaccounted for. Hence, the potential effects of the experimental intervention cannot be completely dismissed.
2.7. Significant Taxonomic Changes from Baseline to Interventional Period
Taxonomic changes from baseline to the interventional period were statistically assessed using ANCOM. Data for all statistically significant changes with a W value of 5 or above are listed in
Table 5. Associated changes were compared from the baseline to the total interventional period unless otherwise specified. These results show statistically significant taxonomic changes (from baseline) detected down to a species level for 7 participants—P01, P02, P03, P07, P08, P09 and P10.
In P01, a marked increase in the Hungatella genus with a W value of 40 was revealed and fewer marked changes across other genera and families were observed. In P02, decreases in Anaerococcus, Corynebacterium, and Finegoldia genera were detected with W values ranging between 48 and 64. Large declines in Dialister (W = 94) and Gastranaerophilales (W = 86) were observed in P03. A large decrease in Erysipelatoclostridiaceae (W = 102) was displayed in P07. In P08, there was a decrease in the Eubacterium siraeum group (W = 35) with lower significant changes in other bacterial genera. P09 revealed large increases in multiple bacterial genera, including Eubacterium siraeum, (W = 38). This participant also showed smaller increases in Eubacterium hallii group and Stomatobaculum (W = 5 for each) and further smaller increases in Anaerococcus, Ruminococcus, Megasphaera, Solobacterium (W = 7–8) in either low or high RBAC doses. P10 showed a large decrease in Prevotella (W = 20) and smaller changes across other groups. Some changes were only detected during the washout stage as observed in P01.
3. Discussion
This basic scientific study aimed to explore possible changes in the gut microbial composition in response to dietary RBAC supplementation in healthy adults. Our results revealed that alpha and beta diversity were not associated with the experimental phase, interventional period, RBAC dosage, or time point. This suggests that the dietary supplementation did not significantly moderate the gut microbiota composition. However, the absence of the supplementation effect on the gut microbiota composition could be affected by a couple of factors. Firstly, the unknown species are not accounted for in alpha diversity measurements. This indicates that the true richness and evenness of the microbial environment was not fully examined in the present study. Consequently, the results on alpha diversity may be misrepresented [
47]. Secondly, the finding that participant is a significant determinant for alpha and beta diversity suggests that the individual microbiome requires separate assessment alongside the explanatory factors during RBAC supplementation on gut microbiota.
The present research findings revealed differences in the alpha and beta diversity of the gut microbiota within individual participants. The gut microbial composition presents a profile that is distinct for the individual, with moderating factors such as genetics, environment, and lifestyle [
48]. Previous research on twins indicated heritable common gut microbiota, with cohabitating twins sharing the highest number of strains [
49]. However, differences in the microbiota between twins were also shown in previous studies, indicating that non-genetic factors are also at play [
48,
49]. Since genetics and environment are known factors influencing gut microbiota composition, it was not unexpected that the microbial composition between participants significantly differed in the present study. Compounding these factors is the individual lifestyle, which further impacts microbiota diversity among participants [
50].
The present results also revealed significant association between alpha (Shannon’s evenness) and beta diversity with alcohol consumption, cigarette smoking, and diet (Australian Recommended Food Score, ARFS). Alcohol consumption has been shown to affect gut microbiota and has been linked to substantial losses in diversity, including reductions in Bacteroidetes and Firmicutes [
51]. Alcohol consumption also correlates with decreased connectivity of the microbial network and subsequent alteration of gut microbiome composition [
28]. Cigarette smoking causes taxonomic changes in the gut microbiota, specifically decreased Bacteroidetes and increased Firmicutes, due to nicotine exposure [
52]. Moreover, after smoking cessation, increased alpha diversity of gut microbiota was observed [
27]. Due to the the toxic effects on gut mucosa, cigarette smoking has also been linked to gut microbial dysbiosis [
52]. The present study identified lifestyle, including diet and subsequent ARFS, alcohol consumption, and cigarette smoking, as important factors that impact gut microbiota in healthy adults.
Five of the six participants in the present study with the highest ARFS scores revealed significant changes in beta diversity. Although there is limited research on how the ARFS is associated with the gut microbiome, Aslam et al. [
53] reported that higher ARFS correlated with the consumption of a more diverse diet, with differences shown in the beta diversity compared to limited diets with lower ARFS. Hence, the association of ARFS with changes in beta diversity may be explained by the differences between the consumption of core foods (e.g., grains and water) and discretionary foods (e.g., fried products and packaged sweets) in the diet [
54]. The increased consumption of core foods and low discretionary foods also shows a higher proportion of complex carbohydrates, which benefits gut microbiota. This may explain the significant changes observed in the beta diversity for participants with a higher ARFS in the present study [
54].
Age group as an explanatory factor was revealed to be associated with significant changes in beta diversity. Considerable shifts in the microbiome were previously shown during infancy, puberty, to the later stage of life (>75 years of age) [
21]. The stability of the microbiome is reached during adulthood (18–25 years of age) and remains relatively constant until approximately 75 years of age when a loss of diversity occurs [
21]. However, Odamaki et al. [
23] observed significant differences between adult clusters at the ages of 33 (cluster 1) and 42 (cluster 2), with higher levels of Bacteroidetes observed in the younger age group (cluster 1). This is consistent with the significant differences in beta diversity observed in the present study for participants ≤30 years and >30 years of age.
Associations between sex, and alpha and beta diversity were observed. Sex is a known modulator of the gut microbiome through the actions of sex hormones [
55]. This is supported by recent studies showing that sex is a prime contributor to microbial diversity with increased alpha diversity observed in females compared to males [
56,
57]. Furthermore, sex has also been linked to responses of the gut microbiota to diet, anti-microbial effects, and obesity [
55,
58]. The present findings affirm that age and sex significantly influence the gut microbiota composition within healthy adults.
Dietary RBAC supplementation was shown to change the abundance of beneficial bacteria, including
Lactobacillus,
Roseburia, and
Ruminococcaceae, although these changes were not observed in all participants. Some participants exhibited trends in taxonomic changes during the interventional period. For example, participant 9 (P09) showed increases across multiple genera. These beneficial bacteria play a vital role in gut health through the production of SCFAs as essential energy sources for colonic enterocytes [
12,
14], the provision of vitamins [
10], and the possession of anti-inflammatory properties [
13]. This finding suggests that dietary RBAC supplementation may be used as a prebiotic for regulating these beneficial bacteria.
The increases in beneficial bacteria observed in some participants may also be associated with the ARFS. Diets with less diversity have been associated with lower bacterial variety [
59]. Of the four participants who exhibited taxonomic changes in beneficial bacteria, three had the lowest ARFS. Hence, this finding suggests a potential association between RBAC supplementation responses and ARFS. This indicates that RBAC supplementation may influence the growth of the beneficial gut microbiota of people with diets that are lower in core foods and higher in discretionary foods. In addition to increased beneficial bacteria, there were also reductions in bacteria known to be opportunistic pathogens, including
Anaerococcus and
Corynebacterium [
60,
61]. However, these taxonomic changes are difficult to interpret due to the difference in gut microbiota between individuals and how rapidly the gut microbiome can change in response to daily diet and lifestyle factors [
50].
In six participants, significant changes in beta diversity were associated with RBAC dosage, experimental phase, and/or interventional period. Previous research has indicated that the composition and diversity of the gut microbiome can determine whether participants respond to interventions, including dietary modification for health-related purposes [
62]. This is further extended to other interventions, such as treatments for cervical cancer and responsiveness to exercise for pre-diabetes treatment [
62,
63,
64]. The present study used multiple analyses to classify participants into two groups, responders and non-responders to RBAC supplementation [
65]. Four participants (P02, P03, P07, and P09) observed significant changes in all three classification criteria of individual beta diversity, weighted unifrac EMPeror plots, and taxonomic analysis, thus making them responders to RBAC supplementation. Two participants (P04 and P08) revealed changes in two criteria; hence, they were probable responders to RBAC supplementation. However, further supplementation and analysis is required to determine whether these participants can be classified as responders. Two participants (P01 and P10) only showed microbial taxonomic changes and therefore were unlikely to be responders to the supplementation. Further, two participants (P05 and P06) showing no statistical changes across the analyses were considered non-responders to the RBAC supplementation.
A limitation of this study is the small sample size. The number of recruited participants was constrained by the limitation of available resources (time and money). While the results demonstrated the potential modulation effect of RBAC supplementation on the gut microbiome, we caution against generalising the findings and drawing any definitive inference of the impact on the population. Further research with a larger sample size is required. As demonstrated in the present study, research aiming to investigate the effects of dietary RBAC supplementation on gut microbiota may require controlling for individual characteristics as confounding factors, such as age. Also, individualised doses according to body weight may be more appropriate to assess the impact of supplementation and dosage on gut microbiota. Future studies should also investigate the potential effects of RBAC supplementation on the gut microbiome at a molecular level, such as the impact on different SCFAs at the intestinal lumens. A further limitation of this study is the short time frame of the intervention. An extended period of supplementation may reveal further changes in the gut microbiota composition. Maintaining a food journal by participants to track the dietary intake and physical activity could be correlated with gut microbiota composition [
50].


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}