


m-Terphenylamines, Acting as Selective COX-1 Inhibitors, Block Microglia Inflammatory Response and Exert Neuroprotective Activity

 ,
,  , ,
, ,  ,
,  , ,
, ,  and
and 
Abstract
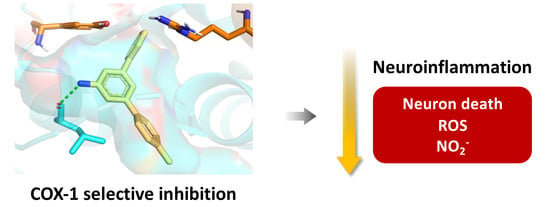
1. Introduction
2. Results and Discussion
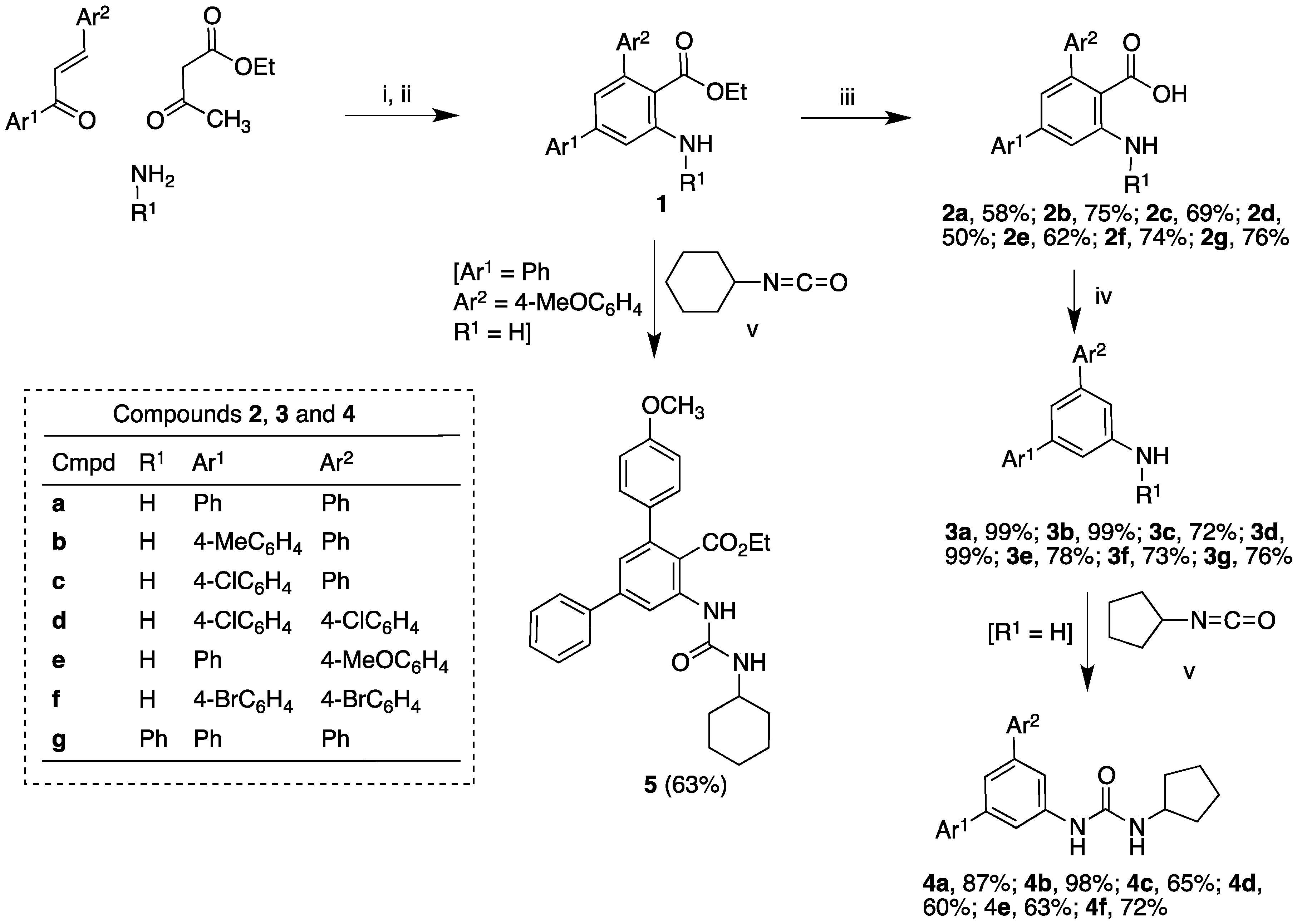
2.1. Synthesis
2.2. COX Inhibition Studies
- (a)
- Amino esters in 1 lack significant activity, although some of them (1c and 1d) slightly inhibited COX-1 at 50 μM.
- (b)
- The corresponding amino acids in 2 do not achieve significant inhibition levels either, although they are slightly more active. They seem to be selective towards COX-1.
- (c)
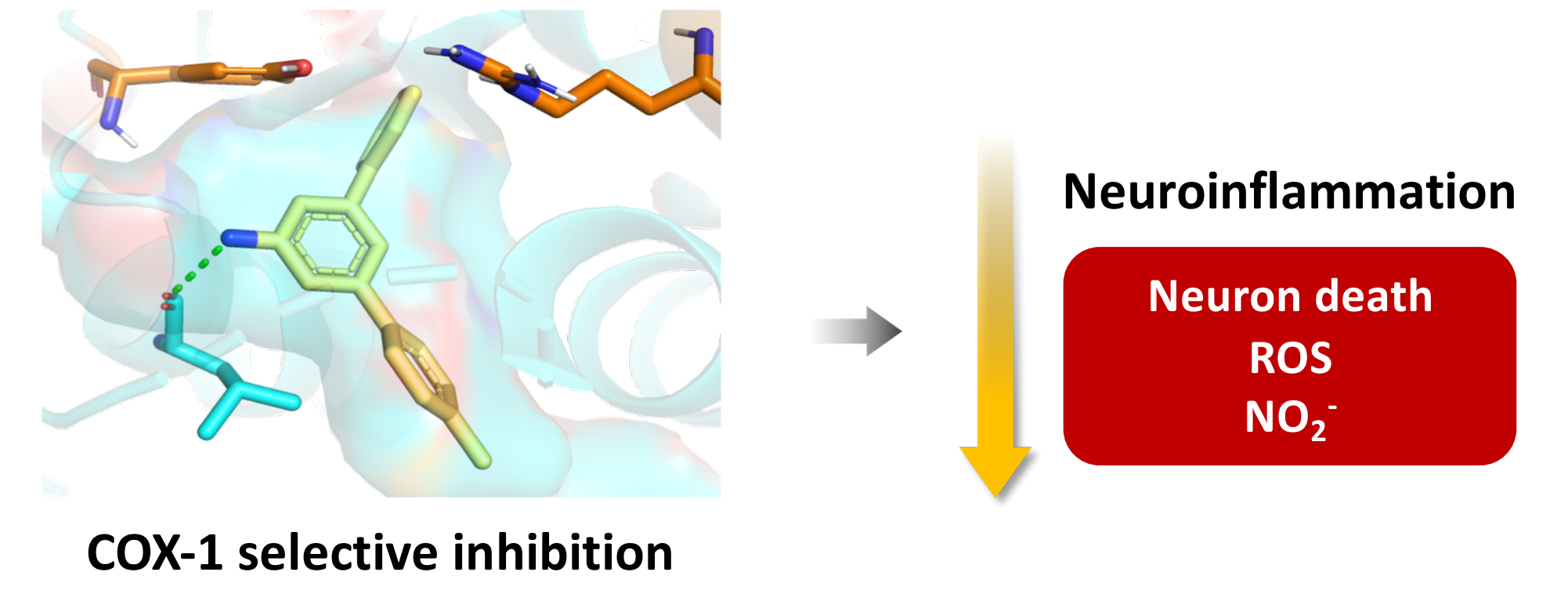
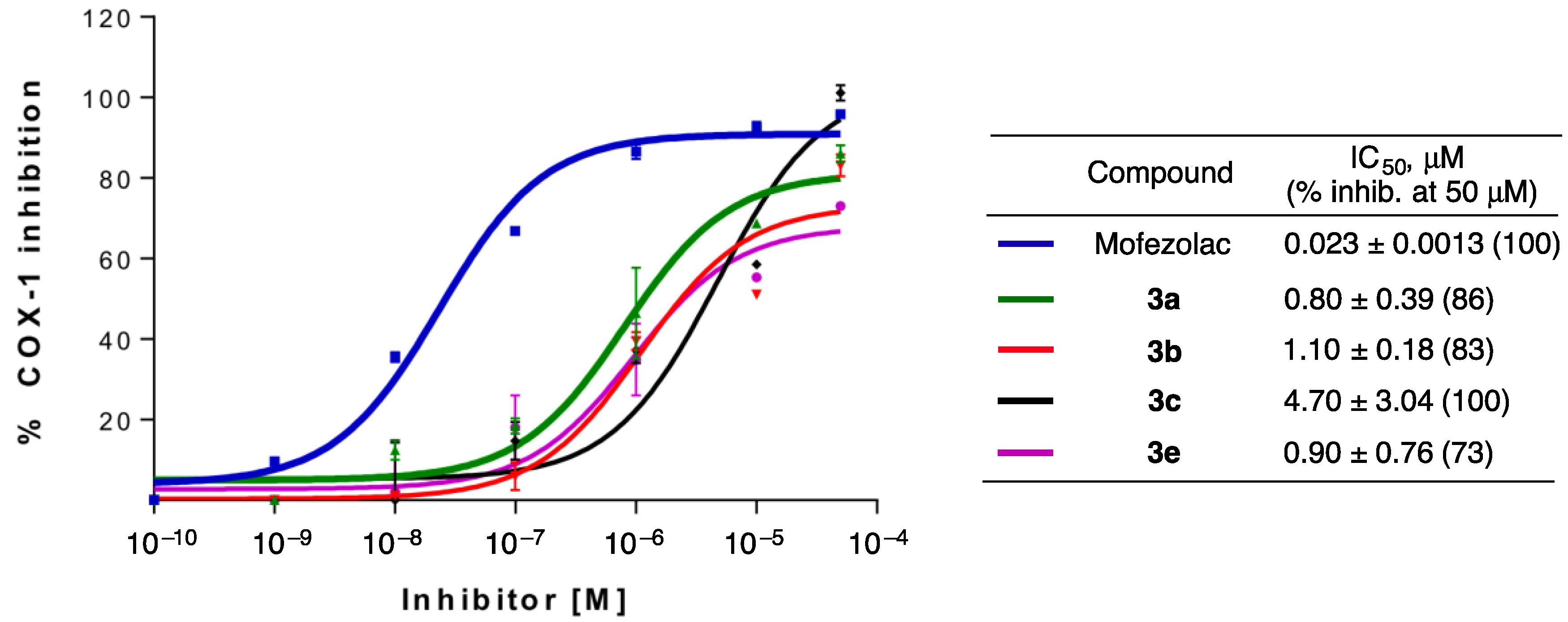
- Amino-m-terphenyls in 3 were the most interesting derivatives, and most of them showed COX-1 inhibition at IC50 values in the μM range. Moreover, three of these compounds (3b, 3c, and 3e) showed a complete selectivity towards COX-1, and a fourth (3a) showed a COX-1/COX-2 inhibition ratio around 7. COX-1 inhibition and COX-1/COX-2 selectivity are not connected with the electron density of the aromatic rings since similar results have been obtained for compounds bearing no substitution (3a), electron-withdrawing groups (3c, 3d) and electron-releasing groups (3e). This level of selectivity had not been previously observed for this type of compound and is similar to or better than the one found in other families of COX-1 inhibitors currently being developed, such as diarylisoxazoles [33].
- (d)
- Urea derivatives in 4 and 5 are essentially inactive. The only example with some activity (compound 4f) did not show COX-1 selectivity.
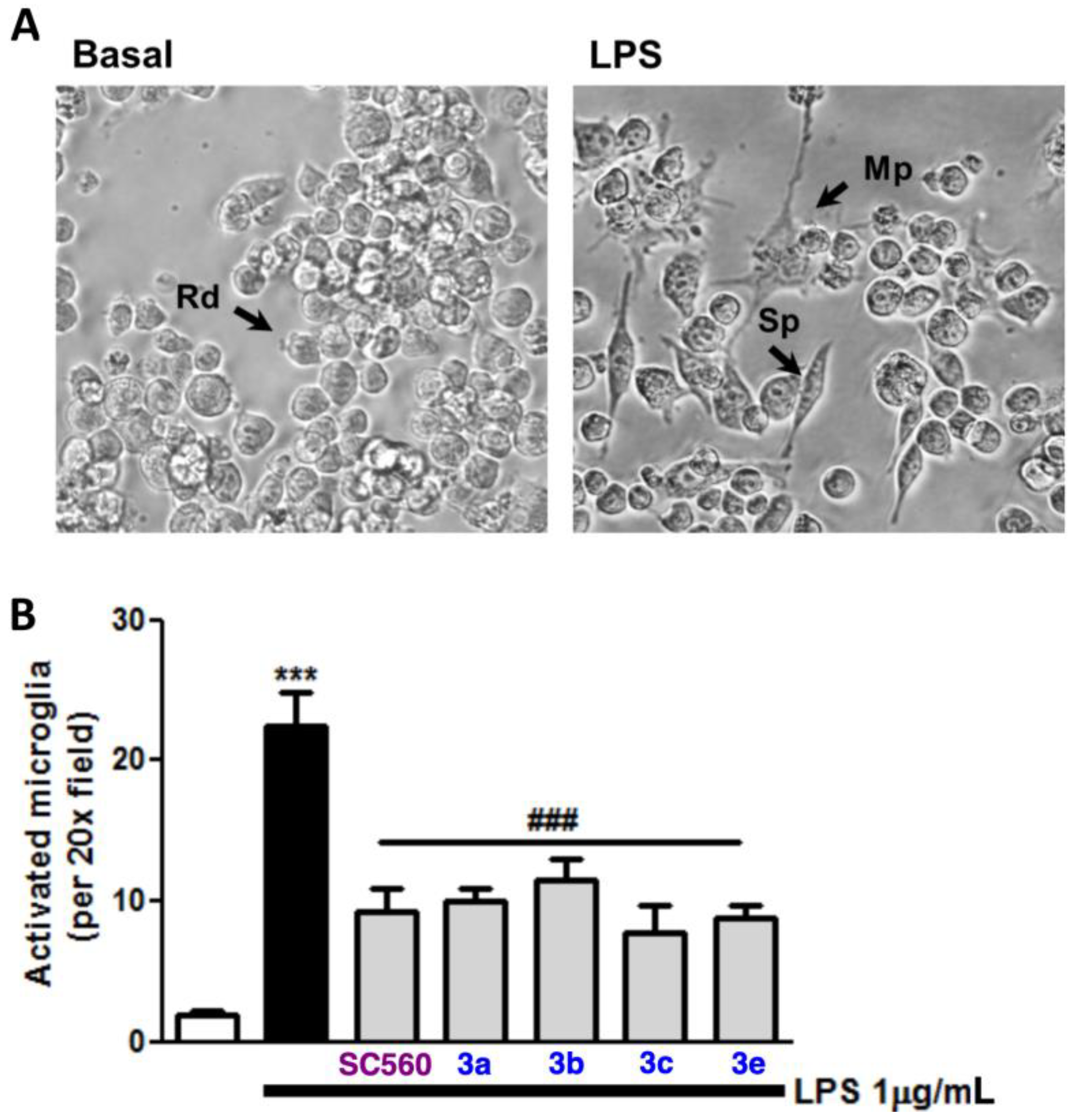
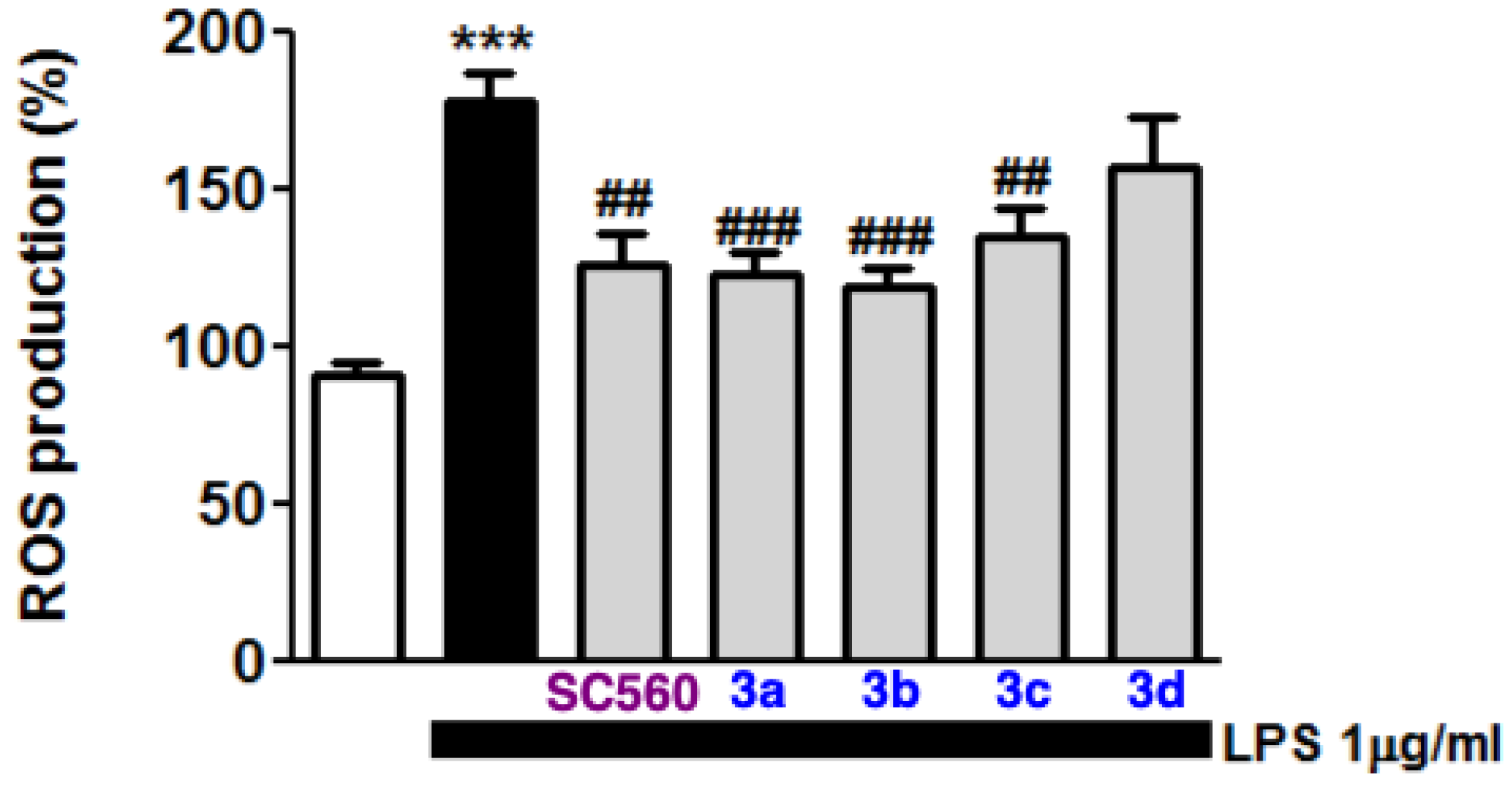
2.3. Blockade of Microglia Inflammatory and Oxidative Responses
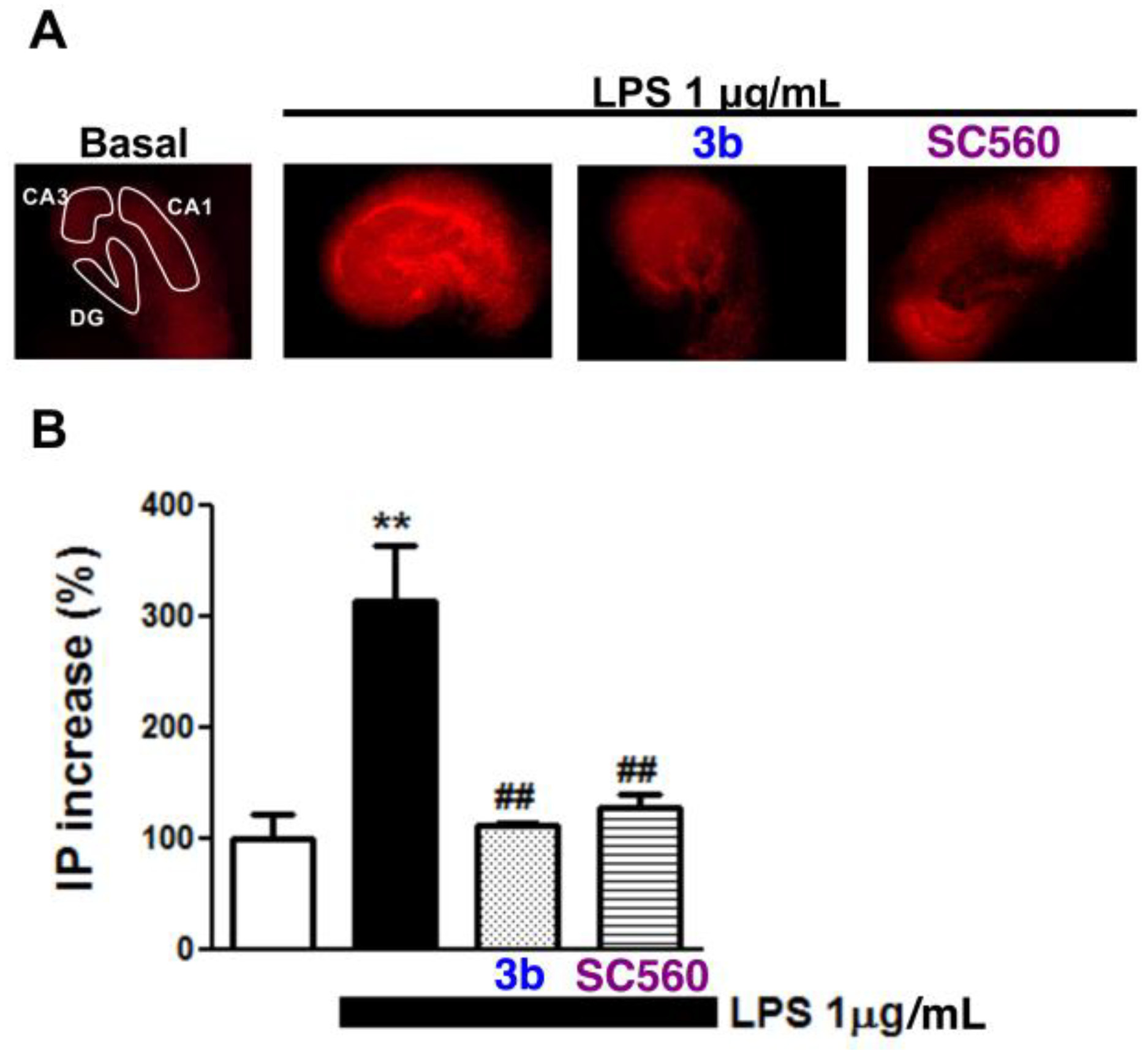
2.4. Compound Protects against LPS Challenge in Organotypic Hippocampal Slices
2.5. In Silico Prediction of the ADME Properties of Compounds
3. Materials and Methods
3.1. General Experimental Information
3.2. General Procedure for the Synthesis of Compounds by Saponification of Anthranilic Esters
- 6-Amino-2,4-diphenylbenzoic acid (2a). Prepared from ethyl 6-amino-2,4-diphenylbenzoate 1a (305 mg, 1.2 mmol). Yield: 161 mg (58%), as a pale brown solid. Mp: 162–163 °C. Elemental analysis (%) calcd for C19H15NO2 (M = 289.33): C, 78.87; H, 5.23; N, 4.84; found: C, 78.83; H, 5.27; N, 4.82. IR νmax (film): 3481, 3376, 3024, 1666, 1596, 1560 cm−1. 1H NMR (250 MHz, MeOD) δ 7.59 (d, J = 6.9 Hz, 2H), 7.46–7.23 (m, 10H), 7.03 (d, J = 1.7 Hz, 1H), 6.80 (d, J = 1.7 Hz, 1H). 13C NMR (63 MHz, MeOD) δ 173.1, 149.9, 145.3, 144.9, 144.3, 141.7, 129.8, 129.3, 128.9, 128.8, 128.0, 127.9, 119.4, 116.1, 114.7.
- 6-Amino-2-phenyl-4-(4-tolyl)benzoic acid (2b). Prepared from ethyl 6-amino-2-phenyl-4-(4-tolyl) benzoate 1b (305 mg, 1.2 mmol). Yield: 218 mg (75%), as an off-white solid. Mp: 164–165 °C. Elemental analysis (%) calcd for C20H17NO2 (M = 303.35): C, 79.19; H, 5.65; N, 4.62; found: C, 79.23; H, 5.62; N, 4.58. IR νmax (film): 3485, 3377, 3022, 2857, 1684, 1603 cm−1. 1H NMR (250 MHz, DMSO) δ 7.54 (d, J = 8.1 Hz, 2H), 7.44–7.32 (m, 5H), 7.27 (d, J = 8.4 Hz, 3H), 6.88 (d, J = 1.4 Hz, 1H), 2.33 (s, 3H). 13C NMR (63 MHz, DMSO) δ 169.6, 145.6, 143.2, 142.3, 142.2, 137.6, 136.5, 129.6, 128.1, 128.1, 127.0, 126.6, 118.7, 115.8, 114.3, 20.8.
- 6-Amino-2-phenyl-4-(4-chlorophenyl)benzoic acid (2c). Prepared from ethyl 6-amino-2-phenyl-4-(4-chlorophenyl) benzoate 1c (338 mg, 1.2 mmol). Yield: 268 mg (69%), as a pale brown solid. Mp: 229–230 °C. Elemental analysis (%) calcd for C19H14ClNO2 (M = 323.77): C, 70.48; H, 4.36; N, 4.33; found: C, 70.53; H, 4.32; N, 4.29. IR νmax (film): 3444, 3400, 2923, 2851, 1692 cm−1. 1H NMR (250 MHz, DMSO) δ 7.74–7.64 (m, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.45–7.32 (m, 3H), 7.29 (s, 1H), 6.96 (d, J = 1.3 Hz, 1H). 13C NMR (63 MHz, DMSO) δ 169.4, 150.9, 144.6, 143.2, 141.8, 141.1, 138.1, 133.0, 129.0, 128.6, 128.2, 128.1, 127.2, 119.3, 114.9.
- 6-Amino-2,4-di-(4-chlorophenyl)benzoic acid (2d). Prepared from ethyl 6-amino-2,4-di-(4-chlorophenyl) benzoate 1d (371 mg, 1.2 mmol). Yield: 215 mg (50%), as a pale brown solid. Mp: 178–179 °C. Elemental analysis (%) calcd for C19H13Cl2NO2 (M = 358.22): C, 63.71; H, 3.66; N, 3.91; found: C, 63.68; H, 3.72; N, 3.86. IR νmax (film): 3496, 3371, 1657, 1605, 1581, 1491 cm−1. 1H NMR (250 MHz, MeOD) δ 7.53 (d, J = 8.3 Hz, 2H), 7.42–7.33 (m, 3H), 7.33–7.23 (m, 4H), 6.99 (s, 1H), 6.70 (s, 1H). 13C NMR (63 MHz, MeOD) δ 172.1, 150.7, 144.7, 143.9, 143.1, 140.0, 135.0, 133.9, 130.8, 129.9, 129.5, 129.0, 118.8, 114.9, 114.5.
- 6-Amino-2-phenyl-4-(4-methoxyphenyl)benzoic acid (2e). Prepared from ethyl 6-amino-2-(4-methoxyphenyl)-2-phenylbenzoate 1e (334 mg, 1.2 mmol). Yield: 238 mg (62%), as a pale-yellow solid. Mp: 195–196 °C. Elemental analysis (%) calcd for C20H17NO3 (M = 319.35): C, 75.22; H, 5.37; N, 4.39; found: C, 75.18; H, 5.35; N, 4.42. IR νmax (film): 3484, 3372, 1647, 1605, 1581 cm−1. 1H NMR (250 MHz, DMSO) δ 7.58 (d, J = 8.7 Hz, 2H), 7.40–7.26 (m, 5H), 7.05–6.96 (m, 3H), 6.68 (d, J = 1.4 Hz, 1H), 3.79 (s, 3H). 13C NMR (63 MHz, DMSO) δ 170.2, 159.3, 149.0, 143.3, 142.8, 142.1, 132.0, 128.0, 128.0, 127.8, 126.8, 116.4, 114.4, 112.9, 112.3, 55.2.
- 6-Amino-2,4-di-(4-bromophenyl)benzoic acid (2f). Prepared from ethyl 6-amino-2,4-di-(4-bromophenyl) benzoate 1f (456 mg, 1.2 mmol). Yield: 397 mg (74%), as an off-white solid. Mp: 239–240 °C. Elemental analysis (%) calcd for C19H13Br2NO2 (M = 447.12): C, 51.04; H, 2.93; N, 3.13; found: C, 51.07; H, 2.89; N, 3.18. IR νmax (film): 2922, 2851, 1674, 1590, 1492 cm−1. 1H NMR (250 MHz, DMSO) δ 7.75–7.49 (m, 7H), 7.31 (d, J = 8.5 Hz, 3H), 6.93 (d, J = 1.2 Hz, 1H). 13C NMR (63 MHz, DMSO) δ 169.1, 150.9, 142.2, 141.3, 141.3, 138.3, 132.0, 131.0, 130.3, 129.4, 128.9, 121.8, 120.6, 113.8, 101.9.
- 6-Phenylamino-2,4-diphenylbenzoic acid (2g). Prepared from ethyl 6-phenylamino-2,4-diphenyl-2,3-dihydro benzoate 1g (378 mg, 1.2 mmol). Yield: 333 mg (76%), as a pale-yellow solid. Mp: 182–183 °C. Elemental analysis (%) calcd for C25H19NO2 (M = 365.42): C, 82.17; H, 5.24; N, 3.83; found: C, 82.13; H, 5.29; N, 3.85. IR νmax (film): 3374, 3055, 3029, 2924, 1656, 1589, 1557 cm−1. 1H NMR (250 MHz, CDCl3) δ 7.45 (d, J = 1.7 Hz, 1H), 7.41 (d, J = 1.4 Hz, 1H), 7.40 (d, J = 1.6 Hz, 1H), 7.33–7.23 (m, 7H), 7.23–7.16 (m, 4H), 7.12 (d, J = 7.9 Hz, 2H), 6.95 (t, J = 7.3 Hz, 1H), 6.90 (d, J = 1.5 Hz, 1H). 13C NMR (63 MHz, CDCl3) δ 174.56, 146.33, 145.39, 144.90, 142.57, 141.37, 140.10, 129.59, 128.90, 128.30, 128.16, 127.38, 127.30, 123.20, 121.48, 120.97, 113.01.
3.3. General Procedure for the Synthesis of m-Terphenylanilines by Decarboxylation of Compounds
- 5′-Amino-m-terphenyl (3a). Prepared from 6-amino-2,4-diphenylbenzoic acid 2a (289 mg, 1.0 mmol). Yield: 243 mg (99%), as a white solid. Mp: 108–109 °C; lit [55], 107–109 °C. Elemental analysis (%) calcd for C18H15N (M = 245.32): C, 88.13; H, 6.16; N, 5.71; found: C, 88.16; H, 6.14; N, 5.65. IR νmax (film): 3454, 3370, 3055, 1595. 1H NMR (250 MHz, DMSO) δ 7.64 (d, J = 7.1 Hz, 4H), 7.45 (t, J = 7.3 Hz, 4H), 7.35 (t, J = 7.2 Hz, 2H), 7.02 (s, 1H), 6.84 (d, J = 1.5 Hz, 2H), 5.32 (s, 2H). 13C NMR (63 MHz, DMSO) δ 149.6, 141.6, 141.1, 128.8, 127.3, 126.7, 113.3, 111.5. These characterization data were coincident with those found in the literature [28,56].
- 4-Methyl-5′-amino-m-terphenyl (3b). Prepared from 6-amino-2-phenyl-4-(4-tolyl) benzoic acid (303 mg, 1.0 mmol) 2b. Yield: 257 mg (99%), as a white solid. Mp: 98–99 °C. Elemental analysis (%) calcd for C19H17N (M = 259.34): C, 87.99; H, 6.61; N, 5.40; found: C, 88.02; H, 6.63; N, 5.38. IR νmax (film): 3418, 3294, 3025, 1594 cm−1. 1H NMR (250 MHz, CDCl3) δ 7.64 (d, J = 6.9 Hz, 2H), 7.55 (d, J = 8.1 Hz, 2H), 7.51–7.42 (m, 2H), 7.41–7.34 (m, 1H), 7.26 (s, 1H), 7.23 (t, J = 1.5 Hz, 1H), 6.93–6.89 (m, 2H), 2.43 (s, 3H). 13C NMR (63 MHz, DMSO) δ 149.6, 141.6, 141.5, 141.2, 138.2, 136.5, 129.4, 128.8, 127.3, 126.7, 126.5, 113.1, 111.3, 20.7. These characterization data were coincident with those found in the literature [28,55].
- 4-Chloro-5′-amino-m-terphenyl (3c). Prepared from 6-amino-2-phenyl-4-(4-chlorophenyl)benzoic acid 2c (324 mg, 1.0 mmol). Yield: 201 mg, (72%), as a white solid. Mp: 96–97 °C. Elemental analysis (%) calcd for C18H14ClN (M = 279.76): C, 77.28; H, 5.04; N, 5.01; found: C, 77.25; H, 5.07; N, 5.04. IR νmax (film): 3460, 3374, 3057, 2923, 1597, 1495 cm−1. 1H NMR (250 MHz, MeOD) δ 7.62 (d, J = 8.4 Hz, 4H), 7.45–7.38 (m, 4H), 7.36–7.30 (m, 1H), 7.12 (t, J = 1.6 Hz, 1H), 6.97 (t, J = 1.6 Hz, 1H), 6.95 (t, J = 1.6 Hz, 1H). 13C NMR (63 MHz, MeOD) δ 149.9, 144.1, 142.8, 142.6, 141.7, 134.2, 129.8, 129.7, 129.5, 128.3, 128.0, 116.7, 114.5, 113.9. These characterization data were coincident with those found in the literature [28,55].
- 4,4″-Dichloro-5′-amino-m-terphenyl (3d). Prepared from 6-amino-2,4-di-(4-chlorophenyl)benzoic acid (358 mg, 1.0 mmol) 2d. Yield: 212 mg (99%), as an off-white solid. Mp: 253–254 °C. Elemental analysis (%) calcd for C18H13Cl2N (M = 314.21): C, 68.81; H, 4.17; N, 4.46; found: C, 68.86; H, 4.14; N, 4.42. IR νmax (film): 3327, 3012, 2957, 1626, 1544, 1512 cm−1. 1H NMR (250 MHz, CDCl3) δ 7.60–7.51 (m, 4H), 7.43 (d, J = 8.7 Hz, 4H), 7.13 (t, J = 1.6 Hz, 1H), 6.88 (d, J = 1.6 Hz, 2H), 3.89 (bs, 2H). 13C NMR (63 MHz, CDCl3) δ 147.4, 142.0, 139.8, 133.6, 129.0, 128.5, 116.6, 113.0. These characterization data were coincident with those found in the literature [28].
- 4-Methoxy-5′-amino-m-terphenyl (3e). Prepared from 6-amino-2-(4-methoxyphenyl)-2-phenylbenzoic acid 2e (319 mg, 1.0 mmol). Yield: 215 mg (78%), as a white solid. Mp: 118–119 °C. Elemental analysis (%) calcd for C19H17NO (M = 275.34): C, 82.88; H, 6.22; N, 5.09; found: C, 82.83; H, 6.26; N, 5.04. IR νmax (film): 3447, 3369, 3030, 1596, 1514 cm−1. 1H NMR (250 MHz, DMSO) δ 7.63 (d, J = 7.2 Hz, 2H), 7.58 (d, J = 8.7 Hz, 2H), 7.44 (t, J = 7.3 Hz, 2H), 7.34 (t, J = 7.2 Hz, 1H), 7.06–6.95 (m, 3H), 6.80 (s, 2H), 5.28 (br s, 2H), 3.79 (s, 3H). 13C NMR (63 MHz, DMSO) δ 158.8, 149.6, 141.6, 141.3, 141.2, 133.4, 128.8, 127.7, 127.2, 126.7, 114.2, 112.9, 111.1, 110.9, 55.2. These characterization data were coincident with those found in the literature [28,55].
- 4,4″-Dibromo-5′-amino-m-terphenyl (3f). Prepared from 6-amino- 2,4-di-(4-bromophenyl)benzoic acid 2f (447 mg, 1.0 mmol). Yield: 294 mg (73%), as an off-white solid. Mp: 163–164 °C. Elemental analysis (%) calcd for C18H13Br2N (M = 403.11): C, 53.63; H, 3.25; N, 3.47; found: C, 53.61; H, 3.26; N, 3.43. IR νmax (film): 3393, 3308, 1598, 1491 cm−1. 1H NMR (250 MHz, DMSO) δ 7.69–7.54 (m, 8H), 7.01 (s, 1H), 6.84 (d, J = 1.4 Hz, 2H), 5.38 (br s, 2H). 13C NMR (63 MHz, DMSO) δ 149.9, 140.4, 140.1, 131.7, 128.8, 120.7, 112.9, 111.5.
- 5′-Phenylamino-m-terphenyl (3g). Prepared from 6-phenylamino- 2,4-diphenylbenzoic acid 2g (365 mg, 1.0 mmol). Yield: 244 mg, (76%), as an off-white solid. Mp: 128–129 °C. Elemental analysis (%) calcd for C24H19N (M = 321.41): C, 89.68; H, 5.96; N, 4.36; found: C, 89.62; H, 5.93; N, 4.37. IR νmax (film): 3401, 3050, 3026, 1594, 1533 cm−1. 1H NMR (250 MHz, CDCl3) δ 7.55 (d, J = 1.6 Hz, 1H), 7.51 (d, J = 6.9 Hz, 2H), 7.44–7.29 (m, 7H), 7.29–7.20 (m, 3H), 7.19–7.11 (m, 3H), 7.01 (d, J = 7.5 Hz, 2H), 6.85 (t, J = 7.3 Hz, 1H), 5.61 (br s, 1H). 13C NMR (63 MHz, CDCl3) δ 143.4, 141.5, 141.0, 140.6, 138.8, 131.5, 130.6, 129.6, 129.5, 129.1, 128.9, 127.7, 127.5, 127.2, 121.4, 120.0, 118.4, 116.1.
- 1-([m-Terphenyl]-5′-yl)-3-cyclopentyl urea (4a). Prepared from 5′-amino-m-terphenyl 3a (172 mg, 0.7 mmol) and cyclopentyl isocyanate (0.12 mL, 1.05 mmol). Yield: 217 mg (87%), as a white solid. Mp: 224–225 °C. Elemental analysis (%) calcd for C24H24N2O (M = 356.46): C, 80.87; H, 6.79; N, 7.86; found: C, 80.92; H, 6.83; N, 7.81. IR νmax (film): 3337, 2919, 2851, 1643, 1559 cm−1. 1H NMR (250 MHz, DMSO) δ 8.50 (s, 1H), 7.69 (d, J = 7.7 Hz, 6H), 7.48 (t, J = 7.3 Hz, 4H), 7.42–7.34 (m, 3H), 6.28 (d, J = 7.1 Hz, 1H), 4.05–3.87 (m, 1H), 1.93–1.77 (m, 2H), 1.73–1.49 (m, 4H), 1.47–1.32 (m, 2H). 13C NMR (63 MHz, DMSO) δ 154.9, 141.7, 141.4, 140.5, 129.0, 127.6, 126.9, 118.2, 115.1, 51.0, 32.8, 23.2. These characterization data were coincident with those found in the literature [27].
- 1-([4-Methyl-m-terphenyl]-5′-yl)-3-cyclopentyl urea (4b). Prepared from 4-methyl-5′-amino-m-terphenyl 3b (182 mg, 0.7 mmol) and cyclopentyl isocyanate (0.12 mL, 1.05 mmol). Yield: 254 mg (98%), as an off-white solid. Mp: 217–218 °C. Elemental analysis (%) calcd for C25H26N2O (M = 370.49): C, 81.05; H, 7.07; N, 7.56; found: C, 81.02; H, 7.11; N, 7.53. IR νmax (film): 3314, 2955, 2857, 1640, 1557 cm−1. 1H NMR (250 MHz, DMSO) δ 8.47 (s, 1H), 7.68 (d, J = 7.1 Hz, 2H), 7.64 (d, J = 1.5 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.48 (t, J = 7.3 Hz, 2H), 7.42–7.34 (m, 2H), 7.28 (d, J = 8.0 Hz, 2H), 6.26 (d, J = 7.1 Hz, 1H), 4.06–3.87 (m, 1H), 1.93–1.77 (m, 2H), 1.72–1.49 (m, 4H), 1.39 (dd, J = 11.5, 6.0 Hz, 2H). 13C NMR (63 MHz, DMSO) δ 154.9, 141.6, 141.4, 141.3, 140.6, 137.6, 136.9, 129.5, 128.9, 127.6, 126.9, 126.7, 117.9, 114.8, 51.0, 32.9, 23.2, 20.7.
- 1-([4-Chloro-m-terphenyl]-5′-yl)-3-cyclopentylurea (4c). Prepared from 4-chloro-5′-amino-m-terphenyl (196 mg, 0.7 mmol) and cyclopentyl isocyanate (0.12 mL, 1.05 mmol). Yield: 178 mg (65%), as a white solid. Mp: 203–204 °C. Elemental analysis (%) calcd for C24H23ClN2O (M = 390.91): C, 73.74; H, 5.93; N, 7.17; found: C, 73.71; H, 6.02; N, 7.15. IR νmax (film): 3323, 2919, 2851, 1639, 1556 cm−1. 1H NMR (250 MHz, DMSO) δ 8.34 (s, 1H), 7.55 (dd, J = 14.6, 5.9 Hz, 6H), 7.35 (dd, J = 16.4, 8.1 Hz, 4H), 7.29–7.18 (m, 2H), 6.13 (d, J = 7.1 Hz, 1H), 3.89–3.72 (m, 1H), 1.78–1.62 (m, 2H), 1.56–1.33 (m, 4H), 1.32–1.14 (m, 2H). 13C NMR (63 MHz, DMSO) δ 154.9, 141.7, 141.5, 140.3, 140.0, 139.3, 132.4, 129.0, 128.9, 128.7, 127.7, 126.9, 118.1, 115.4, 114.9, 51.0, 32.8, 23.2.
- 1-([4,4″-Dichloro-m-terphenyl]-5′-yl)-3-cyclopentylurea (4d). Prepared from 4,4″-dichloro-5′-amino-m-terphenyl (220 mg, 0.7 mmol) and cyclopentyl isocyanate (0.12 mL, 1.05 mmol). Yield: 179 mg (60%), as a pale-yellow solid. Mp: 239–240 °C. Elemental analysis (%) calcd for C24H22Cl2N2O (M = 425.35): C, 67.77; H, 5.21; N, 6.59; found: C, 67.82; H, 5.19; N, 6.63. IR νmax (film): 3258, 3092, 2955, 1728, 1633 cm−1. 1H NMR (250 MHz, DMSO) δ 8.42 (s, 1H), 7.63 (d, J = 8.6 Hz, 4H), 7.59 (d, J = 1.5 Hz, 2H), 7.43 (d, J = 8.5 Hz, 4H), 7.33 (d, J = 2.0 Hz, 2H), 6.21 (d, J = 7.1 Hz, 1H), 3.94–3.78 (m, 1H), 1.86–1.67 (m, 2H), 1.62–1.39 (m, 4H), 1.37–1.20 (m, 2H). 13C NMR (63 MHz, DMSO) δ 155.2, 142.2, 140.5, 139.5, 132.8, 129.2, 129.0, 118.3, 115.6, 51.3, 33.2, 23.6.
- 1-([4-Methoxy-m-terphenyl]-5′-yl)-3-cyclopentylurea (4e). Prepared from 4-methoxy-5′-amino-m-terphenyl (193 mg, 0.7 mmol) and cyclopentyl isocyanate (0.12 mL, 1.05 mmol). Yield: 170 mg (63%), as a white solid. Mp: 208–209 °C. Elemental analysis (%) calcd for C25H26N2O2 (M = 386.49): C, 77.69; H, 6.78; N, 7.25; found: C, 77.73; H, 6.82; N, 7.21. IR νmax (film): 3312, 2954, 1640, 1605, 1556 cm−1. 1H NMR (250 MHz, MeOD) δ 7.66 (t, J = 1.8 Hz, 1H), 7.63 (d, J = 1.1 Hz, 1H), 7.61 (d, J = 2.1 Hz, 1H), 7.60–7.54 (m, 3H), 7.48–7.42 (m, 1H), 7.40 (dd, J = 3.4, 1.8 Hz, 2H), 7.38–7.30 (m, 1H), 4.16–4.01 (m, 1H), 3.83 (s, 3H), 2.08–1.90 (m, 2H), 1.82–1.58 (m, 4H), 1.58–1.42 (m, 2H). 13C NMR (63 MHz, MeOD) δ 160.9, 158.0, 143.7, 143.4, 142.6, 141.9, 134.8, 129.8, 129.1, 128.5, 128.1, 120.4, 117.0, 116.9, 115.2, 55.8, 52.9, 34.2, 24.6.
- 1-([4,4″-Dibromo-m-terphenyl]-5′-yl)-3-cyclopentylurea (4f). Prepared from 4,4″-dibromo-5′-amino-m-terphenyl (282 mg, 0.7 mmol) and cyclopentyl isocyanate (0.12 mL, 1.05 mmol). Yield: 259 mg (72%), as an off-white solid. Mp: 222–223 °C. Elemental analysis (%) calcd for C24H22Br2N2O (M = 514.25): C, 56.05; H, 4.31; N, 5.45; found: C, 56.07; H, 4.35; N, 5.42. IR νmax (film): 3294, 2955, 2860, 1631, 1557 cm−1. 1H NMR (250 MHz, DMSO) δ 8.51 (s, 1H), 7.75–7.59 (m, 10H), 7.42 (s, 1H), 6.30 (d, J = 7.1 Hz, 1H), 4.03–3.88 (m, 1H), 1.93. 13C NMR (63 MHz, DMSO) δ 154.9, 141.9, 140.2, 139.5, 131.8, 129.0, 121.1, 117.9, 115.2, 51.0, 32.8, 23.2.
- Ethyl 6-(3′-cyclohexylureido)-2-(4-methoxyphenyl)-4-phenylbenzoate (5). Prepared from ethyl-6-amino-2-(4-methoxyphenyl)-2-phenylbenzoate (243 mg, 0.7 mmol) and cyclohexyl isocyanate (0.13 mL, 1.05 mmol). Yield: 208 mg (63%), as a white solid. Mp: 234–235 °C. Elemental analysis (%) calcd for C29H32N2O4 (M = 472.58): C, 73.70; H, 6.83; N, 5.93; found: C, 73.67; H, 6.84; N, 5.90. IR νmax (film): 3302, 2926, 2850, 1691, 1644, 1542 cm−1. 1H NMR (250 MHz, CDCl3) δ 8.95 (bs, 1H), 8.60 (d, J = 1.8 Hz, 1H), 7.66 (d, J = 8.8 Hz, 2H), 7.46–7.30 (m, 5H), 7.23 (d, J = 1.8 Hz, 1H), 6.97 (d, J = 8.8 Hz, 2H), 3.93 (q, J = 7.2 Hz, 2H), 3.87 (s, 3H), 3.68 (t, J = 10.5 Hz, 1H), 2.12–1.99 (m, 2H), 1.85–1.60 (m, 3H), 1.53–1.33 (m, 2H), 1.30–1.11 (m, 3H), 0.73 (t, J = 7.2 Hz, 3H). 13C NMR (63 MHz, CDCl3) δ 170.4, 160.2, 155.1, 154.7, 144.4, 144.2, 143.4, 134.6, 132.6, 129.0, 128.5, 128.5, 127.4, 122.8, 116.6, 114.6, 68.2, 61.5, 55.8, 34.1, 26.0, 25.4, 13.4.
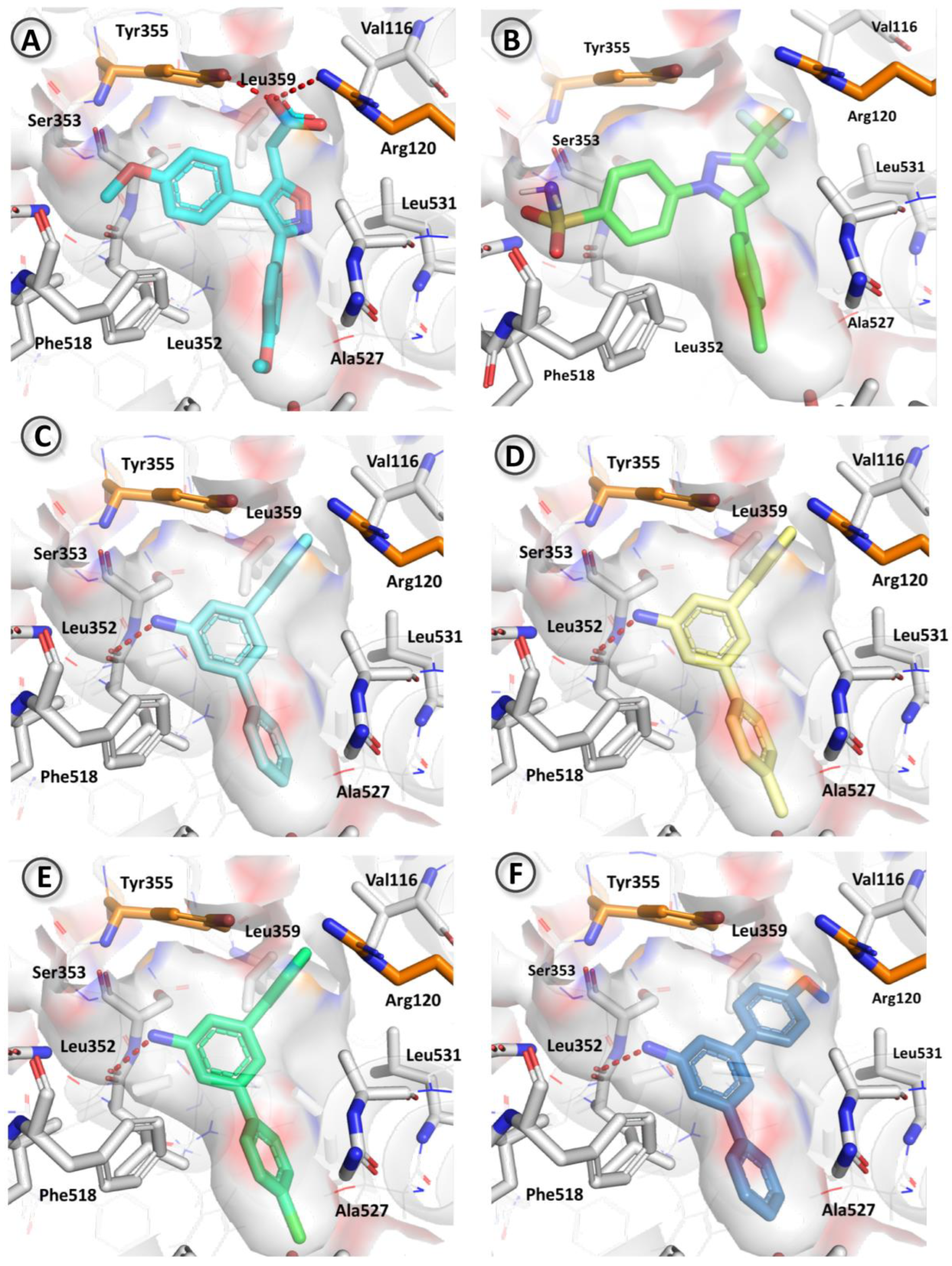
3.4. Docking Studies
3.5. Cyclooxygenase Colorimetric Inhibitor Screening Assay
3.6. Microglia BV2 Cell Line
3.7. Griess Reaction
3.8. ROS Measurement
3.9. Preparation of Organotypic Hippocampal Cultures (OHCs)
3.10. Quantification of Cell Death in OHCs: Propidium Iodide (PI) Uptake
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yagami, T.; Koma, H.; Yamamoto, Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the Central Nervous System. Mol. Neurobiol. 2016, 53, 4754–4771. [Google Scholar] [CrossRef] [PubMed]
- Ghazanfari, N.; van Waarde, A.; Dierckx, R.A.J.O.; Doorduin, J.; de Vries, E.F.J. Is cyclooxygenase-1 involved in neuroinflammation? J. Neurosci. Res. 2021, 99, 2976–2998. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Langenbach, R.; Bosetti, F. Genetic deletion or pharmacological inhibition of cyclooxygenase-1 attenuate lipopol- ysaccharide-induced inflammatory response and brain injury. FASEB J. 2008, 22, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Calvello, R.; Lofrumento, D.D.; Perrone, M.G.; Cianciulli, A.; Salvatore, R.; Vitale, P.; De Nuccio, F.; Giannotti, L.; Nicolardi, G.; Panaro, M.A.; et al. Highly selective cyclooxygenase-1 inhibitors P6 and mofezolac counteract inflammatory state both in vitro and in vivo models of neuroinflammation. Front. Neurol. 2017, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H.K. Neuroinflammation in Alzheimer’s disease: Current progress in molecular signaling and therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Gámez-Tansey, M.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: From pathophysiology to therapeutic strategies. Int. J. Mol. Sci. 2022, 23, 14. [Google Scholar] [CrossRef]
- Tanaka, J. Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system. Neuroimmunol. Neuroinflammat. 2020, 7, 73–91. [Google Scholar] [CrossRef][Green Version]
- Taylor, R.A.; Sansing, L.H. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol. 2013, 2013, 746068. [Google Scholar]
- Jordan, P.; Costa, A.; Specker, E.; Popp, O.; Volkamer, A.; Piske, R.; Obrusnik, T.; Kleissle, S.; Stuke, K.; Rex, A.; et al. Small molecule inhibiting microglial nitric oxide release could become a potential treatment for neuroinflammation. PLoS ONE 2023, 18, e0278325. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Shimamura, M.; Zhou, P.; Casolla, B.; Qian, L.; Capone, C.; Kurinami, H.; Iadecola, C.; Anrather, J. Prostaglandin E2 type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 1207–1214. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Fiebich, B.L. Cyclooxygenase inhibition in ischemic brain injury. Curr. Pharm. Des. 2008, 14, 1401–1418. [Google Scholar] [CrossRef]
- Shukuri, M.; Mawatari, A.; Ohno, M.; Suzuki, M.; Doi, H.; Watanabe, Y.; Onoe, H. Detection of cyclooxygenase-1 in activated microglia during amyloid plaque progression: PET studies in Alzheimer’s disease model mice. J. Nucl. Med. 2016, 57, 291–296. [Google Scholar] [CrossRef]
- Irannejad, H.; Unsal Tan, O.; Ozadali, K.; Dadashpour, S.; Tuylu Kucukkilinc, T.; Ahangar, N.; Ahmadnejad, M.; Emami, S. 1,2-Diaryl-2-hydroxyiminoethanones as dual COX-1 and beta-amyloid aggregation inhibitors: Biological evaluation and in silico study. Chem. Biol. Drug Des. 2015, 85, 494–503. [Google Scholar] [CrossRef]
- Yang, W.; Xiong, G.; Lin, B. Cyclooxygenase-1 mediates neuroinflammation and neurotoxicity in a mouse model of retinitis pigmentosa. J. Neuroinflamm. 2020, 17, 306. [Google Scholar] [CrossRef]
- Masuda, T.; Prinz, M. Microglia: A unique versatile cell in the Central Nervous System. ACS Chem. Neurosci. 2016, 7, 428–434. [Google Scholar] [CrossRef]
- Kenou, B.V.; Manly, L.S.; Rubovits, S.B.; Umeozulu, S.A.; Van Buskirk, M.G.; Zhang, A.S.; Pike, V.W.; Zanotti-Fregonara, P.; Henterand, I.D.; Innis, R.B. Cyclooxygenases as potential PET imaging biomarkers to explore neuroinflammation in dementia. J. Nucl. Med. 2022, 63 (Suppl. S1), 53S–59S. [Google Scholar] [CrossRef]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 inhibitors in cancer: A review of oncology and medicinal chemistry literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef]
- Wallace, J.L.; McKnight, W.; Reuter, B.K.; Vergnolle, N. NSAID-induced gastric damage in rats: Requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 2000, 119, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, T.; Futagami, S.; Tatsuguchi, A.; Suzuki, K.; Shinji, Y.; Kusunoki, M.; Shinoki, K.; Nishigaki, H.; Fujimori, S.; Wada, K.; et al. COX-1 and COX-2 conversely promote and suppress ischemia-reperfusion gastric injury in mice. Scand. J. Gastroenterol. 2008, 40, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, H.; Zheng, X.; Oda, H.; Harada, S.; Sugimoto, Y.; Sasaki, K.; Tai, A. Cyclooxygenase-1-selective inhibitors are attractive candidates for analgesics that do not cause gastric damage. Design and in vitro/in vivo evaluation of a benzamide-type cyclooxygenase-1 selective inhibitor. J. Med. Chem. 2008, 51, 2400–2411. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.G.; Scilimati, A.; Simone, L.; Vitale, P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem. 2011, 17, 3769–3805. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Panella, A.; Scilimati, A.; Perrone, M.G. COX-1 inhibitors: Beyond structure toward therapy. Med. Res. Rev. 2016, 36, 641–671. [Google Scholar] [CrossRef]
- Wenzler, T.; Yang, S.; Patrick, D.A.; Braissant, O.; Ismail, M.A.; Tidwell, R.R.; Boykin, D.W.; Wang, M.Z.; Brun, R. In Vitro and in vivo evaluation of 28DAP010, a novel diamidine for treatment of second-stage African sleeping sickness. Antimicrob. Agents Chemother. 2014, 58, 4452–4463. [Google Scholar] [CrossRef]
- Heitman, L.H.; Narlawar, R.; de Vries, H.; Willemsen, M.N.; Wolfram, D.; Brussee, J.; IJzerman, A.P. Substituted terphenyl compounds as the first class of low molecular weight allosteric inhibitors of the luteinizing hormone receptor. J. Med. Chem. 2009, 52, 2036–2042. [Google Scholar] [CrossRef]
- Bauer, J.D.; Foster, M.S.; Hugdahl, J.D.; Burns, K.L.; May, S.W.; Pollock, S.H.; Cutler, H.G.; Cutler, S.J. Synthesis and pharmacological evaluation of m-terphenyl amines as cyclooxygenase inhibitors. Med. Chem. Res. 2006, 16, 119–129. [Google Scholar] [CrossRef]
- Sridharan, V.; Menéndez, J.C. Two-step, stereocontrolled synthesis of densely functionalized cyclic β-aminoesters containing four stereocenters, based on a new CAN-catalyzed sequential three-component reaction. Org. Lett. 2008, 10, 4303–4306. [Google Scholar] [CrossRef]
- Rocchi, D.; González, J.F.; Gómez-Carpintero, J.; González-Ruiz, V.; Martín, M.A.; Sridharan, V.; Menéndez, J.C. Three-component synthesis of a library of m-terphenyl derivatives with embedded β-aminoester moieties. ACS Comb. Sci. 2018, 20, 722–731. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Urea derivatives in modern drug discovery and medicinal chemistry. J. Med. Chem. 2019, 63, 2751–2788. [Google Scholar] [CrossRef]
- Stumpe, M.C.; Grubmüller, H. Interaction of urea with amino acids: Implications for urea-induced protein denaturation. J. Am. Chem. Soc. 2007, 129, 16126–16131. [Google Scholar] [CrossRef]
- Vitale, P.; Perrone, M.G.; Malerba, P.; Lavecchia, A.; Scilimati, A. Selective COX-1 inhibition as a target of theranostic novel diarylisoxazoles. Eur. J. Med. Chem. 2014, 74, 606–618. [Google Scholar] [CrossRef]
- Miciaccia, M.; Belviso, B.D.; Iaselli, M.; Cingolani, G.; Ferorelli, S.; Cappellari, M.; Loguercio Polosa, P.; Perrone, M.G.; Caliandro, R.; Scilimati, A. Three-dimensional structure of human cyclooxygenase (hCOX)-1. Sci. Rep. 2021, 11, 4312. [Google Scholar] [CrossRef]
- Cingolani, G.; Panella, A.; Perrone, M.G.; Vitale, P.; Di Mauro, G.; Fortuna, C.G.; Armen, R.S.; Ferorelli, S.; Smith, W.L.; Scilimati, A. Structural basis for selective inhibition of cyclooxygenase-1 (COX-1) by diarylisoxazoles mofezolac and 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6). Eur. J. Med. Chem. 2017, 138, 661–668. [Google Scholar] [CrossRef]
- Rossi-George, A.; Guo, C.J.; Oakes, B.L.; Gow, A.J. Copper modulates the phenotypic response of activated BV2 microglia through the release of nitric oxide. Nitric Oxide 2012, 27, 201–209. [Google Scholar] [CrossRef]
- Lampiasi, N.; Fodera, D.; D’Alessandro, N.; Cusimano, A.; Azzolina, A.; Tripodo, C.; Florena, A.M.; Minervini, M.I.; Notarbartolo, M.; Montalto, G.; et al. The selective cyclooxygenase-1 inhibitor SC-560 suppresses cell proliferation and induces apoptosis in human hepatocellular carcinoma cells. Int. J. Mol. Med. 2006, 17, 245–252. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272. [Google Scholar] [CrossRef]
- Cizkova, D.; Devaux, S.; Le Marrec-Croq, F.; Franck, J.; Slovinska, L.; Blasko, J.; Rosocha, J.; Spakova, T.; Lefebvre, C.; Fournier, I.; et al. Modulation properties of factors released by bone marrow stromal cells on activated microglia: An in vitro study. Sci. Rep. 2014, 4, 7514. [Google Scholar] [CrossRef] [PubMed]
- Calvello, R.; Panaro, M.A.; Carbone, M.L.; Cianciulli, A.; Perrone, M.G.; Vitale, P.; Malerba, P.; Scilimati, A. Novel selective COX-1 inhibitors suppress neuroinflammatory mediators in LPS-stimulated N13 microglial cells. Pharmacol. Res. 2012, 65, 137–148. [Google Scholar] [CrossRef]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; López, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef]
- Fruhauf, P.K.; Ineu, R.P.; Tomazi, L.; Duarte, T.; Mello, C.F.; Rubin, M.A. Spermine reverses lipopolysaccharide-induced memory deficit in mice. J. Neuroinflamm. 2015, 12, 3. [Google Scholar] [CrossRef]
- Delpech, J.C.; Madore, C.; Joffre, C.; Aubert, A.; Kang, J.X.; Nadjar, A.; Laye, S. Transgenic increase in n-3/n-6 fatty acid ratio protects against cognitive deficits induced by an immune challenge through decrease of neuroinflammation. Neuropsychopharmacology 2015, 40, 525–536. [Google Scholar] [CrossRef]
- Delpech, J.C.; Thomazeau, A.; Madore, C.; Bosch-Bouju, C.; Larrieu, T.; Lacabanne, C.; Remus-Borel, J.; Aubert, A.; Joffre, C.; Nadjar, A.; et al. Dietary n-3 PUFAs deficiency increases vulnerability to inflammation-induced spatial memory impairment. Neuropsychopharmacology 2015, 40, 2774–2787. [Google Scholar] [CrossRef]
- Yang, L.; Wang, M.; Guo, Y.Y.; Sun, T.; Li, Y.J.; Yang, Q.; Zhang, K.; Liu, S.B.; Zhao, M.G.; Wu, Y.M. Systemic inflammation induces anxiety disorder through CXCL12/CXCR4 pathway. Brain Behav. Immun. 2016, 56, 352–362. [Google Scholar] [CrossRef]
- Park, S.J.; Jung, H.J.; Son, M.S.; Jung, J.M.; Kim, D.H.; Jung, I.H.; Cho, Y.B.; Lee, E.H.; Ryu, J.H. Neuroprotective effects of INM-176 against lipopolysaccharide-induced neuronal injury. Pharmacol. Biochem. Behav. 2012, 101, 427–433. [Google Scholar] [CrossRef]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for evaluating chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Qi, S.; Shi, K.; Gao, H.; Liu, Q.; Wang, H. Synthesis and fluorescence properties of 5,7-diphenylquinoline and 2,5,7-triphenylquinoline derived from m-terphenylamine. Molecules 2007, 12, 988–996. [Google Scholar] [CrossRef]
- Wang, S.K.; You, X.; Zhao, D.-Y.; Mou, N.-J.; Luo, Q.-L. Iodine-promoted Semmler–Wolff reactions: Step-economic access to meta-substituted primary anilines via aromatization. Chem. Eur. J. 2017, 23, 11757–11760. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2012, 31, 455–461. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Parada, E.; Buendia, I.; Navarro, E.; Avendaño, C.; Egea, J.; López, M.G. Microglial HO-1 induction by curcumin provides antioxidant, antineuroinflammatory, and glioprotective effects. Mol. Nutr. Food Res. 2015, 59, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Etrl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar]
- Delaney, J.S. Prediction of aqueous solubility and partition coefficient optimized by a genetic algorithm-based descriptor selection method. J. Chem. Inf. Model. 2004, 44, 1000–1005. [Google Scholar]
- Saina, A.; Zoete, V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 2016, 11, 1117–1121. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | oCOX-1; IC50 (µM) (% Inhibition at 50 µM) a | hCOX-2; IC50 (µM) (% Inhibition at 50 µM) |
|---|---|---|---|
| 1 | Mofezolac | 0.023 ± 0.0013 (100) | >50 (42) |
| 2 | Celecoxib | >50 (40) | 0.24 (95) |
| 3 | 1a | >50 | >50 |
| 4 | 1b | >50 | NM (6) |
| 5 | 1c | NM (11) | >50 |
| 6 | 1d | NM (11) | NM (5) |
| 7 | 1e | NM (9) | >50 |
| 8 | 1f | >50 | >50 |
| 9 | 1g | >50 | >50 |
| 10 | 2a | NM (32) | NM (3) |
| 11 | 2b | NM (46) | NM (2) |
| 12 | 2c | >50 | NM (4) |
| 13 | 2d | >50 | NM (13) |
| 14 | 2e | >50 | NM (20) |
| 15 | 2f | NM (8) | NM (12) |
| 16 | 2g | NM (7) | NM (13) |
| 17 | 3a | 0.80 ± 0.39 (86) | 5.4 (70) |
| 18 | 3b | 1.10 ± 0.18 (83) | >50 |
| 19 | 3c | 4.70 ± 3.04 (100) | >50 |
| 20 | 3d | NM (36] | >50 |
| 21 | 3e | 0.90 ± 0.76 (73) | NM (8) |
| 22 | 3f | >50 | >50 |
| 23 | 3g | >50 | >50 |
| 24 | 4a | >50 | >50 |
| 25 | 4b | >50 | >50 |
| 26 | 4c | >50 | >50 |
| 27 | 4d | >50 | >50 |
| 28 | 4e | >50 | >50 |
| 29 | 4f | NM (10) | NM (30) |
| 30 | 5 | >50 | >50 |
| Compound | Binding Energy (kcal/mol) |
|---|---|
| Mofezolac | 7.97 |
| Celecoxib | 5.06 |
| 3a | 6.68 |
| 3b | 6.68 |
| 3c | 6.54 |
| 3e | 5.54 |
| Treatment | Concentration | Nitrite Release after 24 h | Nitrite Release after 48 h |
|---|---|---|---|
| Basal | 0 | 100 | 100 |
| LPS | 1 μg/mL | 150 ± 8 ### | 188 ± 15 ### |
| SC-560 | 100 nM | 114 ± 9 * | 135 ± 8 * |
| 3a | 1 μM | 130 ± 14 | 145 ± 17 |
| 3b | 1 μM | 123 ± 8 | 140 ± 14 |
| 3c | 1 μM | 135 ± 6 | 181 ± 9 |
| 3e | 1 μM | 124 ± 9 | 144 ± 15 |
| 3a | 3 μM | 110 ± 7 * | 119 ± 10 ** |
| 3b | 3 μM | 100 ± 4 *** | 123 ± 10 ** |
| 3c | 3 μM | 117 ± 8 | 163 ± 5 |
| 3e | 3 μM | 110 ± 6 ** | 133 ± 13 * |
| 3a | 10 μM | 122 ± 10 | 120 ± 15 ** |
| 3b | 10 μM | 105 ± 6 ** | 128 ± 10 * |
| 3c | 10 μM | 110 ± 6 * | 138 ± 7 |
| 3e | 10 μM | 103 ± 9 ** | 131 ± 13 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocchi, D.; González, J.F.; Martín-Cámara, O.; Perrone, M.G.; Miciaccia, M.; Scilimati, A.; Decouty-Pérez, C.; Parada, E.; Egea, J.; Menéndez, J.C. m-Terphenylamines, Acting as Selective COX-1 Inhibitors, Block Microglia Inflammatory Response and Exert Neuroprotective Activity. Molecules 2023, 28, 5374. https://doi.org/10.3390/molecules28145374
Rocchi D, González JF, Martín-Cámara O, Perrone MG, Miciaccia M, Scilimati A, Decouty-Pérez C, Parada E, Egea J, Menéndez JC. m-Terphenylamines, Acting as Selective COX-1 Inhibitors, Block Microglia Inflammatory Response and Exert Neuroprotective Activity. Molecules. 2023; 28(14):5374. https://doi.org/10.3390/molecules28145374
Chicago/Turabian StyleRocchi, Damiano, Juan F. González, Olmo Martín-Cámara, Maria Grazia Perrone, Morena Miciaccia, Antonio Scilimati, Celine Decouty-Pérez, Esther Parada, Javier Egea, and J. Carlos Menéndez. 2023. "m-Terphenylamines, Acting as Selective COX-1 Inhibitors, Block Microglia Inflammatory Response and Exert Neuroprotective Activity" Molecules 28, no. 14: 5374. https://doi.org/10.3390/molecules28145374
APA StyleRocchi, D., González, J. F., Martín-Cámara, O., Perrone, M. G., Miciaccia, M., Scilimati, A., Decouty-Pérez, C., Parada, E., Egea, J., & Menéndez, J. C. (2023). m-Terphenylamines, Acting as Selective COX-1 Inhibitors, Block Microglia Inflammatory Response and Exert Neuroprotective Activity. Molecules, 28(14), 5374. https://doi.org/10.3390/molecules28145374