
Valence and Core Photoelectron Spectra of Aqueous

 , , and
, , and
Abstract
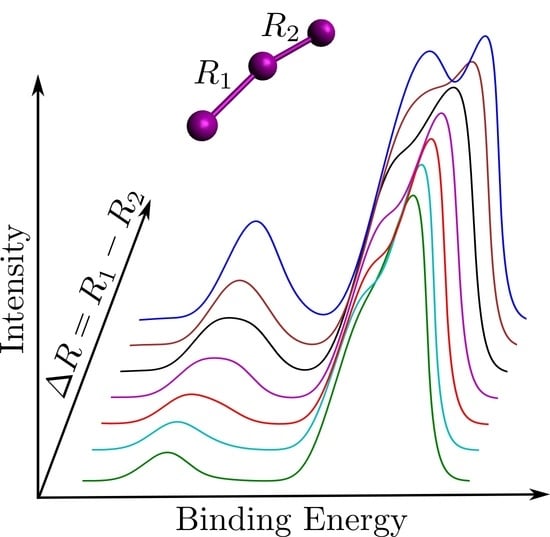
1. Introduction
2. Methods
2.1. Considered Structures
2.2. Multi-Reference Calculations
- Val_Full: All nine valence orbitals, forming and bonding and anti-bonding as well as non-bonding MOs, are included in the RAS2 subspace. Full CI (FCI) is performed within this subspace, see Figure 2. This active space allows the 3h2p configurations to be included in addition to the 1h and 2h1p ones, which are of main interest here. This active space can be also denoted as (16/15 e; RAS1: 0, RAS2: 9, RAS3:0; 0h, FCI, 0p), meaning that the ionized states have 15 electrons instead of 16.
- Val_Medium: It is essentially the same as Val_Full but the orbital was put to the RAS3 space, allowing for only one electron there, i.e., (16/15 e; RAS1: 0, RAS2: 8, RAS3:1; 0h, FCI, 1p). Effectively, since only doubly-occupied orbitals enter RAS2, it only includes 1h and 2h1p configurations. This active space allows the reproduction of the key features of the valence spectrum but with lower effort.
- N_Full: To access the N-edges, the fifteen orbitals were put into the RAS1 space allowing for a single hole. The rest of the active space is the same as in Val_Full, thus making it a (46/45 e; RAS1: 15, RAS2: 9, RAS3:0; 1h, FCI, 0p) space. The results for a similar active space, M_Full, were obtained only for the spectrum of the Sym structure with symmetry without PCM. A large number of states and unstable Restricted Active Space Second-Order Perturbation Theory (RASPT2) calculations, that should be performed and verified for each irreducible representation, make the results extremely computationally demanding and unreliable. Thus, the associated results are not suitable for systematic comparison but are briefly discussed here for consistency.
- N_Medium: The reduction of the active space is the same as between Val_Full and Val_Medium resulting in a (46/45 e; RAS1: 15, RAS2: 8, RAS3:1; 1h, FCI, 1p) space.
- N_Small: In this case, only three combinations orbitals, , , and , were included into RAS2, constituting a (34/33 e; RAS1: 15, RAS2: 3, RAS3:0; 1h, FCI, 0p) active space. This active space has been used before in Ref. [21] and includes only those orbitals for which the SU excitation has a “dipole-allowed” nature. Although this seems restrictive, it reproduces the features of N-edge PES [21].
- N_VerySmall: This is a particularly small AS, where two occupied orbitals are put in RAS2 and the unoccupied is placed in RAS3 with one electron allowed.
- M_Full:, M_Medium, M_Small The active spaces for the M-edge are full analogues of those for the N-edge, with the difference that instead of the orbitals, they include the ones.
2.3. Experiments
3. Results
3.1. General PES Assignment
3.2. Effect of Geometry
3.3. Effect of PCM
3.4. Comparison of the Active Spaces and Inclusion of Dynamic Correlation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| BE | Binding Energy |
| CASPT2 | Complete Active Space Second-Order Perturbation Theory |
| FCI | Full CI |
| MD | Molecular Dynamics |
| MO | Molecular Orbital |
| MS-RASPT2 | Multi-State RASPT2 |
| PES | Photoelectron Spectrum |
| PCM | Polarized Continuum Model |
| RASPT2 | Restricted Active Space Second-Order Perturbation Theory |
| RASSCF | Restricted Active Space Self–Consistent Field |
| SOC | Spin–Orbit Coupling |
| SU | Shake-Up |
| SS-RASPT2 | Single-State RASPT2 |
| XMS-RASPT2 | X Multi-State RASPT2 |
References
- Hüfner, S. Photoelectron Spectroscopy: Principles and Applications, 3rd ed.; Advanced Texts in Physics; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Thürmer, S.; Malerz, S.; Trinter, F.; Hergenhahn, U.; Lee, C.; Neumark, D.M.; Meijer, G.; Winter, B.; Wilkinson, I. Accurate Vertical Ionization Energy and Work Function Determinations of Liquid Water and Aqueous Solutions. Chem. Sci. 2021, 12, 10558–10582. [Google Scholar] [CrossRef] [PubMed]
- Winter, B.; Faubel, M. Photoemission from Liquid Aqueous Solutions. Chem. Rev. 2006, 106, 1176–1211. [Google Scholar] [CrossRef] [PubMed]
- Winter, B. Liquid Microjet for Photoelectron Spectroscopy. Nucl. Instrum. Methods Phys. Res. A 2009, 601, 139–150. [Google Scholar] [CrossRef]
- Ptasińska, S.; Stypczyńska, A.; Nixon, T.; Mason, N.J.; Klyachko, D.V.; Sanche, L. X-ray Induced Damage in DNA Monitored by X-ray Photoelectron Spectroscopy. J. Chem. Phys. 2008, 129, 08B604. [Google Scholar] [CrossRef] [PubMed]
- Pokapanich, W.; Ottosson, N.; Svensson, S.; Öhrwall, G.; Winter, B.; Björneholm, O. Bond Breaking, Electron Pushing, and Proton Pulling: Active and Passive Roles in the Interaction between Aqueous Ions and Water as Manifested in the O 1s Auger Decay. J. Phys. Chem. B 2012, 116, 3–8. [Google Scholar] [CrossRef]
- Sakane, H.; Mitsui, T.; Tanida, H.; Watanabe, I. XAFS Analysis of Triiodide Ion in Solutions. J. Synchrotron Radiat. 2001, 8, 674–676. [Google Scholar] [CrossRef]
- Josefsson, I.; Eriksson, S.K.; Ottosson, N.; Öhrwall, G.; Siegbahn, H.; Hagfeldt, A.; Rensmo, H.; Björneholm, O.; Odelius, M. Collective Hydrogen-Bond Dynamics Dictates the Electronic Structure of Aqueous I. Phys. Chem. Chem. Phys. 2013, 15, 20189. [Google Scholar] [CrossRef]
- Jena, N.K.; Josefsson, I.; Eriksson, S.K.; Hagfeldt, A.; Siegbahn, H.; Björneholm, O.; Rensmo, H.; Odelius, M. Solvent-Dependent Structure of the I Ion Derived from Photoelectron Spectroscopy and Ab Initio Molecular Dynamics Simulations. Chem. Eur. J. 2015, 21, 4049–4055. [Google Scholar] [CrossRef]
- Banin, U.; Ruhman, S. Ultrafast Photodissociation of I3. Coherent Photochemistry in Solution. J. Chem. Phys. 1993, 98, 4391–4403. [Google Scholar] [CrossRef]
- Ashkenazi, G.; Banin, U.; Bartana, A.; Kosloff, R.; Ruhman, S. Quantum Description of the Impulsive Photodissociation Dynamics of I in Solution. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 229–315. [Google Scholar] [CrossRef]
- Vala, J.; Kosloff, R.; Harvey, J.N. Ab Initio and Diatomics in Molecule Potentials for I, I2, I, and I3. J. Chem. Phys. 2001, 114, 7413–7423. [Google Scholar] [CrossRef]
- Ledbetter, K.; Biasin, E.; Nunes, J.P.F.; Centurion, M.; Gaffney, K.J.; Kozina, M.; Lin, M.F.; Shen, X.; Yang, J.; Wang, X.J.; et al. Photodissociation of Aqueous I Observed with Liquid-Phase Ultrafast Mega-Electron-Volt Electron Diffraction. Struct. Dyn. 2020, 7, 064901. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Photoelectrochemical Cells. Nature 2001, 414, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Conversion of Sunlight to Electric Power by Nanocrystalline Dye-Sensitized Solar Cells. J. Photochem. Photobiol. A Chem. 2004, 164, 3–14. [Google Scholar] [CrossRef]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Grätzel, M.; Gerbaldi, C. Unveiling Iodine-Based Electrolytes Chemistry in Aqueous Dye-Sensitized Solar Cells. Chem. Sci. 2016, 7, 4880–4890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.S.; Lynden-Bell, R.M. Solvent-Induced Symmetry Breaking. Phys. Rev. Lett. 2003, 90, 185505. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, J.H.; Kim, J.; Nozawa, S.; Sato, T.; Tomita, A.; Ichiyanagi, K.; Ki, H.; Kim, J.; Adachi, S.i.; et al. Solvent-Dependent Molecular Structure of Ionic Species Directly Measured by Ultrafast X-Ray Solution Scattering. Phys. Rev. Lett. 2013, 110, 165505. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.S.; Kochetov, V.; Hein, D.; Bokarev, S.I.; Wilkinson, I. Probing the Molecular Structure of Aqueous Triiodide via X-ray Photoelectron Spectroscopy and Correlated Electron Phenomena. Phys. Chem. Chem. Phys. 2022, 24, 15540–15555. [Google Scholar] [CrossRef]
- Zanni, M.T.; Greenblatt, B.J.; Davis, A.V.; Neumark, D.M. Photodissociation of Gas Phase I Using Femtosecond Photoelectron Spectroscopy. J. Chem. Phys. 1999, 111, 2991–3003. [Google Scholar] [CrossRef]
- Norell, J.; Grell, G.; Kühn, O.; Odelius, M.; Bokarev, S.I. Photoelectron Shake-Ups as a Probe of Molecular Symmetry: 4d XPS Analysis of in Solution. Phys. Chem. Chem. Phys. 2018, 20, 19916–19921. [Google Scholar] [CrossRef]
- Hwan Kim, K.; Kim, J.; Hyuk Lee, J.; Ihee, H. Topical Review: Molecular Reaction and Solvation Visualized by Time-Resolved X-ray Solution Scattering: Structure, Dynamics, and Their Solvent Dependence. Struct. Dyn. 2014, 1, 011301. [Google Scholar] [CrossRef]
- Eriksson, S.K.; Josefsson, I.; Ottosson, N.; Öhrwall, G.; Björneholm, O.; Siegbahn, H.; Hagfeldt, A.; Odelius, M.; Rensmo, H. Solvent Dependence of the Electronic Structure of I− and I. J. Phys. Chem. B 2014, 118, 3164–3174. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.P.; Visscher, L.; Bolvin, H.; Saue, T.; Knecht, S.; Fleig, T.; Eliav, E. The Electronic Structure of the Triiodide Ion from Relativistic Correlated Calculations: A Comparison of Different Methodologies. J. Chem. Phys. 2010, 133, 064305. [Google Scholar] [CrossRef] [PubMed]
- Veryazov, V.; Malmqvist, P.Å.; Roos, B.O. How to Select Active Space for Multiconfigurational Quantum Chemistry? Int. J. Quant. Chem. 2011, 111, 3329–3338. [Google Scholar] [CrossRef]
- Pierloot, K. The CASPT2 Method in Inorganic Electronic Spectroscopy: From Ionic Transition Metal to Covalent Actinide Complexes. Mol. Phys. 2003, 101, 2083–2094. [Google Scholar] [CrossRef]
- Roos, B.O.; Andersson, K. Multiconfigurational Perturbation Theory with Level Shift—The Cr2 Potential Revisited. Chem. Phys. Lett. 1995, 245, 215–223. [Google Scholar] [CrossRef]
- Roos, B.O.; Andersson, K.; Fülscher, M.P.; Serrano-Andrés, L.; Pierloot, K.; Merchán, M.; Molina, V. Applications of Level Shift Corrected Perturbation Theory in Electronic Spectroscopy. J. Mol. Struct. Theochem. 1996, 388, 257–276. [Google Scholar] [CrossRef]
- Battaglia, S.; Galván, I.F.; Lindh, R. Multiconfigurational Quantum Chemistry: The CASPT2 Method. In Theoretical and Computational Photochemistry; Elsevier: Amsterdam, The Netherlands, 2023; pp. 135–162. [Google Scholar] [CrossRef]
- Johnson, A.E.; Myers, A.B. Solvent Effects in the Raman Spectra of the Triiodide Ion: Observation of Dynamic Symmetry Breaking and Solvent Degrees of Freedom. J. Phys. Chem. 1996, 100, 7778–7788. [Google Scholar] [CrossRef]
- Li Manni, G.; Galván, I.F.; Alavi, A.; Aleotti, F.; Aquilante, F.; Autschbach, J.; Avagliano, D.; Baiardi, A.; Bao, J.J.; Battaglia, S.; et al. The OpenMolcas Web: A Community-Driven Approach to Advancing Computational Chemistry. J. Chem. Theory Comput. 2023. [Google Scholar] [CrossRef]
- Grell, G.; Bokarev, S.I.; Winter, B.; Seidel, R.; Aziz, E.F.; Aziz, S.G.; Kühn, O. Multi-Reference Approach to the Calculation of Photoelectron Spectra Including Spin-Orbit Coupling. J. Chem. Phys. 2015, 143, 074104. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.Å.; Veryazov, V.; Widmark, P.O. Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set. J. Phys. Chem. A 2004, 108, 2851–2858. [Google Scholar] [CrossRef]
- Douglas, M.; Kroll, N.M. Quantum Electrodynamical Corrections to the Fine Structure of Helium. Ann. Phys. 1974, 82, 89–155. [Google Scholar] [CrossRef]
- Hess, B.A. Relativistic Electronic-Structure Calculations Employing a Two-Component No-Pair Formalism with External-Field Projection Operators. Phys. Rev. A 1986, 33, 3742. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Roos, B.O.; Schimmelpfennig, B. The Restricted Active Space (RAS) State Interaction Approach with Spin-Orbit Coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Polarizable Dielectric Model of Solvation with Inclusion of Charge Penetration Effects. J. Chem. Phys. 2001, 114, 5691–5701. [Google Scholar] [CrossRef]
- Lynden-Bell, R.M.; Kosloff, R.; Ruhman, S.; Danovich, D.; Vala, J. Does Solvation Cause Symmetry Breaking in the I Ion in Aqueous Solution? J. Chem. Phys. 1998, 109, 9928–9937. [Google Scholar] [CrossRef]
- Sato, H.; Hirata, F.; Myers, A.B. Theoretical Study of the Solvent Effect on Triiodide Ion in Solutions. J. Phys. Chem. A 1998, 102, 2065–2071. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Rendell, A.; Roos, B.O. The Restricted Active Space Self-Consistent-Field Method, Implemented with a Split Graph Unitary Group Approach. J. Phys. Chem. 1990, 94, 5477–5482. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O.; Sadlej, A.J.; Wolinski, K. Second-Order Perturbation Theory with a CASSCF Reference Function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Forsberg, N.; Malmqvist, P.Å. Multiconfiguration Perturbation Theory with Imaginary Level Shift. Chem. Phys. Lett. 1997, 274, 196–204. [Google Scholar] [CrossRef]
- Granovsky, A.A. Extended Multi-Configuration Quasi-Degenerate Perturbation Theory: The New Approach to Multi-State Multi-Reference Perturbation Theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.Å.; Roos, B.O.; Serrano-Andrés, L. The Multi-State CASPT2 Method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Viefhaus, J.; Scholz, F.; Deinert, S.; Glaser, L.; Ilchen, M.; Seltmann, J.; Walter, P.; Siewert, F. The Variable Polarization XUV Beamline P04 at PETRA III: Optics, Mechanics and Their Performance. Nucl. Instrum. Methods Phys. Res. A 2013, 710, 151–154. [Google Scholar] [CrossRef]
- Malerz, S.; Haak, H.; Trinter, F.; Stephansen, A.B.; Kolbeck, C.; Pohl, M.; Hergenhahn, U.; Meijer, G.; Winter, B. A Setup for Studies of Photoelectron Circular Dichroism from Chiral Molecules in Aqueous Solution. Rev. Sci. Instrum. 2022, 93, 015101. [Google Scholar] [CrossRef] [PubMed]
- Winter, B.; Aziz, E.F.; Hergenhahn, U.; Faubel, M.; Hertel, I.V. Hydrogen Bonds in Liquid Water Studied by Photoelectron Spectroscopy. J. Chem. Phys. 2007, 126, 124504. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, N.; Karashima, S.; Tang, Y.; Horio, T.; Abulimiti, B.; Suzuki, Y.I.; Ogi, Y.; Oura, M.; Suzuki, T. Photoelectron Spectroscopy of Aqueous Solutions: Streaming Potentials of NaX (X= Cl, Br, and I) Solutions and Electron Binding Energies of Liquid Water and X−. J. Chem. Phys. 2014, 140, 174506. [Google Scholar] [CrossRef]
- Arbman, M.; Holmberg, S.; Lundholm, M.; Siegbahn, H.; Gropen, O.; Wahlgren, U. Liquid ESCA Measurements and ECP Calculations on the 3d Spectrum of I. Chem. Phys. 1983, 81, 113–119. [Google Scholar] [CrossRef]
- Koslowski, T.; Vöhringer, P. Is Solvated I Angular? Chem. Phys. Lett. 2001, 342, 141–147. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van Der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Heo, J.; Kim, J.G.; Choi, E.H.; Ki, H.; Ahn, D.S.; Kim, J.; Lee, S.; Ihee, H. Determining the Charge Distribution and the Direction of Bond Cleavage with Femtosecond Anisotropic X-Ray Liquidography. Nat. Commun. 2022, 13, 522. [Google Scholar] [CrossRef]
- Nimmrich, A.; Panman, M.R.; Berntsson, O.; Biasin, E.; Niebling, S.; Petersson, J.; Hoernke, M.; Björling, A.; Gustavsson, E.; Van Driel, T.B.; et al. Solvent-Dependent Structural Dynamics in the Ultrafast Photodissociation Reaction of Triiodide Observed with Time-Resolved X-ray Solution Scattering. J. Am. Chem. Soc. 2023. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Type | Symmetry | r(I–I), Å | , Å | ∠(I–I–I), ° | References |
|---|---|---|---|---|---|---|
| Sym | Symmetric linear | 2.91, 2.91 | 0.00 | 180 | CASPT2 vacuum [23] | |
| Lin | Linear, slight asymmetry | 2.94, 3.09 | 0.15 | 180 | Exp in methanol [18] | |
| MD | Near-Linear | 2.90, 3.10 | 0.20 | 171 | MD simulations [9] | |
| HBent | Bent, slight asymmetry | 2.94, 3.10 | 0.16 | 165 | Intermediate point | |
| Asym | Linear, max asymmetry | 2.82, 3.37 | 0.55 | 180 | MD simulations [8] | |
| Bent | Asymmetric bent | 2.93, 3.38 | 0.45 | 153 | Exp in water [18] |
| Active Space | Doublet | Quartet | Imaginary Shift, a.u. |
|---|---|---|---|
| Val_Full | 240 | – | 0.1 |
| Val_Medium | 72 | – | 0.1 |
| N_Full, M_Full | 1455 (240) | 624 (84) | 0.2 |
| N_Medium, M_Medium | 327 (72) | 148 (28) | 0.2 |
| N_Small, M_Small | 133 (8) | 46 (1) | 0.2 |
| N_VerySmall, M_VerySmall | 81 (6) | 31 (1) | 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kochetov, V.; Ahsan, M.S.; Hein, D.; Wilkinson, I.; Bokarev, S.I.
Valence and Core Photoelectron Spectra of Aqueous
Kochetov V, Ahsan MS, Hein D, Wilkinson I, Bokarev SI.
Valence and Core Photoelectron Spectra of Aqueous
Kochetov, Vladislav, Md Sabbir Ahsan, Dennis Hein, Iain Wilkinson, and Sergey I. Bokarev.
2023. "Valence and Core Photoelectron Spectra of Aqueous
Kochetov, V., Ahsan, M. S., Hein, D., Wilkinson, I., & Bokarev, S. I.
(2023). Valence and Core Photoelectron Spectra of Aqueous