
An Integrated Electrochemical System for Synergistic Cathodic Nitrate Reduction and Anodic Sulfite Oxidation



Abstract
:1. Introduction
2. Results and Discussion
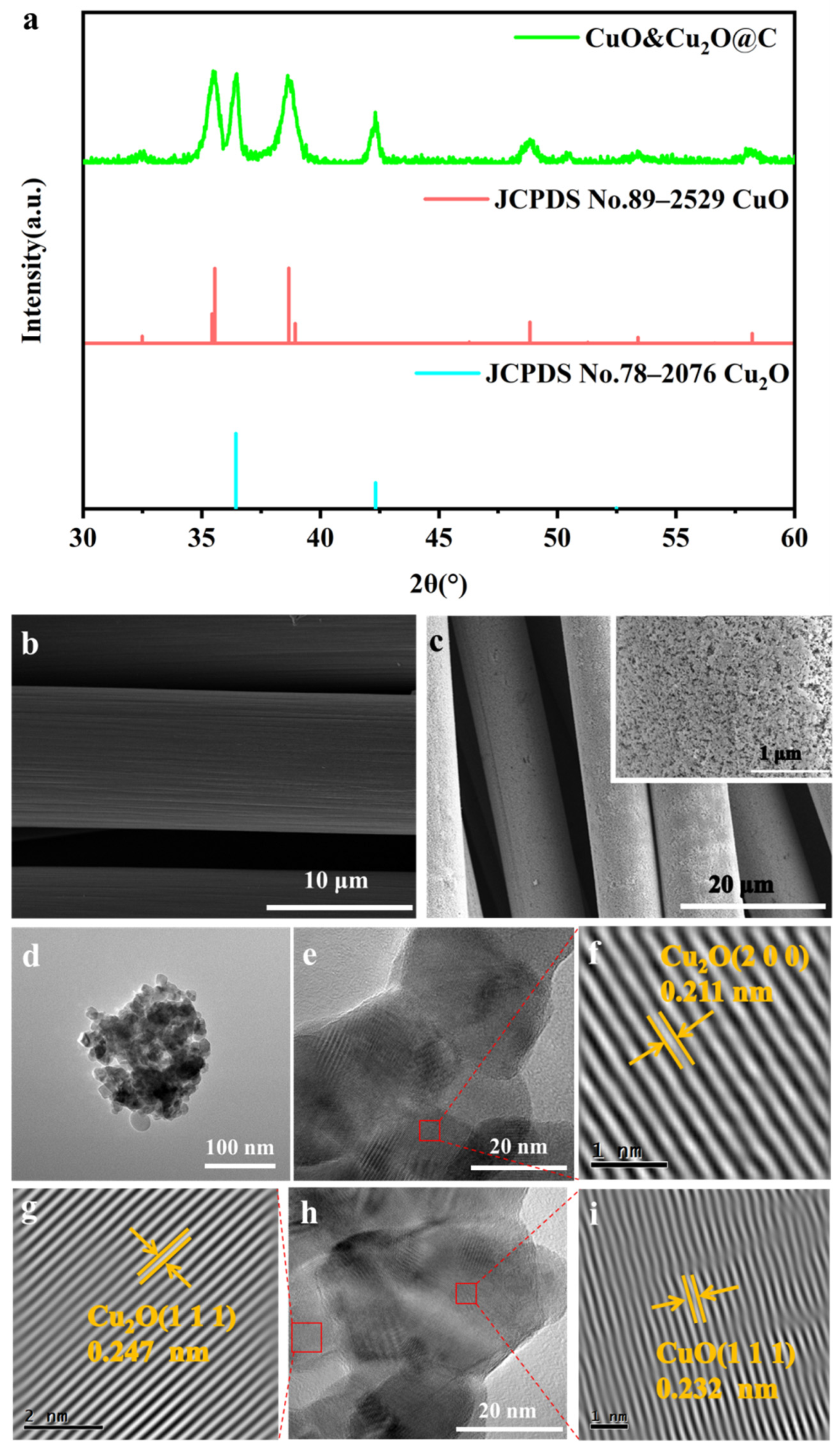
2.1. Characterizations of CuO&Cu2O@C Electrode
2.2. Electrochemical Reduction of Nitrate
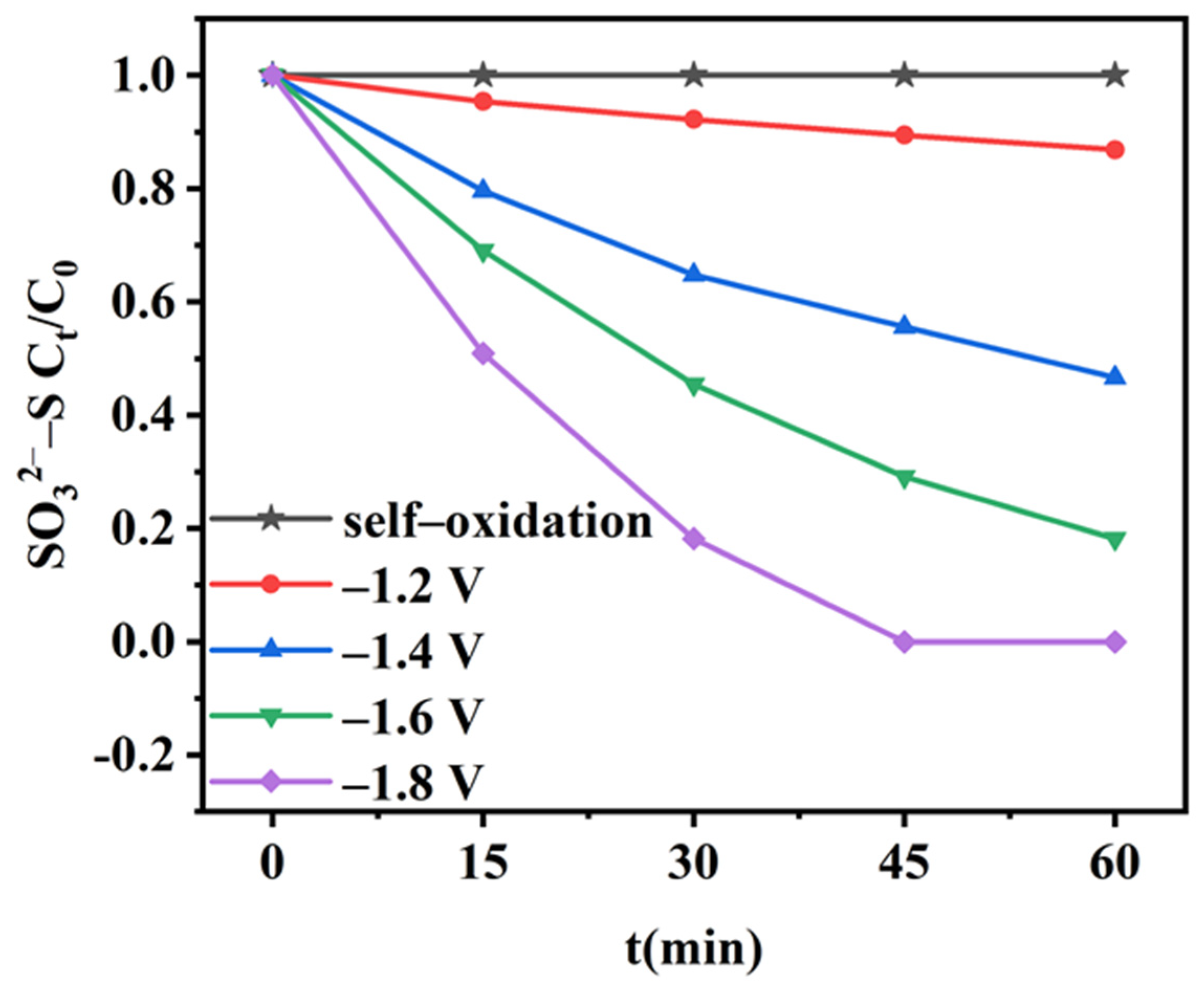
2.3. Self-Oxidation and Anodic Electro-Oxidation of Sulfite
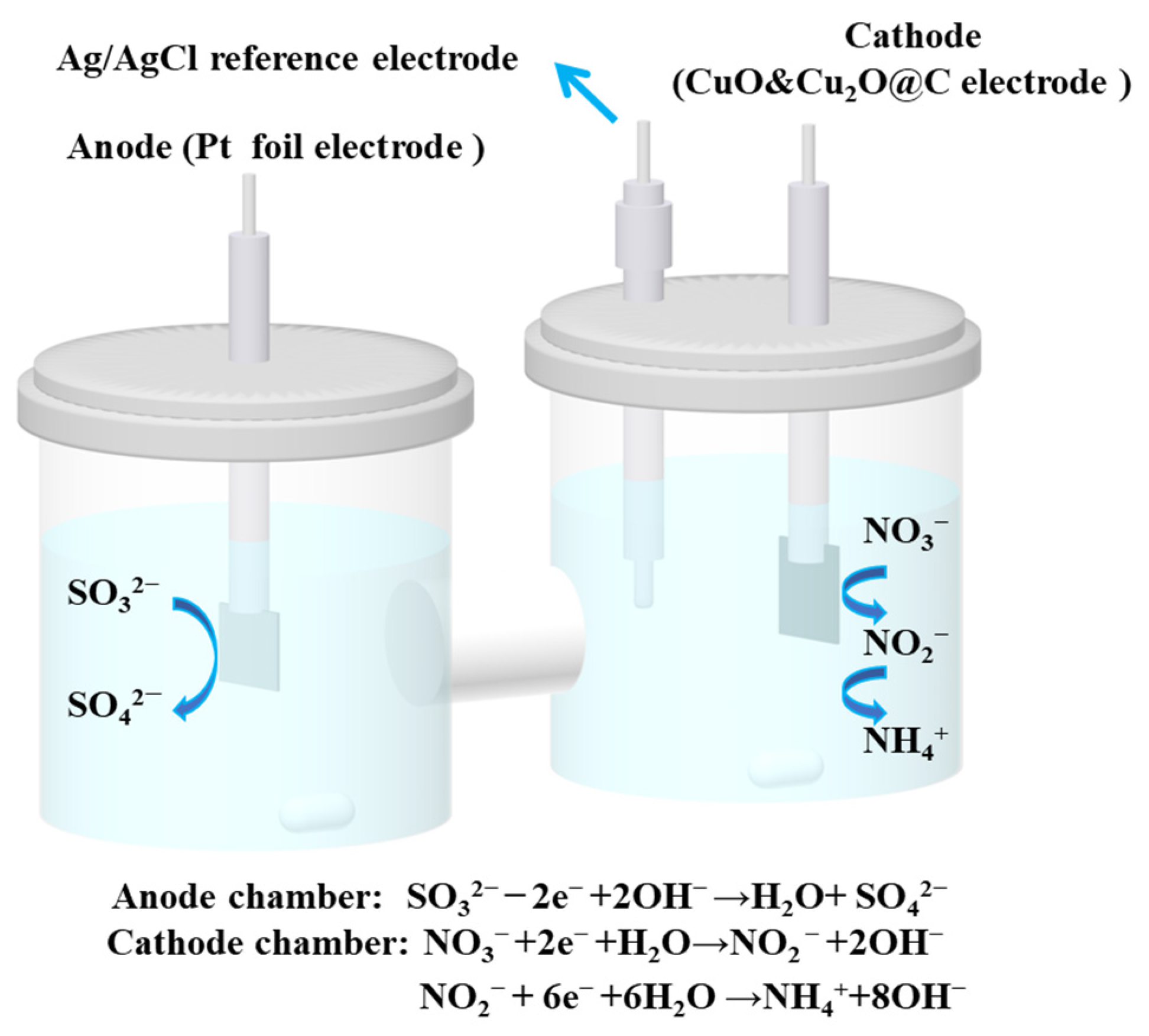
2.4. Integrated Electrochemical System
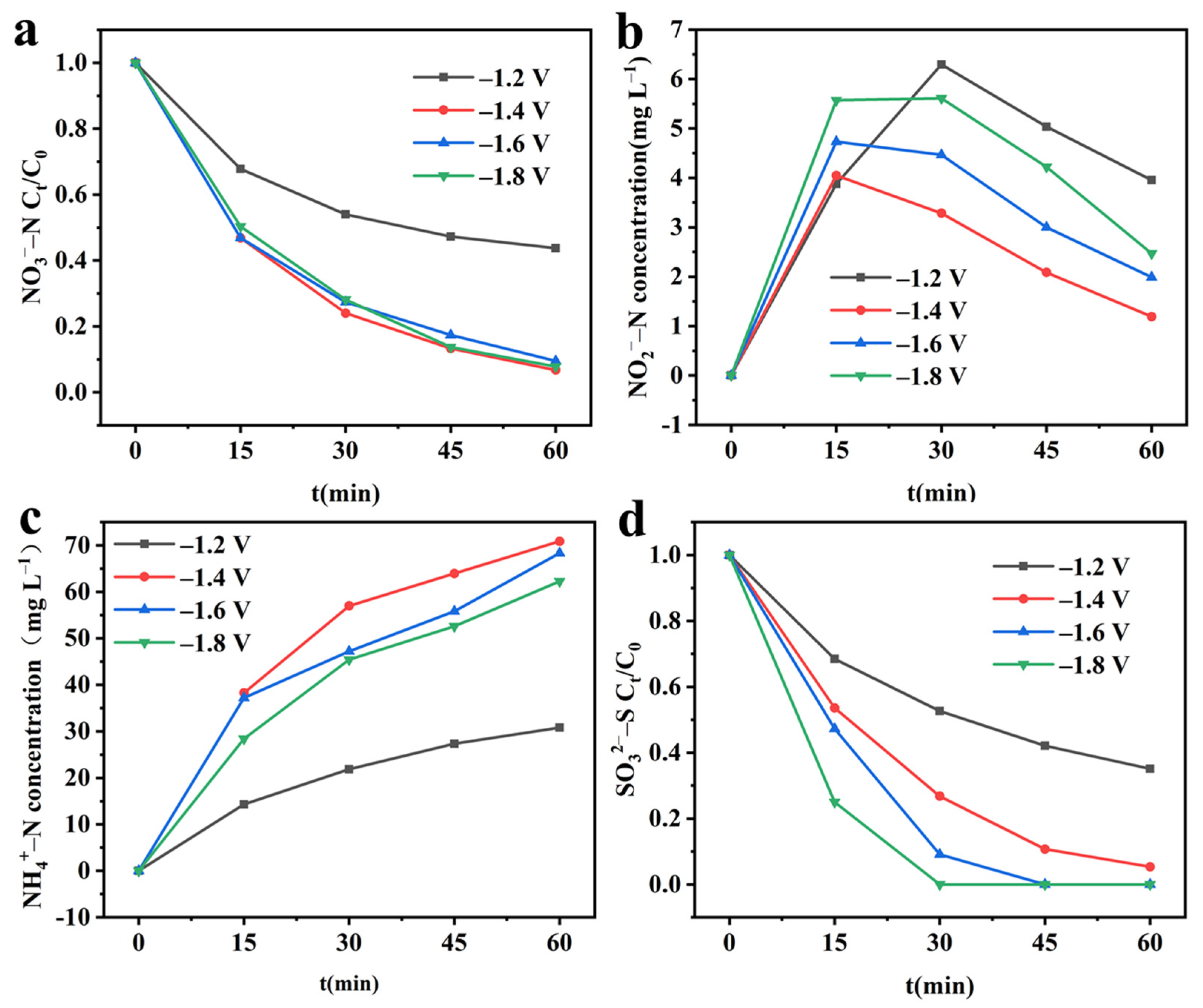
2.4.1. Effect of the Applied Cathode Potentials
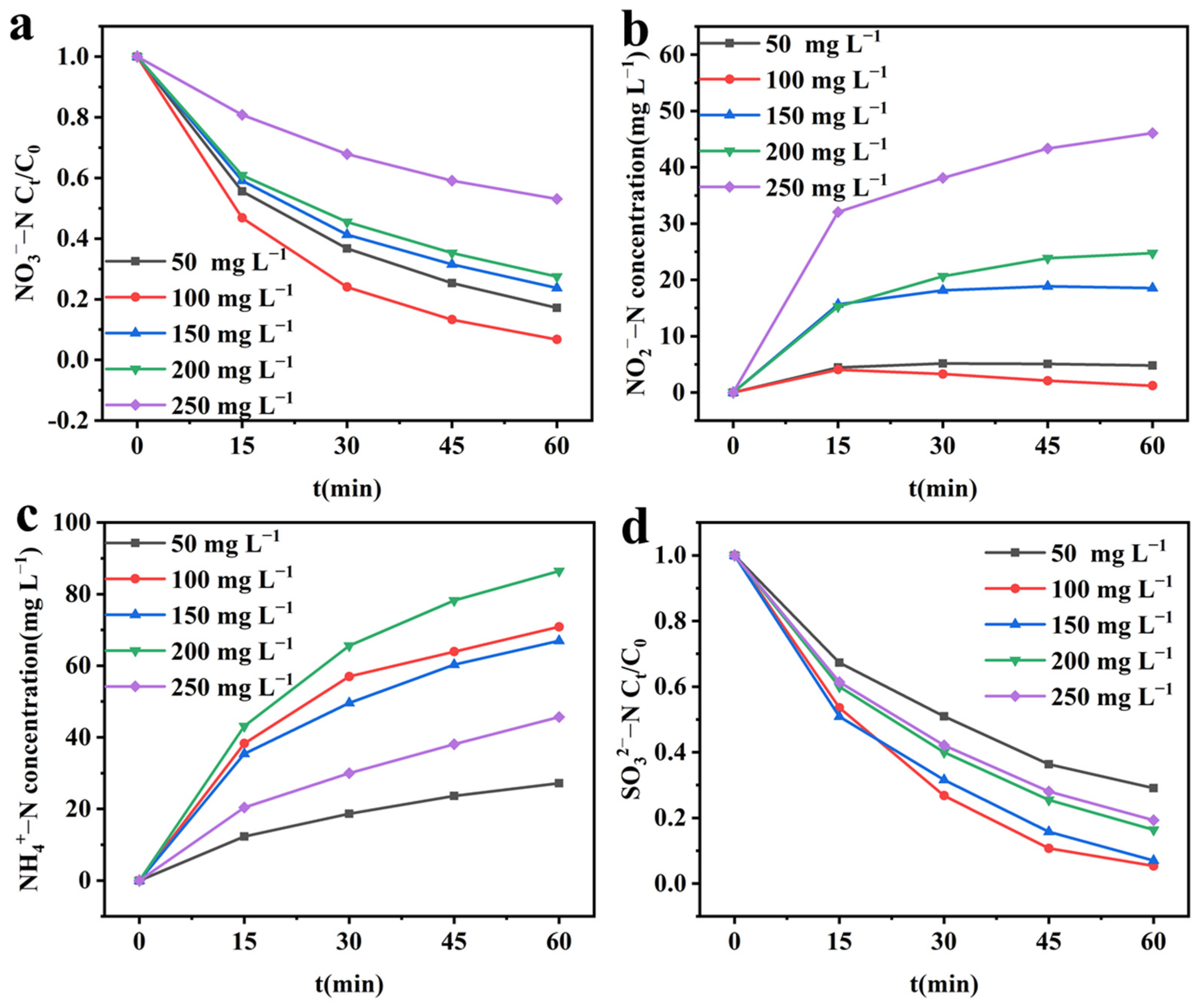
2.4.2. Effect of Initial NO3−–N Concentration
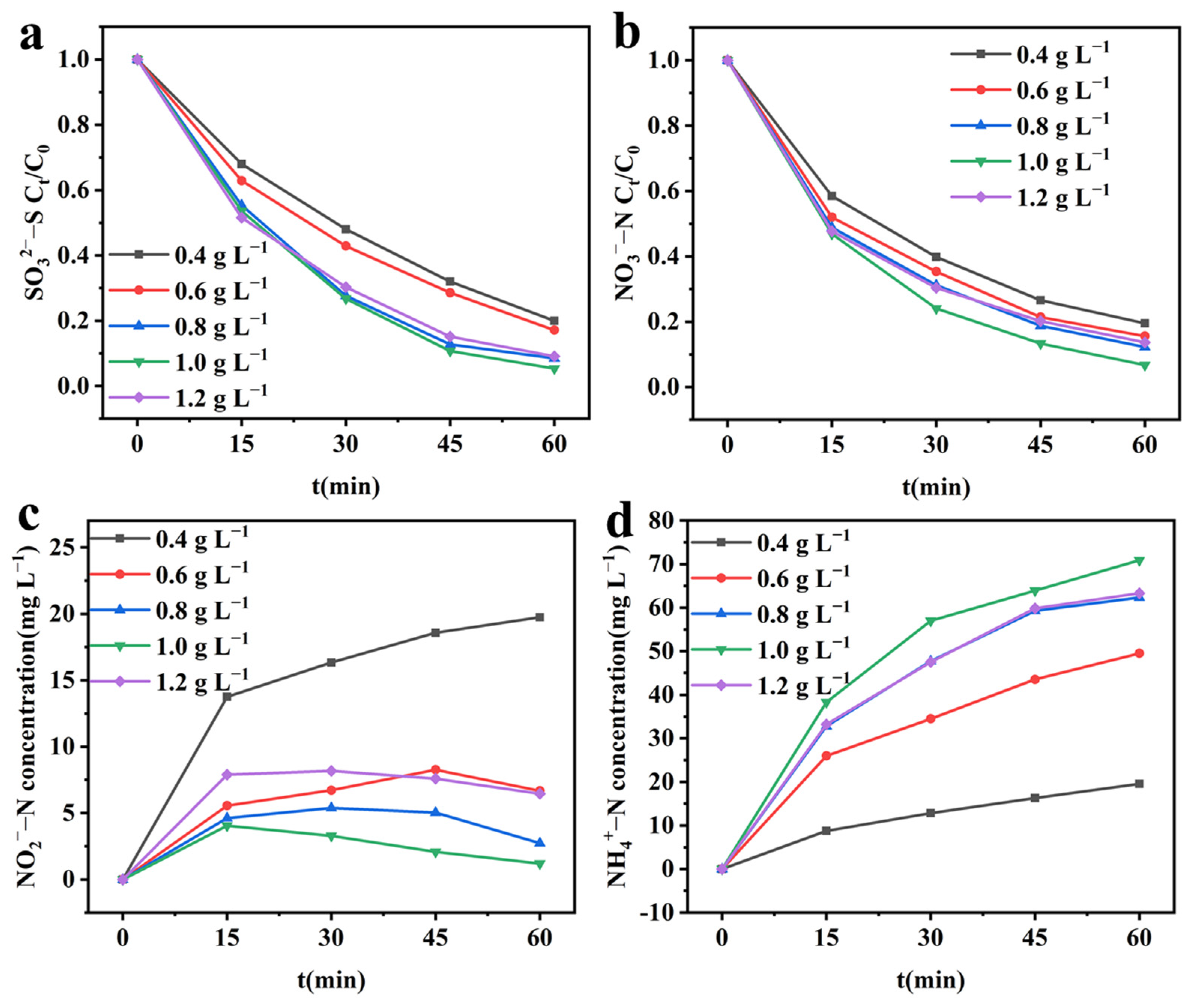
2.4.3. Effect of Initial SO32−–S Concentration on Integrated Electrochemical System
3. Materials and Methods
3.1. Reagents
3.2. Fabrication of CuO&Cu2O@C Electrode
3.3. Characterizations
3.4. Batch Experiments
3.4.1. Single Electrochemical Reduction of Nitrate
3.4.2. Self-Oxidation and Single Electro-Oxidation of Sulfite
3.4.3. Integrated Electrochemical Batch Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chen, M.; Huang, X.; Wang, N.; Wang, T.; Yang, J.; Wei, Y.; Dao, X.; Zhou, L.; Hao, H. Cu2O Nanoparticles Modified BiO2−x Nanosheets for Efficient Electrochemical Reduction of Nitrate-N and Nitrobenzene from Wastewater. Sep. Purif. Technol. 2022, 289, 120728. [Google Scholar] [CrossRef]
- Yao, D.; Tang, C.; Wang, P.; Cheng, H.; Jin, H.; Ding, L.X.; Qiao, S.Z. Electrocatalytic Green Ammonia Production beyond Ambient Aqueous Nitrogen Reduction. Chem. Eng. Sci. 2022, 257, 117735. [Google Scholar] [CrossRef]
- Ren, T.; Ren, K.; Wang, M.; Liu, M.; Wang, Z.; Wang, H.; Li, X.; Wang, L.; Xu, Y. Concave-Convex Surface Oxide Layers over Copper Nanowires Boost Electrochemical Nitrate-to-Ammonia Conversion. Chem. Eng. J. 2021, 426, 130759. [Google Scholar] [CrossRef]
- Yuan, S.; Xue, Y.; Ma, R.; Ma, Q.; Chen, Y.; Fan, J. Advances in Iron-Based Electrocatalysts for Nitrate Reduction. Sci. Total Environ. 2023, 866, 161444. [Google Scholar] [CrossRef]
- Vass, Á.; Kormányos, A.; Kószo, Z.; Endrődi, B.; Janáky, C. Anode Catalysts in CO2 Electrolysis: Challenges and Untapped Opportunities. ACS Catal. 2022, 12, 1037–1051. [Google Scholar] [CrossRef]
- Zou, J.P.; Chen, Y.; Liu, S.S.; Xing, Q.J.; Dong, W.H.; Luo, X.B.; Dai, W.L.; Xiao, X.; Luo, J.M.; Crittenden, J. Electrochemical Oxidation and Advanced Oxidation Processes Using a 3D Hexagonal Co3O4 Array Anode for 4-Nitrophenol Decomposition Coupled with Simultaneous CO2 Conversion to Liquid Fuels via a Flower-like CuO Cathode. Water Res. 2019, 150, 330–339. [Google Scholar] [CrossRef]
- Li, D.; Yang, J.; Lian, J.; Yan, J.; Liu, S. Recent Advances in Paired Electrolysis Coupling CO2 Reduction with Alternative Oxidation Reactions. J. Energy Chem. 2023, 77, 406–419. [Google Scholar] [CrossRef]
- Ali, I.; Barros de Souza, A.; De Laet, S.; Van Eyck, K.; Dewil, R. Anodic Oxidation of Sulfamethoxazole Paired to Cathodic Hydrogen Peroxide Production. Chemosphere 2023, 319, 137984. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, W.; Ding, Y.; Zhao, G.B.; Gao, J.H. Energy-Saving Cathodic Hydrogen Production Enabled by Anodic Oxidation of Aqueous Sodium Sulfite Solutions. Energy Fuels 2020, 34, 9058–9063. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Xu, P.; Li, Q.; Zhang, W.; Cui, S. Selectivity of Transition Metal Catalysts in Promoting the Oxidation of Solid Sulfites in Flue Gas Desulfurization. Appl. Catal. A Gen. 2015, 508, 52–60. [Google Scholar] [CrossRef]
- Wang, L.; Xing, L.; Liu, J.; Qi, T.; Zhang, S.; Ma, Y.; Ning, P. Construction of Lattice-Confined Co-MCM-48 for Boosting Sulfite Oxidation in Wet Desulfuration. Chem. Eng. J. 2021, 407, 127210. [Google Scholar] [CrossRef]
- Han, J.; Cheng, H.; Zhang, L.; Fu, H.; Chen, J. Trash to Treasure: Use Flue Gas SO2 to Produce H2 via a Photoelectrochemical Process. Chem. Eng. J. 2018, 335, 231–235. [Google Scholar] [CrossRef]
- Dubale, A.A.; Ahmed, I.N.; Zhang, Y.J.; Yang, X.L.; Xie, M.H. A Facile Strategy for Fabricating C@Cu2O/CuO Composite for Efficient Photochemical Hydrogen Production with High External Quantum Efficiency. Appl. Surf. Sci. 2020, 534, 147582. [Google Scholar] [CrossRef]
- Du, F.; Cui, G.H.; Yang, B.L.; Zhang, D.S.; Song, R.F.; Li, Z.X. Ingenious Design of One Mixed-Valence Dual-Net Copper Metal-Organic Framework for Deriving Cu2O/CuO Heterojunction with Highly Electrocatalytic Performances from NO3− to NH3. J. Power Sources 2022, 543, 231832. [Google Scholar] [CrossRef]
- Liang, X.; Chen, X.; Xiang, Z.; Yan, R.; Xi, H.; Bian, T.; Zhang, J.; Zhao, J.; Cai, Q.; Wang, H. Design and Synthesis of Surface-Controlled CuOx /RGO Nanocomposites with Unusually High Efficiency in Catalytic Conversion of Organic Reactants in the Presence of NaBH 4. Appl. Surf. Sci. 2018, 459, 716–722. [Google Scholar] [CrossRef]
- Moghanlou, A.O.; Sadr, M.H.; Bezaatpour, A.; Salimi, F.; Yosefi, M. RGO/Cu2O-CuO Nanocomposite as a Visible-Light Assisted Photocatalyst for Reduction of Organic Nitro Groups to Amines. Mol. Catal. 2021, 516, 111997. [Google Scholar] [CrossRef]
- Li, J.; Guo, J.; Zhang, J.; Sun, Z.; Gao, J. Surface Etching and Photodeposition Nanostructures Core-Shell Cu2O@CuO-Ag with S-Scheme Heterojunction for High Efficiency Photocatalysis. Surf. Interfaces 2022, 34, 102308. [Google Scholar] [CrossRef]
- Jiang, G.; Peng, M.; Hu, L.; Ouyang, J.; Lv, X.; Yang, Z.; Liang, X.; Liu, Y.; Liu, H. Electron-Deficient Cuδ+ Stabilized by Interfacial Cu–O-Al Bonding for Accelerating Electrocatalytic Nitrate Conversion. Chem. Eng. J. 2022, 435, 134853. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, K.; Ren, T.; Wang, M.; Wang, Z.; Li, X.; Wang, L.; Wang, H. Ultralow-Content Pd in-Situ Incorporation Mediated Hierarchical Defects in Corner-Etched Cu2O Octahedra for Enhanced Electrocatalytic Nitrate Reduction to Ammonia. Appl. Catal. B Environ. 2022, 306, 121094. [Google Scholar] [CrossRef]
- He, L.; Yao, F.; Zhong, Y.; Tan, C.; Hou, K.; Pi, Z.; Chen, S.; Li, X.; Yang, Q. Achieving High-Performance Electrocatalytic Reduction of Nitrate by N-Rich Carbon-Encapsulated Ni-Cu Bimetallic Nanoparticles Supported Nickel Foam Electrode. J. Hazard. Mater. 2022, 436, 129253. [Google Scholar] [CrossRef]
- Lăboșel, M.A.; Dima, G.D.; Duca, D.A.; Vaszilcsin, N.; Laurentiu Dan, M. Anodic Sulphite Oxidation on Lead Electrode in a Neutral Environment. Mater. Today Proc. 2022, 78, 302–307. [Google Scholar] [CrossRef]
- Zhou, J.; Pan, F.; Yao, Q.; Zhu, Y.; Ma, H.; Niu, J.; Xie, J. Achieving Efficient and Stable Electrochemical Nitrate Removal by In-Situ Reconstruction of Cu2O/Cu Electroactive Nanocatalysts on Cu Foam. Appl. Catal. B Environ. 2022, 317, 121811. [Google Scholar] [CrossRef]
- Yao, F.; Jia, M.; Yang, Q.; Chen, F.; Zhong, Y.; Chen, S.; He, L.; Pi, Z.; Hou, K.; Wang, D.; et al. Highly Selective Electrochemical Nitrate Reduction Using Copper Phosphide Self-Supported Copper Foam Electrode: Performance, Mechanism, and Application. Water Res. 2021, 193, 116881. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, L.; Jiang, H.; Li, F.; Zhao, T.; Zhuo, M.; Chen, Q.; Mao, M.; Xu, Y. Design of Counter Oxidation vs. CO2 Electroreduction for Efficient Formate Production on a Tin Cathode. J. Electroanal. Chem. 2019, 847, 113264. [Google Scholar] [CrossRef]
- Zhu, D.; Li, G.; Yan, X.; Geng, C.; Gao, L. Electrochemical Nitrate Reduction to High-Value Ammonia on Two-Dimensional Molybdenum Carbide Nanosheets for Nitrate-Containing Wastewater Upcycling. Sci. Total Environ. 2023, 878, 163145. [Google Scholar] [CrossRef]
- Bharath, G.; Hai, A.; Rambabu, K.; Kallem, P.; Haija, M.A.; Banat, F.; Theerthagiri, J.; Choi, M.Y. Fabrication of Pd/MnFe2O4 Bifunctional 2-D Nanosheets to Enhance the Yield of HCOOH from CO2 Cathodic Reduction Paired with Anodic Oxidation to CH3OH. Fuel 2022, 311, 122619. [Google Scholar] [CrossRef]
- Chen, M.; Bi, J.; Huang, X.; Wang, T.; Wang, Z.; Hao, H. Bi2O3 Nanosheets Arrays In-Situ Decorated on Carbon Cloth for Efficient Electrochemical Reduction of Nitrate. Chemosphere 2021, 278, 130386. [Google Scholar] [CrossRef]
- Fang, X.; Tan, L.; Luo, H.; Jiang, F.; Chen, H. Efficient Electrochemical Reduction of Nitrate by Bimetallic Cu-Fe Phosphide Derived from Prussian Blue Analogue. Colloids Surf. A Physicochem. Eng. Asp. 2023, 658, 130678. [Google Scholar] [CrossRef]
- Niu, Q.; Yang, S.; Song, Z.; Liu, C.; Li, Z.; Wang, X.; Song, L. Fabrication of Efficient and Robust Fe0/Ni2P/CC Composite and the Employment for Electrochemical Reduction of Nitrate. J. Environ. Chem. Eng. 2021, 9, 106412. [Google Scholar] [CrossRef]
- Enache, A.F.; Dan, M.L.; Muntean, R.; Vaszilcsin, N. New Electrodes Based on Pt-Co Alloys Used as Anodic Materials for Sulphite Oxidation in Alkaline Fuel Cells. IOP Conf. Ser. Mater. Sci. Eng. 2018, 416, 012072. [Google Scholar] [CrossRef]
- Yan, X.; He, L.; Zhang, W.; Chen, W.; Wu, J.; Yang, N.; Cai, X.; Li, L.; Yan, L.; Rao, P. Efficient Ammonia-Nitrogen Removal and Recovery from Wastewater via the Continuous Flat-Sheet Gas-Permeable Membranes Reactor Pretreatment. J. Water Process Eng. 2023, 52, 103571. [Google Scholar] [CrossRef]
- Lee, G.; Kim, K.; Chung, J.; Han, J.I. Electrochemical Ammonia Accumulation and Recovery from Ammonia-Rich Livestock Wastewater. Chemosphere 2021, 270, 128631. [Google Scholar] [CrossRef] [PubMed]
- Chatla, A.; Almanassra, I.W.; Abushawish, A.; Laoui, T.; Alawadhi, H.; Atieh, M.A.; Ghaffour, N. Sulphate Removal from Aqueous Solutions: State-of-the-Art Technologies and Future Research Trends. Desalination 2023, 558, 116615. [Google Scholar] [CrossRef]
- Shi, Y.; Xu, L.; Chen, M.; Yang, B.; Cheng, G.; Wu, C.e.; Miao, Z.; Wang, N.; Hu, X. Fabricating Cu2O-CuO Submicron-Cubes for Efficient Catalytic CO Oxidation: The Significant Effect of Heterojunction Interface. J. Ind. Eng. Chem. 2022, 105, 324–336. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R (NO3−–N)/% | S (NO2−–N)/% | S (NH4+–N)/% | O (SO32−–S)/% | |||||
|---|---|---|---|---|---|---|---|---|
| Single | Integrated | Single | Integrated | Single | Integrate | Single | Integrated | |
| −1.2 V | 51.59 | 56.28 | 7.64 | 6.80 | 55.14 | 53.50 | 13.15 | 64.91 |
| −1.4 V | 91.61 | 93.26 | 1.22 | 1.25 | 78.41 | 74.90 | 53.33 | 94.64 |
| −1.6 V | 84.46 | 90.46 | 1.05 | 2.10 | 67.21 | 73.20 | 81.82 | 100.00 |
| −1.8 V | 82.89 | 92.19 | 0.40 | 2.60 | 61.39 | 66.70 | 100.00 | 100.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, B.; Wang, S.; Guo, X.; Zhao, Y.; Rohani, S. An Integrated Electrochemical System for Synergistic Cathodic Nitrate Reduction and Anodic Sulfite Oxidation. Molecules 2023, 28, 4666. https://doi.org/10.3390/molecules28124666
Cui B, Wang S, Guo X, Zhao Y, Rohani S. An Integrated Electrochemical System for Synergistic Cathodic Nitrate Reduction and Anodic Sulfite Oxidation. Molecules. 2023; 28(12):4666. https://doi.org/10.3390/molecules28124666
Chicago/Turabian StyleCui, Bing, Shizhao Wang, Xiaofu Guo, Yingying Zhao, and Sohrab Rohani. 2023. "An Integrated Electrochemical System for Synergistic Cathodic Nitrate Reduction and Anodic Sulfite Oxidation" Molecules 28, no. 12: 4666. https://doi.org/10.3390/molecules28124666
APA StyleCui, B., Wang, S., Guo, X., Zhao, Y., & Rohani, S. (2023). An Integrated Electrochemical System for Synergistic Cathodic Nitrate Reduction and Anodic Sulfite Oxidation. Molecules, 28(12), 4666. https://doi.org/10.3390/molecules28124666