

Binding Affinity and Mechanisms of Potential Antidepressants Targeting Human NMDA Receptors




Abstract
1. Introduction
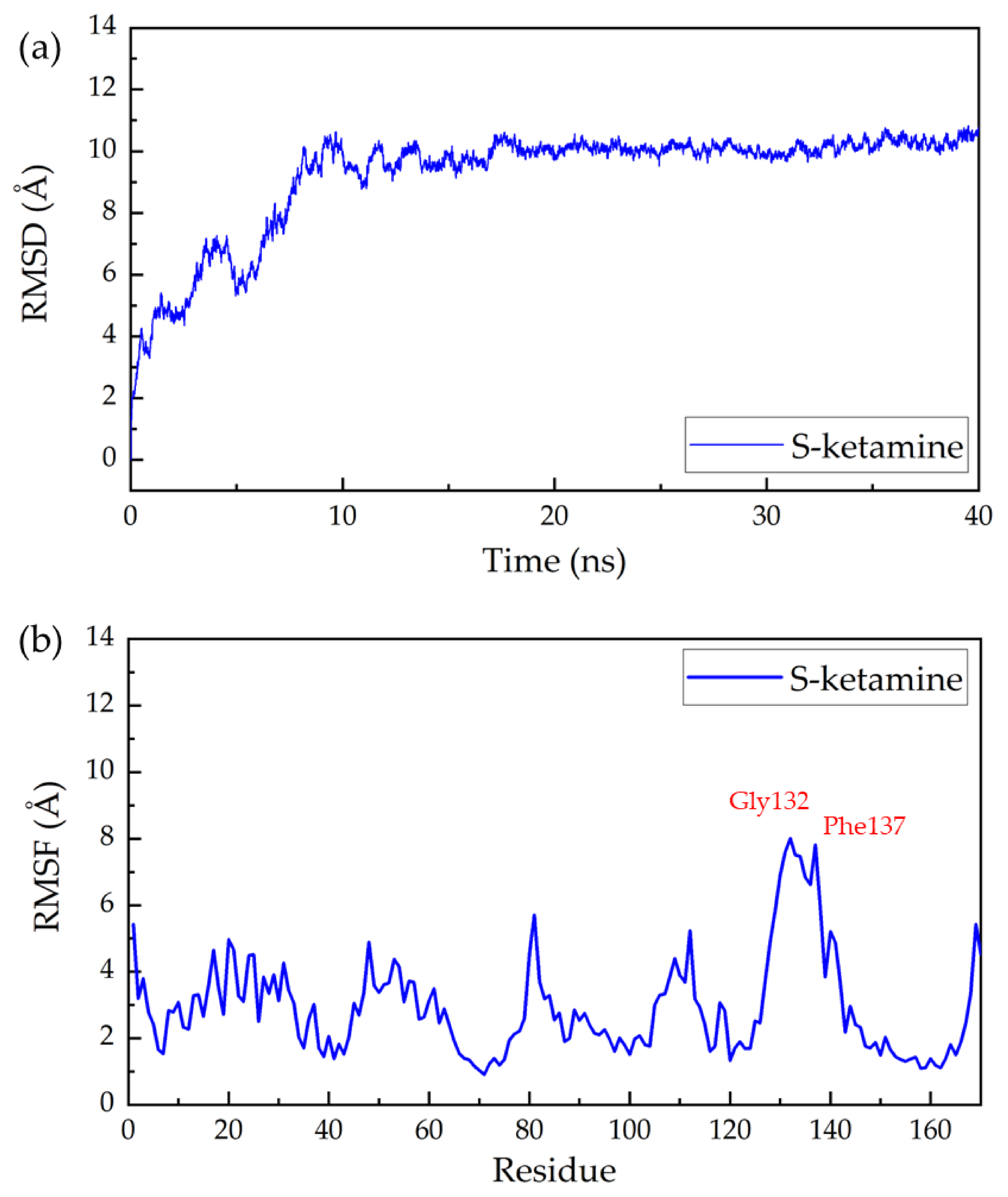
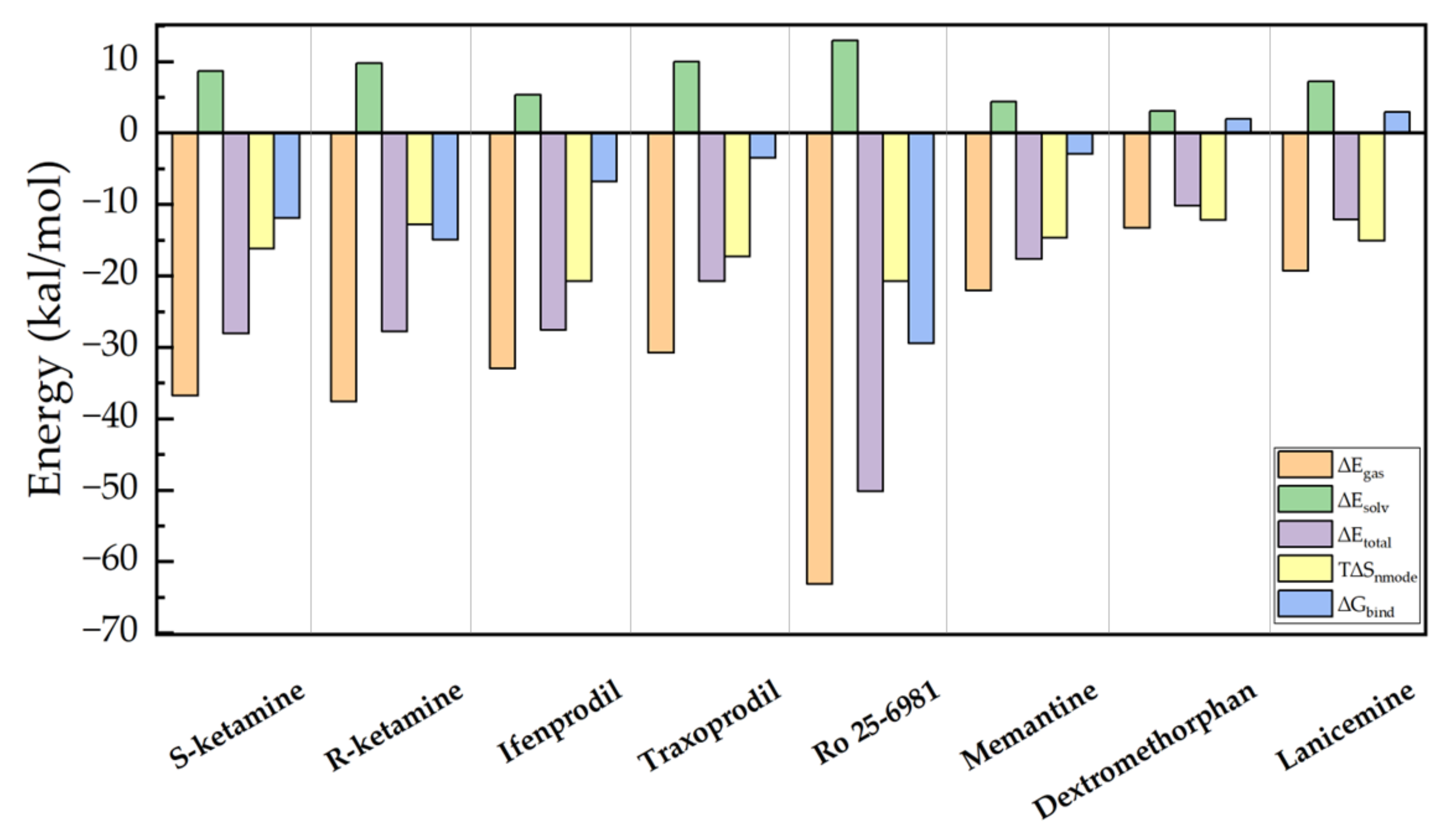
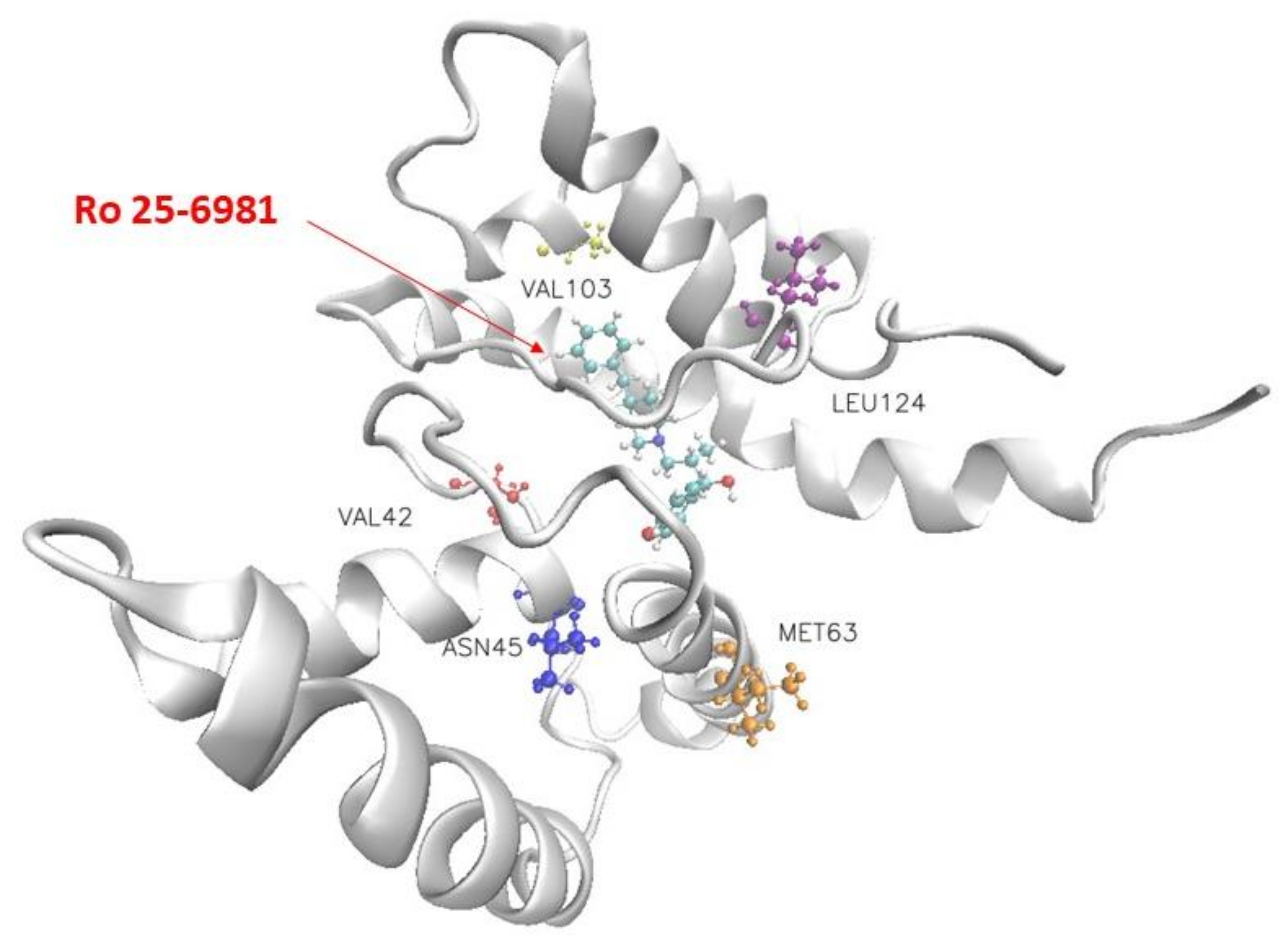
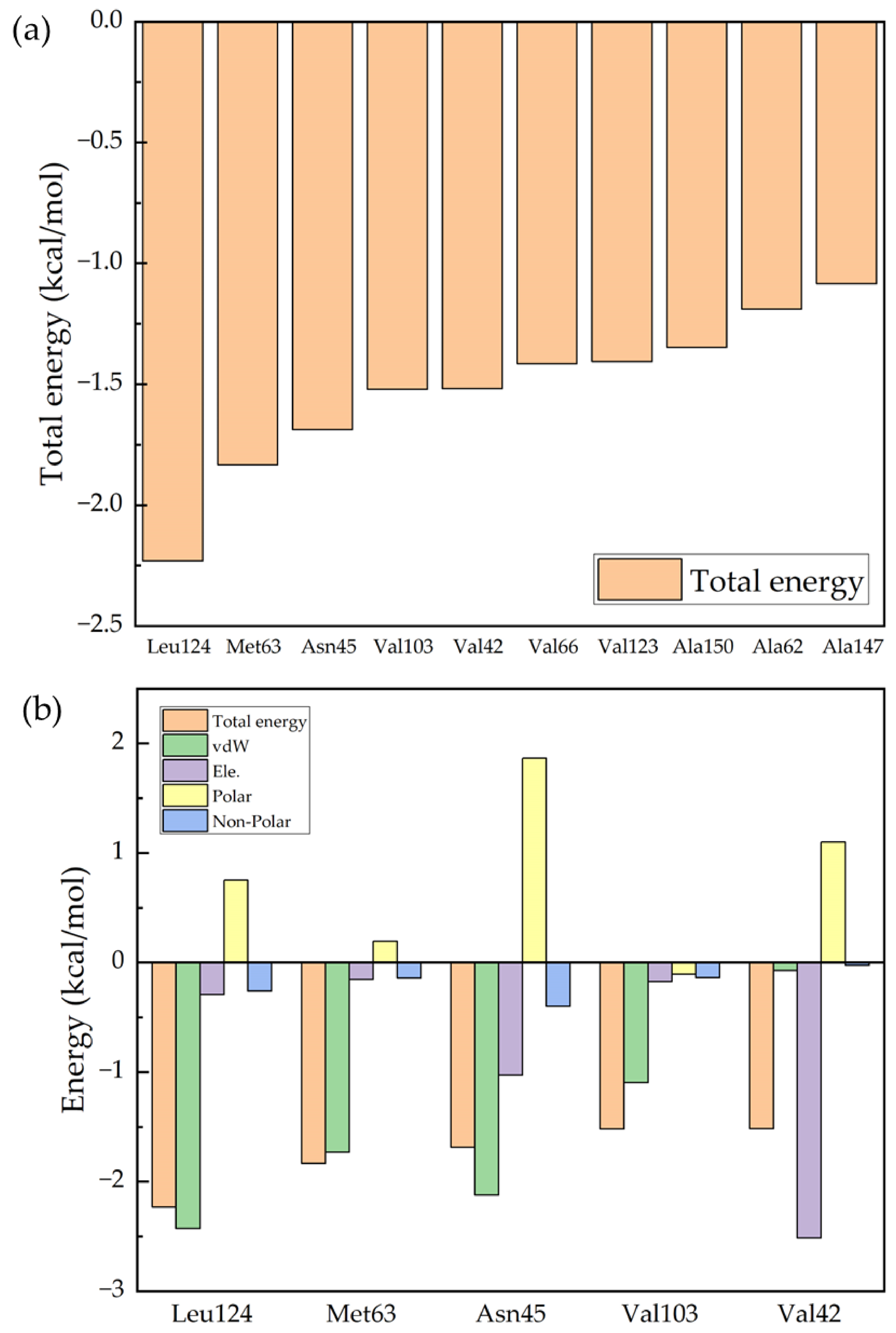
2. Results and Discussion
3. Method
3.1. Protein Structures
3.2. Compounds Selection
3.3. Ligand–Protein Docking
3.4. Molecular Dynamic Simulation
3.5. Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural Basis of Major Depressive Disorder: Beyond Monoamine Hypothesis: Neural Basis of Major Depressive Disorder. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mulinari, S. Monoamine Theories of Depression: Historical Impact on Biomedical Research. J. Hist. Neurosci. 2012, 21, 366–392. [Google Scholar] [CrossRef] [PubMed]
- Newport, D.J.; Carpenter, L.L.; McDonald, W.M.; Potash, J.B.; Tohen, M.; Nemeroff, C.B. Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. AJP 2015, 172, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Furukawa, H. Crystal Structure of a Heterotetrameric NMDA Receptor Ion Channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Lü, W.; Michel, J.C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E. NMDA Receptor Structures Reveal Subunit Arrangement and Pore Architecture. Nature 2014, 511, 191–197. [Google Scholar] [CrossRef]
- Zhu, S.; Stein, R.A.; Yoshioka, C.; Lee, C.-H.; Goehring, A.; Mchaourab, H.S.; Gouaux, E. Mechanism of NMDA Receptor Inhibition and Activation. Cell 2016, 165, 704–714. [Google Scholar] [CrossRef]
- Erreger, K.; Dravid, S.M.; Banke, T.G.; Wyllie, D.J.A.; Traynelis, S.F. Subunit-Specific Gating Controls Rat NR1/NR2A and NR1/NR2B NMDA Channel Kinetics and Synaptic Signalling Profiles. J. Physiol. 2005, 563, 345–358. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, F.; Zhang, T.; Lv, S.; Zhou, L.; Du, D.; Lin, H.; Guo, F.; Luo, C.; Zhu, S. Structural Basis of Ketamine Action on Human NMDA Receptors. Nature 2021, 596, 301–305. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant Effects of Ketamine in Depressed Patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Jelen, L.A.; Young, A.H.; Stone, J.M. Ketamine: A Tale of Two Enantiomers. J. Psychopharmacol. 2021, 35, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hashimoto, K. An Update on Ketamine and Its Two Enantiomers as Rapid-Acting Antidepressants. Expert. Rev. Neurother. 2019, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhang, K.; Pu, Y.; Qu, Y.; Wang, S.; Xiong, Z.; Ren, Q.; Dong, C.; Fujita, Y.; Hashimoto, K. Comparison of Antidepressant and Side Effects in Mice after Intranasal Administration of (R,S)-Ketamine, (R)-Ketamine, and (S)-Ketamine. Pharmacol. Biochem. Behav. 2019, 181, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Jensen, M.Ø.; Jogini, V.; Stein, R.A.; Lee, C.-H.; Mchaourab, H.S.; Shaw, D.E.; Gouaux, E. Mechanism of NMDA Receptor Channel Block by MK-801 and Memantine. Nature 2018, 556, 515–519. [Google Scholar] [CrossRef]
- Vogels, B.A.; Maas, M.A.; Daalhuisen, J.; Quack, G.; Chamuleau, R.A. Memantine, a Noncompetitive NMDA Receptor Antagonist Improves Hyperammonemia-Induced Encephalopathy and Acute Hepatic Encephalopathy in Rats. Hepatology 1997, 25, 820–827. [Google Scholar] [CrossRef]
- Fischer, G.; Mutel, V.; Trube, G.; Malherbe, P.; Kew, J.N.C.; Mohacsi, E.; Heitz, M.P.; Kemp, J.A. Ro 25-6981, a Highly Potent and Selective Blocker of N-Methyl-d-Aspartate Receptors Containing the NR2B Subunit. Characterization in Vitro. J. Pharm. Exp. 1997, 283, 1285–1292. [Google Scholar]
- Loftis, J.M.; Janowsky, A. The N-Methyl-d-Aspartate Receptor Subunit NR2B: Localization, Functional Properties, Regulation, and Clinical Implications. Pharmacol. Ther. 2003, 97, 55–85. [Google Scholar] [CrossRef]
- Kreutzwiser, D.; Tawfic, Q.A. Expanding Role of NMDA Receptor Antagonists in the Management of Pain. CNS Drugs 2019, 33, 347–374. [Google Scholar] [CrossRef]
- Thompson, T.; Whiter, F.; Gallop, K.; Veronese, N.; Solmi, M.; Newton, P.; Stubbs, B. NMDA Receptor Antagonists and Pain Relief: A Meta-Analysis of Experimental Trials. Neurology 2019, 92, e1652–e1662. [Google Scholar] [CrossRef]
- Williams, K. Ifenprodil Discriminates Subtypes of the N-Methyl-D-Aspartate Receptor: Selectivity and Mechanisms at Recombinant Heteromeric Receptors. Mol. Pharm. 1993, 44, 851–859. [Google Scholar]
- Ates-Alagoz, Z.; Adejare, A. NMDA Receptor Antagonists for Treatment of Depression. Pharmaceuticals 2013, 6, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Serafini, G.; Gonda, X.; Rihmer, Z.; Pompili, M.; Girardi, P.; Nasrallah, H.A.; Amore, M. NMDA Receptor Antagonists for Depression: Critical Considerations. Ann. Clin. Psychiatry 2015, 27, 213–220. [Google Scholar] [PubMed]
- Abrusán, G.; Marsh, J.A. Ligand-Binding-Site Structure Shapes Allosteric Signal Transduction and the Evolution of Allostery in Protein Complexes. Mol. Biol. Evol. 2019, 36, 1711–1727. [Google Scholar] [CrossRef]
- Feng, X.; Li, F.; Ding, M.; Zhang, R.; Shi, T.; Jiang, W. Molecular Dynamic Simulation: Structural Insights of Multi-Stranded Curdlan in Aqueous Solution. Carbohydr. Polym. 2021, 261, 117844. [Google Scholar] [CrossRef]
- Bashford, D.; Case, D.A. Generalized Born Models of Macromolecular Solvation Effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [Google Scholar] [CrossRef]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A.; Brooks, C.L., III. Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The Normal-Mode Entropy in the MM/GBSA Method: Effect of System Truncation, Buffer Region, and Dielectric Constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef]
- Fukumoto, K.; Toki, H.; Iijima, M.; Hashihayata, T.; Yamaguchi, J.; Hashimoto, K.; Chaki, S. Antidepressant Potential of (R)-Ketamine in Rodent Models: Comparison with (S)-Ketamine. J. Pharm. Exp. 2017, 361, 9–16. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Hashimoto, K. R (−)-Ketamine Shows Greater Potency and Longer Lasting Antidepressant Effects than S (+)-Ketamine. Pharmacol. Biochem. Behav. 2014, 116, 137–141. [Google Scholar] [CrossRef]
- Lapidus, K.A.B.; Levitch, C.F.; Perez, A.M.; Brallier, J.W.; Parides, M.K.; Soleimani, L.; Feder, A.; Iosifescu, D.V.; Charney, D.S.; Murrough, J.W. A Randomized Controlled Trial of Intranasal Ketamine in Major Depressive Disorder. Biol. Psychiatry 2014, 76, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shirayama, Y.; Zhang, J.-C.; Ren, Q.; Yao, W.; Ma, M.; Dong, C.; Hashimoto, K. R-Ketamine: A Rapid-Onset and Sustained Antidepressant without Psychotomimetic Side Effects. Transl. Psychiatry 2015, 5, e632. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Sanacora, G.; Smith, M.A.; Pathak, S.; Su, H.-L.; Boeijinga, P.H.; McCarthy, D.J.; Quirk, M.C. Lanicemine: A Low-Trapping NMDA Channel Blocker Produces Sustained Antidepressant Efficacy with Minimal Psychotomimetic Adverse Effects. Mol. Psychiatry 2014, 19, 978–985. [Google Scholar] [CrossRef]
- Taylor, C.P.; Traynelis, S.F.; Siffert, J.; Pope, L.E.; Matsumoto, R.R. Pharmacology of Dextromethorphan: Relevance to Dextromethorphan/Quinidine (Nuedexta®) Clinical Use. Pharmacol. Ther. 2016, 164, 170–182. [Google Scholar] [CrossRef]
- Yurkewicz, L.; Weaver, J.; Bullock, M.R.; Marshall, L.F. The Effect of the Selective NMDA Receptor Antagonist Traxoprodil in the Treatment of Traumatic Brain Injury. J. Neurotrauma 2005, 22, 1428–1443. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Chapter Nineteen—Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. In Methods in Enzymology; Johnson, M.L., Brand, L., Eds.; Computer Methods, Part C; Academic Press: Cambridge, MA, USA, 2011; Volume 487, pp. 545–574. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert. Opin. Drug. Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  |  |
| Ro 25-6981 | S-ketamine | R-ketamine | ifenprodil |
| C22H29NO2 | C13H16ClNO | C13H17Cl2NO | C21H27NO2 |
| CID: 6604887 | CID: 182137 | CID: 9838417 | CID: 3689 |
 |  |  |  |
| traxoprodil | memantine | lanicemine | dextromethorphan |
| C20H25NO3 | C12H21N | C13H14N2 | C18H25NO |
| CID: 219101 | CID:4054 | CID:9794203 | CID: 5360696 |
| Conformations | Egas | Esolv | Etotal | Snmode | Gbind |
|---|---|---|---|---|---|
| Ro 25-6981 | −63.116 | 12.923 | −50.193 | −20.762 | −29.431 |
| R-ketamine | −37.565 | 9.795 | −27.770 | −12.809 | −14.961 |
| S-ketamine | −36.777 | 8.675 | −28.102 | −16.179 | −11.922 |
| ifenprodil | −32.952 | 5.366 | −27.587 | −20.751 | −6.835 |
| Traxoprodil | −30.773 | 9.995 | −20.777 | −17.271 | −3.506 |
| Memantine | −22.044 | 4.409 | −17.635 | −14.673 | −2.962 |
| Dextromethorphan | −13.273 | 3.056 | −10.217 | −12.206 | 1.989 |
| Lanicemine | −19.317 | 7.180 | −12.136 | −15.054 | 2.918 |
| Conformations | Important Residues | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| S-ketamine | Phe88 | Asn160 | Leu92 | Tyr157 | Trp93 |
| R-ketamine | Met63 | Ile64 | Leu124 | Ile153 | Tyr157 |
| ifenprodil | Leu77 | Thr70 | Thr158 | Leu73 | Ala74 |
| traxoprodil | Ile152 | Ala155 | Leu77 | Leu92 | Met151 |
| Ro 25-6981 | Leu124 | Met63 | Asn45 | Val103 | Val42 |
| memantine | Met36 | Val15 | Trp12 | Val19 | Trp40 |
| dextromethorphan | Val96 | Phe149 | Trp146 | Arg81 | Val154 |
| lanicemine | Ala155 | Leu73 | Met151 | Leu77 | Val154 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, S.; Han, Y.; Wei, Z.; Li, J. Binding Affinity and Mechanisms of Potential Antidepressants Targeting Human NMDA Receptors. Molecules 2023, 28, 4346. https://doi.org/10.3390/molecules28114346
Ye S, Han Y, Wei Z, Li J. Binding Affinity and Mechanisms of Potential Antidepressants Targeting Human NMDA Receptors. Molecules. 2023; 28(11):4346. https://doi.org/10.3390/molecules28114346
Chicago/Turabian StyleYe, Simin, Yanqiang Han, Zhiyun Wei, and Jinjin Li. 2023. "Binding Affinity and Mechanisms of Potential Antidepressants Targeting Human NMDA Receptors" Molecules 28, no. 11: 4346. https://doi.org/10.3390/molecules28114346
APA StyleYe, S., Han, Y., Wei, Z., & Li, J. (2023). Binding Affinity and Mechanisms of Potential Antidepressants Targeting Human NMDA Receptors. Molecules, 28(11), 4346. https://doi.org/10.3390/molecules28114346