3.3. Synthesis
3.3.1. General Procedure for the Synthesis of 3-Formylbenzopyran-4-one (2a–2c) [39,40]
A solution of substituted (R = H/OMe) o-hydroxyacetophenone (1a/1b/1c, 0.1 mol) was dissolved in anhydrous DMF (80 mL), placed in a two-neck round bottom flask, and freshly distilled POCl3 (80 mL) was added dropwise through a dropping funnel over 30 min with vigorous stirring in an ice-water bath maintained at 0–5 °C. The reaction mixture was then stirred at 60 °C for 13 h. The reaction was monitored by TLC [ethyl acetate–petroleum ether 50:50] to completion, and the mixture was decomposed by pouring over 500 mL of ice water after the reaction was completed. The resulting residue was collected by filtration, washed with water, and recrystallized from ethanol–water. The 3 formyl-substituted benzopyran-4-ones are listed below.
4-Oxo-4H-1-benzopyran-3-yl-carboxaldehyde (
2a). Yield = 69%;
Rf = 0.72 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3715, 3696, 3058, 2971, 2868, 2845, 2825, 2365, 2341, 2170, 2033, 1979, 1694, 1645, 1613, 1562, 1462, 1418, 1363, 1338, 1311, 1271, 1236, 1192, 1148, 1055, 1032, 1012, 849, 767, 744 and 690 cm
−1; UV/vis (MeOH) λ: 297, 238 nm;
1H NMR (400 MHz, CDCl
3): δ 10.37 (s, 1H, C
HO), 8.53 (s, 1H, H-2), 8.28 (dd,
J = 7.9, 1.5 Hz, 1H, H-5), 7.75 (ddd,
J = 8.7, 7.2, 1.7 Hz, 1H, H-7), 7.56–7.46 (m, 2H, H-6 and H-8);
13C NMR (101 MHz, CDCl
3): δ 188.61 (CHO), 175.98 (C-4 C=O), 160.63 (C-2), 156.21 (C-8a), 134.84 (C-7), 126.66(C-6), 126.18 (C-5), 125.32 (C-4a), 120.33 (C-3), 118.63 (C-8); HRMS (ESI-TOF) (
m/
z) C
10H
6O
3 calculated, 197.021, found, 197.140 [M + Na]
+; calculated, 212.995, found, 213.125 [M + K]
+ [
41].
5-Methoxy-4-oxo-4H-1-benzopyran-3-carboxaldehyde (
2b). Yield = 89%;
Rf = 0.31 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3708, 3679, 2970, 2923, 2866, 2844, 2826, 2166, 2074, 2050, 2027, 1977, 1963, 1698, 1653, 1610, 1562, 1539, 1476, 1456, 1434, 1415, 1345, 1272, 1055, 1032, 1013 and 765 cm
−1; UV/vis (MeOH) λ: 315, 256 nm;
1H NMR (400 MHz, CDCl
3): δ 10.35 (s, 1H, CHO), 8.39 (s, 1H, H-2), 7.62 (t,
J = 8.4 Hz, 1H, H-7), 7.07 (dd,
J = 8.4, 0.7 Hz, 1H, H-8), 6.90 (d,
J = 8.4 Hz, 1H, H-6), 4.01 (s, 3H, CH
3);
13C NMR (101 MHz, CDCl
3): δ 189.30 (CHO), 175.97 (C-4 C=O), 160.51 (C-5), 158.88 (C-2), 158.23 (C-8a), 135.08 (C-7), 121.38 (C-3), 115.71 (C-4a), 110.56 (C-6), 108.08 (C-8), 56.72 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
11H
8O
4 calculated, 205.050, found 205.128 [M + H]
+; calculated, 227.032, found, 227.119 [M + Na]
+; and calculated, 243.006, found, 243.100 [M + K]
+ [
40,
42].
6-Methoxy-4-oxo-4H-1-benzopyran-3-carboxaldehyde (
2c). Yield = 51%;
Rf = 0.73 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3710, 3697, 3656, 3635, 2970, 2940, 2923, 2866, 2844, 2826, 2362, 2342, 2165, 2073, 2050, 2025, 1977, 1652, 1481, 1457, 1437, 1345, 1321, 1055, 1032 and 1014 cm
−1; UV/vis (MeOH) λ: 324, 240 nm;
1H NMR (400 MHz, CDCl
3): δ 10.39 (s, 1H, C
HO), 8.52 (s, 1H, H-2), 7.63 (d,
J = 2.9 Hz, 1H, H-5), 7.46 (d,
J = 9.1 Hz, 1H, H-8), 7.31 (dd,
J = 9.1, 3.0 Hz, 1H, H-7), 3.91 (s, 3H, -OCH
3);
13C NMR (101 MHz, CDCl
3): δ 188.91 (CHO), 176.01 (C-4 C=O), 160.36 (C-2), 158.08 (C-6), 151.09 (C-8a), 126.25 (C-4a), 124.60 (C-7), 120.13 (C-8), 119.73 (C-3), 105.58 (C-5), 56.18 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
11H
8O
4 calculated, 205.050, found 205.105 [M + H]
+, and calculated, 227.032, found, 227.093 [M + Na]
+ [
40,
43].
3.3.2. 7-Methoxy-4-oxo-4H-1-benzopyran-3-carboxaldehyde (2d)
7-methoxy-4-oxo-4
H-1-benzopyran-3-carbaldehyde was synthesized according to the literature-reported procedure [
25]. A solution of 2′-hydroxy-4′-methoxyacetophenone 1d (3.8 g, 23 mmol), BF
3-etharate (5.6 mL, 46 mmol), and acetic anhydride (8.6 mL, 92 mmol) was taken in the round bottom flask. The reaction mixture was heated at 100 °C for 1 h. The precipitate was collected and washed 3 times with diethyl ether. The resulting precipitate was filtered to afford difluorodioxaborin as brown-colored crystals. Yield = 98.15%;
1H NMR (400 MHz, CDCl
3) δ 7.63 (d,
J = 9.2 Hz, 1H, H-5), 6.58 (dd,
J = 9.2, 2.4 Hz, 1H, H-6), 6.46 (d,
J = 2.4 Hz, 1H, H-8), 3.93 (s, 3H, -OCH
3), 2.73 (s, 3H, -CH
3). In the second step, POCl
3 (5.6 mL) was added to 20 mL of DMF at 0 °C, followed by the addition of difluorodioxaborin (4.8 g, 22 mmol) in 5 mL DMF. The reaction mixture was stirred at 100 °C for 1 h and then poured on crushed ice (250 g). The mixture was then refrigerated overnight. The resulting precipitate was filtered and washed with water. The crude product was recrystallized from acetone to afford (2d) as brown-colored crystals. Yield = 64%;
Rf = 0.67 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3712, 3696, 3655, 3635, 2969, 2923, 2866, 2844, 2827, 2362, 2342, 2165, 2074, 2027, 1978, 1703, 1653, 1619, 1559, 1528, 1502, 1439, 1363, 1347, 1304, 1277, 1244, 1187, 1141, 1055, 1031, 1015, 935, 841, 800 and 765 cm
−1; UV/vis (MeOH) λ: 291, 247, 241 nm;
1H NMR (400 MHz, CDCl
3): δ 10.37 (s, 1H, C
HO), 8.47 (s, 1H, H-2), 8.20 (d,
J = 8.9 Hz, 1H, H-5), 7.05 (dd,
J = 8.9, 2.4 Hz, 1H, H-6), 6.91 (d,
J = 2.4 Hz, 1H, H-8), 3.93 (s, 3H, -OCH
3);
13C NMR (101 MHz, CDCl
3): δ 189.06 (CHO), 175.47 (C-4 C=O), 165.05 (C-2), 160.39 (C-7), 158.13 (C-8a), 127.69 (C-5), 120.40 (C-3), 118.98 (C-8a), 115.68 (C-6), 101.27 (C-8), 56.17 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
11H
8O
4 calculated, 205.050, found 205.147 [M + H]
+, calculated, 227.032, found, 227.140 [M + Na]
+; and calculated, 243.006, found, 243.121 [M + K]
+ [
40].
3.3.3. General Procedure for the Synthesis of 3-(Hydroxymethyl) Substituted-4-oxo-4H-1-benzopyrane (3a–3d) [43]
A mixture of 4-oxo-4H-1-benzopyran-3-yl-carboxaldehyde (2a, 5 g, 28.7 mmol) or methoxy-substituted 4-oxo-4H-1-benzopyran-3-yl-carboxaldehyde (2b/2c/2d, 5 g, 24.5 mmol), basic alumina (50 g) and 2-propanol (300 mL) were taken in a round bottom flask. The reaction mixture was heated at 75 °C for 4 h. The reaction was monitored by TLC [ethyl acetate–petroleum ether 50:50] to completion, and then the reaction mixture was filtered by pouring over celite. The solvent was evaporated in vacuum, and the resulting precipitate was purified by column chromatography to afford 3a–3d as yellow solid. The substituted 3-(hydroxymethyl) benzopyran-4-ones are listed below:
3-(Hydroxymethyl)-4-oxo-4H-1-benzopyrane (
3a). Yield = 52%;
Rf = 0.41 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3359, 3088, 2952, 2723, 2367, 2343, 1635, 1605, 1571, 1466, 1425, 1407, 1350, 1315, 1271, 1234, 1216, 1187, 1162, 1109, 1062, 1025, 974, 946, 927, 847, 805, 758, 724, 708, 686 and 632 cm
−1; UV/vis (MeOH) λ: 296, 249 nm;
1H NMR (400 MHz, CDCl
3): δ 8.22 (dd,
J = 8.0, 1.7 Hz, 1H, H-5), 7.95 (s, 1H, H-2), 7.69 (ddd,
J = 8.7, 7.1, 1.7 Hz, 1H, H-7), 7.55–7.36 (m, 2H, H-6 and H-8), 4.59 (s, 1H, -C
H2OH), 2.85 (brs, 1H, OH);
13C NMR (101 MHz, CDCl
3): δ 178.65 (C-4 C=O), 156.75 (C-8a), 152.88 (C-2), 134.10 (C-7), 125.75 (C-5), 125.45 (C-6), 123.98 (C-4a), 123.40 (C-3), 118.39 (C-8), 58.89 (CH
2OH); HRMS (ESI-TOF) (
m/
z) C
10H
8O
3 calculated, 199.037, found 199.109 [M + Na]
+ [
43].
3-(Hydroxymethyl)-5-methoxy-4-oxo-4H-1-benzopyrane (
3b). Yield = 33%;
Rf = 0.07 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3698, 3679, 3662, 3427, 2969, 2922, 2866, 2844, 2827, 2361, 2341, 2074, 2027, 1644, 1605, 1566, 1547, 1526, 1512, 1475, 1436, 1347, 1297, 1269, 1182, 1160, 1055, 1032, 1013, 893, 804 and 694 cm
−1; UV/vis (MeOH) λ: 315, 254, 226 nm;
1H NMR (400 MHz, CDCl
3): δ 7.79 (s, 1H, H-2), 7.56 (t,
J = 8.4 Hz, 1H, H-7), 7.02 (d,
J = 8.5 Hz, 1H, H-8), 6.81 (d,
J = 8.3 Hz, 1H, H-6), 4.50 (s, 2H, -
CH2OH), 3.99 (s, 3H, -OCH
3), 3.25 (s, 1H, -OH);
13C NMR (101 MHz, CDCl
3): δ 178.85 (C-4 C=O), 160.05 (C-5), 158.78 (C-8a), 150.90 (C-2), 134.27 (C-7), 124.32 (C-3), 114.64 (C-4a), 110.40 (C-6), 106.44 (C-8), 59.20 (CH
2OH), 56.56 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
11H
10O
4 calculated, 207.066, found 207.061 [M + H]
+; calculated, 229.048, found, 229.043 [M + Na]
+; and calculated, 245.022, found, 245.017 [M + K]
+ [
44].
3-(Hydroxymethyl)-6-methoxy-4-oxo-4H-1-benzopyrane (
3c). Yield = 43%;
Rf = 0.32 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3697, 3679, 3656, 3635, 3618, 3404, 2922, 2867, 2845, 2363, 2342, 2165, 2074, 2050, 1637, 1609, 1581, 1546, 1527, 1512, 1485, 1458, 1438, 1395, 1375, 1344, 1322, 1269, 1246, 1200, 1150, 1102, 1065, 1031, 1015, 891, 823, 766 and 707 cm
−1; UV/vis (MeOH) λ: 324, 239, 231 nm;
1H NMR (400 MHz, CDCl
3): δ 7.96 (s, 1H, H-2), 7.49 (d,
J = 3.0 Hz, 1H, H-5), 7.36 (d,
J = 9.2 Hz, 1H, H-8), 7.22 (dd,
J = 9.2, 3.1 Hz, 1H, H-7), 4.50 (s, 2H, -C
H2OH), 3.84 (s, 3H, -OCH
3);
13C NMR (101 MHz, CDCl
3): δ 178.24 (C-4 C=O), 157.05 (C-6), 153.41 (C-2), 151.64 (C-8a), 124.29 (C-4a), 124.22 (C-7), 122.68 (C-3), 119.74 (C-8), 104.48 (C-5), 57.47 (CH
2OH), 55.89 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
11H
10O
4 calculated, 207.0656, found 207.194 [M + H]
+; and calculated, 229.048, found, 229.190 [M + Na]
+ [
43,
44].
3-(Hydroxymethyl)-7-methoxy-4H-1-benzopyran-4-one (
3d). Yield = 12%;
Rf = 0.28 [ethyl acetate–petroleum ether 50:50]; IR (neat) ν
max: 3713, 3696, 3655, 3635, 3618, 3403, 2922, 2867, 2845, 2363, 2342, 2165, 2073, 2030, 1977, 1641, 1605, 1565, 1546, 1524, 1496, 1442, 1400, 1376, 1344, 1319, 1275, 1242, 1206, 1174, 1095, 1055, 1032 and 1014 cm
−1; UV/vis (MeOH) λ: 280, 247 nm;
1H NMR (400 MHz, CDCl
3): δ 8.12 (d,
J = 8.9 Hz, 1H, H-5), 7.86 (s, 1H, H-2), 6.99 (dd,
J = 8.9, 2.4 Hz, 1H, H-6), 6.84 (d,
J = 2.4 Hz, 1H, H-8), 4.56 (s, 2H, -OCH
2-), 3.91 (s, 3H, -OCH
3);
13C NMR (101 MHz, CDCl
3): δ 178.07 (C-4 C=O), 164.46 (C-7), 158.61 (C-8a), 152.29 (C-2), 127.12 (C-5), 123.16 (C-3), 117.90 (C-4a), 114.99 (C-6), 100.40 (C-8), 59.01 (CH
2OH), 55.99 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
11H
10O
4 calculated, 229.048, found 228.949 [M + Na]
+; and calculated, 245.022, found, 246.102 [M + K]
+ [
44].
3.3.4. General Procedure for the Synthesis of Substituted (E)-3-(4-Oxo-4H-1-benzopyran-3-yl)acrylic Acid (4a–4d) [45]
A mixture of 4-oxo-4H-1-benzopyran-3-yl-carboxaldehyde (2a, 5 g, 28.7 mmol) or methoxy-substituted 4-oxo-4H-1-benzopyran-3-yl-carboxaldehyde (2b/2c/2d, 5 g, 24.5 mmol), malonic acid (6.2 g, 28.7 mmol for 2a/5.3 g, 24.5 mmol for 2b/2c/2d), and pyridine (20 mL) was heated at 110 °C. After an hour, additional malonic acid (3.1 g, 14.36 mmol for 2a/2.6 g, 12.23 mmol for 2b/2c/2d) was added to the reaction mixture. After 30 min, the solvent was evaporated in vacuum and the resulting precipitate was recrystallized from acetone to afford 4a–d.
(E)-3-(4-Oxo-4H-1-benzopyran-3-yl)acrylic acid (
4a). Yield = 74%;
Rf = 0.69 [ethyl acetate]; IR (neat) ν
max: 3364, 2955, 2919, 2848, 2666, 2367, 2343, 1659, 1611, 1562, 1547, 1464, 1425, 1410, 1360, 1323, 1300, 1249, 1015, 987, 858, 784, 760, 710, 684, 657 and 614 cm
−1; UV/vis (MeOH) λ: 289, 266 nm;
1H NMR (400 MHz, DMSO): δ 12.34 (brs, 1H, -COO
H), 8.87 (s, 1H, H-2), 8.13 (dd,
J = 8.0, 1.5 Hz, 1H, H-5), 7.84 (ddd,
J = 8.7, 7.1, 1.7 Hz, 1H, H-7), 7.70 (dd,
J = 8.4, 0.6 Hz, 1H, H-8), 7.54 (ddd,
J = 8.1, 7.2, 1.1 Hz, 1H, H-6), 7.43 (d,
J = 15.9 Hz, 1H, H-1′), 7.12 (d,
J = 15.9 Hz, 1H, H-2′);
13C NMR (101 MHz, DMSO): δ 175.32 (C-4 C=O), 167.78 (C-3′ C=O), 159.83 (C-2), 155.12 (C-8a), 135.90 (C-1′), 134.56 (C-7), 126.08 (C-8), 125.45 (C-6), 123.51 (C-4a), 121.35 (C-5), 118.54 (C-3), 118.14 (C-2′); HRMS (ESI-TOF) (
m/
z) C
12H
8O
4 calculated, 239.032, found 239.179 [M + Na]
+; and calculated, 255.006, found, 255.163 [M + K]
+ [
46,
47].
(E)-3-(5-Methoxy-4-oxo-4H-1-benzopyran-3-yl)acrylic acid (
4b). Yield = 24%;
Rf = 0.31 [ethyl acetate]; IR (neat) ν
max: 3698, 3679, 3656, 3634, 3399, 2941, 2867, 2843, 2363, 2342, 2075, 1642, 1604, 1472, 1437, 1408, 1358, 1269, 1178, 1068, 1031, 1015, 872, 812, 776, 689, and 650 cm
−1; UV/vis (MeOH) λ: 299, 254 nm;
1H NMR (400 MHz, MeOD): δ 8.42 (s, 1H, H-2), 7.68 (t,
J = 8.4 Hz, 1H, H-7), 7.48 (d,
J = 15.9 Hz, 1H, H-1′), 7.09 (m, 3H, H-2′, H-8 and H-6), 3.95 (s, 3H, -OCH
3);
13C NMR (101 MHz, MeOD): δ 177.60 (C-4 C=O), 170.53 (C-3′ C=O), 161.46 (C-5), 159.06 (C-8a), 158.62 (C-2), 137.48 (C-1′), 136.11 (C-7), 122.05 (C-3), 121.11 (C-2′), 115.25 (C-4a), 111.15 (C-6), 108.63 (C-8), 56.75 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
13H
10O
5 calculated, 247.061, found 247.214 [M + H]
+; and calculated, 269.043, found, 269.211 [M + Na]
+ [
37].
(E)-3-(6-Methoxy-4-oxo-4H-1-benzopyran-3-yl)acrylic acid (
4c). Yield = 81%;
Rf = 0.72 [ethyl acetate]; IR (neat) ν
max: 3696, 3679, 3655, 3634, 3346, 3064, 2970, 2866, 2844, 2362, 2342, 2165, 2074, 2050, 1695, 1646, 1611, 1564, 1547, 1484, 1454, 1436, 1421, 1395, 1374, 1343, 1320, 1296, 1212, 1197, 1159, 1055, 1031, 1014, 941, 867, 830, 791, 726, 678 and 610 cm
−1; UV/vis (MeOH) λ: 245 nm;
1H NMR (400 MHz, DMSO): δ 12.37 (s, 1H, -COOH), 8.83 (s, 1H, H-2), 7.65 (d,
J = 9.1 Hz, 1H, H-8), 7.47–7.40 (m, 3H, H-2′, H-7 and H-5), 7.10 (d,
J = 15.9 Hz, 1H, H-1′), 3.86 (s, 3H, -OCH
3);
13C NMR (101 MHz, DMSO): δ 174.96 (C-4 C=O), 167.81 (C-3′ C=O), 159.51 (C-2), 156.97 (C-6), 149.86 (C-8a), 136.02 (C-2′), 124.24 (C-4a), 123.53 (C-3), 121.11 (C-7), 120.14 (C-8), 117.31 (C-1′), 105.22 (C-5), 55.79 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
13H
10O
5 calculated, 247.061, found 247.215 [M + H]
+; calculated, 269.043, found, 269.211 [M + Na]
+; and calculated, 285.017, found, 285.195 [M + K]
+ [
48].
(E)-3-(7-Methoxy-4-oxo-4H-1-benzopyran-3-yl)acrylic acid (
4d). Yield = 75%;
Rf = 0.66 [ethyl acetate]; IR (neat) ν
max: 3696, 3678, 3655, 3634, 3345, 2947, 2866, 2843, 2362, 2341, 2164, 2073, 2032, 1701, 1652, 1620, 1563, 1546, 1524, 1496, 1439, 1420, 1345, 1317, 1277, 1247, 1180, 1096, 1055, 1030, 1015, 836, 684, 671, 654, 551 and 503 cm
−1; UV/vis (MeOH) λ: 266 nm;
1H NMR (400 MHz, DMSO): δ 12.23 (s, 1H, -COOH), 8.76 (s, 1H, H-2), 7.99 (d,
J = 8.9 Hz, 1H, H-5), 7.38 (d,
J = 15.9 Hz, 1H, H-2′), 7.15 (d,
J = 2.3 Hz, 1H, H-8), 7.11–7.06 (m, 2H, H-7 and H-1′), 3.89 (s, 3H, -OCH
3);
13C NMR (101 MHz, DMSO): δ 174.55 (C-4 C=O), 167.80 (C-3′ C=O), 164.01 (C-7), 159.29 (C-2), 156.94 (C-8a), 135.96 (C-2′), 126.86 (C-5), 121.20 (C-1′), 118.01 (C-3), 117.25 (C-4a), 115.22 (C-6), 100.85 (C-8), 56.20 (OCH
3); HRMS (ESI-TOF) (
m/
z) C
13H
10O
5 calculated, 247.0606, found 247.082 [M + H]
+; calculated, 269.043, found, 269.065 [M + Na]
+; and calculated, 285.017, found, 285.041 [M + K]
+ [
49].
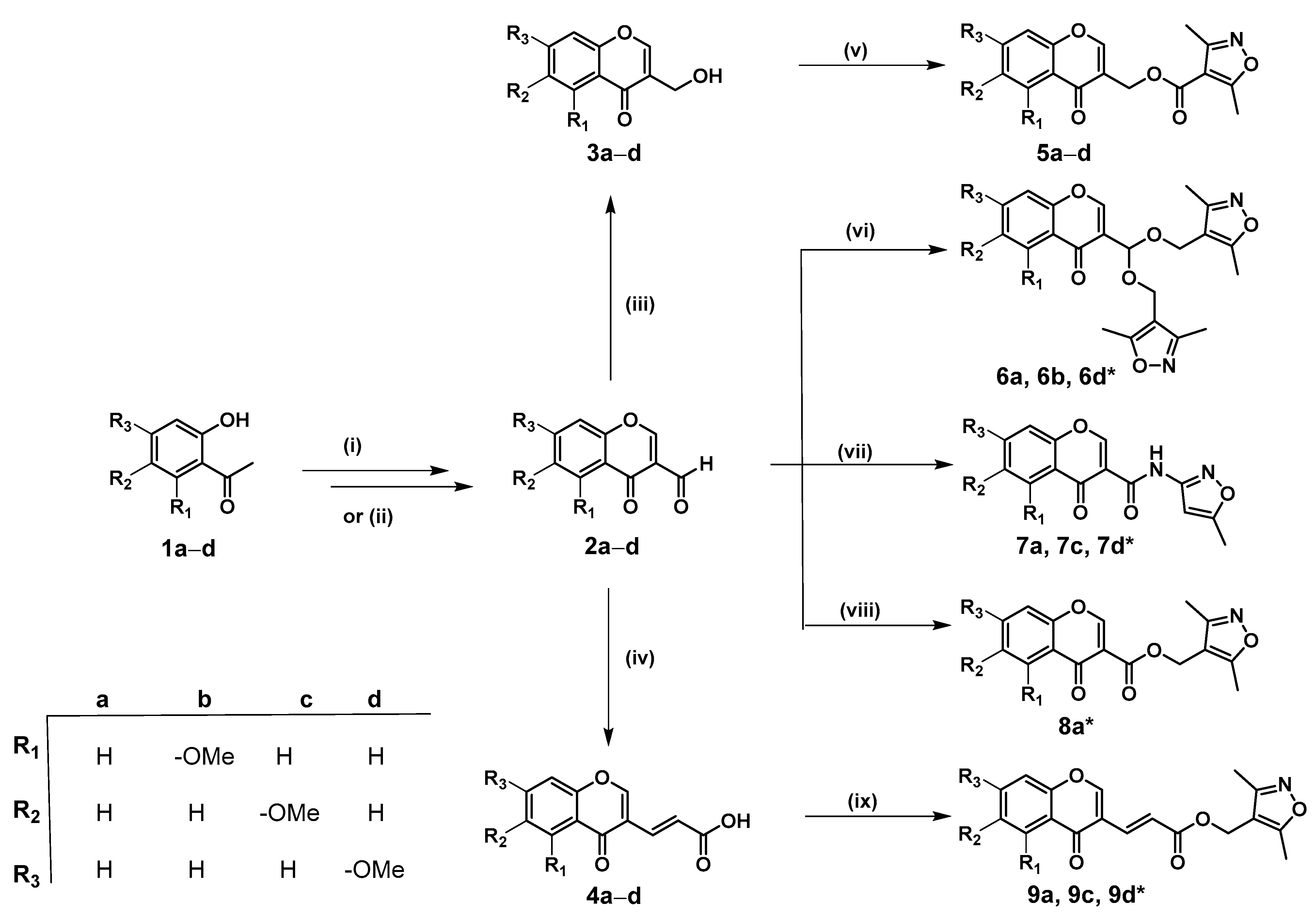
3.3.5. General Procedure for the Synthesis of Conjugates 5a–d
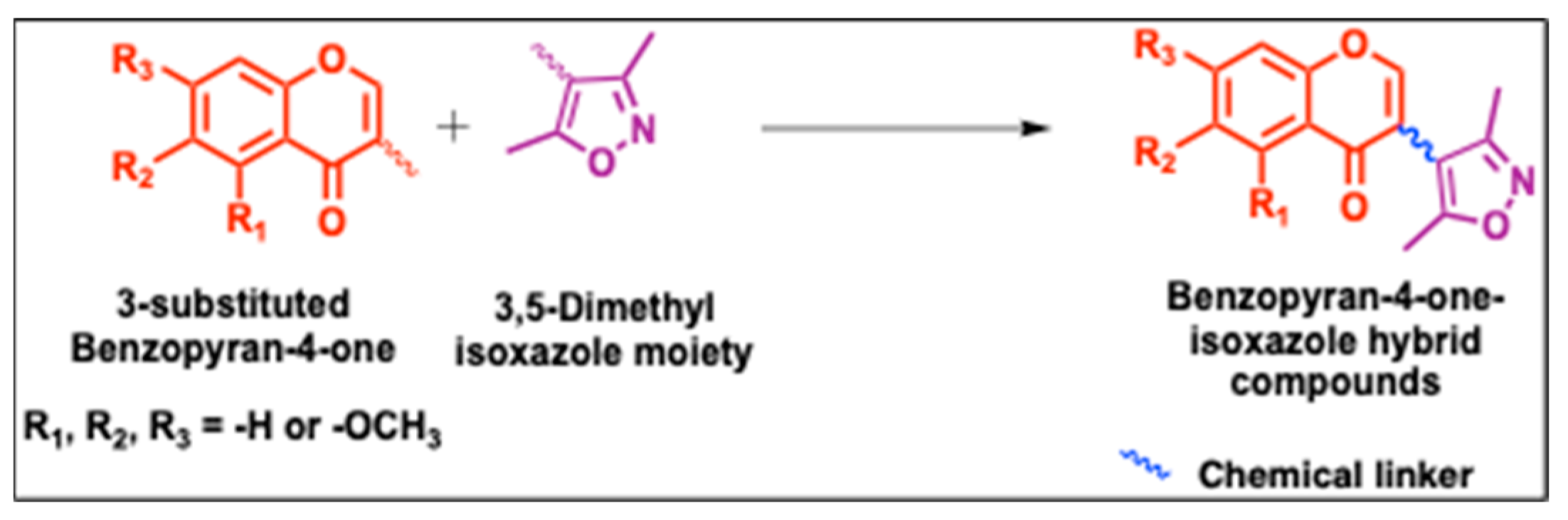
A mixture of 3,5-dimethylisoxazole-4-carboxylic acid (1.0 g, 7 mmol), EDCI (2 g, 10.6 mmol), and DMF (10 mL) was stirred at 0 °C. After 30 min, DMAP (catalytic) was added, and the reaction was stirred for another 10 min. Then, 3-(hydroxymethyl)-4H-1-benzopyran-4-one (3a, 0.62 g, 3.5 mmol) or methoxy substituted 3-(hydroxymethyl)-4H-1-benzopyran-4-one (3b/3c/3d, 0.73 g, 3.5 mmol) was added, and the reaction was stirred overnight. TLC [ethyl acetate–petroleum ether 50:50] was used to monitor the reaction to completion. After completion of the reaction, the solvent was evaporated in a vacuum, and the resulting precipitate was dissolved in ethyl acetate and washed 3 times with water. The solvent was dried over MgSO4 and evaporated, and the residue was purified via column chromatography to afford 5a–5d as white to off-white solids.
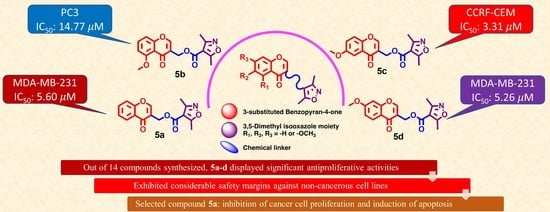
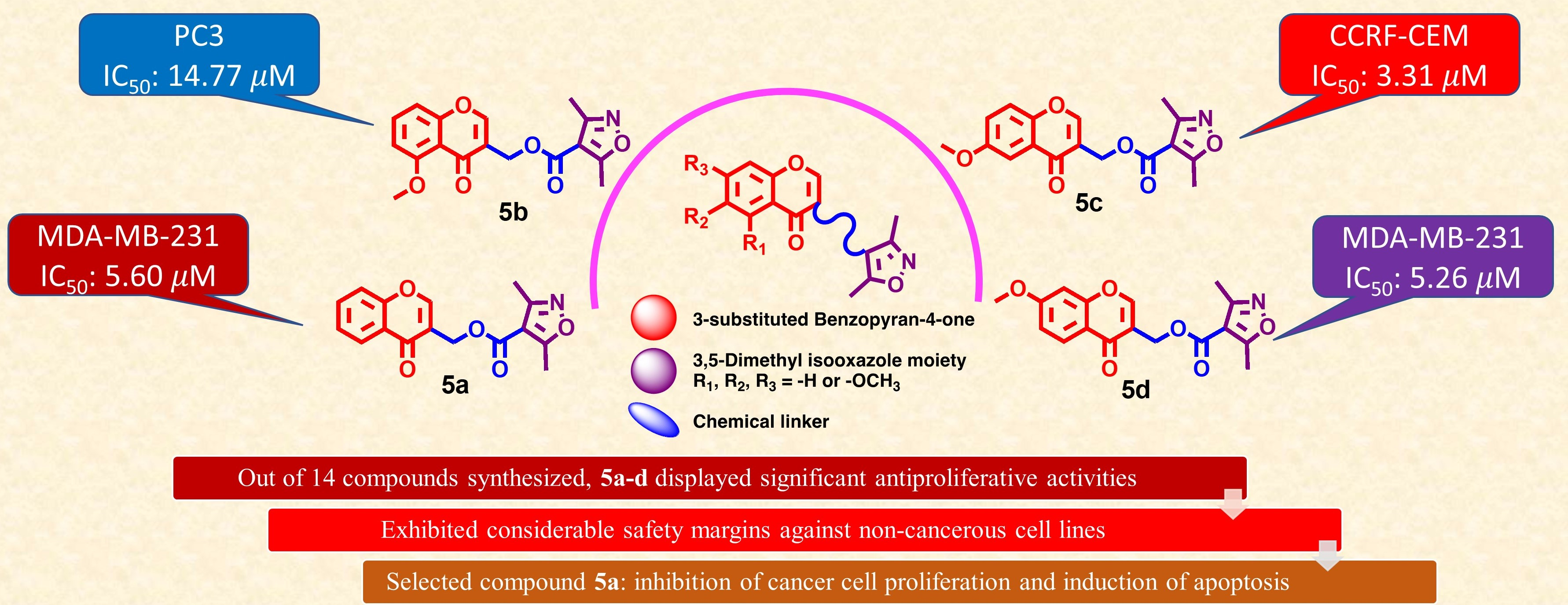
(4-Oxo-4H-1-benzopyran-3-yl)methyl 3,5-dimethylisoxazole-4-carboxylate (5a). Yield = 71%; Rf = 0.79 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 2954, 2923, 2867, 2384, 2349, 2298, 2249, 2215, 2167, 2147, 2032, 1995, 1976, 1961, 1900, 1715, 1652, 1610, 1575, 1500, 1464, 1425, 1407, 1381, 1348, 1299, 1253, 1217, 1190, 1168, 1146, 1104, 1055, 1032, 1013, 953, 914, 874, 849, 802, 760, 705, 689, 658 and 627 cm−1; UV/vis (MeOH) λ: 293, 245 nm; 1H NMR (400 MHz, CDCl3): δ 8.25 (dd, J = 8.0, 1.5 Hz, 1H, H-5), 8.15 (s, 1H, H-2), 7.70 (ddd, J = 8.7, 7.1, 1.7 Hz, 1H, H-7), 7.53–7.36 (m, 2H, H-6 and H-8), 5.22 (s, 2H. –COOCH2), 2.63 (s, 3H, -CH3), 2.41 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 176.69 (C-4 C=O), 175.75 (C-5′), 162.56 (C-3′ C=O), 160.10 (C-3′′), 156.61 (C-8a), 156.29 (C-2), 134.19 (C-7), 126.11 (C-5), 125.73 (C-6), 124.27 (C-4a), 119.45 (C-3), 118.41 (C-8), 108.65 (C-4′), 58.36 (C-1′), 13.59 (CH3), 11.92 (CH3); HRMS (ESI-TOF) (m/z) C16H13NO5 calculated, 322.0691, found 322.0622 [M + Na]+; and calculated, 338.0431, found, 338.0361 [M + K]+.
(5-Methoxy-4-oxo-4H-1-benzopyran-3-yl)methyl 3,5-dimethylisoxazole-4-carboxylate (5b). Yield = 25%; Rf = 0.32 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3697, 3679, 3656, 3635, 2923, 2867, 2845, 2362, 2342, 2165, 2075, 2027, 1717, 1656, 1607, 1574, 1546, 1524, 1476, 1459, 1438, 1408, 1377, 1357, 1298, 1273, 1240, 1186, 1107, 1055, 1032, 1014, 922, 850, 810, 778, 742 and 701 cm−1; UV/vis (MeOH) λ: 314, 254, 224 nm; 1H NMR (400 MHz, CDCl3): δ 8.00 (s, 1H, H-2), 7.56 (t, J = 8.4 Hz, 1H, H-7), 7.02 (dd, J = 8.5, 0.8 Hz, 1H, H-8), 6.83 (d, J = 8.3 Hz, 1H, H-6), 5.18 (s, 2H, -OCH2-), 3.98 (s, 3H, -OCH3), 2.64 (s, 3H, -CH3), 2.42 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 176.20 (C-4 C=O), 175.71 (C-5′′), 162.64 (C-3′), 160.23, 160.14 (C-5 and C-8a), 158.64 (C-3′′), 154.36 (C-2), 134.26 (C-7), 120.49 (C-3), 114.92 (C-4a), 110.31 (C-8), 108.68 (C-4′′), 106.84 (C-6), 58.36 (C-1′), 56.62 (OCH3), 13.66 (CH3), 11.99 (CH3); HRMS (ESI-TOF) (m/z) C17H15NO6 calculated, 329.0899, found 329.0978 [M]+; calculated, 352.0797, found, 352.0741 [M + Na]+; and calculated, 368.4063, found, 368.0479 [M + K]+.
(6-Methoxy-4-oxo-4H-1-benzopyran-3-yl)methyl 3,5-dimethylisoxazole-4-carboxylate (5c). Yield = 32%; Rf = 0.82 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3712, 3696, 3679, 3656, 3635, 2923, 2867, 2844, 2362, 2342, 2164, 2073, 2030, 1978, 1714, 1643, 1610, 1547, 1525, 1484, 1457, 1437, 1403, 1371, 1345, 1320, 1300, 1249, 1203, 1173, 1152, 1107, 1055, 1017, 900, 866, 840, 796, 774, 734, 702 and 627 cm−1; UV/vis (MeOH) λ: 320, 248 nm; 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 1H, H-2), 7.60 (d, J = 3.1 Hz, 1H, H-5), 7.41 (d, J = 9.2 Hz, 1H, H-8), 7.28 (dd, J = 9.2, 3.1 Hz, 1H, H-7), 5.23 (s, 2H, -OCH2-), 3.90 (s, 3H, -OCH3), 2.64 (s, 3H, -CH3), 2.42 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 176.57 (C-4 C=O), 175.74 (C-5′′), 162.59 (C-3′ C=O), 160.11 (C-3′′), 157.38 (C-6), 156.07 (C-2), 151.48 (C-8a), 124.89 (C-4a), 124.29 (C-7), 119.83 (C-8), 118.59 (C-3), 108.68 (C-4′′), 105.08 (C-5), 58.45 (C-1′), 56.11 (OCH3), 13.59 (CH3), 11.93 (CH3); HRMS (ESI-TOF) (m/z) C17H15NO6 calculated, 330.0978, found, 330.0932 [M + H]+; calculated, 352.0797, found, 352.0744 [M + Na]+; and calculated, 368.4063, found, 368.0484 [M + K]+.
(7-Methoxy-4-oxo-4H-1-benzopyran-3-yl)methyl 3,5-dimethylisoxazole-4-carboxylate (5d). Yield = 63%; Rf = 0.77 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3711, 3696, 3679, 3655, 3634, 2970, 2923, 2866, 2844, 2826, 2362, 2341, 2165, 2073, 2031, 1979, 1562, 1545, 1524, 1511, 1495, 1477, 1457, 1345, 1322, 1055, 1032 and 1013 cm−1; UV/vis (MeOH) λ: 295, 248, 240 nm; 1H NMR (400 MHz, CDCl3): δ 8.15 (d, J = 8.9 Hz, 1H, H-5), 8.07 (s, 1H, H-2), 7.00 (dd, J = 8.9, 2.4 Hz, 1H, H-6), 6.85 (d, J = 2.4 Hz, 1H, H-8), 5.21 (s, 2H, -OCH2-), 3.91 (s, 3H, -OCH3), 2.63 (s, 3H, -CH3), 2.42 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 176.00 (C-4 C=O), 175.74 (C-5′′), 164.50 (C-7), 162.60 (C-3′ C=O), 160.13 (C-3′′), 158.42 (C-8a), 155.81 (C-2), 127.51 (C-5), 119.32 (C-3), 118.15 (C-4a), 115.01 (C-6), 108.70 (C-4′′), 100.58 (C-8), 58.37 (C-1′), 56.03 (OCH3), 13.60 (CH3), 11.94 (CH3); HRMS (ESI-TOF) (m/z) C17H15NO6 calculated, 330.0978, found 330.0929 [M + H]+; calculated, 352.0797, found, 352.0742 [M + Na]+; and calculated, 368.4063, found, 368.0480 [M + K]+.
3.3.6. General Procedure for the Synthesis of Conjugates 6a, 6b, and 6d
Conjugates
6a,
6b, and
6d were prepared by the general procedure adopted from the literature report [
50]. A mixture of 4-oxo-4
H-1-benzopyran-3-yl-carboxaldehyde (
2a, 0.5 g, 2.9 mmol) or methoxy-substituted 4-oxo-4
H-1-benzopyran-3-yl-carboxaldehyde (
2b/
2c/
2d, 0.58 g, 2.9 mmol), (3,5-Dimethyl-isoxazol-4-yl)-methanol (0.80 g, 6.3 mmol), InF
3 (24 mg, 1.43 mmol, 5 mol%) and Toluene (5 mL) was taken in a round bottom flask fitted with a reflux condenser. The mixture was refluxed for 5 h (125 °C). After completion of the reaction, the reaction mixture was filtered to remove the catalyst and washed with toluene. The solvent was then evaporated in a vacuum, and the resulting precipitate was purified via column chromatography on basic alumina to afford
6a,
6b, and
6d as white solids. Conjugate
6c was not obtained during the reaction as Starting Material
2c was left unreacted.
3-(Bis((3,5-dimethylisoxazol-4-yl)methoxy)methyl)-4H-1-benzopyran-4-one (6a). Yield = 13%; Rf = 0.48 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 2925, 1650, 1611, 1574, 1465, 1425, 1386, 1344, 1312, 1265, 1217, 1178, 1146, 1079, 1026, 883, 848, 818, 761, 741, 698, 674 and 640 cm−1; UV/vis (MeOH) λ: 303, 297, 227 nm; 1H NMR (400 MHz, CDCl3): δ 8.23 (dd, J = 8.0, 1.5 Hz, 1H, H-5), 8.13 (s, 1H, H-2), 7.70 (ddd, J = 8.7, 7.1, 1.7 Hz, 1H, H-7), 7.59–7.36 (m, 2H, H-6 and H-8), 5.83 (s, 1H, H-1′), 4.43 (dd, J = 35.3, 11.9 Hz, 2H, H-6′′), 2.36 (s, 6H, 2xCH3), 2.26 (s, 6H, 2xCH3); 13C NMR (101 MHz, CDCl3): δ 176.12 (C-4 C=O), 167.58 (C-5′′), 159.86 (C-3′′), 156.40 (C-8a), 154.93 (C-2), 134.23 (C-7), 126.01 (C-6), 125.78 (C-5), 124.23 (C-4a), 121.56 (C-3), 118.38 (C-8), 110.78 (C-4′′), 95.73 (C-1), 58.19 (C-6′′), 11.19 (CH3), 10.28 (CH3); HRMS (ESI-TOF) (m/z) C22H22N2O6 calculated, 433.1376, found 433.1302 [M + Na]+; and calculated, 449.1115, found, 449.1039 [M + K]+.
3-(Bis((3,5-dimethylisoxazol-4-yl)methoxy)methyl)-5-methoxy-4H-1-benzopyran-4-one (6b). Yield = 11%; Rf = 0.12 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3708, 3679, 2969, 2923, 2866, 2844, 2827, 2350, 2322, 2165, 2073, 2029, 1976, 1653, 1606, 1572, 1540, 1475, 1456, 1432, 1375, 1346, 1324, 1293, 1270, 1211, 1162, 1054, 1032, 101, 886, 803, 776, 736, 700 and 677 cm−1; UV/vis (MeOH) λ: 315, 255, 225 nm; 1H NMR (400 MHz, CDCl3): δ 7.96 (s, 1H, H-2), 7.56 (t, J = 8.4 Hz, 1H, H-7), 7.02 (dd, J = 8.5, 0.8 Hz, 1H, H-8), 6.83 (d, J = 8.3 Hz, 1H, H-6), 5.78 (s, 1H, H-1′), 4.41 (dd, J = 43.3, 11.9 Hz, 4H, H-6′′), 3.98 (s, 3H, -OCH3), 2.36 (s, 6H, 2X –CH3), 2.25 (s, 6H, 2X –CH3); 13C NMR (101 MHz, CDCl3): δ 175.86 (C-4 C=O), 167.52 (C-5′′), 160.20 (C-5), 159.87 (C-3′′), 158.39 (C-8a), 152.99 (C-2), 134.26 (C-7), 122.94 (C-3), 114.86 (C-4a), 110.92 (C-4′′), 110.31 (C-6), 106.89 (C-8), 96.03 (C-1′), 58.58 (C-6′′), 56.64 (OCH3), 11.21 (CH3), 10.29 (CH3); HRMS (ESI-TOF) (m/z) C23H24N2O7 calculated, 441.1662, found 441.1602 [M + H]+; calculated, 463.1481, found, 463.1481 [M + Na]+; and calculated, 479.1221, found, 479.1156 [M + K]+.
3-(Bis((3,5-dimethylisoxazol-4-yl)methoxy)methyl)-7-methoxy-4H-1-benzopyran-4-one (6d). Yield = 10%; Rf = 0.42 [ethyl acetate–petroleum ether 50:50]; IR (kbr) νmax: 3711, 3696, 3678, 3655, 2970, 2923, 2866, 2844, 2826, 2362, 2341, 2165, 2074, 2028, 1978, 1544, 1524, 1511, 1476, 1457, 1345, 1323, 1054, 1032 and 1013 cm−1; UV/vis (MeOH) λ: 295, 248, 240, 223 nm; 1H NMR (400 MHz, CDCl3): δ 8.12 (d, J = 8.9 Hz, 1H, H-5), 8.04 (d, J = 0.7 Hz, 1H, H-2), 6.99 (dd, J = 8.9, 2.4 Hz, 1H, H-6), 6.84 (d, J = 2.4 Hz, 1H, H-8), 5.82 (d, J = 0.7 Hz, 1H, H-1′), 4.42 (dd, J = 37.6, 11.9 Hz, 4H, H-6′′), 3.90 (s, 3H, -OCH3), 2.35 (s, 6H, 2X-CH3), 2.26 (s, 6H, 2X -CH3); 13C NMR (101 MHz, CDCl3): δ 175.45 (C-4 C=O), 167.56 (C-7), 164.50 (C-5′′), 159.87 (C-3′′), 158.19 (C-8a), 154.45 (C-2), 127.36 (C-5), 121.41 (C-3), 118.05 (C-4a), 115.08 (C-6), 110.83 (C-4′′), 100.50 (C-1′), 95.82 (C-8), 58.18 (C-6′′), 56.02 (OCH3), 11.18 (CH3), 10.26 (CH3); HRMS (ESI-TOF) (m/z) C23H24N2O7 calculated, 441.1662, found 441.1594 [M + H]+; calculated, 463.1481, found, 463.1410 [M + Na]+; and calculated, 479.1221, found, 479.1145 [M + K]+.
3.3.7. General Procedure for the Synthesis of Conjugates 7a, 7c, and 7d
Conjugates
7a,
7c, and
7d were prepared via the general procedure adopted from the literature report [
51]. A mixture of 4-oxo-4
H-1-benzopyran-3-yl- carboxaldehyde (
2a, 0.5 g, 2.9 mmol) or methoxy substituted 4-oxo-4
H-1-benzopyran-3-yl- carboxaldehyde (
2b/
2c/
2d, 0.59 g, 2.9 mmol), NBS (0.61 g, 3.44 mmol), and AIBN (20 mg) in CCl
4 (25 mL) was taken in a round bottom flask and refluxed for 1 h. The mixture was then cooled in ice-water bath, followed by the addition of 3-amino-5-methyl isoxazole (0.56 g, 5.74 mmol). After stirring the reaction mixture for 30 min at room temperature, the mixture was diluted with CH
2Cl
2 (100 mL), washed with water, dried with MgSO
4, and the solvent evaporated. The resulting precipitate was purified by column chromatography to afford
7a,
7c, and
7d as solid products. For Conjugate
7b, Starting Material
2b reacted and gave a very complicated reaction as indicated by TLC, and Product
7b could not be isolated in pure form.
N-(5-Methylisoxazol-3-yl)-4-oxo-4H-1-benzopyran-3-carboxamide (7a). Yield = 31%; Rf = 0.69 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3207, 3146, 3074, 2956, 2922, 2869, 1689, 1634, 1614, 1567, 1548, 1465, 1436, 1397, 1377, 1346, 1313, 1268, 1252, 1206, 1177, 1142, 1123, 1104, 1054, 1033, 1013, 939, 912, 887, 862, 832, 806, 764, 718 and 644 cm−1; UV/vis (MeOH) λ: 295, 242 nm; 1H NMR (400 MHz, CDCl3): δ 11.80 (s, 1H, NH), 9.03 (s, 1H, H-2), 8.35 (dd, J = 8.0, 1.7 Hz, 1H, H-5), 7.80 (ddd, J = 8.7, 7.1, 1.7 Hz, 1H, H-7), 7.67–7.45 (m, 2H, H-6 and H-8), 6.75 (s, 1H, CH), 2.43 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3): δ 176.96 (C-4 C=O), 169.95 (C-1′ C=O), 163.24 (C-2), 161.10 (C-5), 157.71 (C-3′′), 156.24 (C-8a), 135.21 (C-7), 126.86, 126.64 (C6 and C-5), 124.16 (C-4a), 118.61 (C-8), 115.28 (C-3), 97.16 (C-4′′), 12.84 (CH3); HRMS (ESI-TOF) (m/z) C14H10N2O4 calculated, 271.0719, found 271.0676 [M + H]+; calculated, 293.0538, found, 293.0490 [M + Na]+; and calculated, 309.0279, found, 309.0230 [M + K]+.
6-Methoxy-N-(5-methylisoxazol-3-yl)-4-oxo-4H-1-benzopyran-3-carboxamide (7c). Yield = 25%; Rf = 0.76 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3697, 3679, 3662, 3573, 2969, 2922, 2866, 2844, 2826, 2361, 2341, 2165, 2073, 2050, 1977, 1690, 1611, 1560, 1546, 1512, 1478, 1457, 1434, 1374, 1346, 1307, 1262, 1160, 1142, 1054, 1032, 1014 and 722 cm−1; UV/vis (MeOH) λ: 326, 237 nm; 1H NMR (400 MHz, CDCl3): δ 11.84 (s, 1H, -NH-), 9.00 (s, 1H, H-2), 7.66 (d, J = 3.0 Hz, 1H, H-5), 7.52 (d, J = 9.2 Hz, 1H, H-8), 7.36 (dd, J = 9.2, 3.1 Hz, 1H, H-7), 6.75 (s, 1H, CH), 3.94 (s, 3H, -OCH3), 2.43 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 176.75 (C-4 C=O), 169.95 (C-5′′), 162.82 (C-2), 161.31 (C-1′), 158.18 (C-6), 157.77 (C-3′′), 151.14 (C-8a), 125.23 (C-7), 124.96 (C-4a), 120.01 (C-8), 114.52 (C-3), 105.55 (C-5), 97.17 (C-4′′), 56.24 (OCH3), 12.82 (CH3); HRMS (ESI-TOF) (m/z) C15H12N2O5 calculated, 323.0644, found 323.0599 [M + Na]+; and calculated, 339.0383, found, 339.0330 [M + K]+.
7-Methoxy-N-(5-methylisoxazol-3-yl)-4-oxo-4H-1-benzopyran-3-carboxamide (7d). Yield = 73%; Rf = 0.65 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3712, 3696, 3678, 3655, 2922, 2867, 2846, 2362, 2341, 2164, 2031, 1690, 1652, 1615, 1560, 1547, 1508, 1437, 1377, 1304, 1277, 1204, 1154, 1092, 1055, 1031, 1015, 940, 889, 849, 794, 763, 735, 671, 638, 610, 564 and 505 cm−1; UV/vis (MeOH) λ: 293, 255, 247, 227 nm; 1H NMR (400 MHz, CDCl3): δ 11.91 (s, 1H, -NH-), 8.94 (s, 1H, H-2), 8.23 (d, J = 9.0 Hz, 1H, H-5), 7.09 (dd, J = 9.0, 2.4 Hz, 1H, H-6), 6.95 (d, J = 2.4 Hz, 1H, H-8), 6.73 (d, J = 0.5 Hz, 1H, -CH-), 3.94 (s, 3H, -OCH3), 2.43 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 176.23 (C-4 C=O), 169.90 (C-5″), 165.28 (C-7), 162.75 (C-2), 161.33 (C-1′), 158.17, 157.76 (C-8a and C-3″), 127.99 (C-5), 117.79 (C-4a), 116.14 (C-6), 115.16 (C-3), 100.77 (C-8), 97.17 (C-4″), 56.23 (OCH3), 12.82 (CH3); HRMS (ESI-TOF) (m/z) C15H12N2O5 calculated, 301.0824, found 301.0776 [M + H]+; calculated, 323.0644, found, 323.0591 [M + Na]+; and calculated, 339.0383, found, 339.0330 [M + K]+.
3.3.8. Synthesis of Conjugate 8a
Conjugate
8a was prepared via the general procedure adopted from the literature report [
51]. A mixture of 4-oxo-4
H-1-benzopyran-3-yl- carboxaldehyde (
2a, 0.5 g, 2.9 mmol), NBS (0.61 g, 3.44 mmol), and AIBN (20 mg) in CCl
4 (25 mL) was taken in a round bottom flask and refluxed for 1 h. The mixture was then cooled in an ice-water bath, followed by the addition of (3,5-dimethyl-isoxazol-4-yl)-methanol (0.73 g, 5.74 mmol). After stirring the reaction mixture for 30 min at room temperature, the mixture was diluted with CH
2Cl
2 (100 mL), washed with water, dried with MgSO
4, and the solvent evaporated. The resulting precipitate was purified by column chromatography to afford
8a as white solid. For the Conjugates
8b,
8c, and
8d, Starting Materials
2b,
2c, and
2d reacted and gave very complicated reactions, as indicated by TLC′s, and Products
8b,
8c, and
8d could not be isolated in pure form.
(3,5-Dimethylisoxazol-4-yl)methyl 4-oxo-4H-1-benzopyran-3-carboxylate (8a). Yield = 29%; Rf = 0.5 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3055, 2957, 2923, 2867, 2350, 2082, 1705, 1655, 1614, 1569, 1508, 1463, 1426, 1383, 1339, 1311, 1291, 1263, 1201, 1151, 1115, 1103, 1085, 1056, 1033, 971, 957, 938, 900, 873, 855, 813, 791, 771, 750, 739, 686, 675 and 634 cm−1; UV/vis (MeOH) λ: 293, 225 nm; 1H NMR (400 MHz, CDCl3): δ 8.62 (s, 1H, H-2), 8.26 (dd, J = 8.0, 1.5 Hz, 1H, H-5), 7.70 (ddd, J = 8.7, 7.1, 1.7 Hz, 1H, H-7), 7.50–7.44 (m, 2H, H-6 and H-8), 5.13 (s, 2H, -OCH2), 2.48 (s, 3H, CH3), 2.37 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3): δ 173.29 (C-4 C=O), 168.80 (C-1′ C=O), 163.31 (C-3′′), 162.12 (C-2), 159.97 (C-5′′), 155.69 (C-8a), 134.45 (C-7), 126.69, 126.52 (C-6 and C-5), 125.20 (C-4a), 118.30 (C-8), 115.97 (C-3), 109.67 (C-4′′), 56.07 (C-6′′ OCH2), 11.34 (CH3), 10.26 (CH3); HRMS (ESI-TOF) (m/z) C16H13NO5 calculated, 300.0872, found 300.0825 [M + H]+; calculated, 322.0691, found, 322.0640 [M + Na]+; and calculated, 338.0431, found, 338.0378 [M + K]+.
3.3.9. General Procedure for the Synthesis of Conjugates 9a, 9c, and 9d
A mixture of (E)-3-(4-oxo-4H-1-benzopyran-3-yl) acrylic acid (4a, 0.5 g, 2.3 mmol) or methoxy substituted (E)-3-(4-oxo-4H-1-benzopyran-3-yl) acrylic acid (4b/4c/4d, 0.57 g, 2.3 mmol), EDCI (1.33 g, 6.9 mmol) and DMF (10 mL) were stirred at 0 °C. After 30 min, a catalytic amount of DMAP was added and stirred for an additional 10 min. Then, (3,5-dimethyl-isoxazol-4-yl)-methanol (0.35 g, 2.8 mmol) was added, and the reaction was stirred overnight. After completion of the reaction, the solvent was evaporated under vacuum, and the resulting precipitate was dissolved in ethyl acetate and washed 3 times with water. The solvent was dried over MgSO4, evaporated, and the residue was purified via column chromatography to afford 9a, 9c, and 9d as solid products.
(3,5-Dimethylisoxazol-4-yl)methyl€-3-(4-oxo-4H-1-benzopyran-3-yl)acrylate (9a). Yield = 2%; Rf = 0.71 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3698, 3679, 3656, 2954, 2922, 2867, 2846, 2362, 2341, 1713, 1658, 1616, 1564, 1464, 1406, 1376, 1356, 1309, 1290, 1265, 1245, 1216, 1155, 1054, 1032, 1013, 857, 762 and 687 cm−1; UV/vis (MeOH) λ: 290, 267 nm; 1H NMR (400 MHz, CDCl3): δ 8.27 (dd, J = 8.0, 1.6 Hz, 1H, H-5), 8.11 (s, 1H, H-2), 7.70 (ddd, J = 8.6, 7.1, 1.7 Hz, 1H, H-7), 7.50–7.43 (m, 2H, H-6 and H-8), 7.35 (q, J = 15.8 Hz, 2H, H-1′ and H-2′), 5.01 (s, 2H, -CH2-OCO-), 2.45 (s, 3H, CH3), 2.31 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3): δ 176.04 (C-4 C=O), 168.52 (C-3′ C=O), 167.24 (C-3″), 159.96 (C-5″), 157.91 (C-2), 155.64 (C-8a), 136.45 (C-1′), 134.26 (C-7), 126.47, 126.10 (C-5 and C-6), 124.32 (C-4a), 121.56 (C-8), 119.26 (C-3), 118.27 (C-2′), 110.04 (C-4″), 55.30 (C-6″ OCH2), 11.28 (CH3), 10.21 (CH3); HRMS (ESI-TOF) (m/z) C18H15NO5 calculated, 326.1028, found 326.0973 [M + H]+; calculated, 348.0848, found, 348.0788 [M + Na]+; and calculated, 364.0587, found, 364.0527 [M + K]+.
(3,5-Dimethylisoxazol-4-yl)met€(E)-3-(6-methoxy-4-oxo-4H-1-benzopyran-3-yl)acrylate (9c). Yield = 8%; Rf = 0.78 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3712, 3696, 3679, 3655, 3634, 3618, 3593, 3571, 3379, 2968, 2923, 2867, 2845, 2362, 2342, 2163, 2072, 2030, 1978, 1711, 1656, 1609, 1566, 1546, 1525, 1512, 1483, 1457, 1436, 1401, 1369, 1341, 1287, 1245, 1198, 1165, 1145, 1101, 1055, 1032, 1012, 954, 866, 839, 823, 727, 671 and 603 cm−1; UV/vis (MeOH) λ: 244, 225 nm; 1H NMR (400 MHz, CDCl3): δ 8.10 (s, 1H, H-2), 7.62 (d, J = 3.1 Hz, 1H, H-5), 7.41 (dd, J = 11.8, 4.7 Hz, 2H, H-1′ and H-2′), 7.33–7.27 (m, 2H, H-7 and H-8), 5.01 (s, 2H, -OCH2), 3.91 (s, 3H, -OCH3), 2.46 (s, 3H, -CH3), 2.32 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 175.90 (C-4 C=O), 168.53 (C-3′ C=O), 167.31 (C-3″), 159.99 (C-5″), 157.69 (C-6), 157.67 (C-2), 150.52 (C-8a), 136.68 (C-1′), 125.03 (C-4a), 124.28 (C-2′), 121.30 (C-7), 119.71 (C-8), 118.45 (C-3), 110.08 (C-4″), 105.55 (C-5), 56.13 (OCH3), 55.32 (C-6′′ OCH2), 11.30 (CH3), 10.23 (CH3); HRMS (ESI-TOF) (m/z) C19H17NO6 calculated, 356.1134, found 356.1086 [M + H]+; calculated, 378.0954, found, 378.0904 [M + Na]+; and calculated, 394.0693, found, 394.0643 [M + K]+.
(3,5-Dimethylisoxazol-4-yl)€hyl(E)-3-(7-methoxy-4-oxo-4H-1-benzopyran-3-yl)acrylate (9d). Yield = 8%; Rf = 0.68 [ethyl acetate–petroleum ether 50:50]; IR (neat) νmax: 3751, 3710, 3696, 3678, 3655, 3634, 2970, 2923, 2866, 2844, 2826, 2362, 2341, 2165, 2073, 2029, 1978, 1544, 1523, 1511, 1495, 1476, 1457, 1345, 1322, 1055, 1032 and 1013 cm−1; UV/vis (MeOH) λ: 293, 255, 223 nm; 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.9 Hz, 1H, H-5), 8.03 (s, 1H, H-2), 7.34 (q, J = 15.8 Hz, 2H, H-1′ and H-2′), 7.01 (dd, J = 8.9, 2.4 Hz, 1H, H-6), 6.85 (d, J = 2.4 Hz, 1H, H-8), 5.00 (s, 2H, -OCH2-), 3.91 (s, 3H, -OCH3), 2.45 (s, 3H, -CH3), 2.31 (s, 3H, -CH3); 13C NMR (101 MHz, CDCl3): δ 175.43 (C-4 C=O), 168.52 (C-3′ C=O), 167.31 (C-3″), 164.55 (C-7), 159.99 (C-5″), 157.49 (C-2), 157.43 (C-8a), 136.62 (C-1′), 127.88 (C-5), 121.41 (C-2′), 119.22 (C-3), 118.18 (C-4a), 115.30 (C-6), 110.08 (C-4″), 100.52 (C-8), 56.08 (OCH3), 55.28 (C-6″ OCH2), 11.29 (CH3), 10.21 (CH3); HRMS (ESI-TOF) (m/z) C19H17NO6 calculated, 356.1134, found 356.1084 [M + H]+; calculated, 378.0954, found, 378.0899 [M + Na]+; and calculated, 394.0693, found, 394.0636 [M + K]+.



 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}