
Salvianolic Acid B Inhibits Ferroptosis and Apoptosis during Myocardial Ischemia/Reperfusion Injury via Decreasing the Ubiquitin-Proteasome Degradation of GPX4 and the ROS-JNK/MAPK Pathways

 , ,
, , 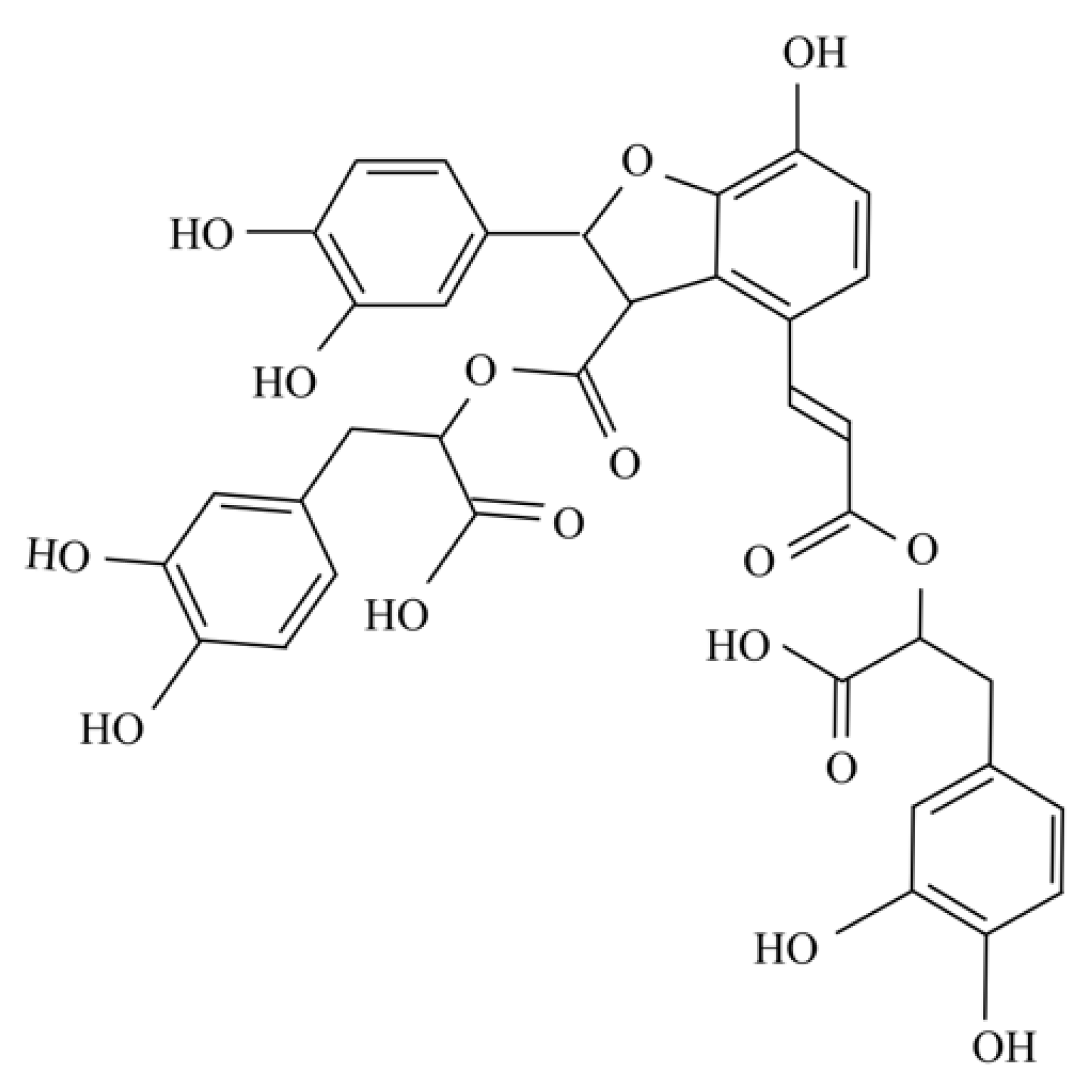{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
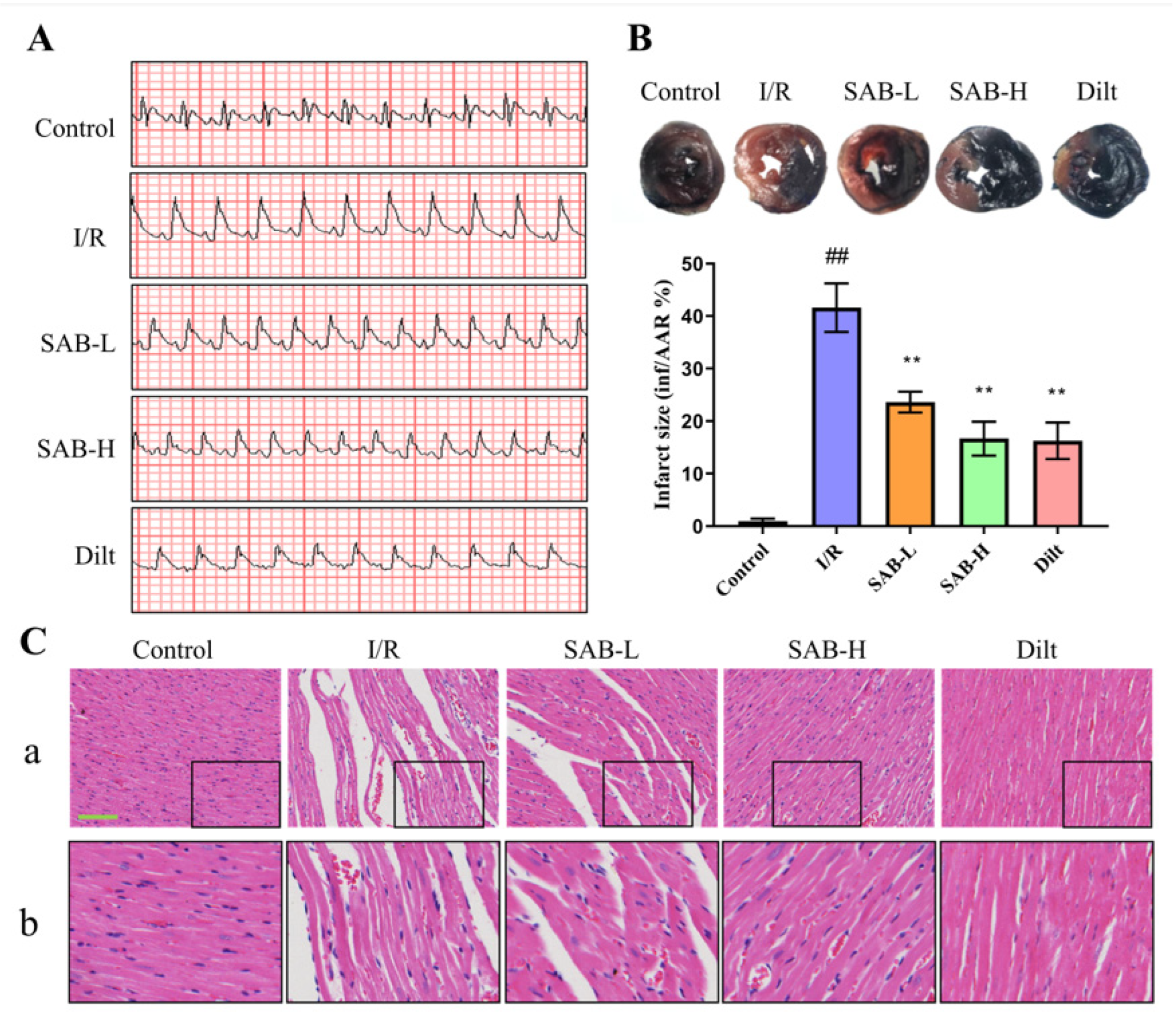
2.1. SAB Protects Hearts from Myocardial I/R Injury in Rats
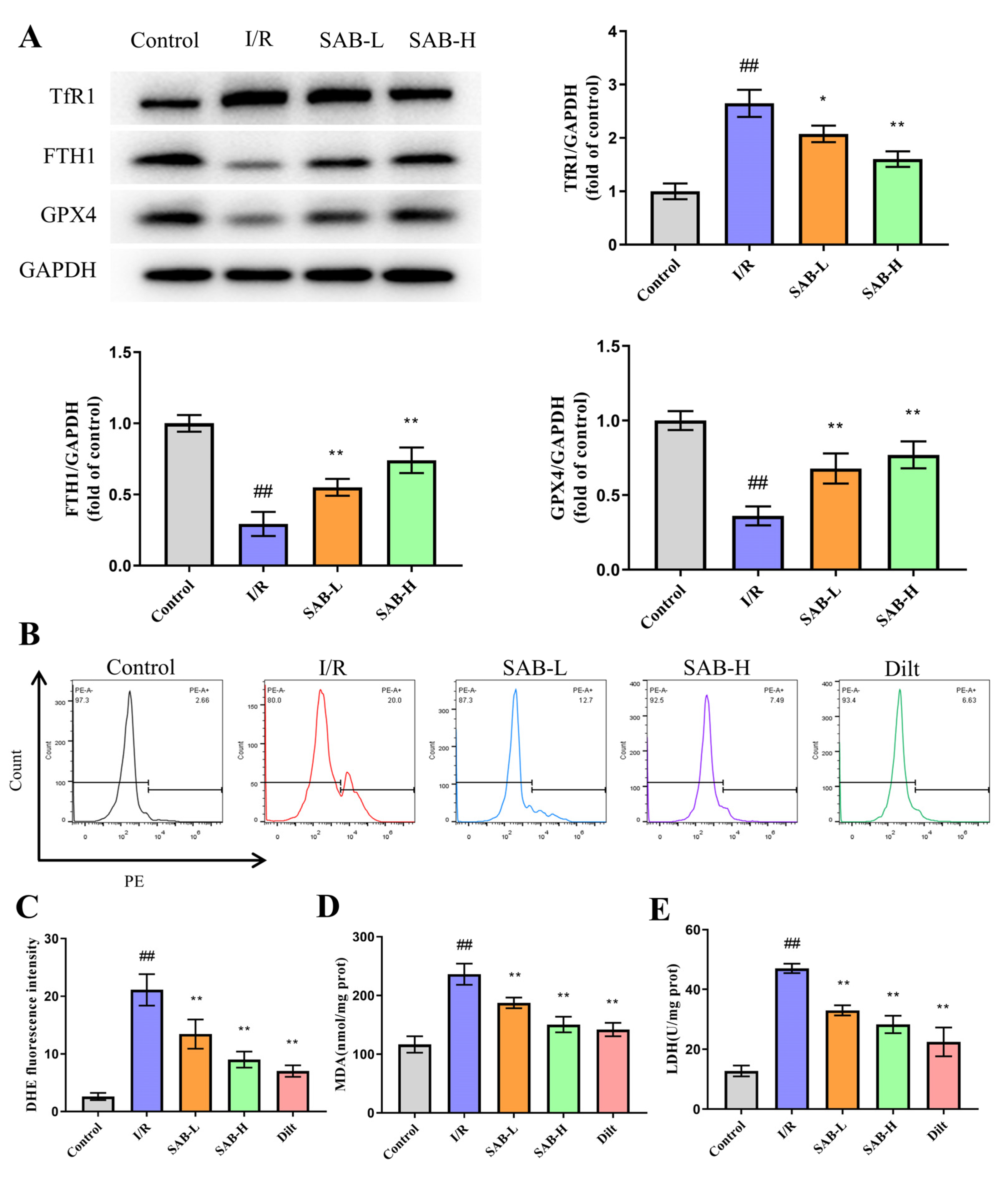
2.2. SAB Inhibits I/R-Induced Ferroptosis in the Infarcted Heart
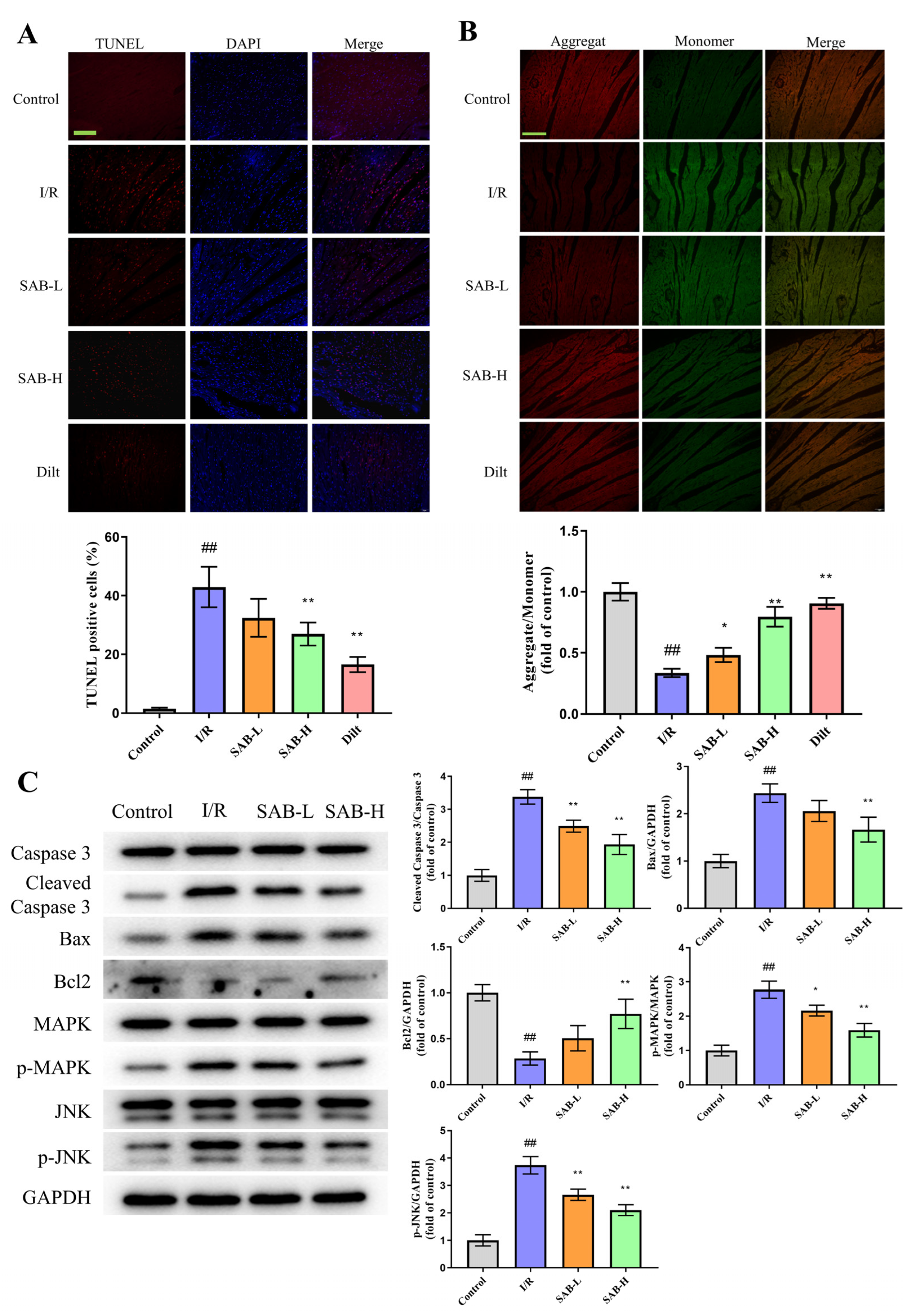
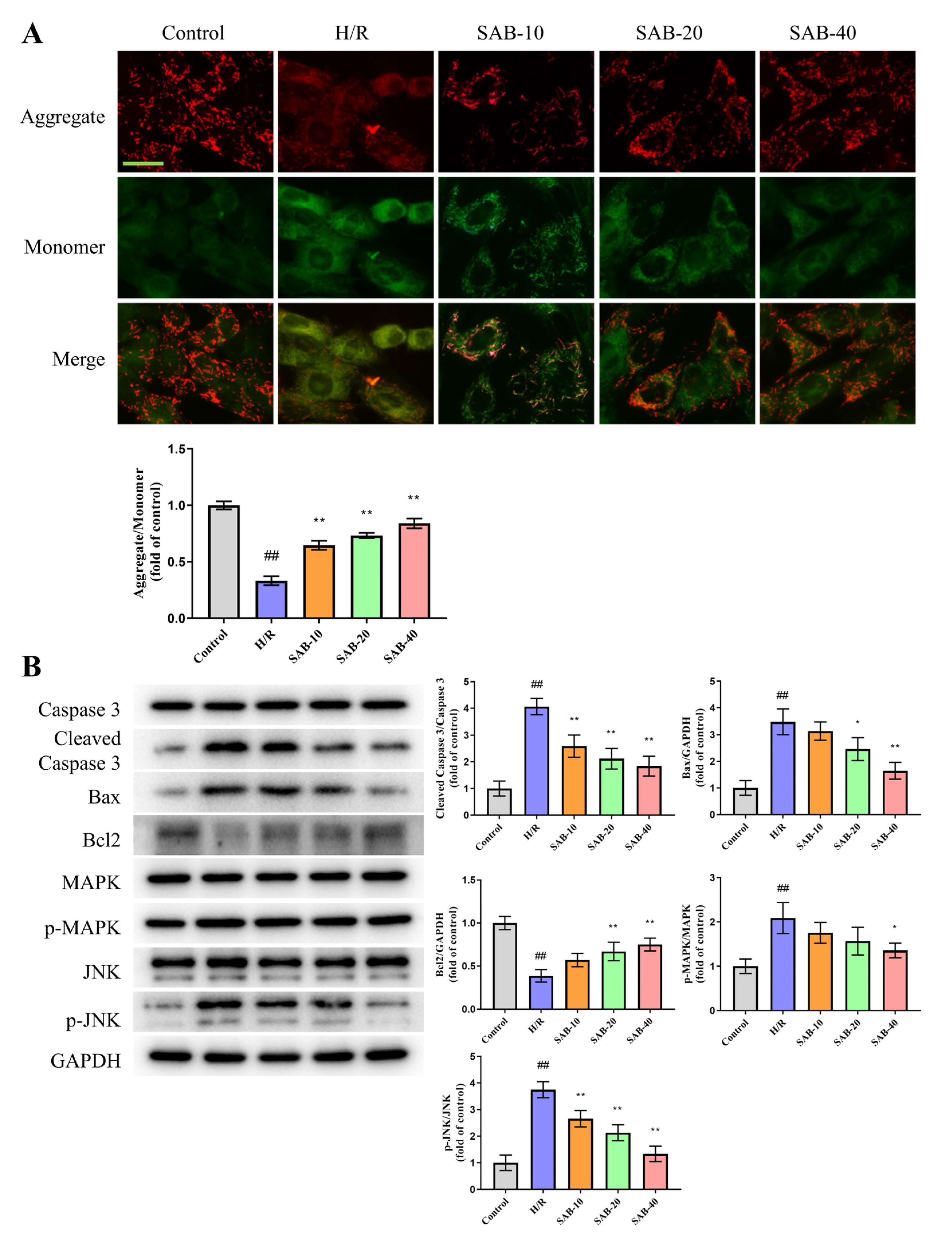
2.3. SAB Decreases I/R-Induced Apoptosis in the Infarcted Heart
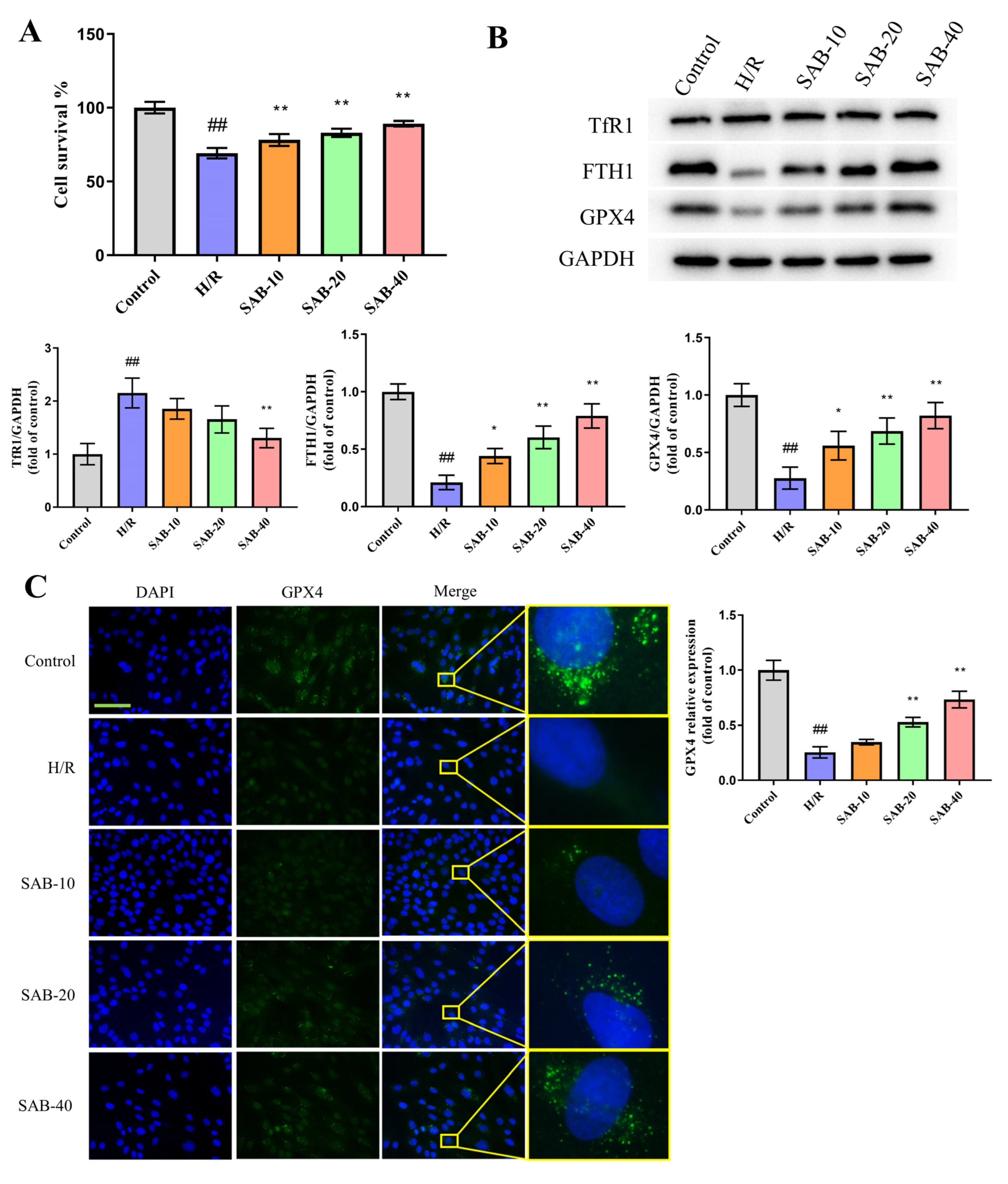
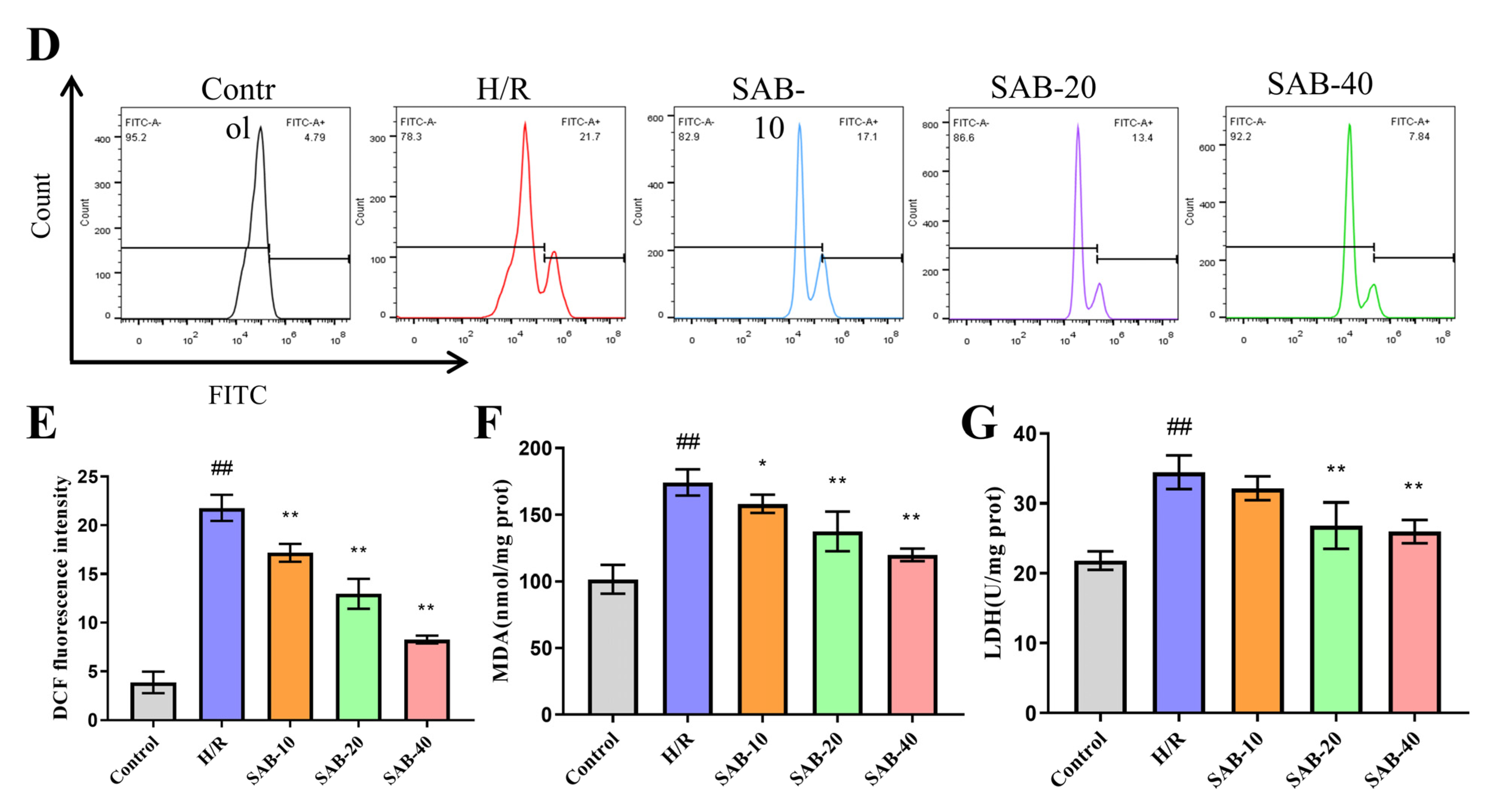
2.4. SAB Prevents H/R Injury in H9c2 Cells through Regulating Ferroptosis and Apoptosis
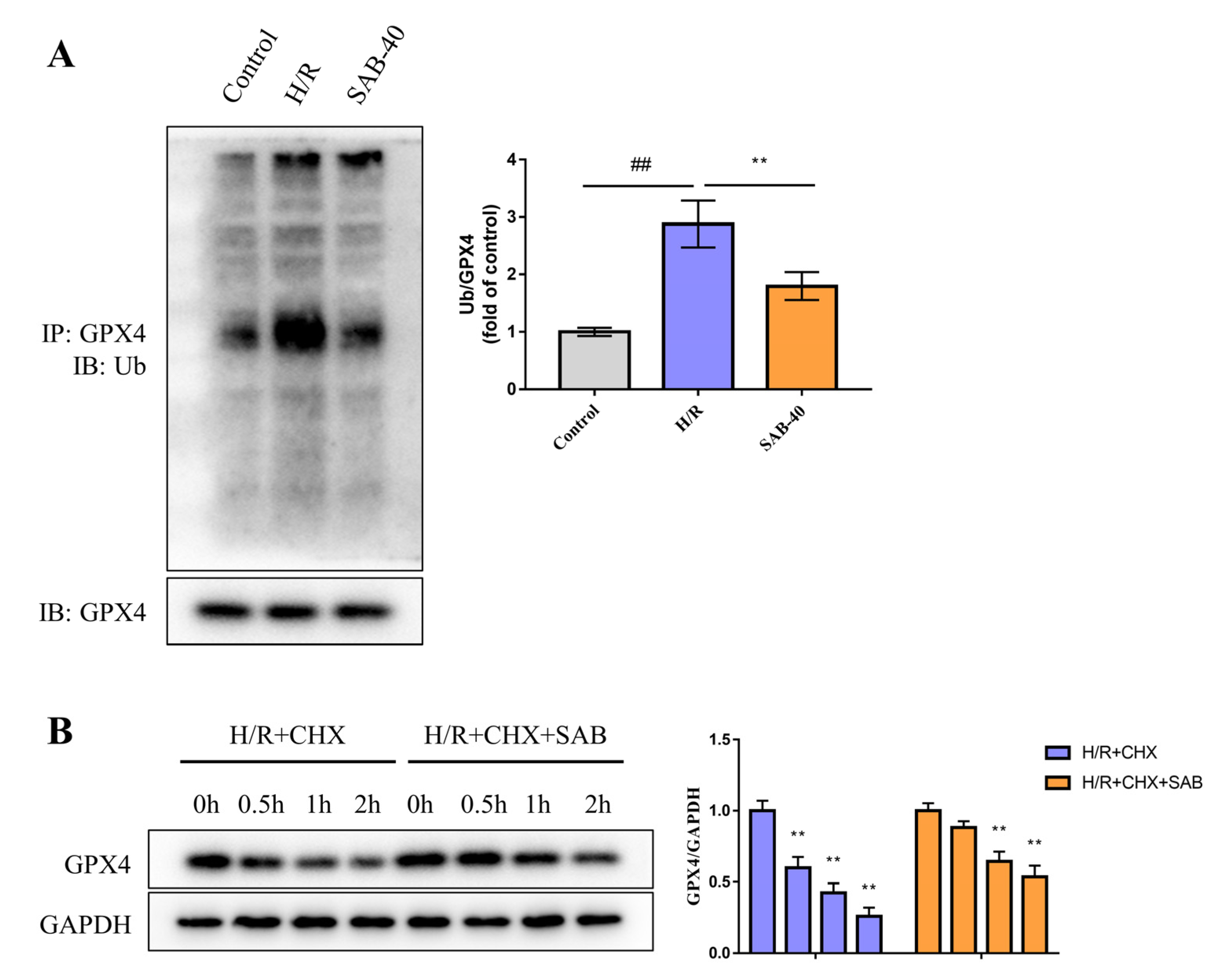
2.5. SAB Decreases the Ubiquitin-Proteasome Degradation of GPX4 in H/R-Stimulated H9c2 Cells
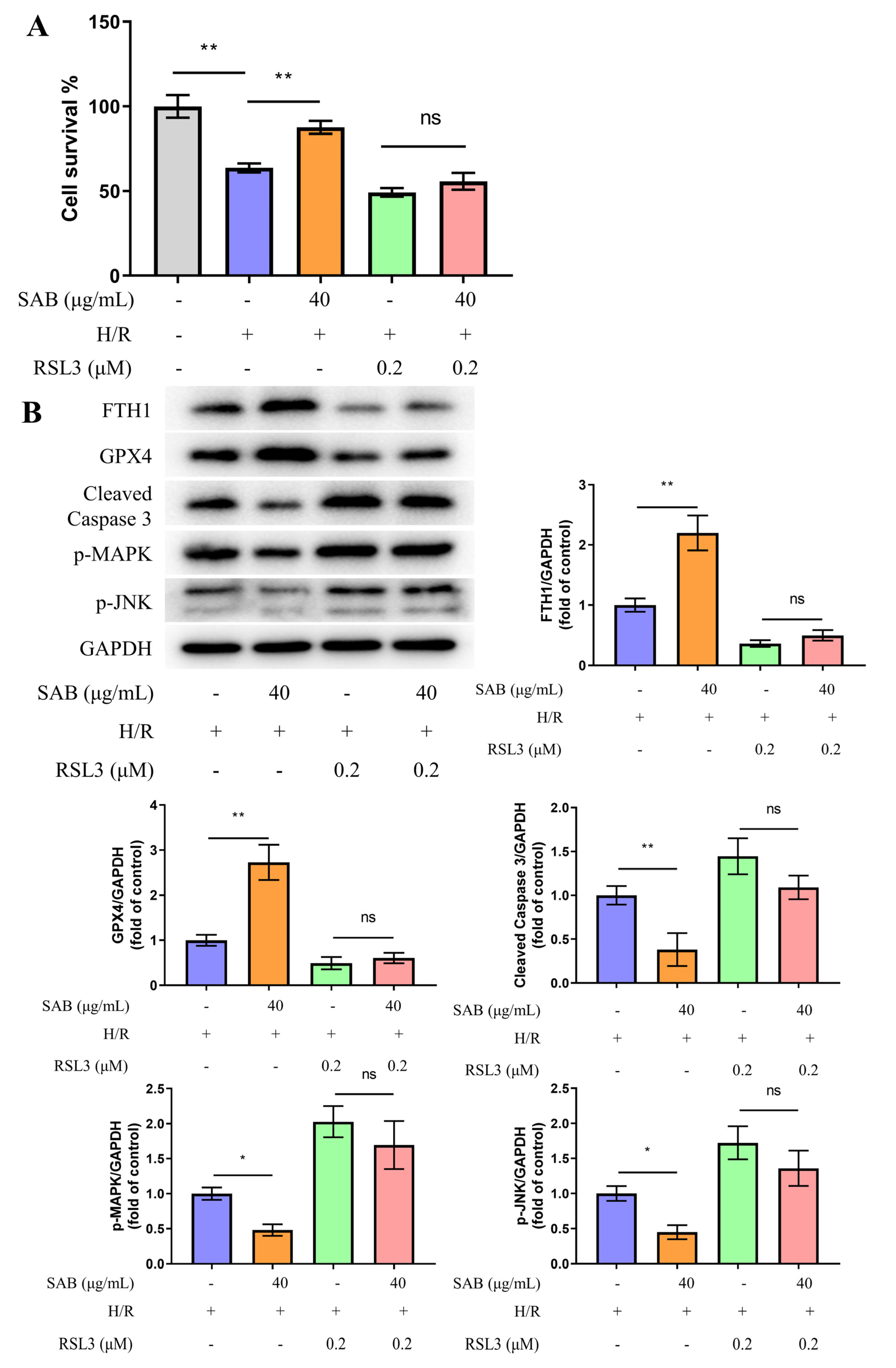
2.6. GXP4 Inhibition Abolishes the Protective Effect of SAB in H9c2 Cells Stimulated with H/R
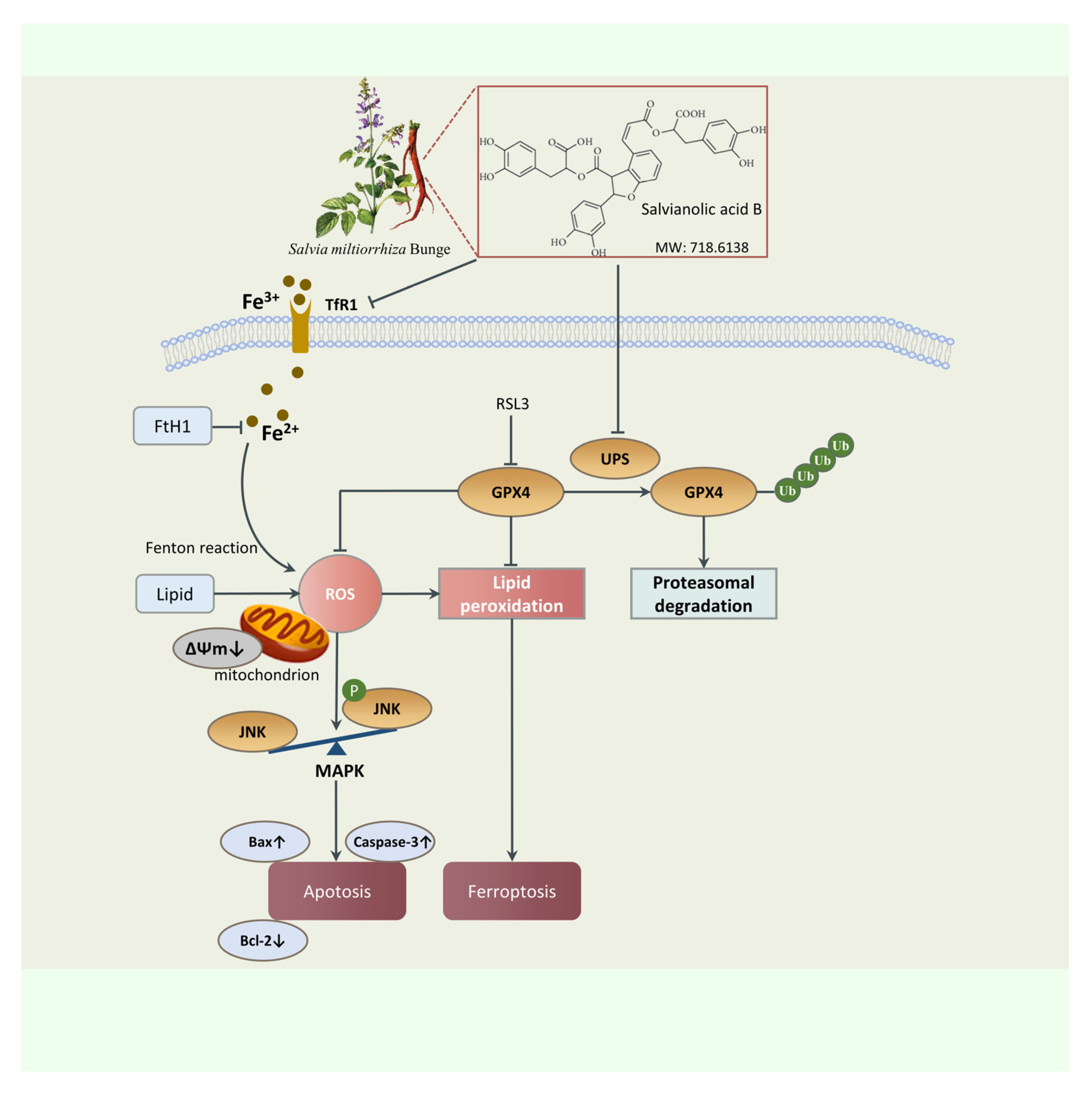
3. Discussion
4. Materials and Methods
4.1. Animals and Grouping
4.2. Myocardial I/R Surgery
4.3. The Culture of Cells and the Establishment of H/R Model
4.4. Measurement of Infarct Size
4.5. Histopathological Examination
4.6. Western Blot Analysis
4.7. Co-Immunoprecipitation
4.8. Detection of ROS Production
4.9. Measurements of Malondialdehyde (MDA) and Lactate Dehydrogenase (LDH) Levels
4.10. TUNEL Assay
4.11. Mitochondrial Membrane Potential (MMP, ΔΨm) Detection
4.12. The MTT Assay
4.13. Immunofluorescence
4.14. Cycloheximide Chase Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Guo, B.; Cao, J.; Liu, Y.; Wang, Y.; Qian, Y.; Chen, G.; Zhu, W. Cardiac Protection of a Novel Lupane-Type Triterpenoid from Injuries Induced by Hypoxia–Reperfusion. Int. J. Mol. Sci. 2022, 23, 9473. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Cebova, M.; Pechanova, O. Protective Effects of Polyphenols against Ischemia/Reperfusion Injury. Molecules 2020, 25, 3469. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Q.; Li, J.; Yao, Y.; Sun, W.; Zhang, Z.; Qi, H.; Chen, Z.; Liu, J.; Zhao, D.; et al. Panax ginseng against myocardial ischemia/reperfusion injury: A review of preclinical evidence and potential mechanisms. J. Ethnopharmacol. 2023, 300, 115715. [Google Scholar] [CrossRef]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Higa, J.K.; Shimada, B.K.; Horiuchi, K.M.; Suhara, T.; Kobayashi, M.; Woo, J.D.; Aoyagi, H.; Marh, K.S.; Kitaoka, H.; et al. Protective effects of the mechanistic target of rapamycin against excess iron and fer-roptosis in cardiomyocytes. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H659–H668. [Google Scholar] [CrossRef] [PubMed]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Shen, Y.; Shen, X.; Wang, S.; Zhang, Y.; Wang, Y.; Ding, Y.; Shen, J.; Zhao, J.; Qin, H.; Xu, Y.; et al. Protective effects of Salvianolic acid B on rat ferroptosis in myocardial infarction through upregulating the Nrf2 signaling pathway. Int. Immunopharmacol. 2022, 112, 109257. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.-Y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Yu, D.; Li, M.; Tian, Y.; Liu, J.; Shang, J. Luteolin inhibits ROS-activated MAPK pathway in myocardial ischemia/reperfusion injury. Life Sci. 2015, 122, 15–25. [Google Scholar] [CrossRef]
- Chang, W.T.; Bow, Y.D.; Fu, P.J.; Li, C.Y.; Wu, C.Y.; Chang, Y.H.; Teng, Y.N.; Li, R.N.; Lu, M.C.; Liu, Y.C.; et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Car-cinoma Cells and Involves the ROS and MAPK Pathways. Oxid. Med. Cell. Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef]
- Jung, I.; Kim, H.; Moon, S.; Lee, H.; Kim, B. Overview of Salvia miltiorrhiza as a Potential Therapeutic Agent for Various Diseases: An Update on Efficacy and Mechanisms of Action. Antioxidants 2020, 9, 857. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wu, Y.; Lian, J.; Yu, N.; An, T.; Wang, T.; Bao, X.; Mo, F.; Zhao, D.; Yang, X.; et al. Effects of Salvianolic acid B on RNA expression and co-expression network of lncRNAs in brown adipose tissue of obese mice. J. Ethnopharmacol. 2021, 278, 114289. [Google Scholar] [CrossRef]
- Ali, M.; Khan, T.; Fatima, K.; Ali, Q.U.A.; Ovais, M.; Khalil, A.T.; Ullah, I.; Raza, A.; Shinwari, Z.K.; Idrees, M. Selected hepatoprotective herbal medicines: Evidence from ethnomedicinal applications, animal models, and possible mechanism of actions. Phytother. Res. 2018, 32, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, G. Danshen (Salvia miltiorrhiza Bunge): A prospective healing sage for cardiovascular diseases. Curr. Pharm. Des. 2017, 23, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-Y.; Ming, Q.-L.; Rahman, K.; Han, T.; Qin, L.-P. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 2015, 13, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Y.; Wang, S.; Zhao, X.; Zhao, L.; Wang, Y. Salvianolic acid B and ferulic acid synergistically promote angiogenesis in HUVECs and zebrafish via regulating VEGF signaling. J. Ethnopharmacol. 2022, 283, 114667. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, W.; Qiu, H.; Zou, D.; Cai, H.; Chen, Q.; Zheng, C.; Xu, D. Salvianolic acid B protects against myocardial ischaemia-reperfusion injury in rats via inhibiting high mobility group box 1 protein expression through the PI3K/Akt signalling pathway. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1527–1539. [Google Scholar] [CrossRef]
- Lu, B.; Li, J.; Gui, M.; Yao, L.; Fan, M.; Zhou, X.; Fu, D. Salvianolic acid B inhibits myocardial I/R-induced ROS generation and cell apoptosis by regulating the TRIM8/GPX1 pathway. Pharm. Biol. 2022, 60, 1458–1468. [Google Scholar] [CrossRef]
- Wang, J.; Xiong, X.; Feng, B. Cardiovascular Effects of Salvianolic Acid B. Evid.-Based Complement. Altern. Med. 2013, 2013, 247948. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zeng, G.; Wang, T.; Ren, H.; An, H.; Lian, C.; Liu, J.; Guo, L.; Li, W. Astragaloside IV Ameliorates Myocardial Infarction Induced Apoptosis and Restores Cardiac Function. Front. Cell Dev. Biol. 2021, 9, 671255. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Q.; Yuan, F.; Dong, H.; Zhang, H.; Geng, R.; Qi, Y.; Xiong, X.; Chen, Q.; Liu, B. RND2 attenuates apoptosis and autophagy in glioblastoma cells by targeting the p38 MAPK signalling pathway. J. Exp. Clin. Cancer Res. 2020, 39, 174. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Jin, M.; Shi, C.; Li, T.; Wu, Y.; Hu, C.; Huang, G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomed. Pharmacother. 2020, 129, 110282. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, Y.; Fu, W.; Yu, X.; Sui, D. Combination of the ginsenosides Rb3 and Rb2 exerts protective effects against myocardial ischemia reperfusion injury in rats. Int. J. Mol. Med. 2020, 45, 519–531. [Google Scholar] [CrossRef]
- Wagniart, P.; Ferguson, R.J.; Chaitman, B.R.; Achard, F.; Benacerraf, A.; Delanguenhagen, B.; Morin, B.; Pasternac, A.; Bourassa, M.G. Increased exercise tolerance and reduced electrocardiographic ischemia with diltiazem in patients with stable angina pectoris. Circulation 1982, 66, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Stone, P.H. Calcium antagonists for Prinzmetal’s variant angina, unstable angina and silent myocardial ischemia: Therapeutic tool and probe for identification of pathophysiologic mechanisms. Am. J. Cardiol. 1987, 59, 101B–115B. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lu, W.; Wu, G.; Lv, L.; Chen, W.; Huang, L.; Wu, X.; Xu, N.; Wu, Y. Cardioprotective effects of combined therapy with diltiazem and superoxide dismutase on myocardial ischemia-reperfusion injury in rats. Life Sci. 2017, 183, 50–59. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK 2/VDAC 1 Disassociation-Involved mPTP Opening. J. Am. Heart Assoc. 2017, 6, e005328. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.; da Cruz Junho, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Shen, W.; Ma, S.; Chen, W.; Qi, R. Rosa rugosa flavonoids alleviate myocardial ischemia reperfusion injury in mice by suppressing JNK and p38 MAPK. Microcirculation 2017, 24, e12385. [Google Scholar] [CrossRef]
- Conrad, M.; Friedmann Angeli, J.P. Glutathione peroxidase 4 (Gpx4) and ferroptosis: What’s so special about it. Mol. Cell. Oncol. 2015, 2, e995047. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Cai, L.; Wang, S.; Wang, J.; Chen, B. Baicalin Prevents Myocardial Ischemia/Reperfusion Injury Through Inhibiting ACSL4 Mediated Ferroptosis. Front. Pharmacol. 2021, 12, 628988. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Qiao, S.; Zhao, J.; Liu, Y.; Li, Q.; Wei, Z.; Dai, Q.; Kang, L.; Xu, B. miRNA-181a over-expression in mesenchymal stem cell-derived exosomes influenced inflammatory response after myocardial ischemia-reperfusion injury. Life Sci. 2019, 232, 116632. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Huang, Q.; Li, S.; Zhou, Y.; Li, Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-κB pathway in vivo and in vitro. Redox Biol. 2018, 15, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Zhang, Q.; Sheng, Z.; Li, Y.; Lu, H.H. Ciliary neurotrophic factor (CNTF) protects myocardial cells from oxygen glucose deprivation (OGD)/re-oxygenation via activation of Akt-Nrf2 signaling. Cell. Physiol. Biochem. 2018, 51, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Mou, S.-Q.; Li, W.-J.; Zhang, N.; Zhou, Z.-Y.; Ding, W.; Bian, Z.-Y.; Liao, H.-H. Resveratrol Inhibits Ischemia-Induced Myocardial Senescence Signals and NLRP3 Inflammasome Activation. Oxid. Med. Cell. Longev. 2020, 2020, 2647807. [Google Scholar] [CrossRef]
- Xie, S.; Deng, W.; Chen, J.; Wu, Q.Q.; Li, H.; Wang, J.; Wei, L.; Liu, C.; Duan, M.; Cai, Z.; et al. Andro-grapholide Protects Against Adverse Cardiac Remodeling After Myocardial Infarction through Enhancing Nrf2 Signaling Pathway. Int. J. Biol. Sci. 2020, 16, 12–26. [Google Scholar] [CrossRef]
- Boulghobra, D.; Grillet, P.-E.; Laguerre, M.; Tenon, M.; Fauconnier, J.; Fança-Berthon, P.; Reboul, C.; Cazorla, O. Sinapine, but not sinapic acid, counteracts mitochondrial oxidative stress in cardiomyocytes. Redox Biol. 2020, 34, 101554. [Google Scholar] [CrossRef]
- Gallo, S.; Spilinga, M.; Albano, R.; Ferrauto, G.; Di Gregorio, E.; Casanova, E.; Balmativola, D.; Bonzano, A.; Boccaccio, C.; Sapino, A.; et al. Activation of the MET receptor attenuates doxorubicin-induced cardiotoxicity in vivo and in vitro. Br. J. Pharmacol. 2020, 177, 3107–3122. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Hernández-Jiménez, E.; Avendaño-Ortiz, J.; Toledano, V.; Varela-Serrano, A.; Fernández-Navarro, I.; Casitas, R.; Carpio, C.; Aguirre, L.A.; García-Río, F.; et al. Obstructive Sleep Apnea Monocytes Exhibit High Levels of Vascular Endothelial Growth Factor Secretion, Augmenting Tumor Progression. Mediators Inflamm. 2018, 2018, 7373921. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Mao, C.; Zhang, C.; Zhang, M.; Gong, J.; Wang, X. Salvianolic Acid B Inhibits Ferroptosis and Apoptosis during Myocardial Ischemia/Reperfusion Injury via Decreasing the Ubiquitin-Proteasome Degradation of GPX4 and the ROS-JNK/MAPK Pathways. Molecules 2023, 28, 4117. https://doi.org/10.3390/molecules28104117
Xu X, Mao C, Zhang C, Zhang M, Gong J, Wang X. Salvianolic Acid B Inhibits Ferroptosis and Apoptosis during Myocardial Ischemia/Reperfusion Injury via Decreasing the Ubiquitin-Proteasome Degradation of GPX4 and the ROS-JNK/MAPK Pathways. Molecules. 2023; 28(10):4117. https://doi.org/10.3390/molecules28104117
Chicago/Turabian StyleXu, Xiaojin, Chenhan Mao, Chengbo Zhang, Meng Zhang, Jianbin Gong, and Xindong Wang. 2023. "Salvianolic Acid B Inhibits Ferroptosis and Apoptosis during Myocardial Ischemia/Reperfusion Injury via Decreasing the Ubiquitin-Proteasome Degradation of GPX4 and the ROS-JNK/MAPK Pathways" Molecules 28, no. 10: 4117. https://doi.org/10.3390/molecules28104117
APA StyleXu, X., Mao, C., Zhang, C., Zhang, M., Gong, J., & Wang, X. (2023). Salvianolic Acid B Inhibits Ferroptosis and Apoptosis during Myocardial Ischemia/Reperfusion Injury via Decreasing the Ubiquitin-Proteasome Degradation of GPX4 and the ROS-JNK/MAPK Pathways. Molecules, 28(10), 4117. https://doi.org/10.3390/molecules28104117