2. Results and Discussion
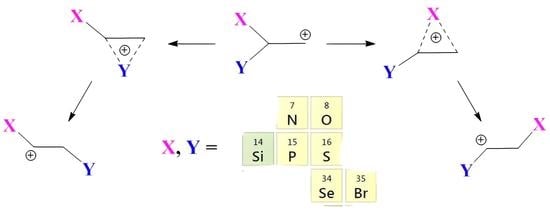
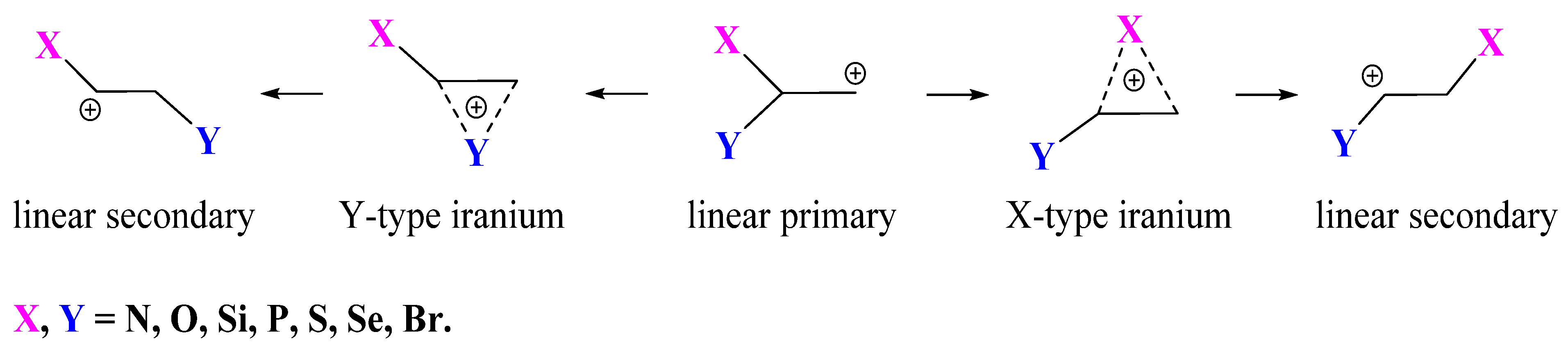
In the present paper, the problem of anchimeric assistance is studied from a new perspective, namely, by investigation of the relative ability of different groups attached to the same carbon to stabilize the adjacent primary carbocationic center either by anchimeric assistance via the formation of the X- or Y-type iranium cation or via full migration to this carbon atom, forming linear secondary cations, as shown in
Figure 1.
For this, high-level theoretical calculations were performed for all 21 combinations of substituents X and Y, including both global and local minima corresponding to the iranium and linear cations presented in
Figure 1. In order to avoid possible isomerization via hydrogen migration from the XH or YH group, all heteroatoms were protected by methyl groups (OMe, SMe, SeMe, NMe
2, PMe
2, and SiMe
3), which allowed for the analysis of “pure” anchimeric assistance or migration of X or Y. Among the issues addressed in this study are the dependence of the type of the migrating group and the degree of its shift towards the carbocationic center on the nature of the migrating and remaining groups X and Y, the reasons for the formation of local (kinetically controlled) and global (thermodynamically controlled) products on the potential energy surface (PES), and the relative effectiveness of anchimeric assistance versus stabilization by direct conjugation with the lone pair of a heteroatom.
The main factors affecting the degree of anchimeric assistance are the covalent radius of the interacting group and its polarizability. The larger the covalent radius, the longer the distance at which the migrating group interacts effectively with the carbocationic center. The larger the polarizability, the easier it is to induce a dipole, and hence the larger the binding energy. The ability of a specific group to stabilize the existing or incipient charge on the adjacent carbon atom is usually estimated by comparing the rates of the reactions of the parent and substituted compounds. By contrast, in the present work, the relative abilities of different groups to stabilize the positive charge are estimated from the relative stabilities of the structures formed by full or incomplete transfer of one of two substituents in the same molecule. This raises four main questions that are answered in this paper: (i) that of which group migrates spontaneously during geometry optimization; (ii) that of which factors are responsible for the migration of a particular group; (iii) that of which stabilization is preferable: anchimeric assistance with the partially shifted group X or direct conjugation with the lone pair on the non-migrating group Y; and (iv) that of the structure of the formed cations (covalently bound iranium ions, anchimerically assisted ions, or π-complexes with practically intact double bonds). The primary cations in
Figure 1 are inherently unstable and in all cases are stabilized by group X or Y to either iranium or secondary cations. Below, the transformations of primary cations with all possible combinations of groups X and Y will be considered.
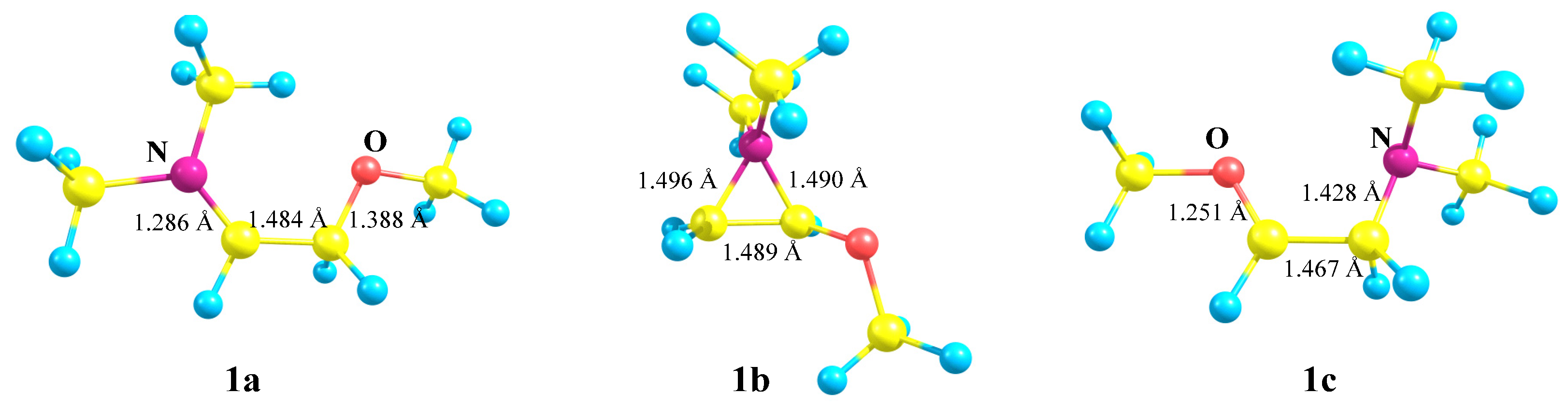
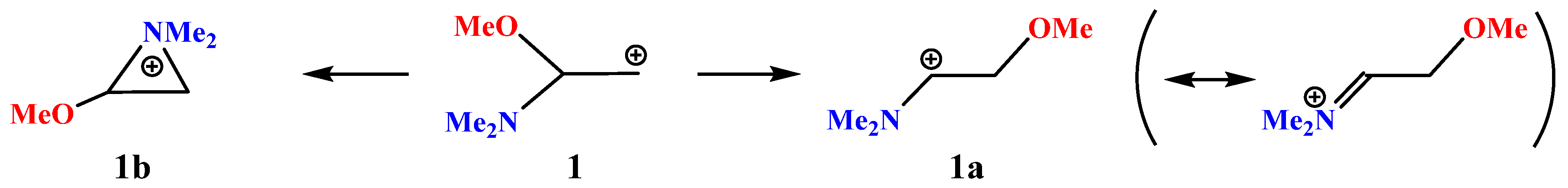
X = NMe2, Y = OMe (1). The neighboring amino group participation may result in the formation of the rearranged products via the formation of the anchimerically assisted aziridinium ion. For example, as applied to our case, the amino group wins the competition with iodine, as proved by the intramolecular ring opening of the preliminarily formed halogen-assisted cation [
37]. The result of geometry optimization of cation
1 depends on the relative orientations of the Me–N–Me and N–C–CH
2 planes. If they are perpendicular, that is, if the
p-orbital and the nitrogen lone pair are eclipsed (lie in the same plane), the aziranium cation
1b is formed; for all other conformations, full migration of the methoxy group is observed, resulting in the iminium cation
1a, as shown in
Scheme 1.
No isomeric oxiranium cation analogous to
1b could be located. However, linear oxenium cation
1c was found to be at a local minimum on the PES (
Figure 2). The relative energies increased in the order
1a (0) <
1b (10.0) <
1c (28.5 kcal/mol) (see
Supplementary Materials).
The geometries of the located minima are given in
Figure 2. Strong N and O stabilization of the secondary cations
1a and
1c is evidenced by ~0.14 Å shortening of the N–C
+ and O–C
+ bonds. In both
1a and
1c, the C–C bond length is close to that of the ordinary bond, suggesting their existence as iminium (
1a) or oxenium (
1c) cations. The 0.017 Å longer C–C bond in
1a relative to that in
1c is indicative of stronger N versus O stabilization. The lengths of the C–N bonds in
1a and in the almost equilateral triangle of
1b fall in the range of normal ordinary C–N bonds.
Therefore, the relative orientation of the C–N bond and the nitrogen lone pair with respect to the empty p-orbital on the carbocation center in 1 has a strong impact on the migration ability of the NMe2 group.
The Mulliken charge distribution in the aziranium cation 1b is strongly polarized: qN = +0.621, qCH2 = −0.683, and qCH = +0.101, suggesting the covalent nature of the N–CH bond and the electrovalent nature of the N–CH2 bond.
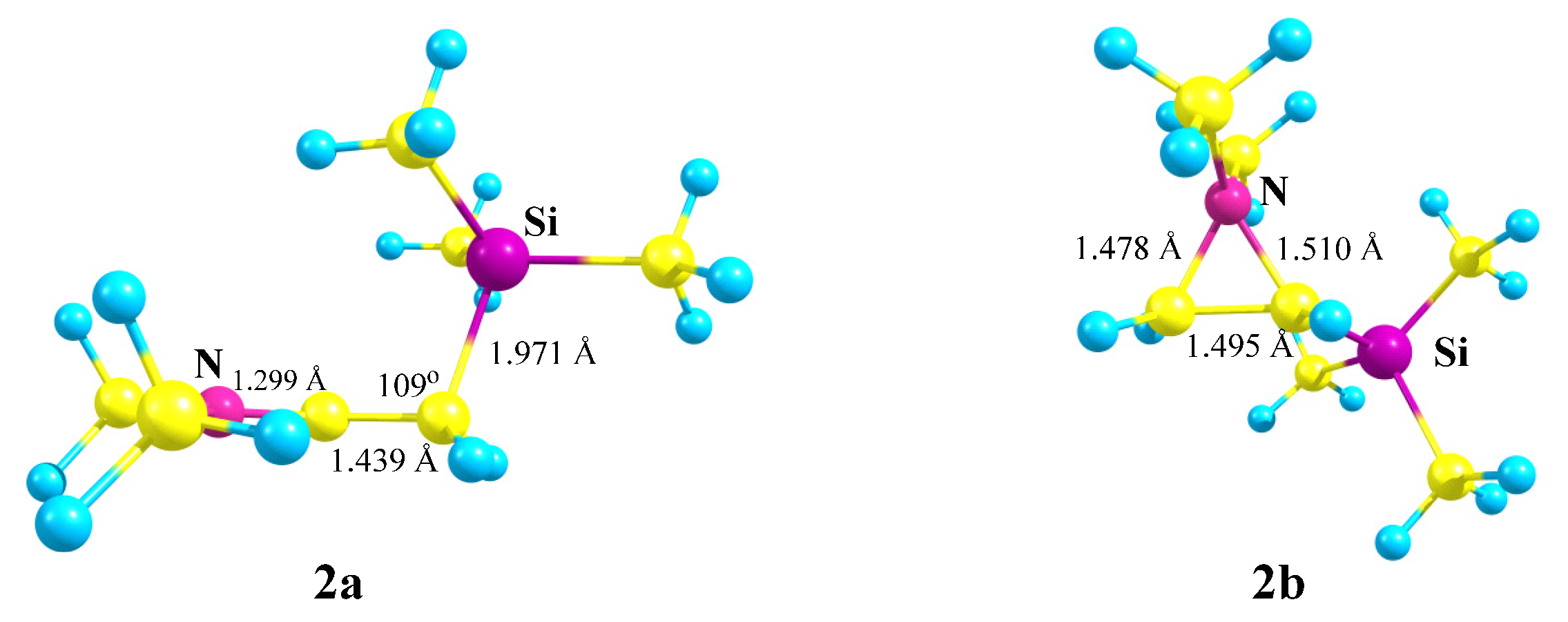
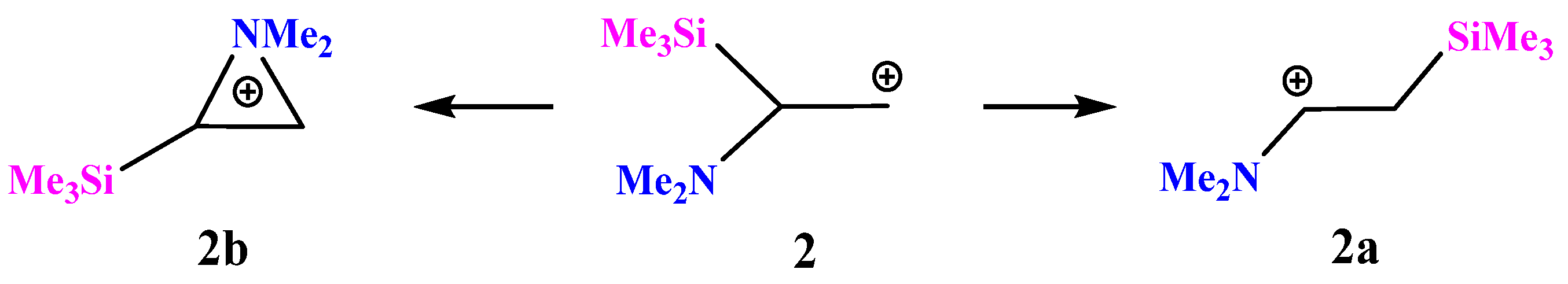
X = NMe2, Y = SiMe3 (2). For the same reasons, optimization of cation
2 gives rise to the iminium cation
2a via the migration of the Me
3Si group for all initial conformations except the one that has both the Me
2N–C bond and the nitrogen lone pair in the same plane with the cationic
p-orbital. In the latter case, aziranium cation
2b was located on the PES as the local minimum lying 20.8 kcal/mol above the global minimum of
2a,
Scheme 2.
The planar structure of the NC3 fragment and the tetrahedral C–C–Si angle of 109° proves the absence of interaction of the silicon atom with the cationic center in 2a. The aziranium triangle in 2b is close to equilateral.
The aziranium cation 2b is polarized more strongly than its methoxy-substituted analogue 1b due to the electron donor effect of the trimethylsilyl group: qN = +0.706, qCH2 = −0.822, and qCH = −0.315. No siliranium cation was found on the PES.
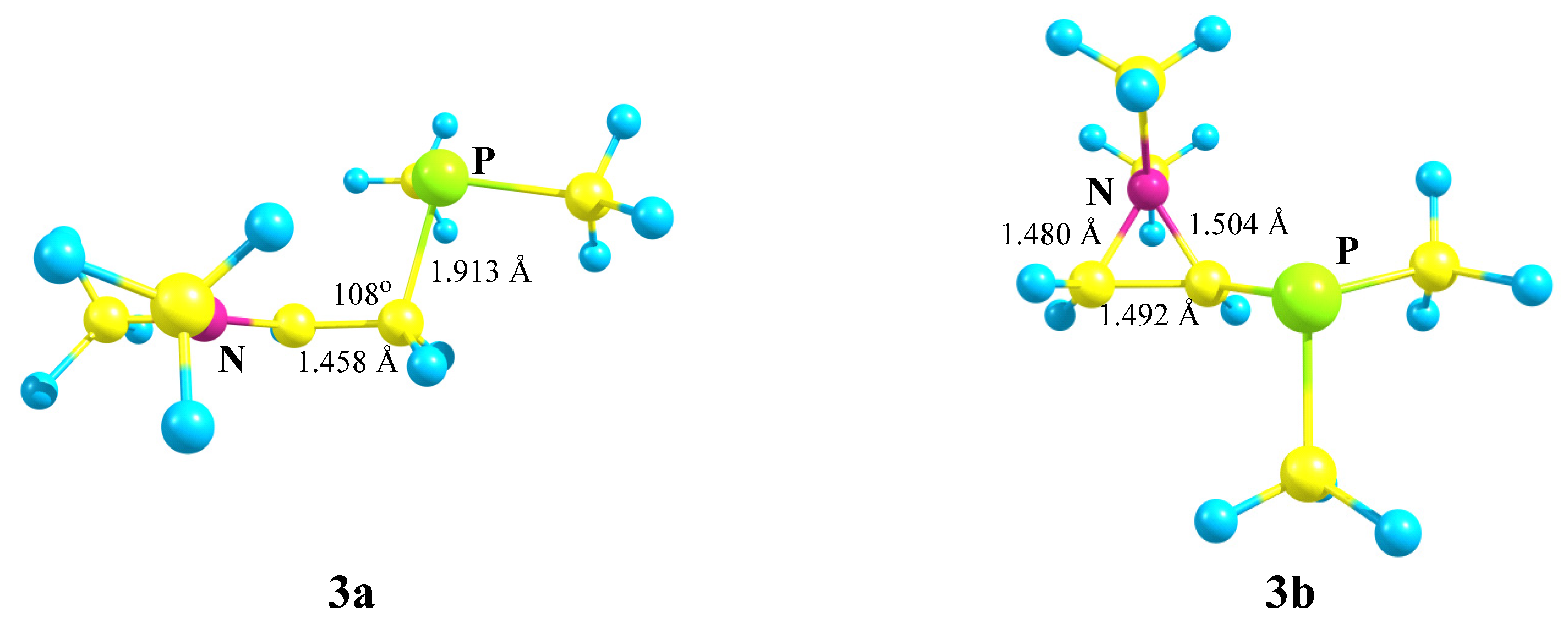
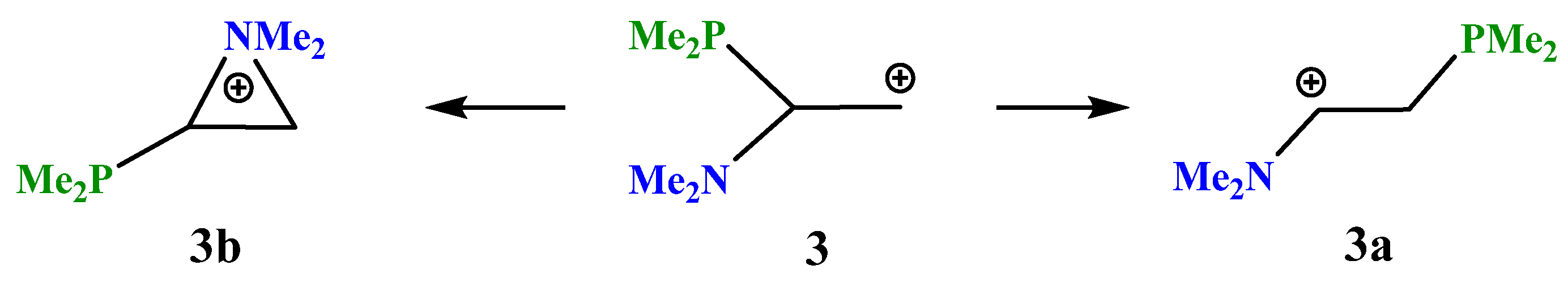
X = NMe2, Y = PMe2 (3). The transformations of cation
3, which has two heteroatoms belonging to the same group of the periodic table (
Scheme 3), are completely similar to those of cation
2, except that the energy difference between the global minimum of the iminium cation
3a and the local minimum of the aziranium cation
3b is notably smaller: 16.1 kcal/mol. Apparently, this is due to the practically equal lengths of all three P–C bonds in
3b (Δ
l~0.01 Å), whereas in its analogue
2b the Si–C bonds with the methyl groups are ~0.06 Å shorter than in the CNC ring carbon atom.
The planar structure of the NC3 fragment and the nearness to the tetrahedral C–C–P angle of 108° proves the iminium structure of cation 3a and the absence of interaction of the phosphorus atom with the cationic center (l(P–C+) = 2.743 Å).
The aziranium cation 3b is polarized even more strongly than its trimethylsilyl-substituted analogue 2b: qN = +0.762, qCH2 = −0.858, and qCH = 0.302. Note that, unlike 2b, the CH carbon in 3b is charged positively, apparently, due to the higher electronegativity of phosphorus compared to silicon. In line with this, the N–C bond between the oppositely charged atoms in 3b (1.480 Å) is shorter than that between the likely charged ones (1.504 Å). No phosphiranium cation was found on the PES.
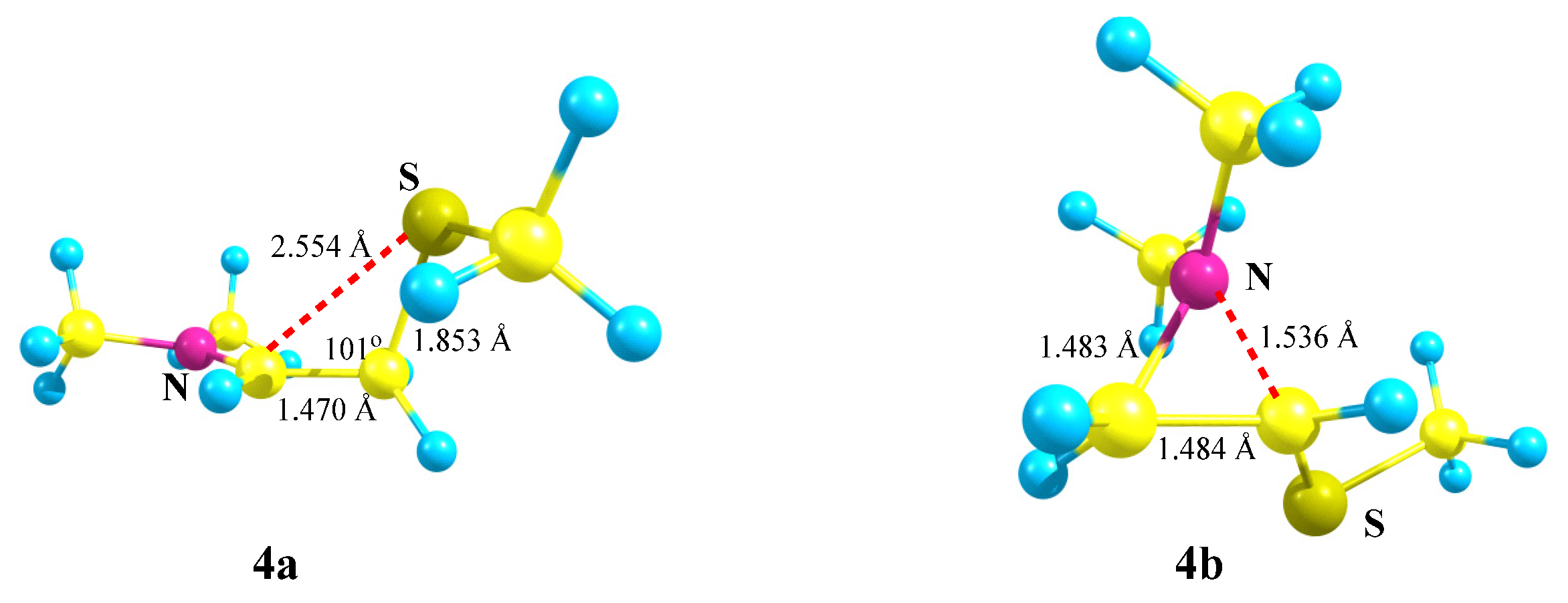
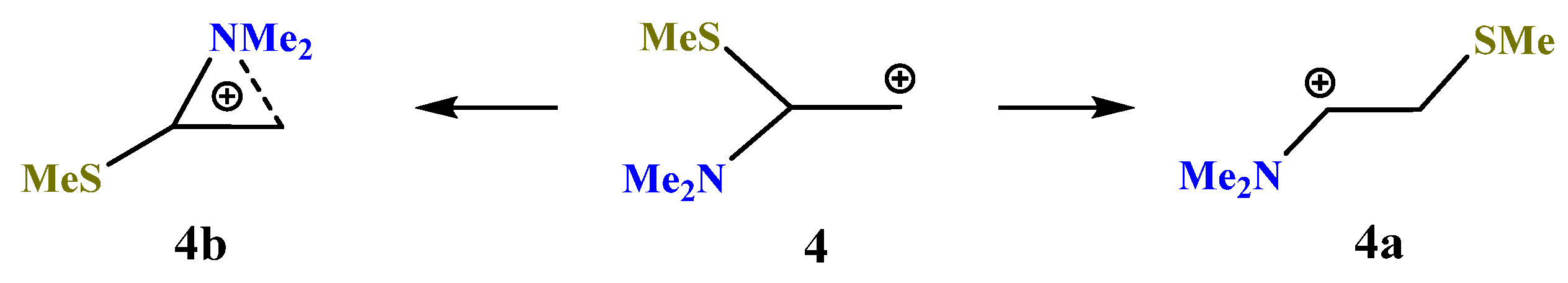
X = NMe2, Y = SMe (4). Moving further along the third row of the periodic table (Si → P → S) results in a further decrease in the energy gap between the global
4a (linear iminium cation) and local
4b (aziranium cation) minima on the PES (
Scheme 4). As above, the primary cation
4 is optimized to the aziranium cation
4b only for the starting conformation of
4 with the
p-orbital, C–N bond and the lone pair on nitrogen lying in one plane. The value of Δ
E = 12.1 kcal/mol is minimal in the series of all Me
2N-containing carbocations (
1)–(
6).
Note the appearance of the signs of anchimeric assistance of the sulfur atom in
4a, as evidenced by a smaller C–C–S angle of 101° in
4a as compared to the tetrahedral angles of C–C–Si and C–C–P in
2a and
3a (
Figure 3,
Figure 4 and
Figure 5). The non-covalent C–S distance in
4a is 2.554 Å, which is larger than the covalent bond length but much less than the sum of the van der Waals radii (3.5 Å) [
38]. Note, also, the less symmetrical structure of the CNC triangle in
4b, in particular, the larger difference in the angles and, especially, the C–N distances in the ring (0.053 Å).
The Mulliken charge on nitrogen and the polarization of the aziranium cation 4b (qN = +0.687, qCH2 = −0.769, and qCH = +0.121) are somewhat higher than those in its oxygen analogue 1b but lower than those in 2b and 3b.
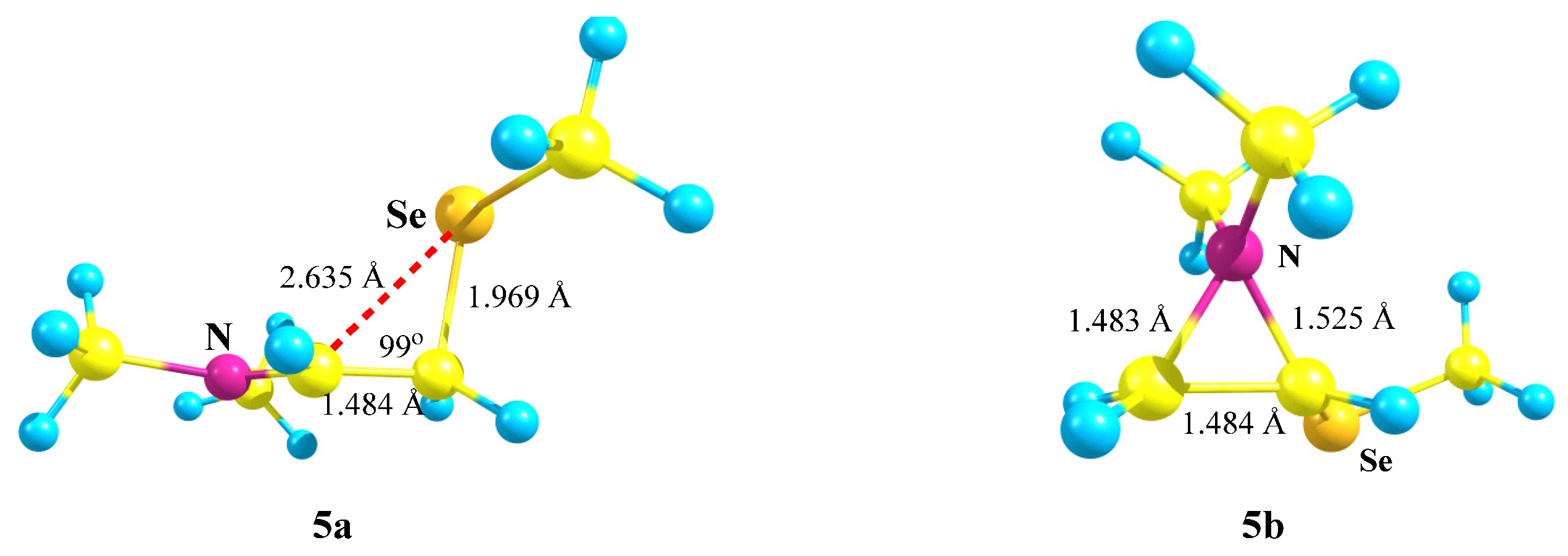
X = NMe2, Y = SeMe (5). Introduction of a heavier chalcogen, selenium, in place of sulfur, has a small effect on the structure and energetics of the corresponding carbocations (
Scheme 5,
Figure 6). The comparison of the covalent and non-covalent distances and bond angles allows the conclusion that the anchimeric assistance in the aziranium cation
5b must be somewhat stronger than in its analogue
4b. However, the calculated energy difference of 15.0 kcal/mol between
5a and
5b is slightly larger than that between
4a and
4b (12.1 kcal/mol). Apparently, this is due to the more diffuse electron density on the selenium atom that weakens the resonance stabilization of the cationic center by selenium with respect to the sulfur atom.
A weaker resonance stabilization by the selenium atom can also be clearly seen by comparison of the atomic charges in the aziranium cations 4b (vide supra) and 5b (qN = +0.715, qCH2 = −0.764, and qCH = −0.220). A larger contraction of the S–CH with respect to the S–Me bond in 4b (0.064 Å) compared to that of the Se–CH with respect to the Se–Me bond in 5b (0.053 Å) is fully consistent with the weaker resonance of the SeMe group and explains the negative charge qCH in 5b and its positive value in 4b.
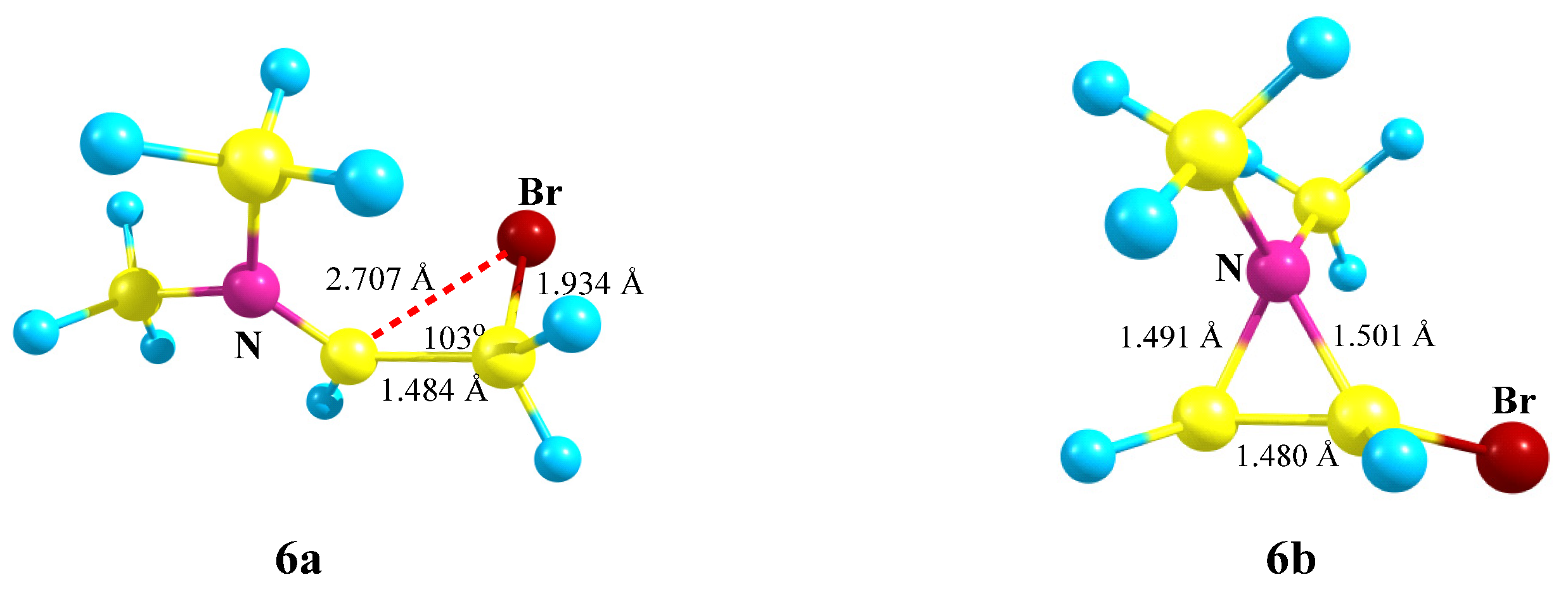
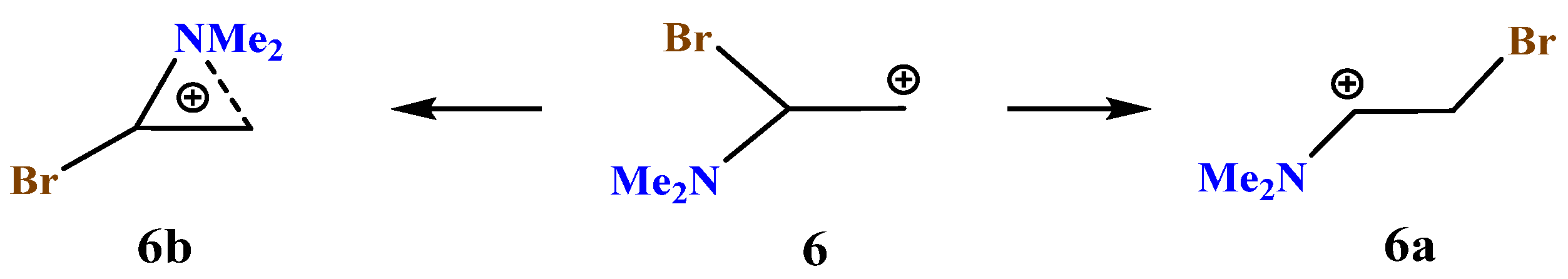
X = NMe2, Y = Br (6). The carbocation
6 potential energy surface profile and the structure of the global and local minima on it (
Scheme 6,
Figure 7) are very close to those of carbocation
5.
The iminium ion 6a lies lower in energy than the aziranium ion 6b by 15.7 kcal/mol, which is very close to the energy gap for their closest analogues 5a and 5b. The charge on the nitrogen atom is the lowest among the above-considered aziranium cations (qN = +0.500). The N–CH2 bond of 1.491 Å is slightly shorter than the N–CHBr bond (1.501 Å), in agreement with a higher electron density for the CH2 carbon (qCH2 = −0.692, qCH = −0.260).
To summarize the results obtained for the amino-containing carbocations (1)–(6), one can conclude that the Me2N group migrates spontaneously towards the carbocationic center to form the corresponding aziranium ions only in the conformers with the p-orbital, C–N bond and the lone pair on the nitrogen lying in one plane. In all other cases, the second heteroatom is shifted towards the carbocationic center to form the corresponding iminium ions [Me2N=CH–CH2Y]+ without (Y = O, SiMe3, PMe2; tetrahedral CCY angle) or with very slight (Y = SMe, SeMe, Br; CCY angle from 99 to 103°) anchimeric assistance. The local minima lie 12–21 kcal/mol above the corresponding global minima.
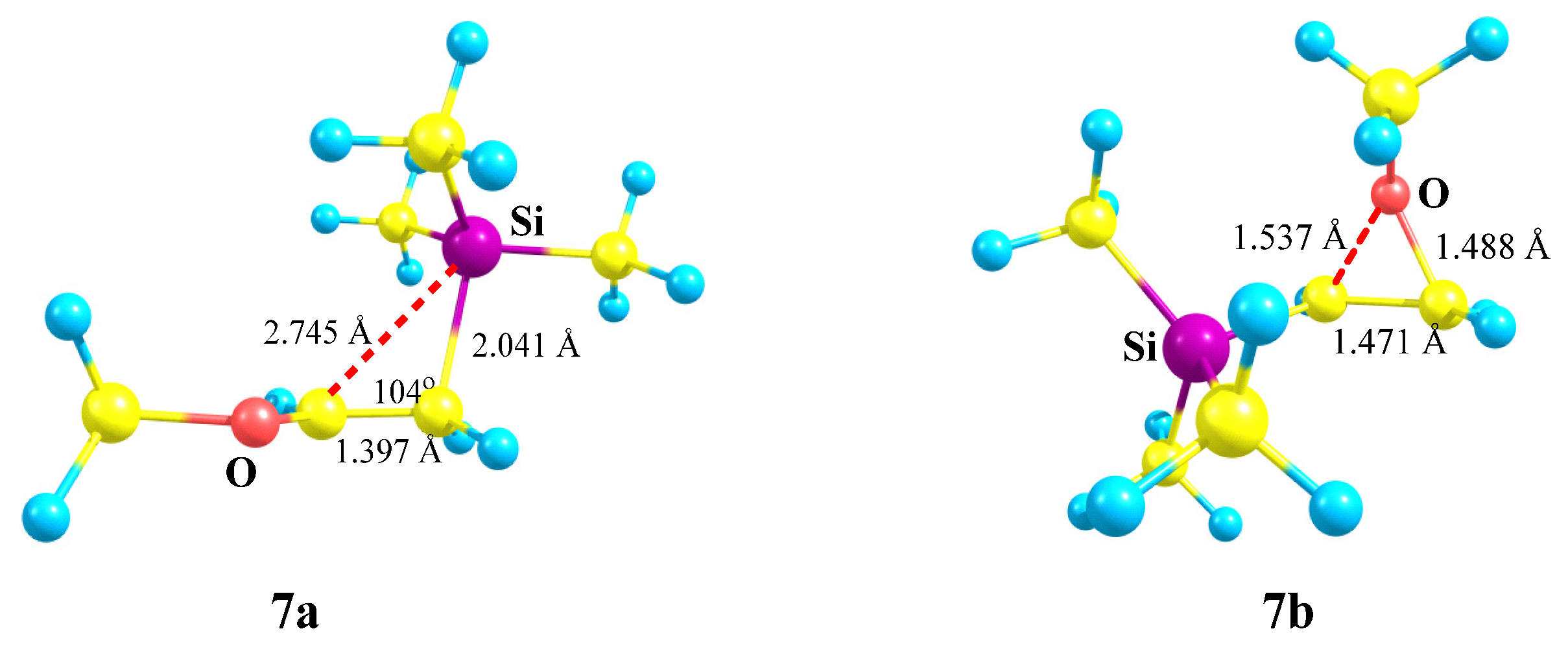
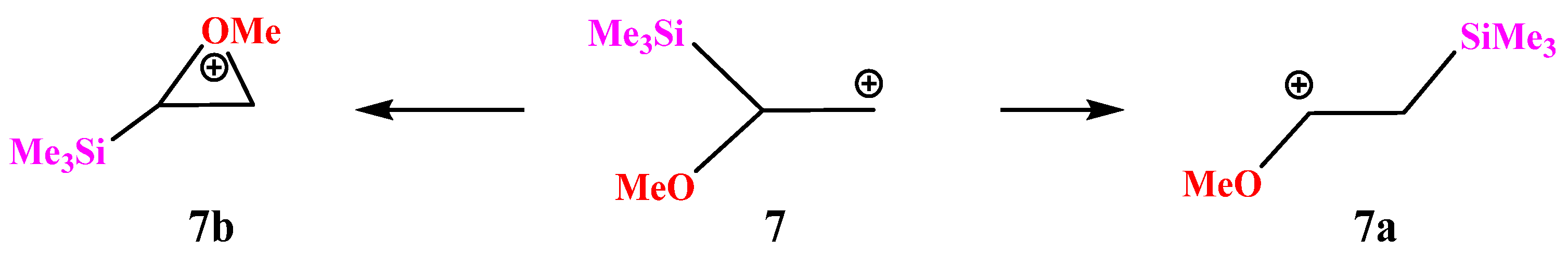
X = OMe, Y = SiMe3 (7). Let us turn to combinations of oxygen with other heteroatoms. With the most electropositive heteroatom, silicon (χ = 1.74), almost full migration of the trimethylsilyl group occurs during geometry optimization, leading to cation
7a (
Scheme 7) for almost all conformations of
7 except the one with the eclipsed MeO–C bond and the
p-orbital. In the latter case, the oxiranium ion
7b is formed as a local minimum on the PES.
The C–C–Si angle in
7a is slightly less than the tetrahedral angle (104°,
Figure 8), and the non-covalent distance C⋯Si is large (2.745 Å) but still less than the sum of the vdW radii (3.8 Å). The C–C bond length in
7a of 1.397 Å is in between those of the double and ordinary bonds, but 0.04 Å shorter than in the nitrogen analogue
2a (1.439 Å,
Figure 3), indicating a weaker conjugation of the OMe versus the NMe
2 group. In the oxiranium ion
7b, the C–C distance is much longer and is close to that for the ordinary bond, indicating strong anchimeric assistance by oxygen. However, ion
7b corresponds to a local minimum on the PES lying as much as 35.2 kcal/mol above the global minimum
7a in energy. This is the maximum difference between the two minima on the PESs of all the studied structures. The much larger value of Δ
E for the
7a/
7b pair than for the
2a/
2b pair could be due to the lower stability of the oxiranium
7b relative to the aziranium ion
2b. Geometrically, it is represented by a slightly shorter C–C bond in
7b as compared with that in
2b (1.471 versus 1.495 Å) and a larger sum for the two C–O bonds in
7b as compared with the two C–N bonds in
2b (3.025 versus 2.988 Å,
Figure 3 and
Figure 8). However, these effects, which are themselves moderate, should be further reduced by a stronger conjugation in the iminium
2a than in the oxenium ion
7a. A more probable reason for a large difference (ΔΔ
E = 14.4 kcal/mol) between the global and local minima is that iminium ions
2a and
7a have principally different electronic structures. While in the aziranium ion
2b the positive charge is localized on the Me
2N group (mostly on nitrogen), in the oxiranium ion
7b it is concentrated on the carbon fragment CHCH
2 (summed with hydrogens), the OMe group bearing negative charge (−0.063 on OMe, −0.409 on oxygen).
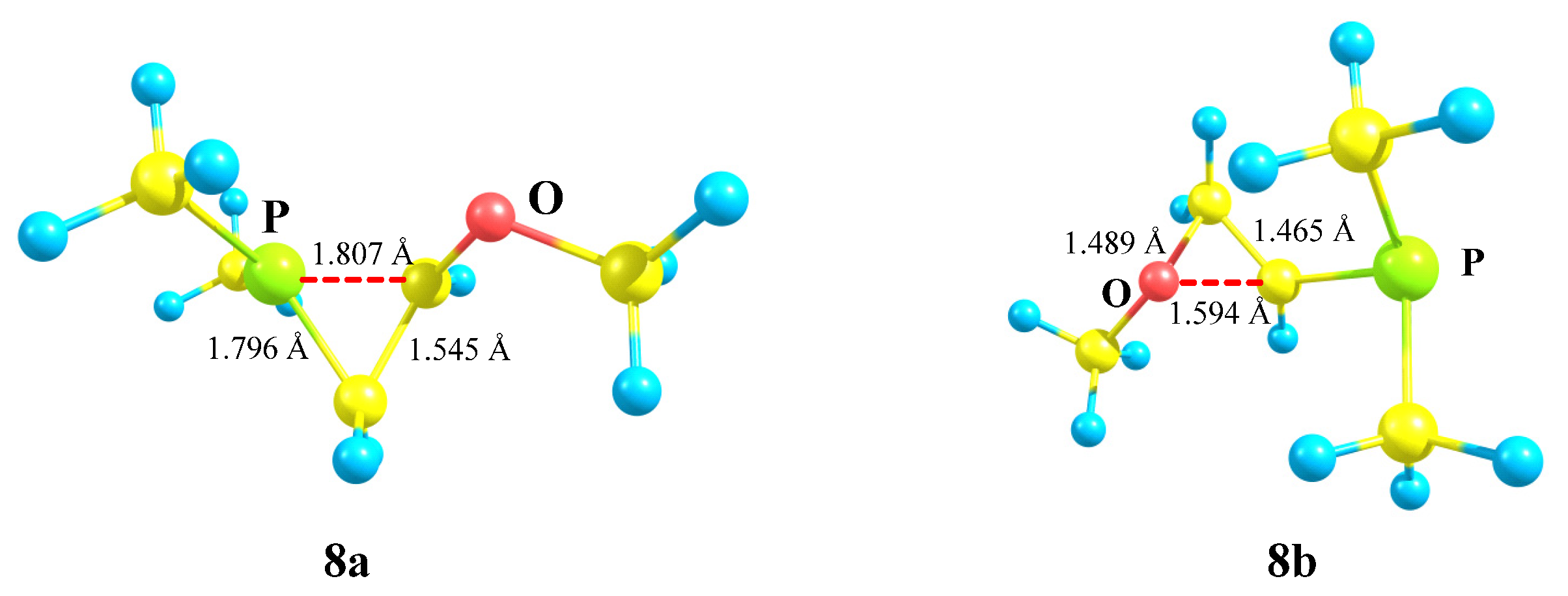
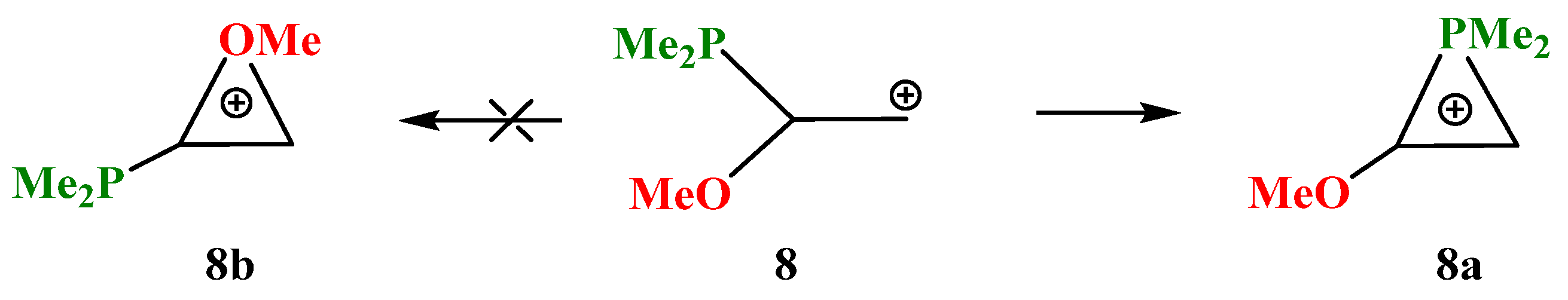
X = OMe, Y = PMe2 (8). The transition from silicon to its closest neighbor, phosphorus, changes the situation dramatically. Even in the most favorable migration conformation for the OMe group in carbocation
8, only the dimethylphosphino group is shifted towards the cationic center and stops at the formation of the anchimerically stabilized phosphiranium cation
8a (
Scheme 8).
The isomeric oxiranium cation
8b was also located on the PES, but only starting with the “preorganized COC triangle”. Note that, unlike all the above-considered structures, no minima on the PES corresponding to open linear cations could be found. The phosphiranium
8a and oxiranium
8b cations look similar, but there is one principal geometrical difference: the C–C bond lengths in the almost isosceles triangle in
8a coincide with that of normal ordinary C–C bonds (1.545 Å), whereas in the COC ring in
8b (1.465 Å) the lengths are intermediate between those of the C–C and C=C bonds (
Figure 9). The charge density distribution is also radically different. The charge on phosphorus
qP in
8a is equal to +1.041, which allows it to be considered as a true phosphiranium ion, whereas in
8b it is −0.399, the largest positive charge of +0.929 (summed with hydrogens) being located on the ring carbon bonded anchimerically with oxygen (
qC = 0.287). Therefore, electronically, cation
8b is similar to
7b, except for the fact that the charges on the ring carbons in the latter are reduced due to the electron-donating effect of the trimethylsilyl group. For the same reasons as discussed for
7b, the oxiranium cation
8b lies high above (28.4 kcal/mol) the phosphiranum cation
8a.
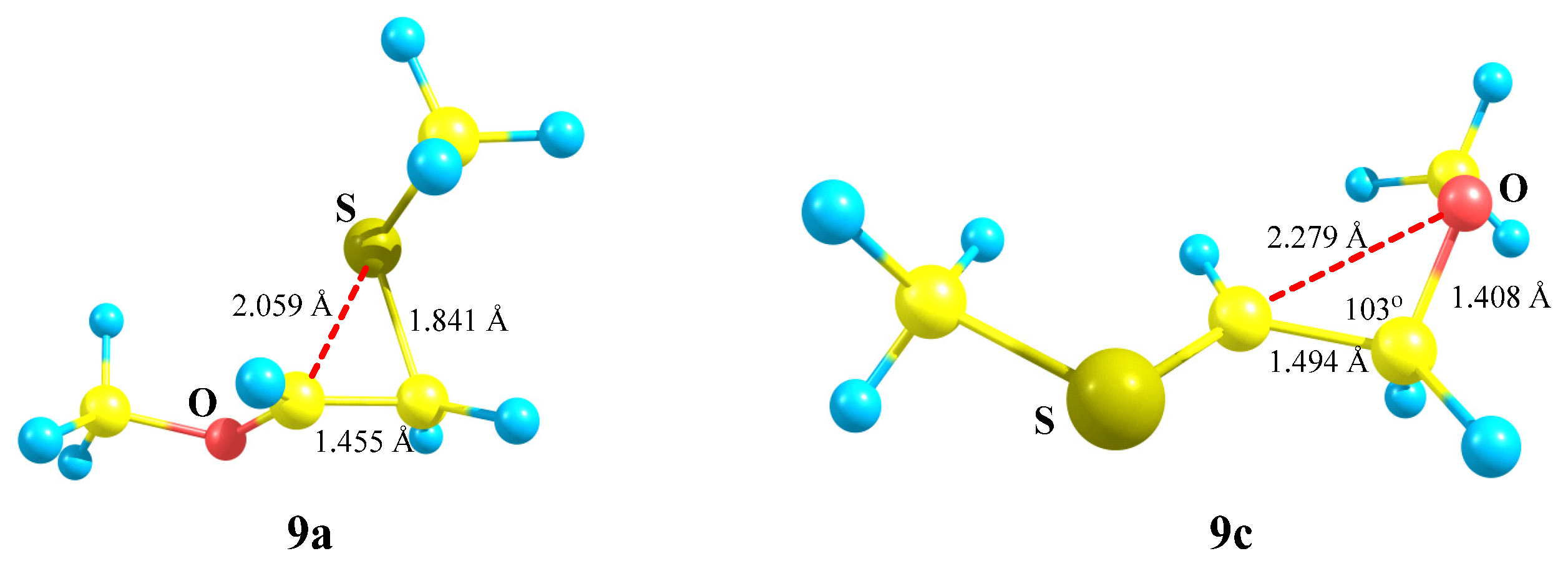
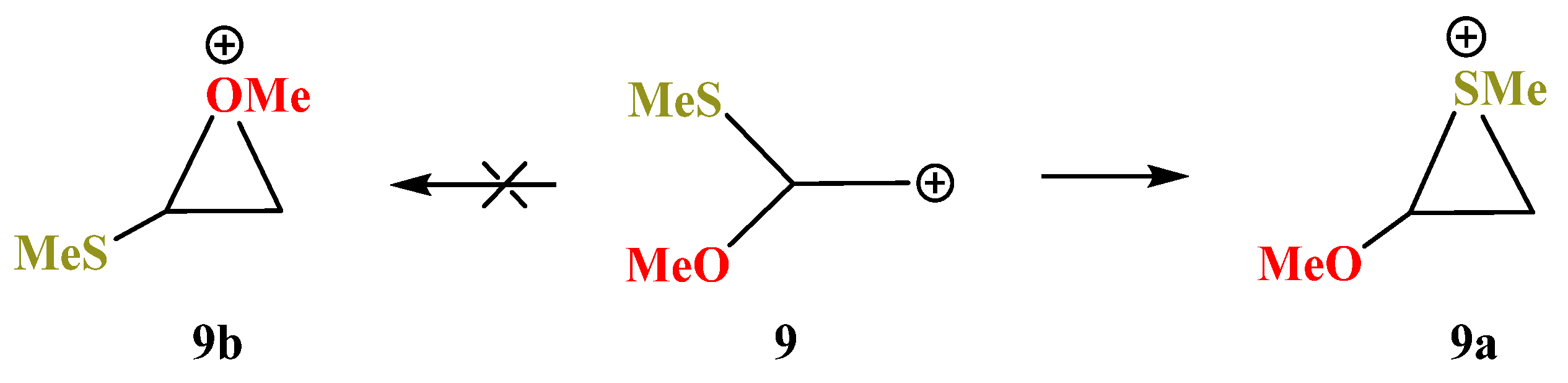
X = OMe, Y = SMe (9). No oxiranium cation
9b could be localized on the PES of cation
9. Even in the conformation with the eclipsed MeO–C bond and
p-orbital, the MeS group shifts towards the cationic center to form the anchimerically stabilized cation
9a (
Scheme 9), which is the global minimum. Moreover, the “preorganized” oxiranium structure
9b suffers the COC ring opening during optimization, resulting in practically linear S-stabilized cation
9c (
Figure 10), which lies 17.0 kcal/mol above the global minimum
9a.
Thiiranium cation
9a has a slightly asymmetrical ring, suggesting a substantial interaction with the sulfur atom, whereas in
9c the interaction of the cationic center with oxygen is insignificant. It follows from a large non-bonded C⋯O distance of 2.279 Å and the C–C–O angle in
9c being only slightly different from the tetrahedral bond angle (
Figure 10).
Noteworthy is the difference in the atomic charges: in the thiiranium cation 9a, the C–C bond is strongly polarized in the direction from the OCH to the CH2S group, the value of Δq = qCH − qCH2 being 0.716, and there is a zero charge on the sulfur atom. In the less stable isomeric cation 9c, the direction of the C–C bond polarization is the same but the degree of polarization is much smaller (Δq = 0.298), and the sulfur atom is positively charged (qS = 0.085). The charge on oxygen is the same (qO = −0.573).
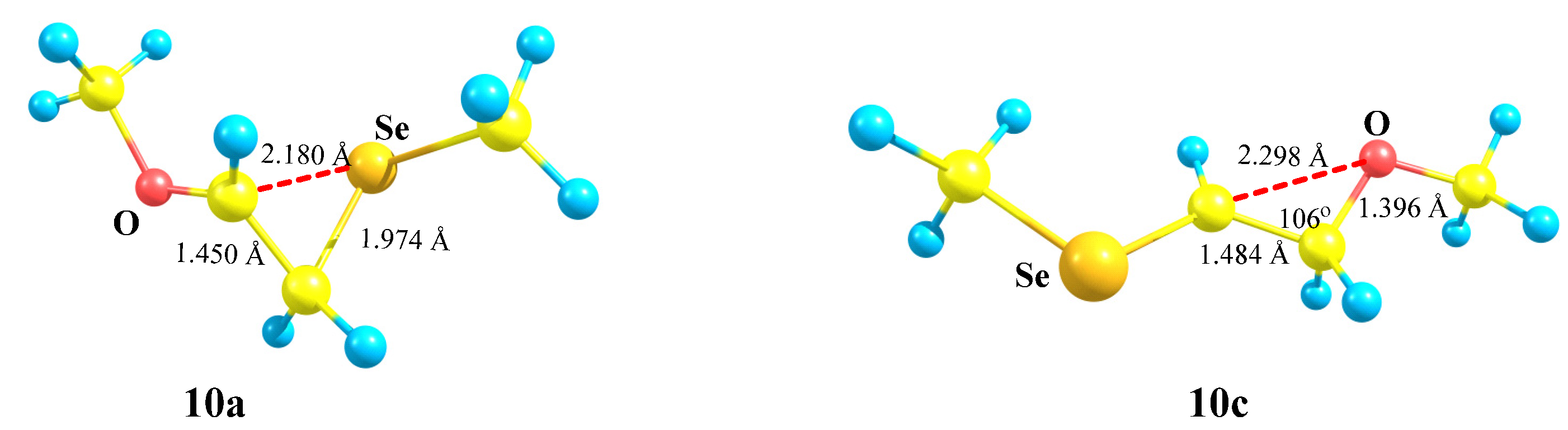
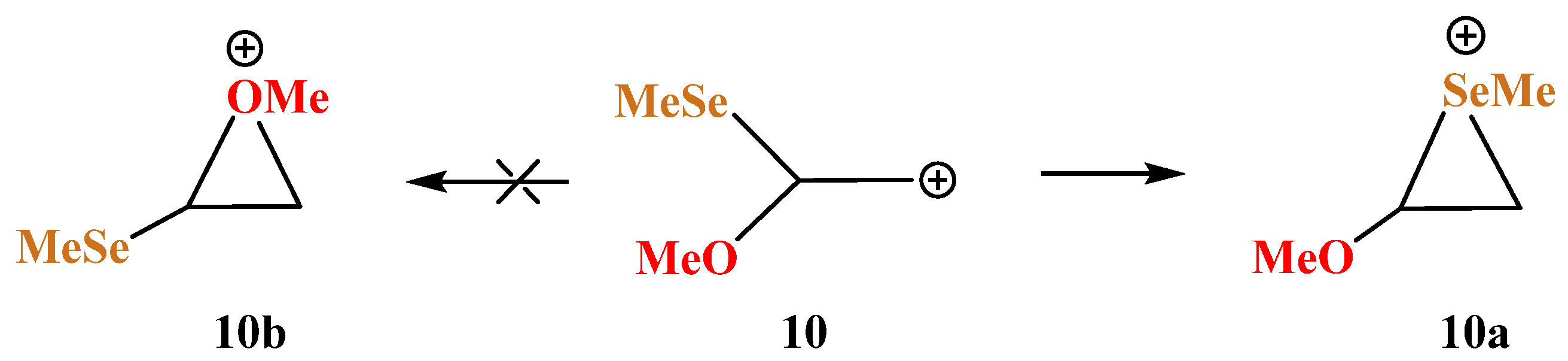
X = OMe, Y = SeMe (10). Selenium, the heavier analogue of sulfur, wins even more in competition with oxygen,
Scheme 10; the seleniranium cation
10a is 22.1 kcal mol more stable than the isomeric
10c. As in the case of
9, neither conformation of cation
10 can be optimized to the oxiranium cation
10b, which, being taken as the starting point, is optimized to the linear cation
10c with the C–C–Se angle even closer to the tetrahedral than in its analogue
9c.
Cation
10a in
Figure 11 is geometrically very similar to
9a in
Figure 10. However, a stronger anchimeric stabilization in
10a becomes evident from the analysis of atomic charges in the two structures. While the electron donation from the sulfur atom estimated as Δ
q(
9c–
9a) is as low as 0.084, the same effect calculated as Δ
q(
10b–
10a) is about three times as large as that, being 0.238, in spite of a slightly larger non-bonded C⋯Se distance and a C–C–Se bond angle. Apparently, this is also due to a more diffuse electron density on the selenium atom, as in the case of cations
5 (vide supra).
The C–C bond in both 10a and 10c is polarized in the same direction as in cation 9, but the values of Δq are substantially larger: 0.850 in 10a and 0.658 in 10c. The electron density on selenium is much lower than in the corresponding sulfur analogues: qSe = 0.192 in 10a versus qS = 0 in 9a and qSe = 0.430 in 10c versus qS = 0.298 in 9c.
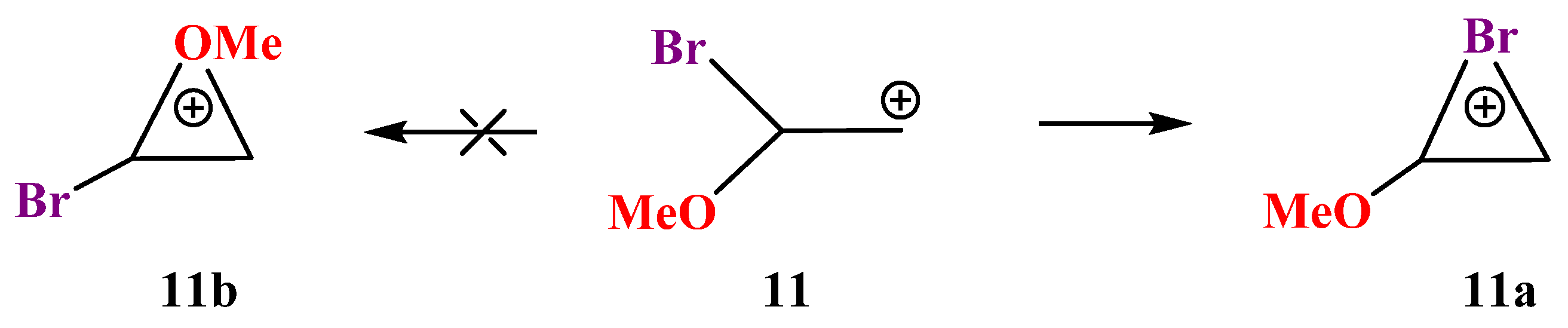
X = OMe, Y = Br (11). Cation
11 behaves similarly to its analogues
9 and
10. The trend of distortion of the most stable Y-stabilized iranium cation and the increase in the energy gap between the global (Y-stabilized) and local (O-stabilized) cations is maintained in going to the heaviest member of the O-containing cations in the series Y = S, Se, Br (
Scheme 11,
Figure 12).
Cation
11a in
Figure 12 is more asymmetric than all its analogues in the series of O-containing cations, except for cation
7a in
Figure 8, which was, apparently, due to the presence of the most electropositive SiMe
3 group in
7a.
Therefore, similar to the amino-containing carbocations (
1)–(
6), no spontaneous migration of the MeO group to the carbocationic center occurs in carbocations (
7)–(
11). However, a partial or complete shift of the MeO group gives rise to the formation of local minima corresponding to the O-anchimerically assisted cations lying 17.0–35.2 kcal/mol above the Y-anchimerically assisted global minima. Moreover, the oxiranium cations are well-known species whose involvement, e.g., in the deoxyfluorination reaction of fluorocarbohydrates, has been shown and reinforced by DFT calculations [
39,
40]. A remarkable feature of the oxygen-containing cations (
7b)–(
11b) is that they can be divided into two types, depending on the C–C–O angle, and have drastically different electronic distributions in the C–C–O fragment as well as different energy gaps between the
a and
c isomers. The former group includes O,S and O,Se cations
9c and
10c, which have C–C–O angles close to tetrahedral angles (103 and 106°) and the lowest values for Δ
E (17.0 and 22.1 kcal/mol). The C–C bonds in
9c and
10c are polarized in the direction from oxygen, as shown in
Figure 13. The second group includes O,Si, O,P, and O,Br cations
7b,
8b, and
11b, which have C–C–O angles of 63, 65, and 60°, respectively, and larger Δ
E values equal to 35.2, 28.4, and 24.3 kcal/mol, respectively. The C–C bonds in them are polarized towards the carbon bearing the covalently bound oxygen atom,
Figure 13.
The oppositely directed polarization in the two types of cations can be explained by substantial binding of the oxygen atom with the second heteroatom-bearing carbon in cations 7b, 8b, and 11b.
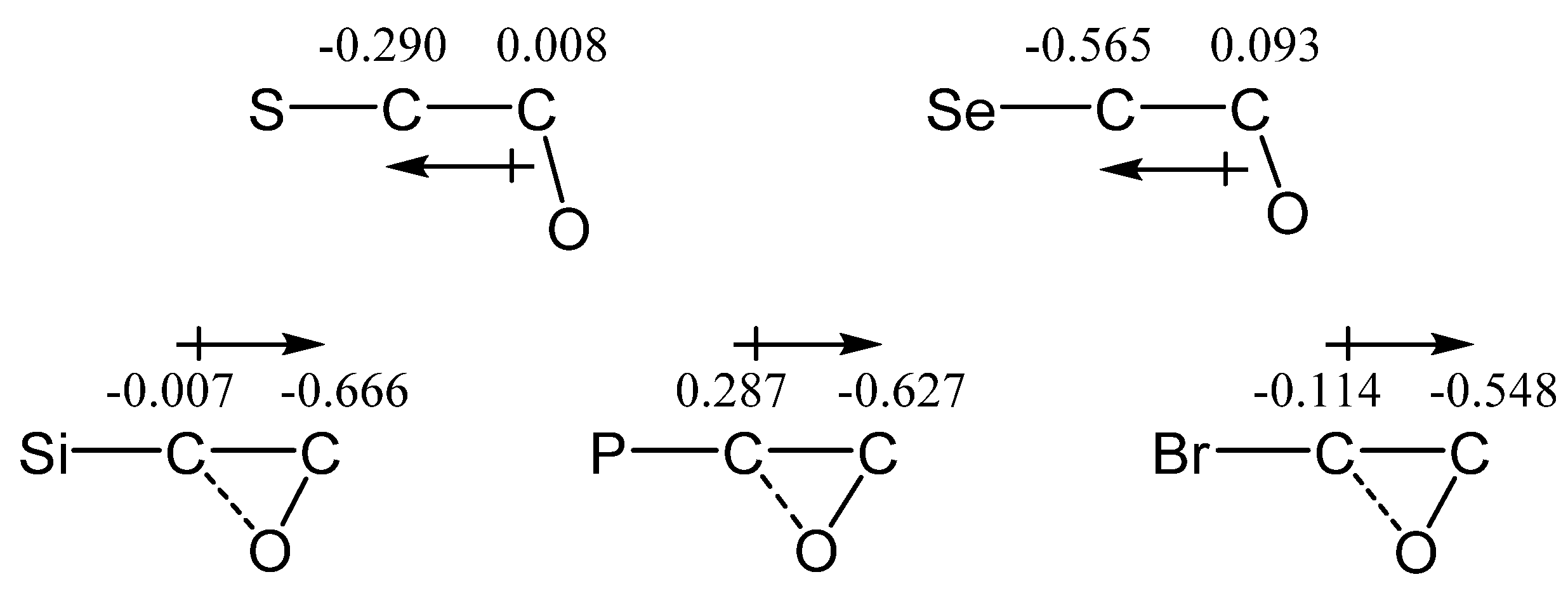
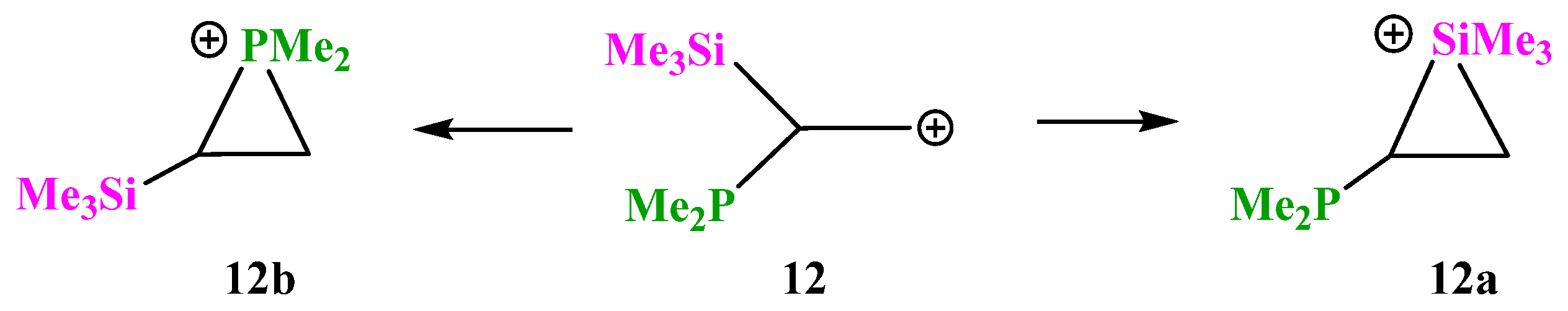
X = SiMe3, Y = PMe2 (12). Cation
12 contains in one molecule a silicon and phosphorus atoms, which are the least electronegative among the studied analogues, both being more electropositive than carbon (χ
Si = 1.8, χ
P = 2.1, and χ
C = 2.5). In the case of
12, as in a number of the above-considered cations containing a group capable of the formation of iranium structures, the P-anchimerically stabilized cation
12b is formed only if the starting conformation has the Me
2P–C and
p-orbital eclipsed. Otherwise, the geometry optimization results in the migration of the more electropositive (and hence less capable of anchimeric assistance) Me
3Si group,
Scheme 12.
However, the phosphiranium cation
12b is the global minimum on the PES lying 20.8 kcal/mol below the local minimum of the siliranium cation
12a, and thus phosphorus wins in the competition with silicon. The C–P and C–C bond lengths in the isosceles CPC triangle in
12b are practically equal to those of ordinary C–P and C–C bonds, proving that it is a true phosphiranium ion (
Figure 14). As for cation
12a, judging from the strongly elongated C–Si bonds and the C–C bond length close to that in alkenes, it is rather a π-complex of the Me
3Si cation with dimethyl(vinyl)phosphine. This is independently proved by a notably larger charge on the Me
3Si group in
12a (
qMe3Si = +0.531) than in
12b (
qMe3Si = +0.281).
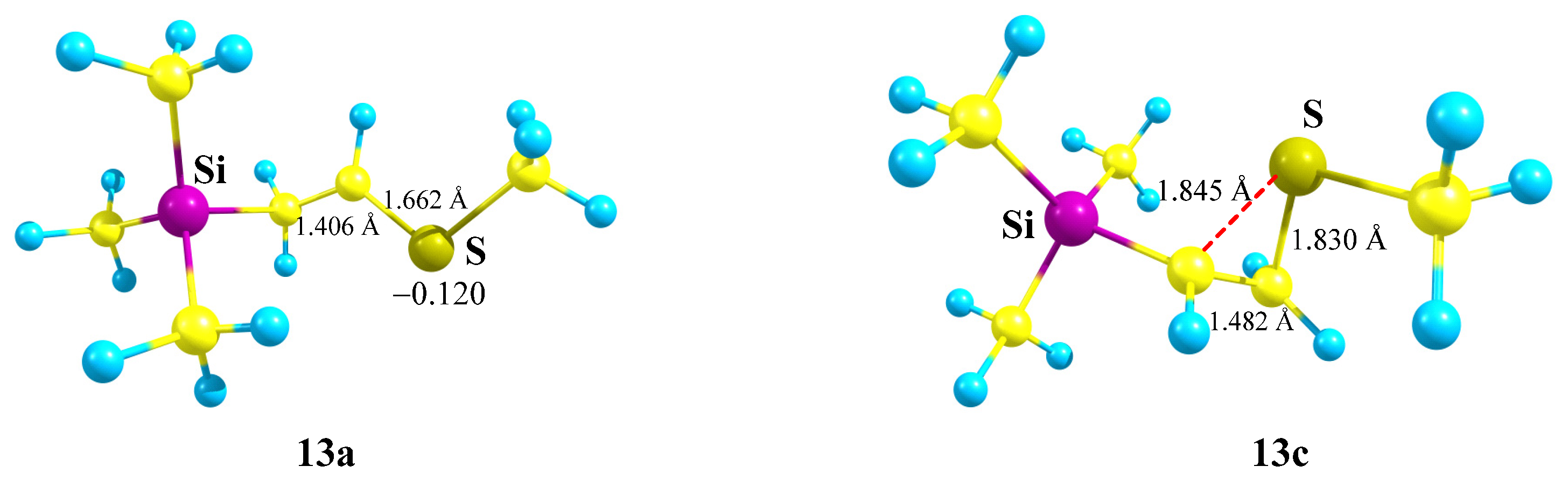
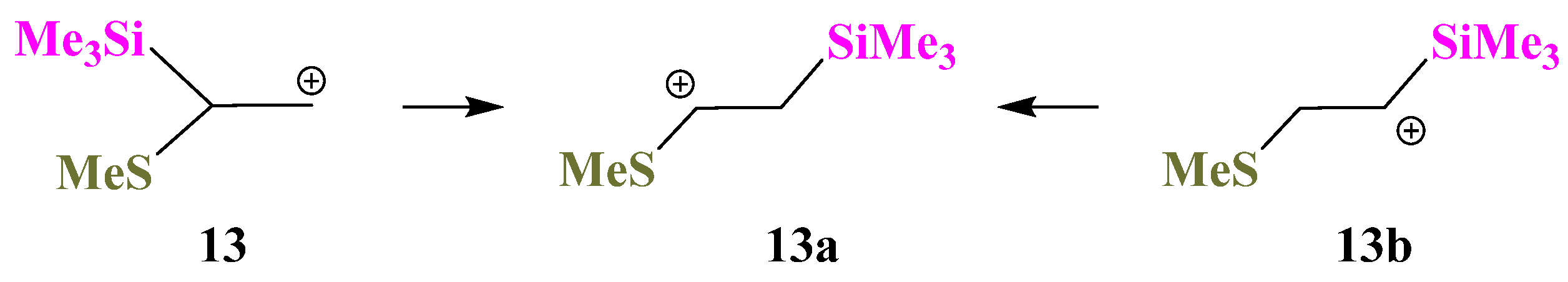
X = SiMe3, Y = Sme (13). Interesting results were obtained for the Si,S-containing cations
13. The same secondary 1-(methylthio)-2-(trimethylsilyl)ethanium cation
13a is formed by geometry optimization of either the primary 2-(methylthio)-2-(trimethylsilyl)ethanium cation
13 or the secondary 2-(methylthio)-1-(trimethylsilyl)ethanium cation
13b,
Scheme 13, and
13a is the global minimum on the PES. The transformation
13 →
13a occurs by migration of the Me
3Si group to the cationic center, whereas the
13b →
13a conversion is the result of the sigmatropic rearrangement with 1,2-hydride shift.
The S-anchimerically stabilized cation
13c is a local minimum on the PES formed only from the most favorable conformation of
13 with the MeS and
p-orbital lying in one plane. Notably, the anchimerically stabilized cation
13c is 2.82 kcal/mol less stable than the linear cation
13a. The structures of both minima are shown in
Figure 15.
The Si–C–C angle in 13a (105°) is close to the tetrahedral angle; the S–C–C angle (124°) is close to trigonal. The structure of cation 13a is close to perpendicular; the Si–C–C–S dihedral angle is 96°. The CCS triangle in 13c is close to isosceles, the lengths of the two C–S bonds evidencing strong anchimeric stabilization by sulfur, which is also proved by the positive charge qS = +0.144 in 13c, in contrast to the negative value of qS = −0.120 in 13a.
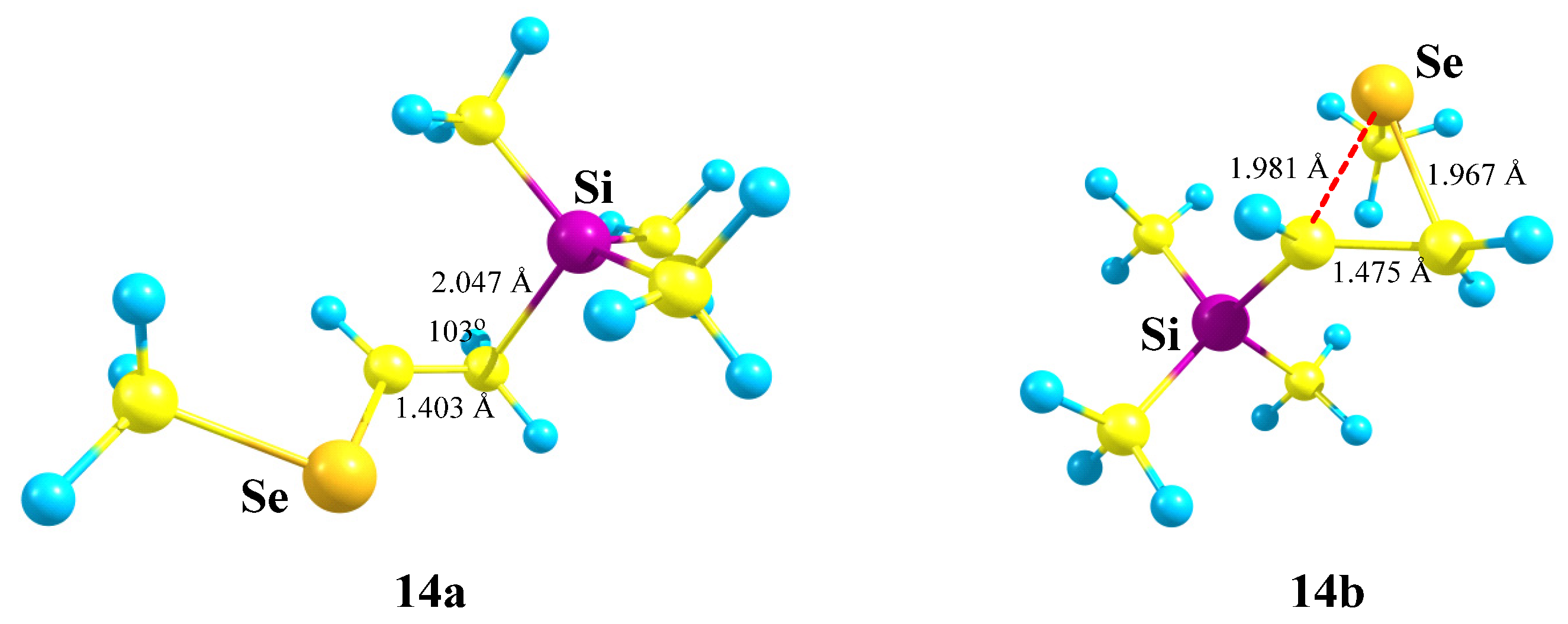
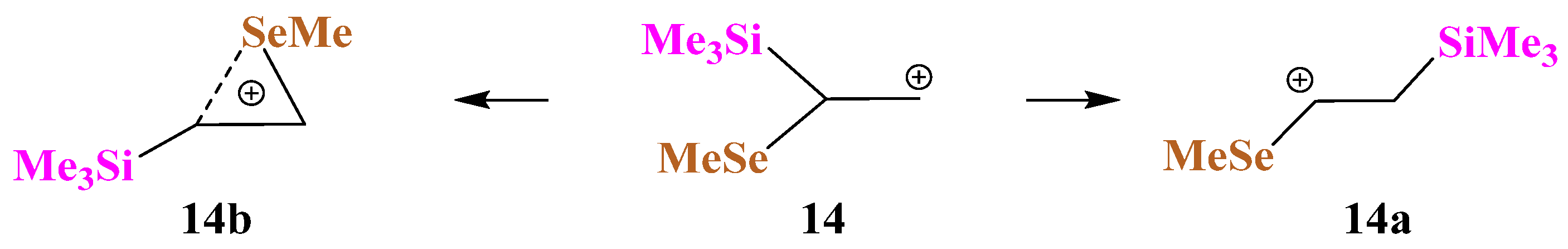
X = SiMe3, Y = SeMe (14). The situation changes when the sulfur atom in cation
13 is replaced by its heavier analogue, selenium, in cation
14. As in the case of Si,S-containing cations, no Si-stabilized cation is formed, even in the form of a π-complex, but the Me
3Si group migrates to the cationic center to form linear cation
14a,
Scheme 14.
As in the case of Si,S-containing cation
13c, the selenium anchimerically stabilized cation
14b was located on the PES by optimization of the conformation of
14 most appropriate for the shift of the MeSe group to the cationic center. Remarkably, the seleniranium cation
14b corresponds to the global minimum, although it lies only 0.9 kcal/mol lower in energy than
14a. The structures of the isomeric cations
14a and
14b are shown in
Figure 16. Note the much shorter C–C bond in
14a compared to that in
14b, indicating a weaker stabilization of the former cationic species, which, probably, is responsible for the energetic preference of the latter.
The inverse energy order of the linear and anchimerically assisted 14 cations with respect to the corresponding Si,S-containing 13 analogues is in agreement with the stronger anchimeric assistance of the selenium atom as compared to that of the sulfur atom.
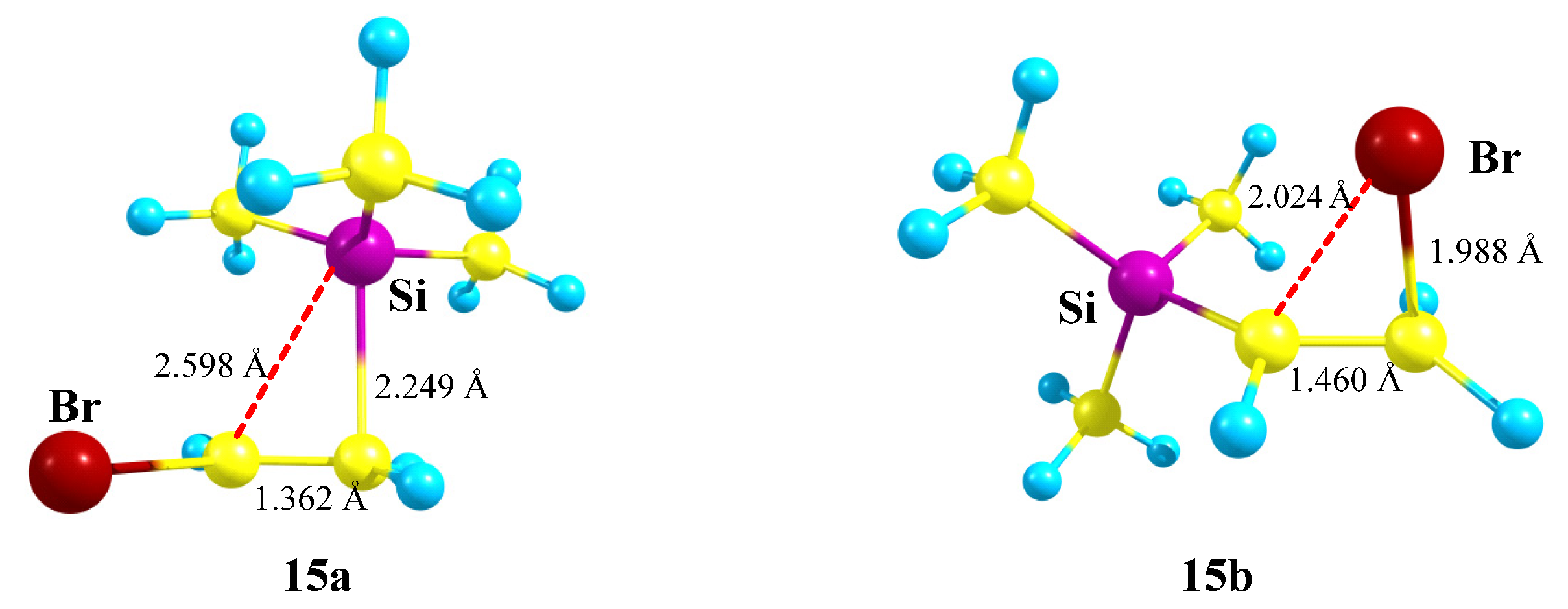
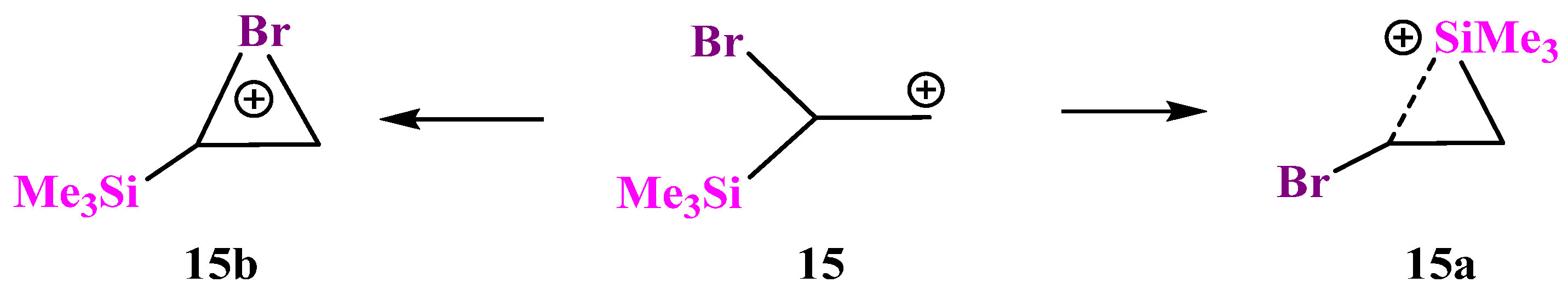
X = SiMe3, Y = Br (15). Similar to the examples above, cation
15 can be stabilized by either the Si atom in the form of a π-complex of the trimethylsilyl cation with the double bond in vinyl bromide or by the Br atom in the form of a bromiranium cation formed from the conformation most appropriate for the bromine shift to the cationic center,
Scheme 15.
The structure of π-complex
15a (
Figure 17) is very similar to that of the Si,P-containing cation
12a (
Figure 14). A drastic difference between the two is that π-complex
15a is 12.2 kcal/mol more stable than the bromiranium cation
15b, whereas π-complex
12a is much less stable than the isomeric phosphiranium ion
12b (vide supra).
The most reasonable explanation for this difference is that stabilization of the phosphiranium ion
12b is much stronger than that of the bromiranium ion
15b, as evidenced by a much longer C–C bond in
12b (1.557 Å) as compared with
15b (1.460 Å). This, in turn, is due to the well-known tendency of phosphorus to form tetracoordinated species, along with the much less pronounced ability of bromine to expand its coordination number to 2, although bromiranium (bromonium) ions have been known for many years [
41,
42,
43,
44] and have been the subject of experimental studies [
45].
Note that the higher stability of cation
15a cannot be assigned to anchimeric assistance by the silicon atom since it has long C⋯Si bonds of 2.25–2.60 Å, a short C–C bond of 1.362 Å, and, as its analogue
12a, is in fact a π-complex of the Me
3Si cation with vinyl bromide. However, stabilization via the formation of a siliranium cation was proposed in the literature [
46]. Thus, its formation via the migration of the Bu
tPh
2Si group in N-tosylazetidines, leading to the corresponding pyrrolidines, was reported [
47]. Notably, the silyl group formation was shown to be non-concerted with the C–N bond cleavage, which is in accordance with our conclusion regarding a π-complex rather than a real Si-anchimerically assisted structure of the “siliranium” ion (vide supra) [
47].
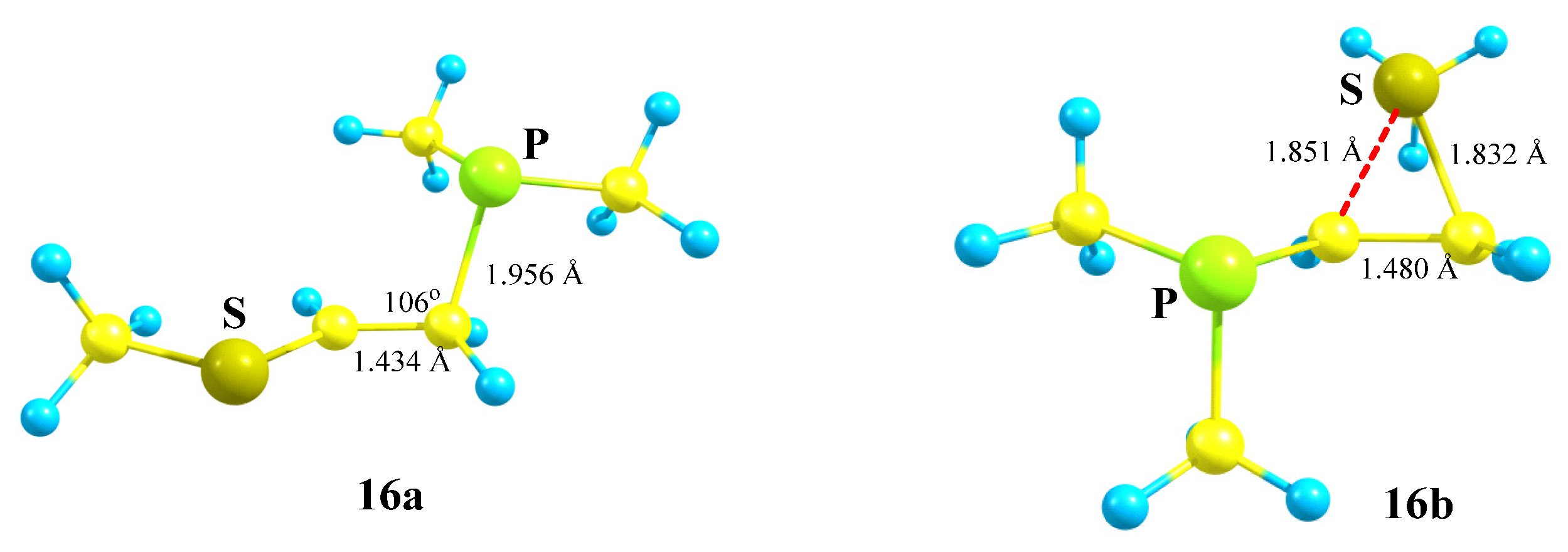
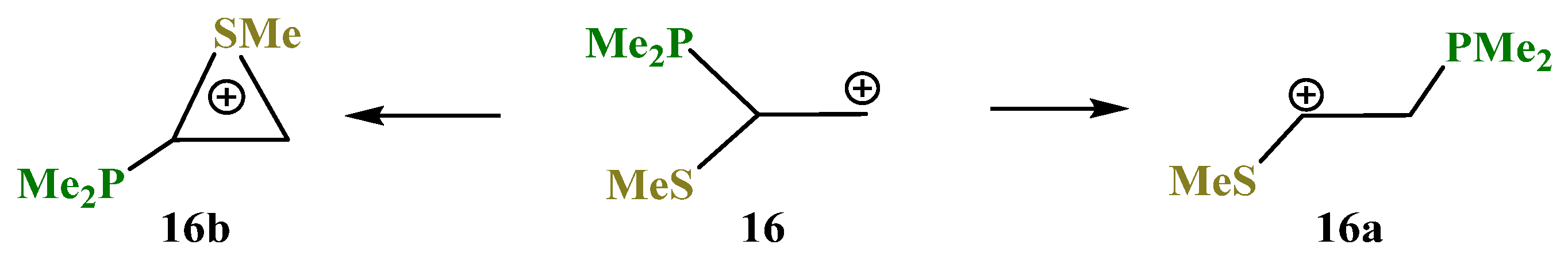
X = PMe2, Y = SMe (16). The phosphorus atom can either fully migrate to the adjacent carbocationic center (N,P cation
3a,
Scheme 3) or form anchimerically stabilized phosphiranium ions (O,P and Si,P cations
8a and
12b,
Scheme 8 and
Scheme 12). The optimization of the conformation of cation
16 with the eclipsed Me
2P–C bond and
p-orbital results in full migration of the Me
2P group and formation of the open cation
16a,
Scheme 16,
Figure 18. The same procedure for the conformation with the eclipsed MeS–C bond and
p-orbital gives rise to the thiiranium cation
16b lying 2.3 kcal/mol lower in energy than
16a, and so the MeS group wins over Me
2P in anchimerical assistance.
Note the opposite charges on the phosphorus and sulfur atoms in 16a and 16b: in the former, the phosphorus atom is charged positively (qP = 0.089) and the sulfur atom negatively (qS = −0.040), whereas in 16b the ratio is vice versa (qP = −0.336, qS = 0.002). The lower electron densities on phosphorous in 16a and on sulfur in 16b are, respectively, due to hyperconjugation with the PMe2 group and the anchimeric assistance by the MeS group.
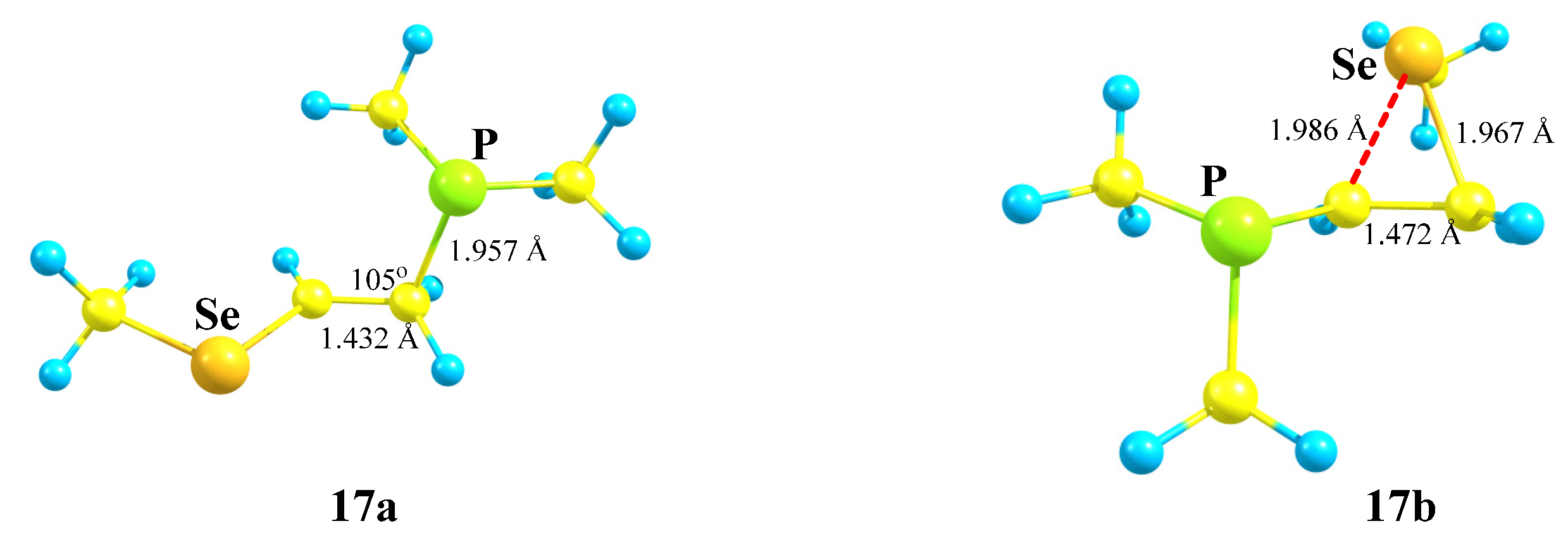
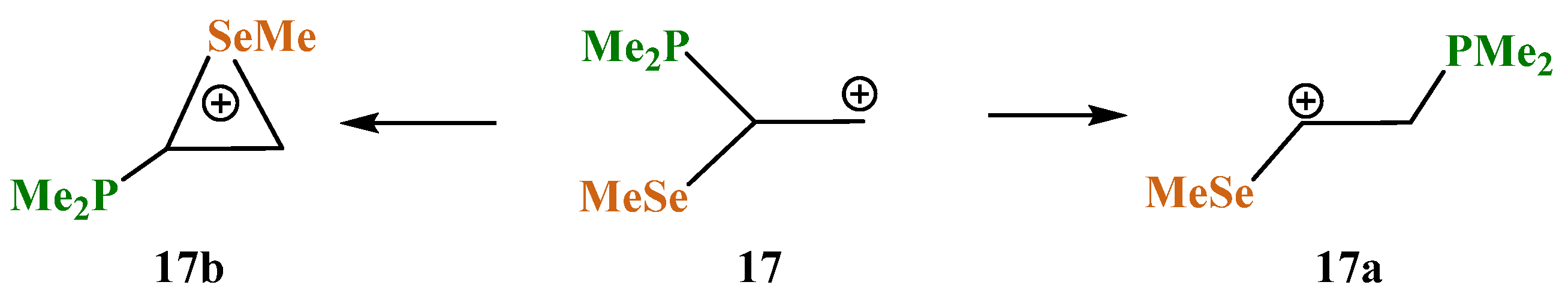
X = PMe2, Y = SeMe (17). The structures and relative energies of the isomeric cations (
Scheme 17,
Figure 19) change only slightly in going to the heavier chalcogen, selenium, except that the energy difference between the anchimerically assisted seleniranium ion
17b and the open 1-methylseleno-2-(dimethylphosphino)ethan-1-ium cation
17a increases to 8.2 kcal/mol, indicating a greater energy gain in the case of selenium as compared to sulfur.
As in the P,S cations 16a and 16b, the phosphorus atom is charged positively in 17a (qP = 0.107) and negatively in 17b (qP = −0.378). The selenium atom is charged positively in both cations, but the electron density on it in 17b is lower (ΔqSe = 0.316 − 0.242 = 0.074), due to the anchimeric assistance being stronger than that of the MeS group.
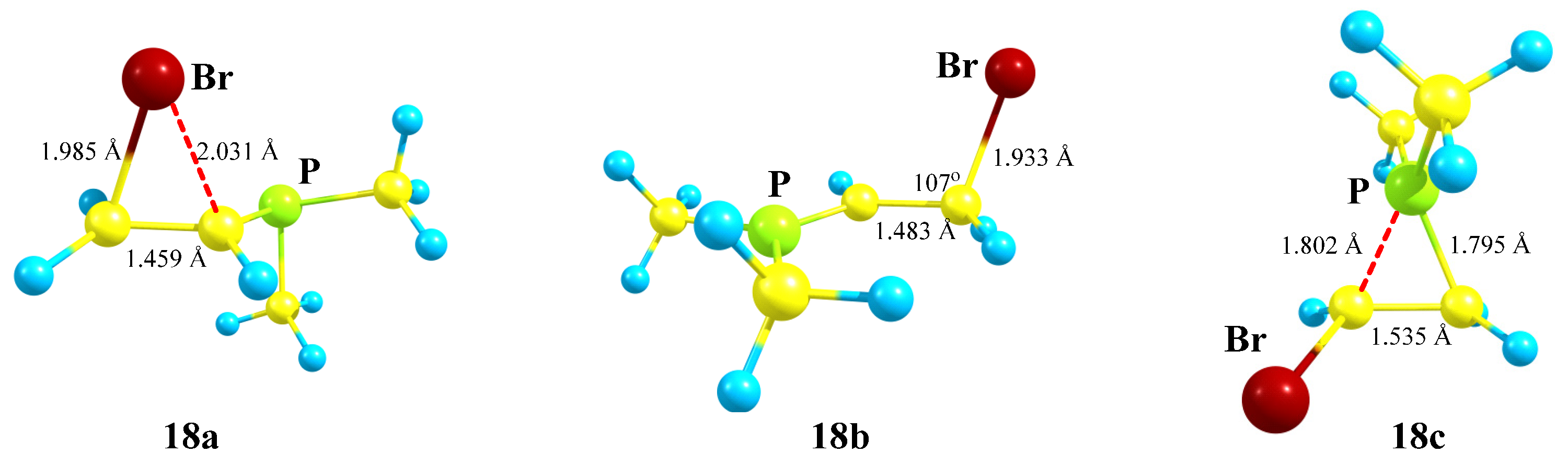
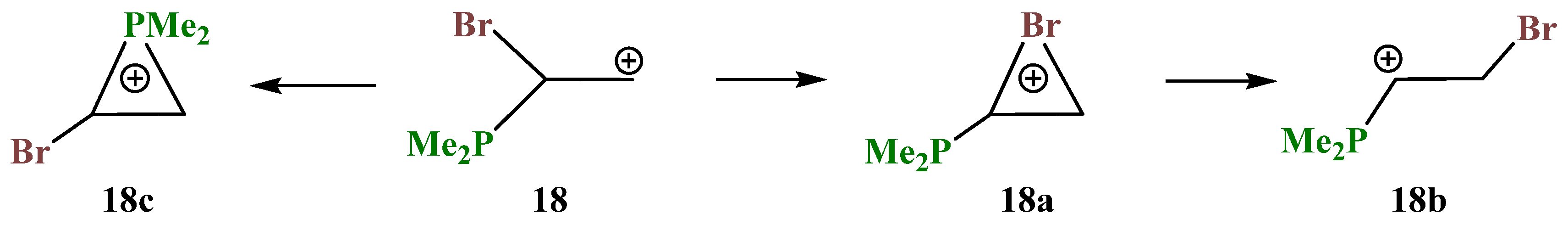
X = PMe2, Y = Br (18). Unlike cations
16 and
17, in which the PMe
2 group fully migrates to the cationic center and in which no phosphiranium ion could be located on the PES, the geometry optimization of cation
18 in the conformation with the eclipsed Me
2P–C bond and the
p-orbital gives rise to phosphiranium ion
18c as the global minimum. Unlike all other cases, two more local minima, bromiranium ion
18a and linear ion
18b, were found on the PES,
Scheme 18.
The phosphiranium ion 18c is energetically most favorable; the next favorable is the open cation 18b, lying 8.2 kcal/mol above 18c; and the least stable is bromiranium ion 18a, lying high above 18c, by 28.9 kcal/mol.
The phosphiranium ion
18c is practically symmetrical (
Figure 20), and the length of the C–C bond in it falls in the range typical for ordinary bonds. The positive charge in
18c is located on the phosphorus atom (
qP = +1.026), and its structure is very similar to the O,P- and Si,P-containing cations
8a and
12b shown in
Figure 9 and
Figure 14. The values for
qP in cation
18b are even larger (
qP = +1.075), which may seem strange, but this is explained by the much shorter P–CH bond (1.647 Å) and a negative charge on the adjacent CH carbon (
qC = −0.628), indicating its dipolar structure: P
+–C
−. The P–CH bonds in
18a and
18c are significantly longer (1.853 and 1.795 and 1.802 Å, respectively). The charges on bromine
qBr in
18b and
18c are slightly negative (−0.04), whereas in
18a the charge is expectedly positive (+0.144).
1-Methyl-1-phenylphosphonium triflate salt was synthesized at the end of the last century and its X-ray crystal structure was determined [
48]. It contains a weakly coordinated triflate counterion and hence an almost “free” phosphiranium cation, which is structurally close to those determined in the present work. Thus, the phosphiranium ring in it is isosceles and the length of the C–P bonds is 1.76 Å (cf.
Figure 9,
Figure 14 and
Figure 20).
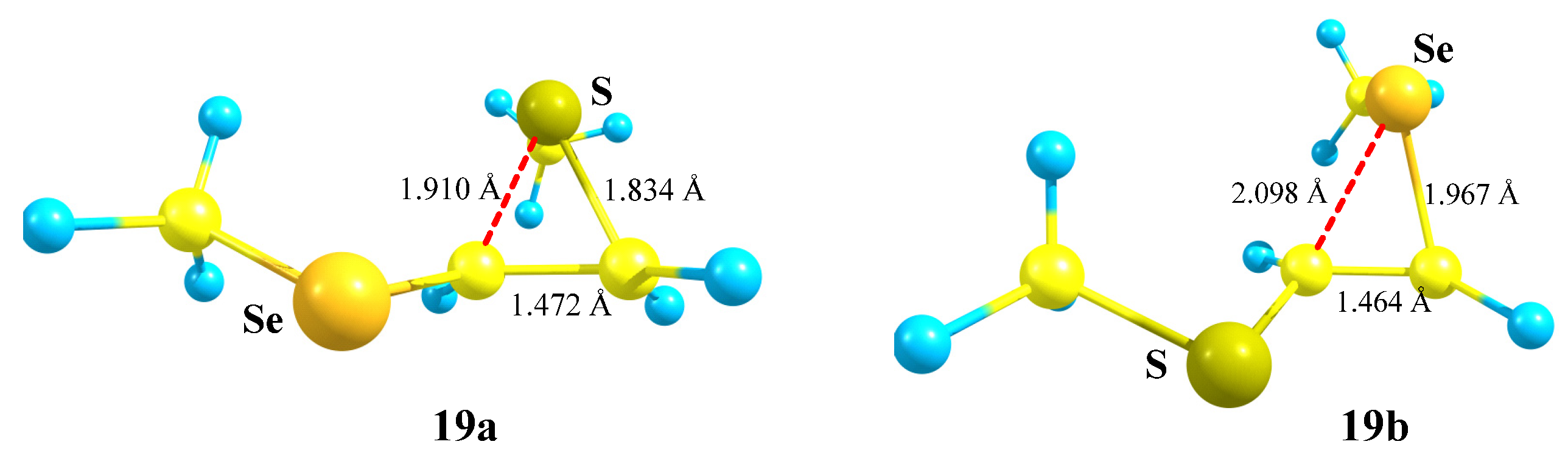
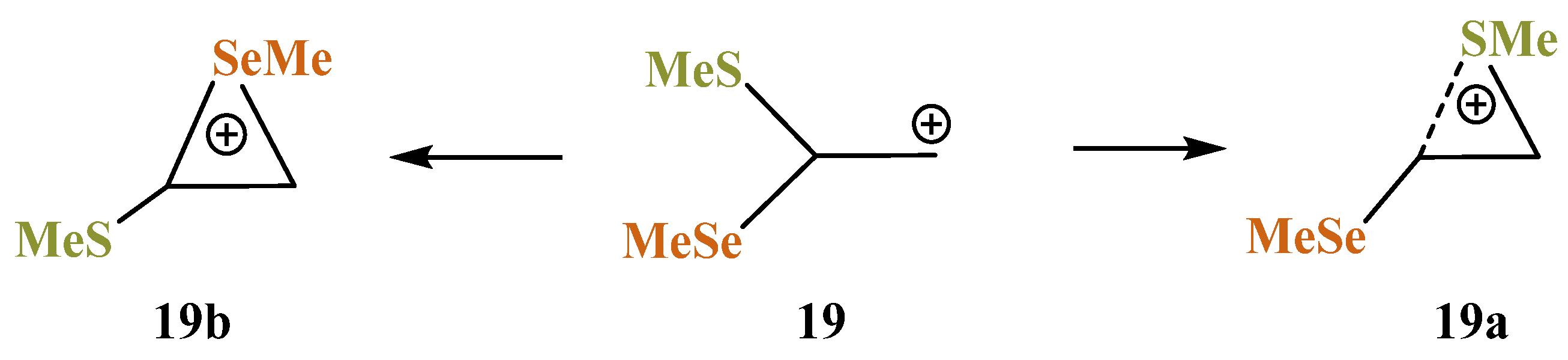
X = SMe, Y = SeMe (19). Of particular interest is the rivalry between the sulfur and selenium atoms in cation
19. The intermediacy of thiiranium and seleniranium ions has been confirmed by extensive studies of the chalcogenofunctionalization reactions of alkenes by Denmark et al. [
49,
50,
51,
52]. One can assume that selenium, as a more polarizable atom capable of stronger anchimeric assistance [
7,
13,
35,
53], would preferably migrate to the cationic center to form the corresponding seleniranium cation. However, the migration of a particular group X is determined by the orientation of the C–X bond with respect to the empty
p-orbital on the carbocation center: either SMe or SeMe migrates, provided that it is eclipsed with the
p-orbital (
Scheme 19).
The isomeric seleniranum cation
19b expectedly turned out to be the global minimum, lying 5.2 kcal/mol below
19a. The structures of the two anchimerically assisted chalcogeniranium cations are presented in
Figure 21.
The chalcogen atoms in the onium rings are expectedly positively charged (qS = +0.167 in 19a, qSe = +0.328 in 19b) but bear the opposite charge, not being involved in anchimeric assistance (qS = −0.284 in 19b, qSe = +0.054 in 19a).
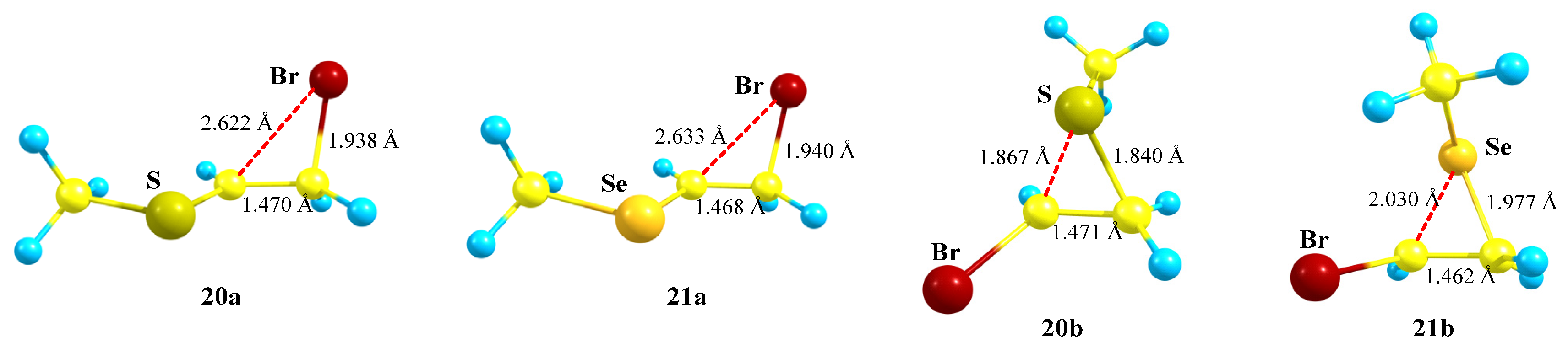
X = SMe, Y = Br (20) and X = SeMe, Y = Br (21). The formation and transformations of haliranium ions were summarized in an excellent review covering the literature published up to 2014 [
54]. Later, the use of haliranium ions as efficient halogenating agents via halogen olefin-to-olefin transfer was reported [
55,
56]. For Y = Br, in the presence of most electronegative second heteroatoms N and O, the geometry optimization results in the migration of the bromine atom (
Scheme 6 and
Scheme 11,
Figure 7 and
Figure 12) to give the most stable cations. The migration of bromine was also observed in the P,Br-containing cation
18 (
Scheme 18), although the formed open cation
18a was only a local minimum. For the most electropositive silicon atom, the Me
3Si group migrated during optimization, the bromiranium ion
15b (
Figure 16) being also a local minimum on the PES. Therefore, the combination of bromine with sulfur and selenium, as the elements with intermediate electronegativity and different polarizability, is of particular interest. In the conformations of cations
20 and
21 with the X–C and
p-orbital eclipsed, the corresponding chalcogeniranium ions
20b and
21b are formed. In all other cases, the shift of bromine was observed with the formation of bromiranium ions
20a and
21a (
Scheme 20).
The chalcogeniranium ions
20b and
21b are the global minima, the isomeric bromiranium ions
20a and
21a being less stable by 1.1 and 8.2 kcal/mol, respectively. Expectedly, the energy difference is notably larger in the case of selenium, clearly demonstrating its higher anchimeric stabilization. Both bromiranium ions are highly asymmetric and have very similar structures,
Figure 22. Interestingly, the positive charge on sulfur in the thiiranium ion
20b (
qS = +0.222) is larger than that in the isomeric bromiranium ion
20a (
qS = +0.114), whereas that on selenium in the seleniranium ion
21b (
qSe = +0.388) is smaller than that in
21a (
qSe = +0.498).
In order to give a compact overview, the results are summarized below in
Table 1.
Note that, among most electropositive substituents Y in the X-stabilized three-membered ring, Y = PMe2 and especially Y = SiMe3 attenuate the demands of anchimeric assistance and decrease the total positive charge on X (qX). It is incorrect to compare the charges on the substituents with different electronegativities, but for those with practically the same electronegativity, SMe and SeMe, the values of qX for X = Se, which are larger by ~0.1–0.2 (depending on Y), provide a semi-quantitative estimation of their anchimeric assistance abilities.














































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
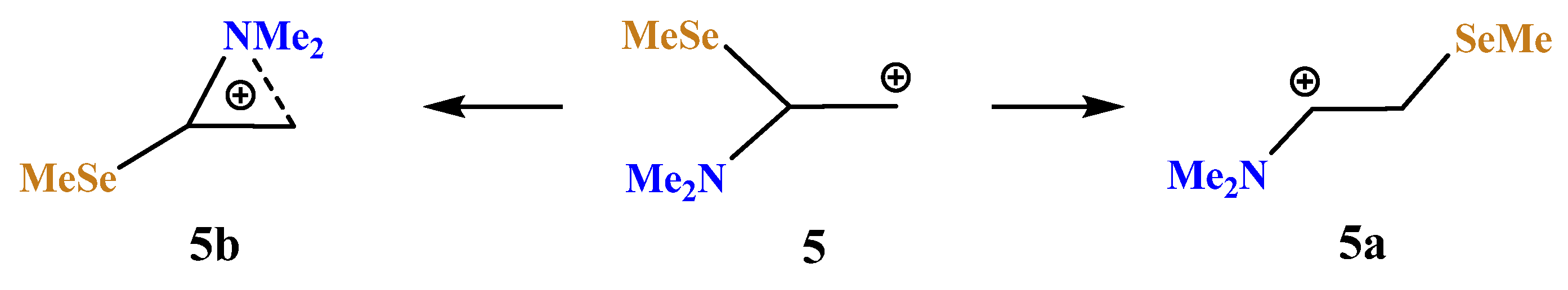{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
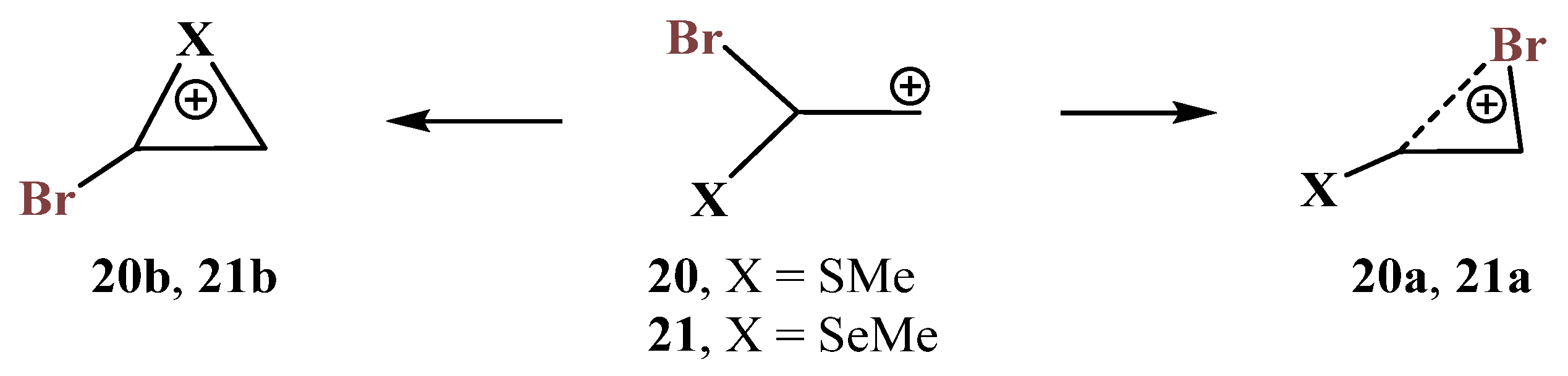{kind=link}
