

Sequential Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Chlorobenzamides/Chemoselective Amide Reduction and Reductive Deuteration to Benzylic Alcohols

 , and
, and 
Abstract
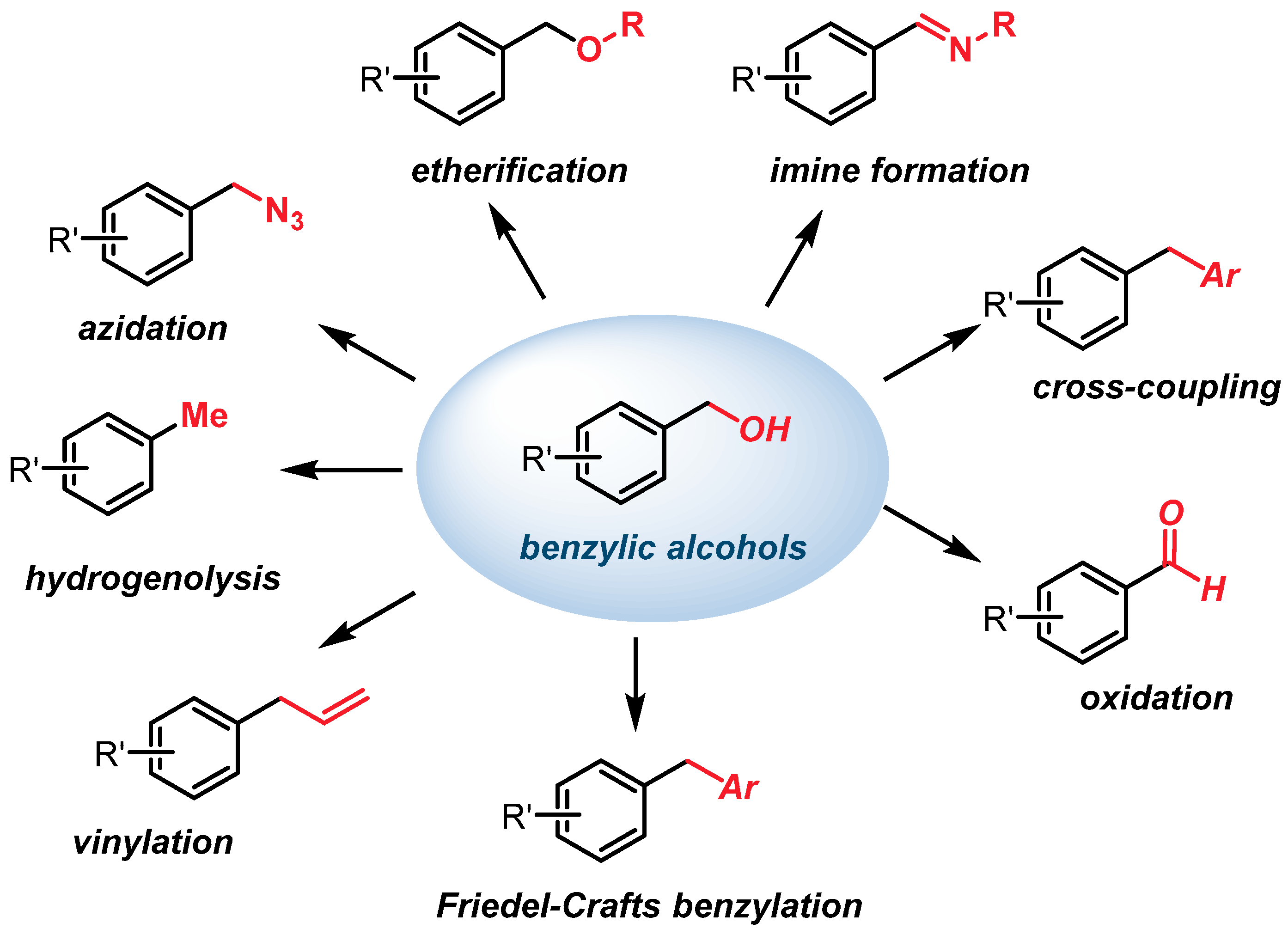
1. Introduction
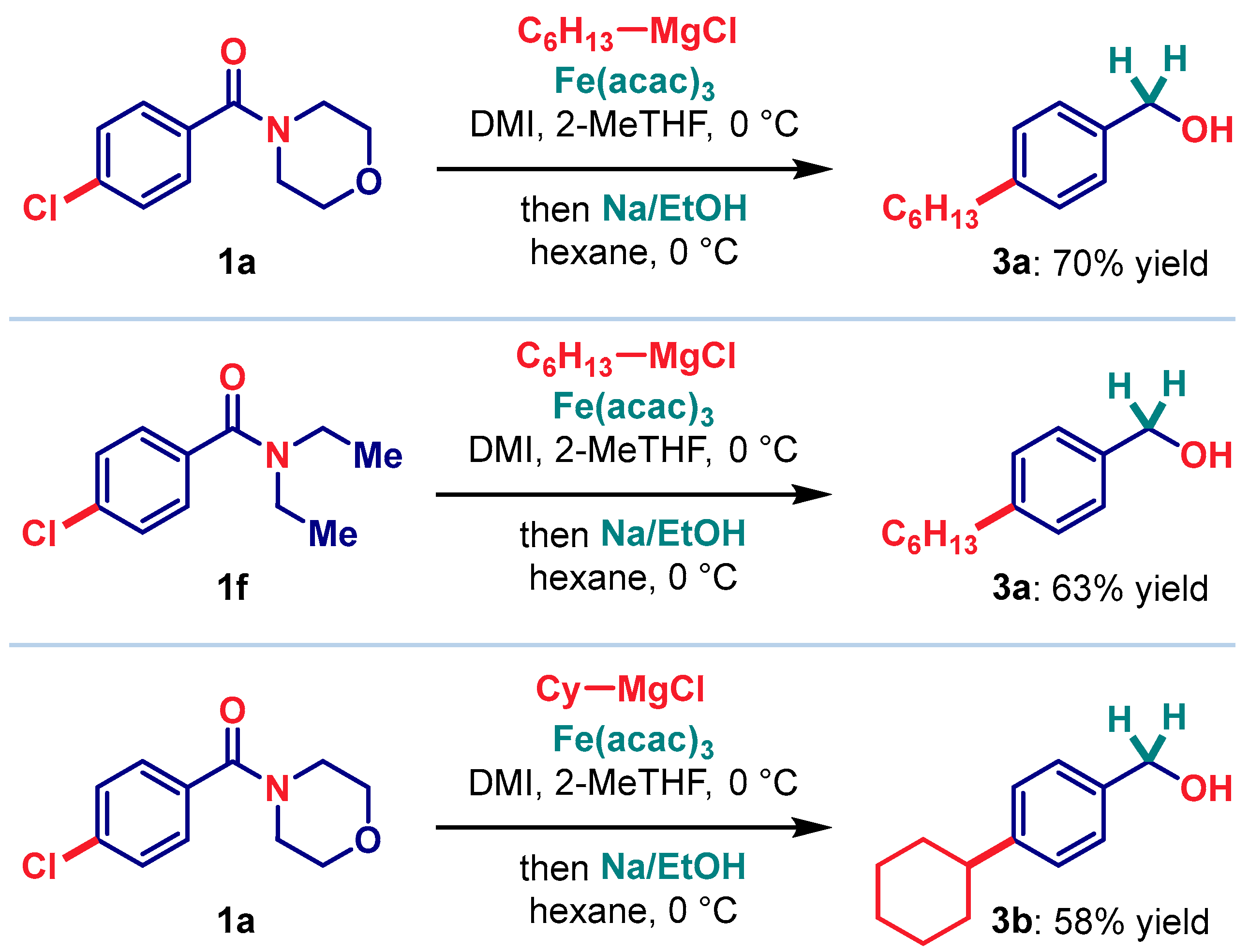
2. Results
3. Discussion
4. Materials and Methods
- Characterization Data of Cross-Coupling Products (Supplementary Materials)
- Characterization Data of Reduction Products (Supplementary Materials)
- Characterization Data of Reductive Deuteration Products (Supplementary Materials)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fürstner, A.; Martin, R. Advances in Iron Catalyzed Cross Coupling Reactions. Chem. Lett. 2005, 34, 624–629. [Google Scholar] [CrossRef]
- Sherry, B.D.; Fürstner, A. The Promise and Challenge of Iron-Catalyzed Cross Coupling. Acc. Chem. Res. 2008, 41, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Czaplik, W.M.; Mayer, M.; Cvengros, J.; Jacobi von Wangelin, A. Coming of Age: Sustainable Iron-Catalyzed Cross-Coupling Reactions. ChemSusChem 2009, 2, 396–417. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Knölker, H.J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Marek, I.; Rappoport, Z. The Chemistry of Organoiron Compounds; Wiley: Weinheim, Germany, 2014. [Google Scholar]
- Plietker, B. Iron Catalysis—Fundamentals and Applications; Springer: Heidelberg, Germany, 2011; Volume 33. [Google Scholar]
- Bauer, E.B. Iron Catalysis II; Springer: Heidelberg, Germany, 2015; Volume 50. [Google Scholar]
- Fürstner, A. Discussion Addendum for: 4-Nonylbenzoic Acid. Org. Synth. 2019, 96, 1–15. [Google Scholar] [CrossRef]
- Legros, J.; Fidegarde, B. Iron-promoted C-C bond formation in the total synthesis of natural products and drugs. Nat. Prod. Rep. 2015, 32, 1541–1555. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C-O Bond Activation: Opportunity for Sustainable Catalysis. ChemSusChem 2017, 10, 3964–3981. [Google Scholar] [CrossRef]
- Fürstner, A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Fürstner, A. Base-Metal Catalysis Marries Utilitarian Aspects with Academic Fascination. Adv. Synth. Catal. 2016, 358, 2362. [Google Scholar] [CrossRef]
- Ludwig, J.R.; Schindler, C.S. Catalyst: Sustainable Catalysis. Chem 2017, 2, 313–316. [Google Scholar] [CrossRef]
- Bakas, N.J.; Neidig, M.L. Additive and Counterion Effects in Iron-Catalyzed Reactions Relevant to C-C Bond Formation. ACS Catal. 2021, 11, 8493–8503. [Google Scholar] [CrossRef] [PubMed]
- Bakas, N.J.; Sears, J.D.; Brennessel, W.W.; Neidig, M.L. A TMEDA–Iron Adduct Reaction Manifold in Iron-Catalyzed C(sp2)−C(sp3) Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2022, 61, e202114986. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.; Zhang, B.; Li, B.; Neidig, M.L.; Byers, J.A. Air-Stable Iron-Based Precatalysts for Suzuki–Miyaura Cross-Coupling Reactions between Alkyl Halides and Aryl Boronic Esters. Org. Process Res. Dev. 2021, 25, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Neate PG, N.; Zhang, B.; Conforti, J.; Brennessel, W.W.; Neidig, M.L. Dilithium Amides as a Modular Bis-Anionic Ligand Platform for Iron-Catalyzed Cross-Coupling. Org. Lett. 2021, 23, 5958–5963. [Google Scholar] [CrossRef]
- Neidig, M.L.; Carpenter, S.H.; Curran, D.J.; DeMuth, J.C.; Fleischauer, V.E.; Iannuzzi, T.E.; Neate PG, N.; Sears, J.D.; Wolford, N.J. Development and Evolution of Mechanistic Understanding in Iron-Catalyzed Cross-Coupling. Acc. Chem. Res. 2019, 52, 140–150. [Google Scholar] [CrossRef]
- Crockett, M.P.; Wong, A.S.; Li, B.; Byers, J.A. Rational Design of an Iron-Based Catalyst for Suzuki–Miyaura Cross-Couplings Involving Heteroaromatic Boronic Esters and Tertiary Alkyl Electrophiles. Angew. Chem. Int. Ed. 2020, 59, 5392–5397. [Google Scholar] [CrossRef]
- Bedford, R.B. How Low Does Iron Go? Chasing the Active Species in Fe-Catalyzed Cross-Coupling Reactions. Acc. Chem. Res. 2015, 48, 1485–1493. [Google Scholar] [CrossRef]
- Cassani, C.; Bergonzini, G.; Wallentin, C.J. Active Species and Mechanistic Pathways in Iron-Catalyzed C–C Bond-Forming Cross-Coupling Reactions. ACS Catal. 2016, 6, 1640–1648. [Google Scholar] [CrossRef]
- Muñoz, S.B., III; Daifuku, S.L.; Sears, J.D.; Baker, T.M.; Carpenter, S.H.; Brennessel, W.W.; Neidig, M.L. The N-Methylpyrrolidone (NMP) Effect in Iron-Catalyzed Cross-Coupling with Simple Ferric Salts and MeMgBr. Angew. Chem. Int. Ed. 2018, 57, 6496–6500. [Google Scholar] [CrossRef]
- Sears, J.D.; Muñoz, S.B.; Daifuku, S.L.; Shaps, A.A.; Carpenter, S.H.; Brennessel, W.W.; Neidig, M.L. The Effect of β-Hydrogen Atoms on Iron Speciation in Cross-Couplings with Simple Iron Salts and Alkyl Grignard Reagents. Angew. Chem. Int. Ed. 2019, 58, 2769–2773. [Google Scholar] [CrossRef]
- Fürstner, A.; Martin, R.; Krause, H.; Seidel, G.; Goddard, R.; Lehmann, C.W. Preparation, Structure, and Reactivity of Nonstabilized Organoiron Compounds. Implications for Iron-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc. 2008, 130, 8773–8787. [Google Scholar] [CrossRef] [PubMed]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-Catalyzed Cross-Coupling in the Synthesis of Pharmaceuticals: In Pursuit of Sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116–11128. [Google Scholar] [CrossRef] [PubMed]
- Metal-Catalyzed Cross-Coupling Reactions and More; de Meijere, A., Bräse, S., Oestreich, M., Eds.; Wiley: New York, NY, USA, 2014. [Google Scholar]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef]
- Giri, R.; Thapa, S.; Kafle, A. Palladium- Catalysed, Directed C-H Coupling with Organometallics. Adv. Synth. Catal. 2014, 356, 1395–1411. [Google Scholar] [CrossRef]
- Trost, B.M.; Fleming, I. Comprehensive Organic Synthesis; Pergamon Press: Oxford, UK, 1991. [Google Scholar]
- Hudlicky, M. Reductions in Organic Chemistry; Ellis Horwood: Chichester, UK, 1984. [Google Scholar]
- Seyden-Penne, J. Reductions by Alumino and Borohydrides in Organic Synthesis; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Andersson, P.G.; Munslow, I.J. Modern Reduction Methods; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
- Brown, D.G.; Boström, J. Analysis of Past and Present Synthetic Methodologies in Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef]
- Tanwar, L.; Börgel, J.; Ritter, T. Synthesis of Benzylic Alcohols by C–H Oxidation. J. Am. Chem. Soc. 2019, 141, 17983–17988. [Google Scholar] [CrossRef]
- Szostak, M.; Spain, M.; Eberhart, A.J.; Procter, D.J. Highly Chemoselective Reduction of Amides (Primary, Secondary and Tertiary) to Alcohols using by SmI2/H2O/Amine under Mild Conditions. J. Am. Chem. Soc. 2014, 136, 2268–2271. [Google Scholar] [CrossRef]
- Huq, S.R.; Shi, S.; Diao, R.; Szostak, M. Mechanistic Study of SmI2/H2O and SmI2/Amine/H2O-Promoted Chemoselective Reduction of Aromatic Amides (Primary, Secondary, Tertiary) to Alcohols via Aminoketyl Radicals. J. Org. Chem. 2017, 82, 6528–6540. [Google Scholar] [CrossRef]
- Shi, S.; Nolan, S.P.; Szostak, M. Well-Defined Palladium(II)-NHC (NHC = N-Heterocyclic Carbene) Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective Acyl CO–X (X = N, O) Cleavage. Acc. Chem. Res. 2018, 51, 2589–2599. [Google Scholar] [CrossRef]
- Li, G.; Ma, S.; Szostak, M. Amide Bond Activation: The Power of Resonance. Trends Chem. 2020, 2, 914–928. [Google Scholar] [CrossRef]
- Meng, G.; Zhang, J.; Szostak, M. Acyclic Twisted Amides. Chem. Rev. 2021, 121, 12746–12783. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Rahman, M.; Zamalloa, A.; Feliciano, J.; Szostak, M. Classes of Amides that Undergo Selective N–C Amide Bond Activation: The Emergence of Ground-State Destabilization. J. Org. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Szostak, M. Cyclic Ureas (DMI, DMPU) as Efficient, Sustainable Ligands in Iron-Catalyzed C(sp2)–C(sp3) Coupling of Aryl Chlorides and Tosylates. Green Chem. 2017, 19, 5361–5366. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. 2-Methyltetrahydrofuran: A Green Solvent for Iron-Catalyzed Cross-Coupling Reactions. ChemSusChem 2018, 11, 1290–1294. [Google Scholar] [CrossRef]
- Piontek, A.; Szostak, M. Iron-Catalyzed C(sp2)-C(sp3) Cross-Coupling of Alkyl Grignard Reagents with Polyaromatic Tosylates. Eur. J. Org. Chem. 2017, 48, 7271–7276. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Chlorobenzamides with Alkyl Grignard Reagents: Development of Catalyst System, Synthetic Scope and Application. Adv. Synth. Catal. 2019, 361, 85–95. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C(sp2)−C(sp3) Cross-Coupling of Chlorobenzenesulfonamides with Alkyl Grignard Reagents: Entry to Alkylated Aromatics. J. Org. Chem. 2019, 84, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Podchorodecka, P.; Szostak, M. N-Methylcaprolactam as a Dipolar Aprotic Solvent for Iron-Catalyzed Cross-Coupling Reactions: Matching Efficiency with Safer Reaction Media. ChemCatChem 2019, 11, 1196–1199. [Google Scholar] [CrossRef]
- Bisz, E.; Kardela, M.; Piontek, A.; Szostak, M. Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling at Low Catalyst Loading. Catl. Sci. Technol. 2019, 9, 1092–1097. [Google Scholar] [CrossRef]
- Bisz, E.; Kardela, M.; Szostak, M. Ligand Effect on Iron-Catalyzed Cross-Coupling Reactions: Evaluation of Amides as O-Coordinating Ligands. ChemCatChem 2019, 11, 5733–5737. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-catalyzed C(sp2)-C(sp3) Cross-Coupling of Aryl Chlorobenzoates with Alkyl Grignard Reagents. Molecules 2020, 25, 230. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E. Iron-Catalyzed Cross-Coupling Reactions of Alkyl Grignards with Aryl Chlorobenzenesulfonates. Molecules 2021, 26, 5895. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Koston, M.; Szostak, M. N-Butylpyrrolidone (NBP) as a Non-Toxic Substitute for NMP in Iron-Catalyzed C(sp2)-C(sp3) Cross-Coupling of Aryl Chlorides. Green Chem. 2021, 23, 7515–7521. [Google Scholar] [CrossRef]
- Zhang, B.; Li, H.; Ding, Y.; Yan, Y.; An, J. Reduction and Reductive Deuteration of Tertiary Amides Mediated by Sodium Dispersions with Distinct Proton Donor-Dependent Chemoselectivity. J. Org. Chem. 2018, 83, 6006–6014. [Google Scholar] [CrossRef]
- Han, M.; Ding, Y.; Yan, Y.; Li, H.; Luo, S.; Adijiang, A.; Ling, Y.; An, J. Transition-Metal-Free, Selective Reductive Deuteration of Terminal Alkynes with Sodium Dispersions and EtOD-d1. Org. Lett. 2018, 20, 3010–3013. [Google Scholar] [CrossRef]
- Lei, P.; Ding, Y.; Zhang, X.; Adijiang, A.; Li, H.; Ling, Y.; An, J. A Practical and Chemoselective Ammonia-Free Birch Reduction. Org. Lett. 2018, 20, 3439–3442. [Google Scholar] [CrossRef]
- Ding, Y.; Luo, S.; Ma, L.; An, J. Reductive Cleavage of Unactivated Carbon–Cyano Bonds under Ammonia-Free Birch Conditions. J. Org. Chem. 2019, 84, 15827–15833. [Google Scholar] [CrossRef]
- Ning, L.; Li, H.; Lai, Z.; Szostak, M.; Chen, X.; Dong, Y.; Jin, S.; An, J. Synthesis of α-Deuterated Primary Amines via Reductive Deuteration of Oximes Using D2O as a Deuterium Source. J. Org. Chem. 2021, 86, 2907–2916. [Google Scholar] [CrossRef]
- Fürstner, A.; Leitner, A. Iron-Catalyzed Cross-Coupling Reactions of Alkyl-Grignard Reagents with Aryl Chlorides, Tosylates, and Triflates. Angew. Chem. Int. Ed. 2002, 41, 609–612. [Google Scholar] [CrossRef]
- Fürstner, A.; Leitner, A.; Mendez, M.; Krause, H. Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Leitner, A. A Catalytic Approach to (R)-(+)-Muscopyridine with Integrated “Self-Clearance”. Angew. Chem. Int. Ed. 2003, 42, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; De Souza, D.; Parra-Rapado, L.; Jensen, J.T. Catalysis-Based Total Synthesis of Latrunculin B. Angew. Chem. Int. Ed. 2003, 42, 5358–5360. [Google Scholar] [CrossRef] [PubMed]
- Czaplik, W.M.; Mayer, M.; Jacobi von Wangelin, A. Domino Iron Catalysis: Direct Aryl-Alkyl Cross-Coupling. Angew. Chem. Int. Ed. 2009, 48, 607–610. [Google Scholar] [CrossRef]
- Gülak, S.; Jacobi von Wangelin, A. Chlorostyrenes in Iron-Catalyzed Biaryl Coupling Reactions. Angew. Chem. Int. Ed. 2012, 51, 1357–1361. [Google Scholar] [CrossRef]
- Gärtner, D.; Stein, A.L.; Grupe, S.; Arp, J.; Jacobi von Wangelin, A. Iron-Catalyzed Cross-Coupling of Alkenyl Acetates. Angew. Chem. Int. Ed. 2015, 54, 10545–10549. [Google Scholar] [CrossRef]
- Kuzmina, O.M.; Steib, A.K.; Markiewicz, J.T.; Flubacher, D.; Knochel, P. Ligand-Accelerated Iron- and Cobalt-Catalyzed Cross-Coupling Reactions between N-Heteroaryl Halides and Aryl Magnesium Reagents. Angew. Chem. Int. Ed. 2013, 52, 4945–4949. [Google Scholar] [CrossRef]
- Casitas, A.; Krause, H.; Goddard, R.; Fürstner, A. Elementary Steps in Iron Catalysis: Exploring the Links between Iron Alkyl and Iron Olefin Complexes for their Relevance in C–H Activation and C–C Bond Formation. Angew. Chem. Int. Ed. 2015, 54, 1521–1526. [Google Scholar] [CrossRef]
- Casitas, A.; Rees, J.A.; Goddard, R.; Bill, E.; DeBeer, D.; Fürstner, A. Two Exceptional Homoleptic Iron(IV) Tetraalkyl Complexes. Angew. Chem. Int. Ed. 2017, 56, 10108–10113. [Google Scholar] [CrossRef]
- Åkesson, B. N-Methyl-2-Pyrrolidone; WHO: Geneva, Switzerland, 2001; Available online: https://echa.europa.eu/candidate-list-table (accessed on 28 November 2022).
- Pace, V.; Hoyos, P.; Castoldi, L.; María, P.D.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Monticelli, S.; Castoldi, L.; Murgia, I.; Senatore, R.; Mazzeo, E.; Wackerlig, J.; Urban, E.; Langer, T.; Pace, V. Recent advancements on the use of 2-methyltetrahydrofuran in organometallic chemistry. Monatsh Chem. 2017, 148, 37–48. [Google Scholar] [CrossRef]
- Lucchini, J.J.; Corre, J.; Cremieux, A. Antibacterial activity of phenolic compounds and aromatic alcohols. Res. Microbiol. 1990, 141, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Atzrodt, J.; Derdau, V.; Fey, T.; Zimmermann, J. The Renaissance of H/D Exchange. Angew. Chem. Int. Ed. 2007, 46, 7744–7765. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Deuterated Drugs Draw Heavier Backing. Nat. Rev. Drug Discov. 2016, 15, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.Y.; Nagao, K.; Hoover, A.J.; Hesk, D.; Rivera, N.R.; Colletti, S.L.; Davies, I.W.; Macmillan DW, C. Photoredox-catalyzed deuteration and tritiation of pharmaceutical compounds. Science 2017, 358, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Atzrodt, J.; Derdau, V.; Kerr, W.J.; Reid, M. Deuterium- and Tritium-Labelled Compounds: Applications in the Life Sciences. Angew. Chemie Int. Ed. 2018, 57, 1758–1784. [Google Scholar] [CrossRef]
- Pirali, T.; Serafini, M.; Cargnin, S.; Genazzani, A.A. Applications of Deuterium in Medicinal Chemistry. J. Med. Chem. 2019, 62, 5276–5297. [Google Scholar] [CrossRef]
- Rushworth, P.J.; Hulcoop, D.G.; Fox, D.J. Iron/Tetramethylethylenediamine-Catalyzed Ambient-Temperature Coupling of Alkyl Grignard Reagents and Aryl Chlorides. J. Org. Chem. 2013, 78, 9517–9521. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Shi, F.; Deng, Y. Dehydrogenative amide synthesis from alcohol and amine catalyzed by hydrotalcite-supported gold nanoparticles. Tetrahedron Lett. 2012, 53, 3178–3180. [Google Scholar] [CrossRef]
- Day, G.M.; Howell, O.T.; Metzler, M.R.; Woodgate, P.D. Synthesis and Characterization of Stilbene Derivatives for Possible Incorporation as Smart Additives in Polymers Used as Packaging Films. Aust. J. Chem. 1997, 50, 425–434. [Google Scholar] [CrossRef]
- Bodroux, D.; Thomassin, R. Syntheses in the phenylcyclohexane series. Bull. Soc. Chim. Fr. 1939, 6, 1411–1416. [Google Scholar]
- Andrianome, M.; Häberle, K.; Delmond, B. Allyl-and benzylstannanes, new reagents in terpenic synthesis. Tetrahedron 1989, 45, 1079–1088. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}

| Entry | Fe(acac)3 (mol%) | Ligand | Ligand (mol %) | Time (h) | Yield 2 (%) |
|---|---|---|---|---|---|
| 1 | - | - | - | 18 | <2 |
| 2 | 0.10 | - | - | 18 | 48 |
| 3 | 0.10 | DMI | 200 | 18 | 90 |
| 4 3 | 0.10 | DMI | 200 | 18 | 88 |
| 5 4 | 0.10 | DMI | 200 | 18 | 76 |
| 6 5 | 0.10 | DMI | 200 | 18 | 95 |

| Entry | 2 | Product | Yield (%) |
|---|---|---|---|
| 1 | 2a |  | 92 |
| 2 | 2b |  | 92 |
| 3 2 | 2c |  | 88 |
| 4 | 2d |  | 73 |
| 5 | 2e |  | 95 |
| 6 2 | 2f |  | 98 |
| 7 2 | 2g |  | 98 |
| 8 | 2h |  | 85 |
| 9 | 2i |  | 95 |
| 10 3 | 2j |  | 80 |
| 11 3 | 2k |  | 56 |
| 12 | 2l |  | 68 |

| Entry | Na (equiv) | EtOH (equiv) | Time (min) | Yield 2 (%) |
|---|---|---|---|---|
| 1 | 10 | 30 | 20 | 81 |
| 2 | 10 | 20 | 20 | 72 |
| 3 | 10 | 40 | 20 | 75 |
| 4 | 10 | 50 | 20 | 65 |
| 5 | 5 | 15 | 20 | 68 |
 | ||
 |  |  |
 |  |  |
 |  |  |
 |  |  |
 | ||
 |  |  |
 |  |  |
 |  |  |
 |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisz, E.; Podchorodecka, P.; Li, H.; Ochędzan-Siodłak, W.; An, J.; Szostak, M. Sequential Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Chlorobenzamides/Chemoselective Amide Reduction and Reductive Deuteration to Benzylic Alcohols. Molecules 2023, 28, 223. https://doi.org/10.3390/molecules28010223
Bisz E, Podchorodecka P, Li H, Ochędzan-Siodłak W, An J, Szostak M. Sequential Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Chlorobenzamides/Chemoselective Amide Reduction and Reductive Deuteration to Benzylic Alcohols. Molecules. 2023; 28(1):223. https://doi.org/10.3390/molecules28010223
Chicago/Turabian StyleBisz, Elwira, Pamela Podchorodecka, Hengzhao Li, Wioletta Ochędzan-Siodłak, Jie An, and Michal Szostak. 2023. "Sequential Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Chlorobenzamides/Chemoselective Amide Reduction and Reductive Deuteration to Benzylic Alcohols" Molecules 28, no. 1: 223. https://doi.org/10.3390/molecules28010223
APA StyleBisz, E., Podchorodecka, P., Li, H., Ochędzan-Siodłak, W., An, J., & Szostak, M. (2023). Sequential Iron-Catalyzed C(sp2)–C(sp3) Cross-Coupling of Chlorobenzamides/Chemoselective Amide Reduction and Reductive Deuteration to Benzylic Alcohols. Molecules, 28(1), 223. https://doi.org/10.3390/molecules28010223