
Designing Functionally Substituted Pyridine-Carbohydrazides for Potent Antibacterial and Devouring Antifungal Effect on Multidrug Resistant (MDR) Strains

 ,
,  , ,
, ,  , and
, and 
Abstract
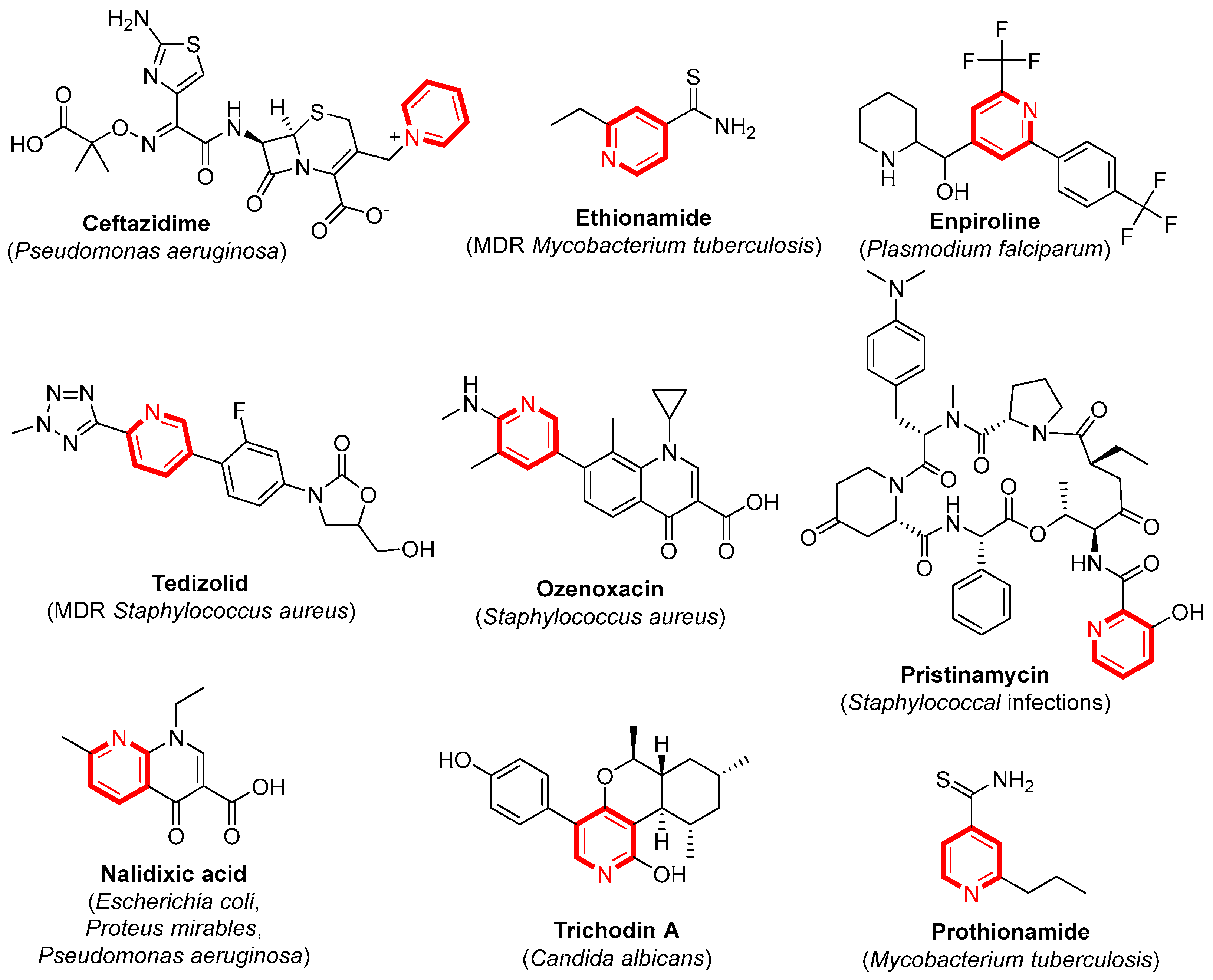
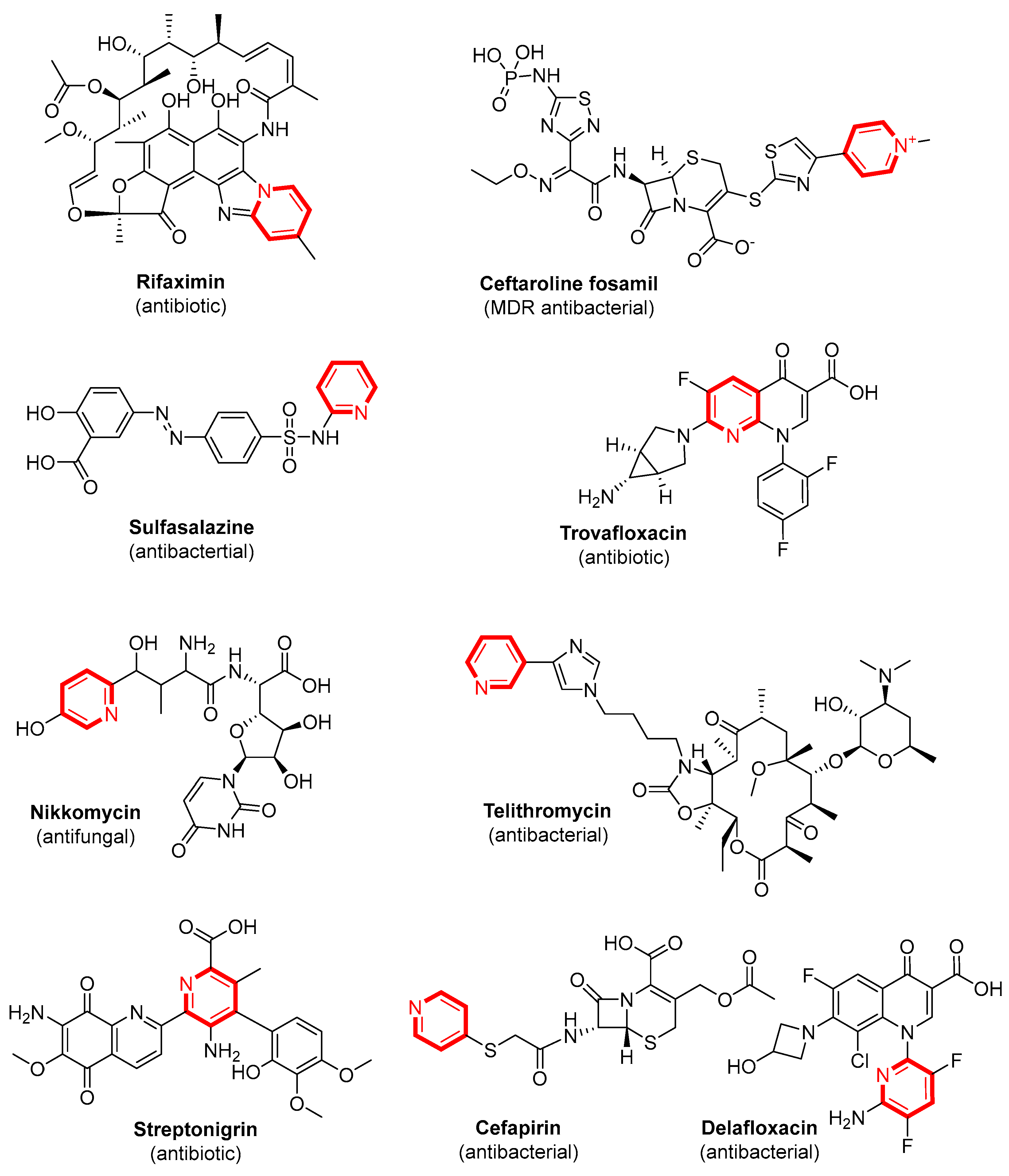
1. Introduction
2. Results and Discussion
2.1. Antifungal Studies
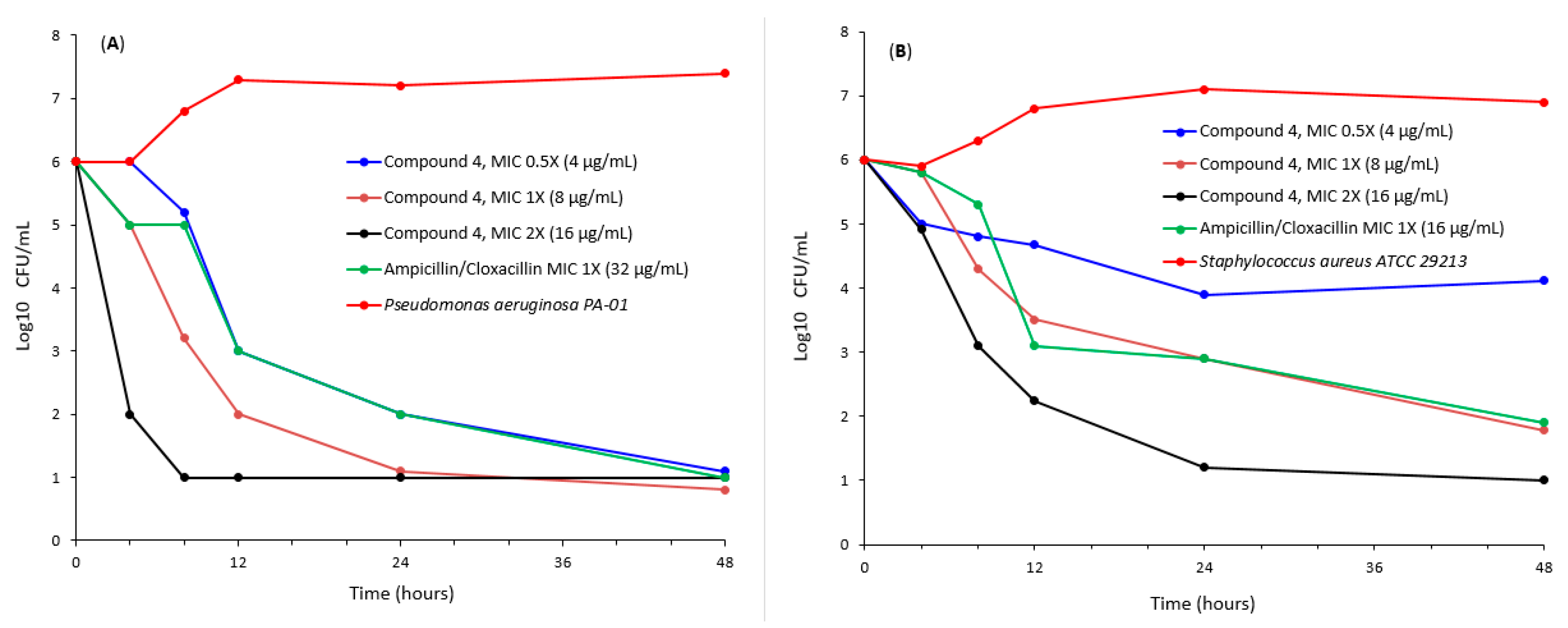
2.2. Antibacterial Studies
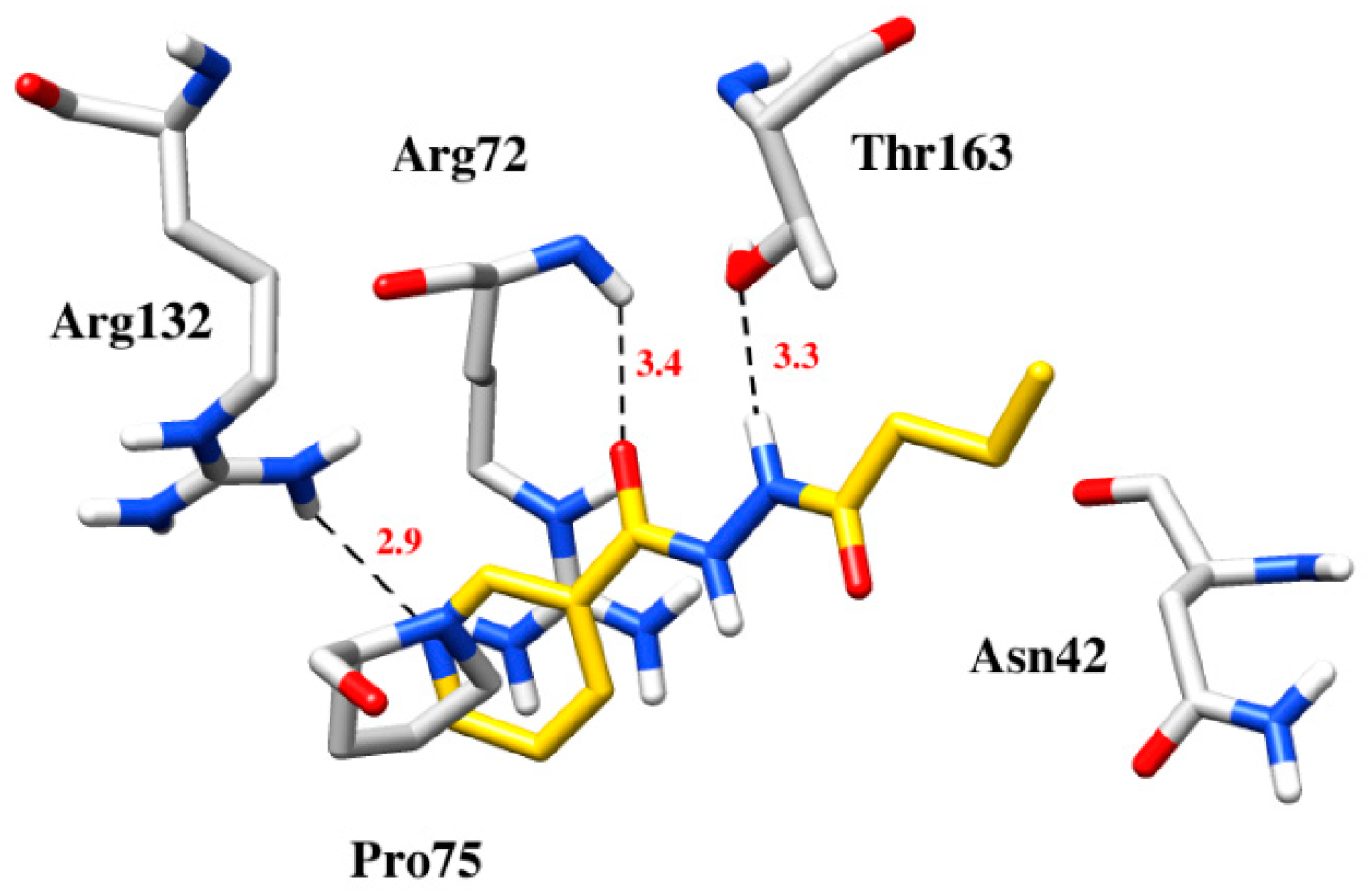
2.3. Computational Studies
3. Materials and Methods
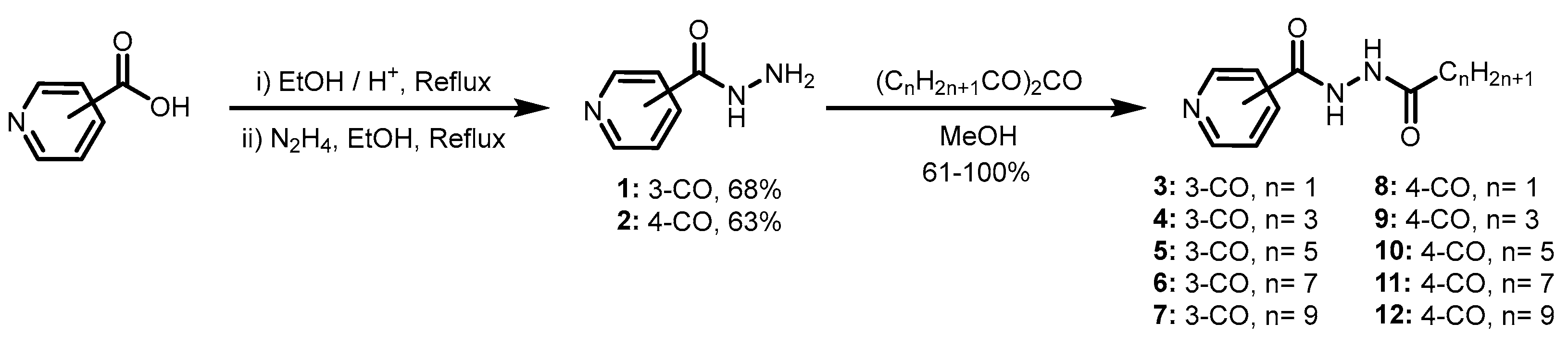
3.1. General Procedure for the Synthesis of Pyridine-3-Carbohydrazides and Pyridine-4-Carbohydrazides Containing Lipophilic Chains
3.1.1. Pridine-3-Carbohydrazide (1)
3.1.2. Pridine-4-Carbohydrazide (2)
3.1.3. N’-Acetylpridine-3-Carbohydrazide (3)
3.1.4. N’-Butyrylpyridine-3-Carbohydrazide (4)
3.1.5. N’-Hexanoylpyridine-3-Carbohydrazide (5)
3.1.6. N’-Octanoylpyridine-3-Carbohydrazide (6)
3.1.7. N’-Decanoylpyridine-3-Carbohydrazide (7)
3.1.8. N’-Acetylpyridine-4-Carbohydrazide (8)
3.1.9. N’-Butyrylpyridine-4-Carbohydrazide (9)
3.1.10. N’-Hexanoylpyridine-4-Carbohydrazide (10)
3.1.11. N’-Octanoylpyridine-4-Carbohydrazide (11)
3.1.12. N’-Decanoylpyridine-4-Carbohydrazide (12)
3.2. Antifungal Studies
3.2.1. Susceptibility Testing
3.2.2. Anti-Fungal Index (AFI)
3.2.3. Minimal Inhibitory Concentration (MIC) of Antifungal Agents
3.2.4. Time Kill Assay of Antifungal Agents
3.2.5. Hemolytic Assay
3.3. Antibacterial Studies
3.3.1. Susceptibility Profiling of Antibacterial Compounds
3.3.2. Minimal Inhibitory Concentration (MIC) of Antibacterial Compounds
3.3.3. Time Kill Assay of Antibacterial Compounds
3.4. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Qin, R.; Wang, P.; Wang, B.; Fu, L.; Batt, S.M.; Besra, G.S.; Wu, C.; Wang, Y.; Huang, H.; Lu, Y.; et al. Identification of Thiophene-Benzenesulfonamide Derivatives for the Treatment of Multidrug-Resistant Tuberculosis. Eur. J. Med. Chem. 2022, 231, 114145. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.M.G.; Hodgkinson, J.T.; Sore, H.F.; Welch, M.; Salmond, G.P.C.; Spring, D.R. Combating Multidrug-Resistant Bacteria: Current Strategies for the Discovery of Novel Antibacterials. Angew. Chemie Int. Ed. 2013, 52, 10706–10733. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Unmet Medical Needs in Antibacterial Therapy. Biochem. Pharmacol. 2006, 71, 991–995. [Google Scholar] [CrossRef]
- Mahajan, D.T.; Masand, V.H.; Patil, K.N.; Hadda, T.B.; Rastija, V. Integrating GUSAR and QSAR Analyses for Antimalarial Activity of Synthetic Prodiginines against Multi Drug Resistant Strain. Med. Chem. Res. 2013, 22, 2284–2292. [Google Scholar] [CrossRef]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular Mechanisms of Drug Resistance and Epidemiology of Multidrug-Resistant Variants of Neisseria Gonorrhoeae. Int. J. Mol. Sci. 2022, 23, 10499. [Google Scholar] [CrossRef]
- Ling, Y.; Hao, Z.-Y.; Liang, D.; Zhang, C.-L.; Liu, Y.-F.; Wang, Y. The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design. Drug Des. Devel. Ther. 2021, 15, 4289–4338. [Google Scholar] [CrossRef]
- Yaqoob, S.; Nasim, N.; Khanam, R.; Wang, Y.; Jabeen, A.; Qureshi, U.; Ul-Haq, Z.; El-Seedi, H.R.; Jiang, Z.-H.; Khan, F.-A. Synthesis of Highly Potent Anti-Inflammatory Compounds (ROS Inhibitors) from Isonicotinic Acid. Molecules 2021, 26, 1272. [Google Scholar] [CrossRef]
- Popiołek, Ł. Updated Information on Antimicrobial Activity of Hydrazide-Hydrazones. Int. J. Mol. Sci. 2021, 22, 9389. [Google Scholar] [CrossRef]
- Mohammadi Ziarani, G.; Fathi Vavsari, V. The Role of Hydrazide Compounds in Asymmetric Synthesis. Tetrahedron Asymmetry 2017, 28, 203–214. [Google Scholar] [CrossRef]
- Popiołek, Ł. Hydrazide-Hydrazones as Potential Antimicrobial Agents: Overview of the Literature since 2010. Med. Chem. Res. 2017, 26, 287–301. [Google Scholar] [CrossRef]
- Ramezanpour, S.; Balalaie, S.; Rominger, F.; Alavijeh, N.S.; Bijanzadeh, H.R. Facile, Efficient and Diastereoselective Synthesis of α-Hydrazine Tetrazoles through a Novel One-Pot Four-Component Reaction. Tetrahedron 2013, 69, 10718–10723. [Google Scholar] [CrossRef]
- Alavijeh, N.S.; Zadmard, R.; Ramezanpour, S.; Balalaie, S.; Alavijeh, M.S.; Rominger, F. Efficient Synthesis of Lower Rim α-Hydrazino Tetrazolocalix[4]Arenes via an Ugi-Azide Multicomponent Reaction. New J. Chem. 2015, 39, 6578–6584. [Google Scholar] [CrossRef]
- Fathi Vavsari, V.; Mohammadi Ziarani, G.; Badiei, A.; Balalaie, S. Application of SBA-Pr-SO3H as a Nanoreactor in the One-Pot Synthesis of Spiroquinazolinones. J. Iran. Chem. Soc. 2016, 13, 1037–1043. [Google Scholar] [CrossRef]
- Maggio, B.; Daidone, G.; Raffa, D.; Plescia, S.; Mantione, L.; Cutuli, V.M.C.; Mangano, N.G.; Caruso, A. Synthesis and Pharmacological Study of Ethyl 1-Methyl-5-(Substituted 3,4-Dihydro-4-Oxoquinazolin-3-Yl)-1H-Pyrazole-4-Acetates. Eur. J. Med. Chem. 2001, 36, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, K.; Ghosh, A.; Basak, A.; Das, G.K. Stabilization of the Transition Structures of Organocatalytic Asymmetric Direct Aldol Reaction in Wet Solvent Free Condition by the Formation of Water Assisted Supramolecular Network: A DFT Study. Comput. Theor. Chem. 2015, 1062, 11–23. [Google Scholar] [CrossRef]
- Murugaiyan, J.; Anand Kumar, P.; Rao, G.S.; Iskandar, K.; Hawser, S.; Hays, J.P.; Mohsen, Y.; Adukkadukkam, S.; Awuah, W.A.; Jose, R.A.M.; et al. Progress in Alternative Strategies to Combat Antimicrobial Resistance: Focus on Antibiotics. Antibiotics 2022, 11, 200. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, S.; Zhao, F.; Gao, C.; Feng, L.; Lv, Z.; Xu, Z.; Wu, X. Isoniazid Derivatives and Their Anti-Tubercular Activity. Eur. J. Med. Chem. 2017, 133, 255–267. [Google Scholar] [CrossRef]
- Nabergoj, S.; Mlinarič-Raščan, I.; Jakopin, Ž. Harnessing the Untapped Potential of Nucleotide-binding Oligomerization Domain Ligands for Cancer Immunotherapy. Med. Res. Rev. 2019, 39, 1447–1484. [Google Scholar] [CrossRef]
- Egorova, A.; Salina, E.G.; Makarov, V. Targeting Non-Replicating Mycobacterium Tuberculosis and Latent Infection: Alternatives and Perspectives (Mini-Review). Int. J. Mol. Sci. 2021, 22, 13317. [Google Scholar] [CrossRef]
- Sharipova, R.R.; Strobykina, I.Y.; Mordovskoi, G.G.; Chestnova, R.V.; Mironov, V.F.; Kataev, V.E. Antituberculosis Activity of Glycosides from Stevia Rebaudiana and Hybrid Compounds of Steviolbioside and Pyridinecarboxylic Acid Hydrazides. Chem. Nat. Compd. 2011, 46, 902–905. [Google Scholar] [CrossRef]
- De, P.; Koumba Yoya, G.; Constant, P.; Bedos-Belval, F.; Duran, H.; Saffon, N.; Daffé, M.; Baltas, M. Design, Synthesis, and Biological Evaluation of New Cinnamic Derivatives as Antituberculosis Agents. J. Med. Chem. 2011, 54, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Ramani, A.V.; Monika, A.; Indira, V.L.; Karyavardhi, G.; Venkatesh, J.; Jeankumar, V.U.; Manjashetty, T.H.; Yogeeswari, P.; Sriram, D. Synthesis of Highly Potent Novel Anti-Tubercular Isoniazid Analogues with Preliminary Pharmacokinetic Evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 2764–2767. [Google Scholar] [CrossRef] [PubMed]
- Bhilare, N.V.; Dhaneshwar, S.S.; Mahadik, K.R. Phenolic Acid-Tethered Isoniazid for Abrogation of Drug-Induced Hepatotoxicity: Design, Synthesis, Kinetics and Pharmacological Evaluation. Drug Deliv. Transl. Res. 2018, 8, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Nayak, N.; Razdan, B.; Bajpai, M. Plumbagin Analogs-Synthesis, Characterization, and Antitubercular Activity. J. Adv. Pharm. Technol. Res. 2014, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.O.; Cantos, J.B.; D’Oca, C.R.M.; Soares, K.L.; Coelho, T.S.; Piovesan, L.A.; Russowsky, D.; da Silva, P.A.; D’Oca, M.G.M. Synthesis and Antimycobacterial Activity of Isoniazid Derivatives from Renewable Fatty Acids. Bioorg. Med. Chem. 2013, 21, 6910–6914. [Google Scholar] [CrossRef]
- Pavan, F.R.; Maia, P.I.d.S.; Leite, S.R.A.; Deflon, V.M.; Batista, A.A.; Sato, D.N.; Franzblau, S.G.; Leite, C.Q.F. Thiosemicarbazones, Semicarbazones, Dithiocarbazates and Hydrazide/Hydrazones: Anti—Mycobacterium Tuberculosis Activity and Cytotoxicity. Eur. J. Med. Chem. 2010, 45, 1898–1905. [Google Scholar] [CrossRef]
- Ramalakshmi, N.; Aruloly, L.; Arunkumar, S.; Ilango, K.; Puratchikody, A. Synthesis and Biological Evaluation of Some Novel Nicotinic Acid Derivatives. Malays. J. Sci. 2009, 28, 197–203. [Google Scholar] [CrossRef][Green Version]
- Aamra, H.; Khan, F.-A.; Jahan, H.; Zafar, M.; Ali, H.; Shaheen, F. Synthesis of Novel Benzimidazole Containing Antimicrobial Peptides (AMPs) with Significant Inhibitory Effect on Multidrug Resistant Strain of Salmonella Typhimurium. Synth. Commun. 2021, 51, 3620–3628. [Google Scholar] [CrossRef]
- Dias, C.; Martins, A.; Pelerito, A.; Oliveira, M.C.; Contino, M.; Colabufo, N.A.; Rauter, A.P. Assessing the Optimal Deoxygenation Pattern of Dodecyl Glycosides for Antimicrobial Activity Against Bacillus Anthracis. Eur. J. Org. Chem. 2019, 2019, 2224–2233. [Google Scholar] [CrossRef]
- Roque-Borda, C.A.; Bento da Silva, P.; Rodrigues, M.C.; Di Filippo, L.D.; Duarte, J.L.; Chorilli, M.; Vicente, E.F.; Garrido, S.S.; Rogério Pavan, F. Pharmaceutical Nanotechnology: Antimicrobial Peptides as Potential New Drugs against WHO List of Critical, High, and Medium Priority Bacteria. Eur. J. Med. Chem. 2022, 241, 114640. [Google Scholar] [CrossRef]
- Ghiano, D.G.; Recio-Balsells, A.; Bortolotti, A.; Defelipe, L.A.; Turjanski, A.; Morbidoni, H.R.; Labadie, G.R. New One-Pot Synthesis of Anti-Tuberculosis Compounds Inspired on Isoniazid. Eur. J. Med. Chem. 2020, 208, 112699. [Google Scholar] [CrossRef] [PubMed]
- Judge, V.; Narasimhan, B.; Ahuja, M.; Sriram, D.; Yogeeswari, P.; De Clercq, E.; Pannecouque, C.; Balzarini, J. Isonicotinic Acid Hydrazide Derivatives: Synthesis, Antimicrobial Activity, and QSAR Studies. Med. Chem. Res. 2012, 21, 1451–1470. [Google Scholar] [CrossRef]
- Rasras, A.J.M.; Al-Tel, T.H.; Al-Aboudi, A.F.; Al-Qawasmeh, R.A. Synthesis and Antimicrobial Activity of Cholic Acid Hydrazone Analogues. Eur. J. Med. Chem. 2010, 45, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, N.; Goh, K.S.; Horgen, L.; Barrow, W.W. Synergistic Activities of Antituberculous Drugs with Cerulenin and Trans-Cinnamic Acid against Mycobacterium Tuberculosis. FEMS Immunol. Med. Microbiol. 1998, 21, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Xaiver, J.J.F.; Krishnasamy, K.; Sankar, C. Synthesis and Antibacterial, Antifungal Activities of Some 2r,4c-Diaryl-3-Azabicyclo[3.3.1]Nonan-9-One-4-Aminobenzoyl Hydrazones. Med. Chem. Res. 2012, 21, 345–350. [Google Scholar] [CrossRef]
- Liu, W.; Yuan, L.; Wang, S. Recent Progress in the Discovery of Antifungal Agents Targeting the Cell Wall. J. Med. Chem. 2020, 63, 12429–12459. [Google Scholar] [CrossRef]
- Brown, G.D. Innate Antifungal Immunity: The Key Role of Phagocytes. Annu. Rev. Immunol. 2011, 29, 1–21. [Google Scholar] [CrossRef]
- Sun, B.; Dong, Y.; An, Y.; Liu, M.; Han, J.; Zhao, L.; Liu, X. Design, Synthesis and Bioactivity Evaluation of Novel Arylalkene-Amide Derivatives as Dual-Target Antifungal Inhibitors. Eur. J. Med. Chem. 2020, 205, 112645. [Google Scholar] [CrossRef]
- Elias, R.; Benhamou, R.I.; Jaber, Q.Z.; Dorot, O.; Zada, S.L.; Oved, K.; Pichinuk, E.; Fridman, M. Antifungal Activity, Mode of Action Variability, and Subcellular Distribution of Coumarin-Based Antifungal Azoles. Eur. J. Med. Chem. 2019, 179, 779–790. [Google Scholar] [CrossRef]
- Sheehan, D.J.; Hitchcock, C.A.; Sibley, C.M. Current and Emerging Azole Antifungal Agents. Clin. Microbiol. Rev. 1999, 12, 40–79. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Kaplancikli, Z.A.; Özdemir, A.; Revial, G.; Güven, K. Synthesis and Antimicrobial Activity of Some 2-(Benzo[d]Oxazol/Benzo[d]Imidazol-2-Ylthio)-N-(9 H-Fluoren-9-Yl)Acetamide Derivatives. Phosphorus. Sulfur. Silicon Relat. Elem. 2007, 182, 639–646. [Google Scholar] [CrossRef]
- Nayak, N.; Ramprasad, J.; Dalimba, U. New INH-Pyrazole Analogs: Design, Synthesis and Evaluation of Antitubercular and Antibacterial Activity. Bioorganic Med. Chem. Lett. 2015, 25, 5540–5545. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Romaine, I.M.; Liu, C.; Murthi, P.; Jones, P.L.; Waterson, A.G.; Sulikowski, G.A.; Zwiebel, L.J. Structure–Activity Relationship of a Broad-Spectrum Insect Odorant Receptor Agonist. ACS Chem. Biol. 2012, 7, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Santos, S.; Ventura, C.; Elvas-Leitão, R.; Santos, L.; Vitorino, S.; Reis, M.; Miranda, V.; Correia, H.F.; Aires-de-Sousa, J.; et al. Design, Synthesis and Biological Evaluation of Novel Isoniazid Derivatives with Potent Antitubercular Activity. Eur. J. Med. Chem. 2014, 81, 119–138. [Google Scholar] [CrossRef]
- Caldara, M.; Belgiovine, C.; Secchi, E.; Rusconi, R. Environmental, Microbiological, and Immunological Features of Bacterial Biofilms Associated with Implanted Medical Devices. Clin. Microbiol. Rev. 2022, 35, e0022120. [Google Scholar] [CrossRef]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the Emerging Threat of Antifungal Resistance to Human Health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef]
- Ghobadi, E.; Saednia, S.; Emami, S. Synthetic Approaches and Structural Diversity of Triazolylbutanols Derived from Voriconazole in the Antifungal Drug Development. Eur. J. Med. Chem. 2022, 231, 114161. [Google Scholar] [CrossRef]
- Marchetti, O.; Moreillon, P.; Glauser, M.P.; Bille, J.; Sanglard, D. Potent Synergism of the Combination of Fluconazole and Cyclosporine in Candida Albicans. Antimicrob. Agents Chemother. 2000, 44, 2373–2381. [Google Scholar] [CrossRef]
- Osterberg, R.E.; Demerlis, C.C.; Hobson, D.W.; McGovern, T.J. Trends in Excipient Safety Evaluation. Int. J. Toxicol. 2011, 30, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.C.; Nelson, C.E.; Yu, S.S.; Beavers, K.R.; Kim, A.J.; Li, H.; Nelson, H.M.; Giorgio, T.D.; Duvall, C.L. Ex Vivo Red Blood Cell Hemolysis Assay for the Evaluation of PH-Responsive Endosomolytic Agents for Cytosolic Delivery of Biomacromolecular Drugs. J. Vis. Exp. 2013, 73, e50166. [Google Scholar] [CrossRef] [PubMed]
- Greco, I.; Molchanova, N.; Holmedal, E.; Jenssen, H.; Hummel, B.D.; Watts, J.L.; Håkansson, J.; Hansen, P.R.; Svenson, J. Correlation between Hemolytic Activity, Cytotoxicity and Systemic in Vivo Toxicity of Synthetic Antimicrobial Peptides. Sci. Rep. 2020, 10, 13206. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef] [PubMed]
- Terreni, M.; Taccani, M.; Pregnolato, M. New Antibiotics for Multidrug-Resistant Bacterial Strains: Latest Research Developments and Future Perspectives. Molecules 2021, 26, 2671. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, M.H.; El-Sherbiny, G.M.; Moghannem, S.A.; Abdelmonem, M.; Elsehemy, I.A.; Metwaly, A.M.; Kalaba, M.H. New Combination Approaches to Combat Methicillin-Resistant Staphylococcus Aureus (MRSA). Sci. Rep. 2021, 11, 4240. [Google Scholar] [CrossRef] [PubMed]
- Glen, K.A.; Lamont, I.L. β-Lactam Resistance in Pseudomonas Aeruginosa: Current Status, Future Prospects. Pathogens 2021, 10, 1638. [Google Scholar] [CrossRef] [PubMed]
- Keniya, M.V.; Sabherwal, M.; Wilson, R.K.; Woods, M.A.; Sagatova, A.A.; Tyndall, J.D.A.; Monk, B.C. Crystal Structures of Full-Length Lanosterol 14α-Demethylases of Prominent Fungal Pathogens Candida Albicans and Candida Glabrata Provide Tools for Antifungal Discovery. Antimicrob. Agents Chemother. 2018, 62, e01134-18. [Google Scholar] [CrossRef]
- Siwek, A.; Stączek, P.; Wujec, M.; Stefańska, J.; Kosikowska, U.; Malm, A.; Jankowski, S.; Paneth, P. Biological and Docking Studies of Topoisomerase IV Inhibition by Thiosemicarbazides. J. Mol. Model. 2011, 17, 2297–2303. [Google Scholar] [CrossRef]
- Lo Monte, F.; Kramer, T.; Gu, J.; Anumala, U.R.; Marinelli, L.; La Pietra, V.; Novellino, E.; Franco, B.; Demedts, D.; Van Leuven, F.; et al. Identification of Glycogen Synthase Kinase-3 Inhibitors with a Selective Sting for Glycogen Synthase Kinase-3α. J. Med. Chem. 2012, 55, 4407–4424. [Google Scholar] [CrossRef]
- Baeva, L.A.; Nugumanov, R.M.; Gataullin, R.R.; Fatykhov, A.A. Reaction of 3-[(Alkylsulfanyl)Methyl]Pentane-2,4-Diones with Nicotinic Hydrazide. Chem. Heterocycl. Compd. 2020, 56, 548–554. [Google Scholar] [CrossRef]
- Fang, T.; Tan, Q.; Ding, Z.; Liu, B.; Xu, B. Pd-Catalyzed Oxidative Annulation of Hydrazides with Isocyanides: Synthesis of 2-Amino-1,3,4-Oxadiazoles. Org. Lett. 2014, 16, 2342–2345. [Google Scholar] [CrossRef]
- Naveen Kumar, H.S.; Parumasivam, T.; Ibrahim, P.; Asmawi, M.Z.; Sadikun, A. Synthesis of Hydrophobic N-Acylated Isonicotinic Acid Hydrazide Derivatives as Potential Enoyl–Acyl Carrier Protein Reductase (InhA) Inhibitors. Med. Chem. Res. 2014, 23, 1267–1277. [Google Scholar] [CrossRef]
- Calvet, H.M.; Yeaman, M.R.; Filler, S.G. Reversible Fluconazole Resistance in Candida Albicans: A Potential in Vitro Model. Antimicrob. Agents Chemother. 1997, 41, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.S.; Galask, R.P.; Messer, S.A.; Hollis, R.J.; Diekema, D.J.; Pfaller, M.A. Antifungal Susceptibilities of Candida Species Causing Vulvovaginitis and Epidemiology of Recurrent Cases. J. Clin. Microbiol. 2005, 43, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Panariello, B.H.D.; Klein, M.I.; Mima, E.G.D.O.; Pavarina, A.C. Fluconazole Impacts the Extracellular Matrix of Fluconazole-Susceptible and -Resistant Candida Albicans and Candida Glabrata Biofilms. J. Oral Microbiol. 2018, 10, 1476644. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J.; Mendez, M.; Kibbler, C.; Erzsebet, P.; Chang, S.C.; Gibbs, D.L.; Newell, V.A.; Finquelievich, J.; Tiraboschi, N.; et al. Candida Guilliermondii, an Opportunistic Fungal Pathogen with Decreased Susceptibility to Fluconazole: Geographic and Temporal Trends from the ARTEMIS DISK Antifungal Surveillance Program. J. Clin. Microbiol. 2006, 44, 3551–3556. [Google Scholar] [CrossRef]
- CLSI, Clinical and Laboratory Standards Institute (formerly NCCLS, National Committee for Clinical and Laboratory Standards). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard M27-A2; National Committee for Clinical and Laboratory Standards: Wayne, PA, USA, 2002; Volume 22. [Google Scholar]
- Montagna, M.T.; Lovero, G.; Coretti, C.; Martinelli, D.; De Giglio, O.; Iatta, R.; Balbino, S.; Rosato, A.; Caggiano, G. Susceptibility to Echinocandins of Candida spp. Strains Isolated in Italy Assessed by European Committee for Antimicrobial Susceptibility Testing and Clinical Laboratory Standards Institute Broth Microdilution Methods. BMC Microbiol. 2015, 15, 106. [Google Scholar] [CrossRef]
- Scorneaux, B.; Angulo, D.; Borroto-Esoda, K.; Ghannoum, M.; Peel, M.; Wring, S. SCY-078 Is Fungicidal against Candida Species in Time-Kill Studies. Antimicrob. Agents Chemother. 2017, 61, e01961-16. [Google Scholar] [CrossRef]
- Alfano, K.M.; Tarasev, M.; Meines, S.; Parunak, G. An Approach to Measuring RBC Haemolysis and Profiling RBC Mechanical Fragility. J. Med. Eng. Technol. 2016, 40, 162–171. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Asagi, T.; Miyoshi-Akiyama, T.; Fujino, T.; Kobayashi, I.; Morita, K.; Kikuchi, Y.; Kuratsuji, T.; Kirikae, T. Multidrug-Resistant Pseudomonas Aeruginosa Strain That Caused an Outbreak in a Neurosurgery Ward and Its Aac(6′)-Iae Gene Cassette Encoding a Novel Aminoglycoside Acetyltransferase. Antimicrob. Agents Chemother. 2005, 49, 3734–3742. [Google Scholar] [CrossRef]
- Grim, C.J.; Kozlova, E.V.; Sha, J.; Fitts, E.C.; van Lier, C.J.; Kirtley, M.L.; Joseph, S.J.; Read, T.D.; Burd, E.M.; Tall, B.D.; et al. Characterization of Aeromonas Hydrophila Wound Pathotypes by Comparative Genomic and Functional Analyses of Virulence Genes. mBio 2013, 4, e00064-13. [Google Scholar] [CrossRef]
- Cockerill, F.R. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 9th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C. glabrata ATCC 2001 | C. parapsilosis ATCC 22019 | C. albicans ATCC 36082 | C. albicans CL1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC (µg/mL) | Inhib. (%) | ZOI (mm) | AFI | MIC (µg/mL) | Inhib. (%) | ZOI (mm) | AFI | MIC (µg/mL) | Inhib. (%) | ZOI (mm) | AFI | MIC (µg/mL) | Inhib. (%) | ZOI (mm) | AFI | |
| Fluconazole | 20 ± 1.33 | 81.88 | 13.0 | - | ≥24 ± 0.83 | 78.31 | 12.4 | - | ≥24 ± 0.83 | 71.29 | 13.4 | - | ≥24 ± 1.05 | 42.19 | 5.0 | - |
| 5 | 16 ± 0.54 | 91.52 | 14.3 * | 1.13 | 12 ± 0.40 | 91.00 | 10.8 * | 1.040 | ≥24 ± 0.54 | 70.58 | 8.2 | 0.95 | ≥24 ± 0.54 | 72.49 | 5.9 | 1.54 |
| 6 | 16 ± 0.42 | 92.57 | 14.7* | 1.33 | 20 ± 0.14 | 91.00 | 12.9 * | 1.04 | 24 ± 0.52 | 87.54 | 12.8 | 1.05 * | ≥24 ± 0.52 | 76.81 | 7.7 | 2.26 * |
| 7 | ≥24 ± 1.40 | 68.78 | 7.0 * | 1.10 | 16 ± 1.40 | 80.23 | 12.9 * | 0.87 | ≥24 ± 0.45 | 23.17 | 14.1 | 0.61 | ≥24 ± 0.45 | 66.79 | 11.3 | 1.18 * |
| 10 | 20 ± 0.81 | 86.30 | 8.3 | 0.63 | 20 ± 0.72 | 80.41 | 11.1 * | 1.17 | ≥24 ± 1.85 | 65.80 | 12.8 | 0.96 | ≥24 ± 1.85 | 57.81 | 6.8 | 1.36 |
| MIC (µg/mL) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ampicillin/ Cloxacillin | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| Proteus mirabilis212a | 32 | - | - | - | 16 | 4 | - | - | 4 | 4 | 16 | - | 4 |
| Pseudomonas aeruginosa PA-01 | 32 | - | - | - | 8 | - | - | - | - | - | - | - | 8 |
| Salmonella typhi XDR ST-CL-15 | >32 | 4 | - | 8 | - | - | 16 | 8 | - | - | - | 16 | - |
| Proteus mirabilis ATCC 12453 | 2 | - | - | - | 16 | 4 | - | - | - | - | - | - | - |
| Pseudomonas aeruginosa ATCC 27853 | 8 | - | - | - | 4 | - | - | - | - | 4 | - | - | - |
| Aeromonas hydrophila ATCC 7966 | 8 | - | - | 2 | - | - | 16 | - | - | - | - | - | - |
| Enterococcus faecalis ATCC 29212 | 4 | - | - | - | - | - | 8 | - | - | - | - | - | - |
| Staphylococcus aureus ATCC 29213 | 16 | - | - | - | 8 | 8 | 2 | 4 | - | - | - | 16 | 16 |
| Compounds | Docking Scores | Hydrogen Bonding | Hydrophobic/π-Interaction |
|---|---|---|---|
| 4 | −6.4 | Arg72 with carbonyl, Thr163 with nitrogen of the hydrazide group, Arg132 with pyridine | Asn42, Arg72and Pro75 formed hydrophobic and π-cation interaction with pyridine and lipophilic chain |
| 5 | −6.2 | Met512 with the nitrogen of Hydrazide group | Leu130, Thr131, Hist382, Ser383 and Phe242 with hydrophobic chain and the pyridine ring, π-π stacking interaction between Tyr127 and the pyridine ring |
| 6 | −6.6 | His382 and Ser383 with Pyridine ring | π-π Stacking interaction between His382 and pyridine ring, hydrophobic interations between Tyr127, Thr131 and Leu130 with lipophilic chain of the compound |
| 7 | −7.1 | His382 and Ser383 with pyridine ring, Ty127 with the nitrogen of hydrazide group | Hydrophobic interactions between Thr131, Tyr127, Leu130, Phe237 and Phe242 with lipophilic chain of the compound |
| 10 | −6.3 | His382 with pyridine functionalized carbonyl, Ser383 with hydrazide | π-Cation interaction with aromatic ring of His382, Tyr127 and Thr131 with lipohilic chain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, F.-A.; Yaqoob, S.; Ali, S.; Tanveer, N.; Wang, Y.; Ashraf, S.; Hasan, K.A.; Khalifa, S.A.M.; Shou, Q.; Ul-Haq, Z.; et al. Designing Functionally Substituted Pyridine-Carbohydrazides for Potent Antibacterial and Devouring Antifungal Effect on Multidrug Resistant (MDR) Strains. Molecules 2023, 28, 212. https://doi.org/10.3390/molecules28010212
Khan F-A, Yaqoob S, Ali S, Tanveer N, Wang Y, Ashraf S, Hasan KA, Khalifa SAM, Shou Q, Ul-Haq Z, et al. Designing Functionally Substituted Pyridine-Carbohydrazides for Potent Antibacterial and Devouring Antifungal Effect on Multidrug Resistant (MDR) Strains. Molecules. 2023; 28(1):212. https://doi.org/10.3390/molecules28010212
Chicago/Turabian StyleKhan, Farooq-Ahmad, Sana Yaqoob, Shujaat Ali, Nimra Tanveer, Yan Wang, Sajda Ashraf, Khwaja Ali Hasan, Shaden A. M. Khalifa, Qiyang Shou, Zaheer Ul-Haq, and et al. 2023. "Designing Functionally Substituted Pyridine-Carbohydrazides for Potent Antibacterial and Devouring Antifungal Effect on Multidrug Resistant (MDR) Strains" Molecules 28, no. 1: 212. https://doi.org/10.3390/molecules28010212
APA StyleKhan, F.-A., Yaqoob, S., Ali, S., Tanveer, N., Wang, Y., Ashraf, S., Hasan, K. A., Khalifa, S. A. M., Shou, Q., Ul-Haq, Z., Jiang, Z.-H., & El-Seedi, H. R. (2023). Designing Functionally Substituted Pyridine-Carbohydrazides for Potent Antibacterial and Devouring Antifungal Effect on Multidrug Resistant (MDR) Strains. Molecules, 28(1), 212. https://doi.org/10.3390/molecules28010212