
Acridine as an Anti-Tumour Agent: A Critical Review
 , ,
, ,  , , and
, , and
Abstract
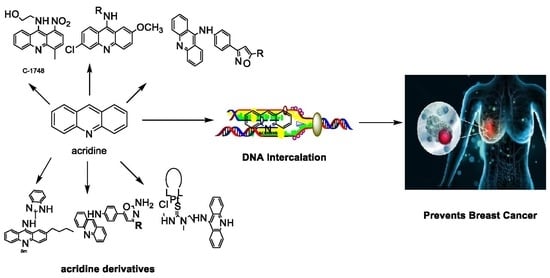
1. Introduction
2. Acridine as an Anti-Tumour Agent
2.1. Acridine
2.2. 9-Amino Acridine
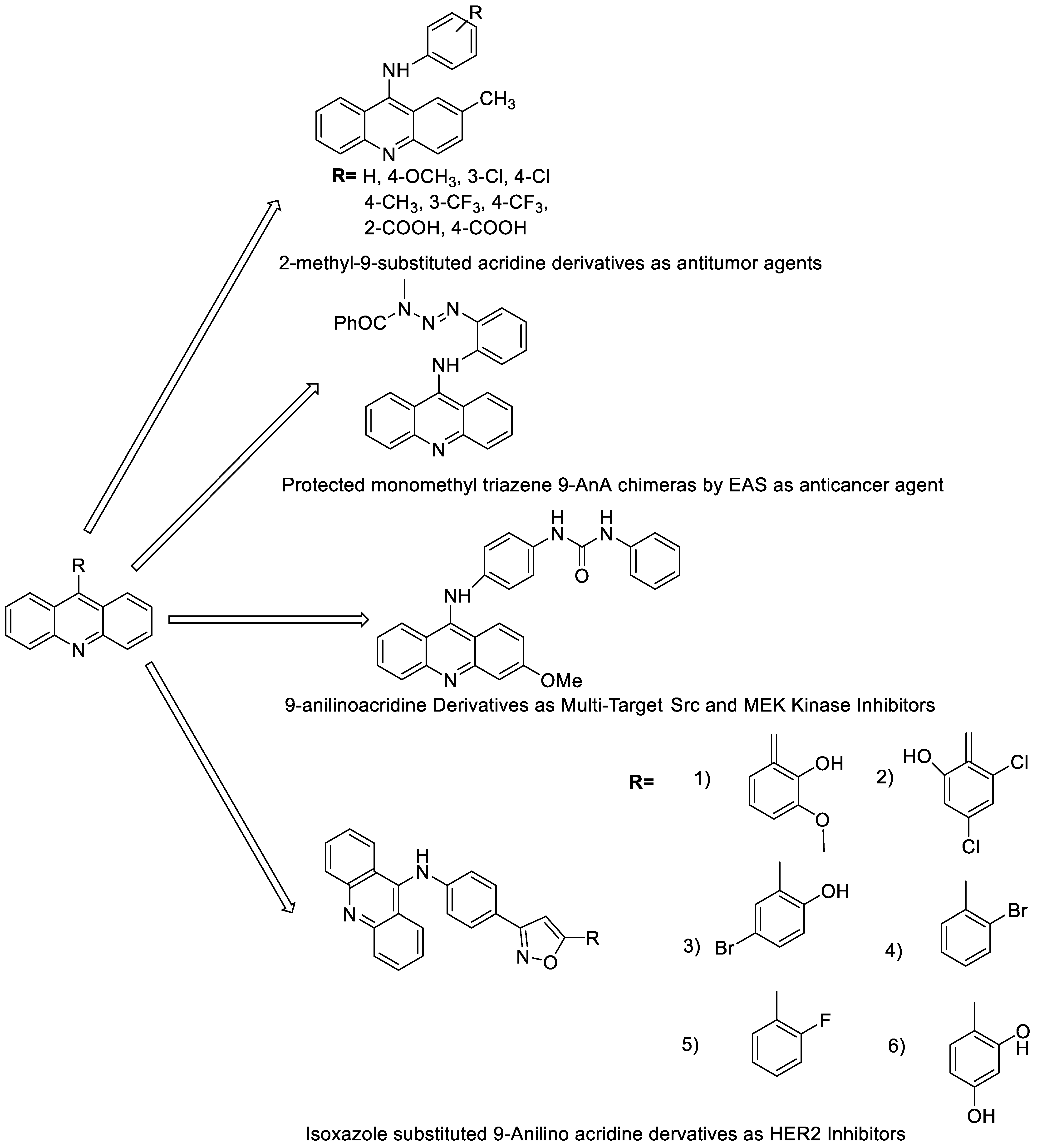
2.3. 9-Anilino Acridines
2.4. Acridine Thiourea Gold
2.5. Acridine-Thiazolidinone
2.6. Acridinone
2.7. Benzimidazole Substituted Acridines
2.8. Benzoacridine
2.9. Imidazoacridinone
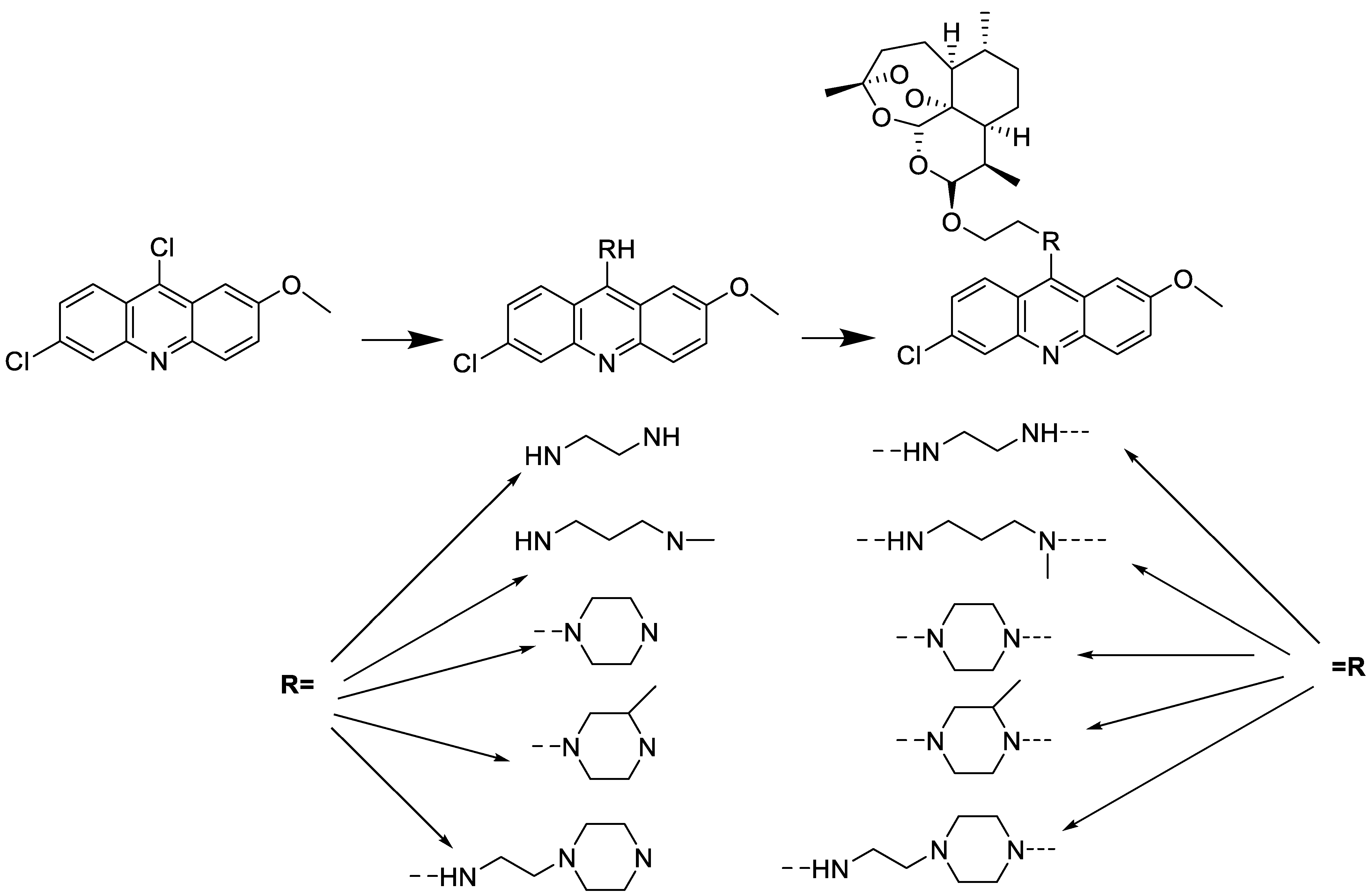
2.10. Nitroacridine
2.11. Oxazine
2.12. Platinum-Acridine Anti-Cancer Agents
2.13. Quinacrine
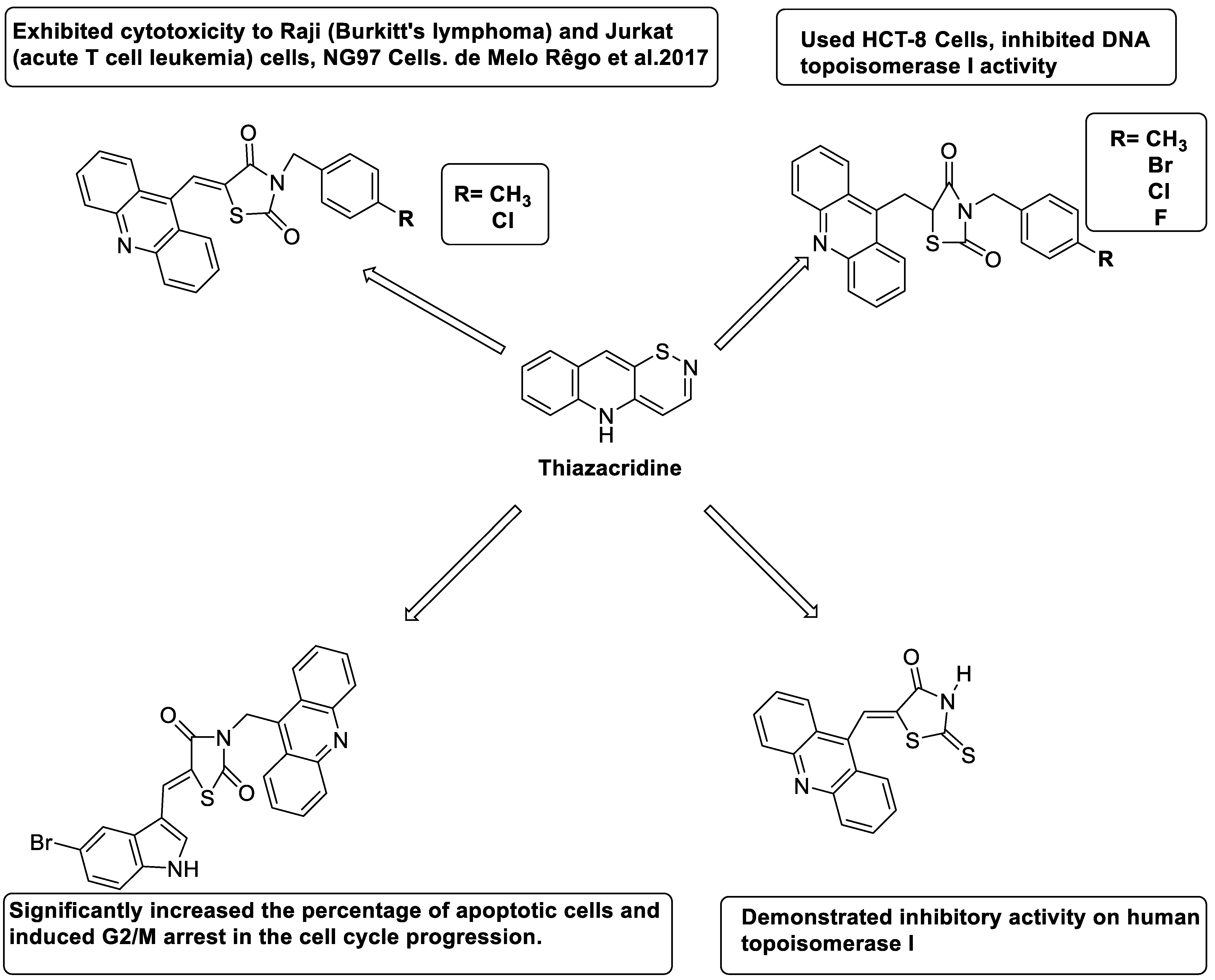
2.14. Thiazacridine
2.15. Azacridine
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cisáriková, A.; Barbieriková, Z.; Janovec, L.; Imrich, J.; Hunáková, L.; Bačová, Z.; Paulíková, H. Acridin-3, 6-dialkyldithiourea hydrochlorides as new photosensitizers for photodynamic therapy of mouse leukaemia cells. Bioorg. Med. Chem. 2016, 24, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Calvillo-Páez, V.; Sotelo-Mundo, R.R.; Leyva-Peralta, M.; Gálvez-Ruiz, J.C.; Corona-Martínez, D.; Moreno-Corral, R.; Escobar-Picos, R.; Höpfl, H.; Juárez-Sánchez, O.; Lara, K.O. Synthesis, spectroscopic, physicochemical and structural characterization of tetrandrine-based macrocycles functionalized with acridine and anthracene groups: DNA binding and anti-proliferative activity. Chem.-Biol. Interact. 2018, 286, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mróz, A.; Ryska, I.; Sominko, H.; Bejrowska, A.; Mazerska, Z. Drug-drug interaction potential of antitumor acridine agent C-1748: The substrate of UDP-glucuronosyltransferases 2B7, 2B17 and the inhibitor of 1A9 and 2B7. Pharmacol. Rep. 2018, 70, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Szymański, P.; Olszewska, P.; Mikiciuk-Olasik, E.; Różalski, A.; Maszewska, A.; Markiewicz, L.; Cuchra, M.; Majsterek, I. Novel tetrahydroacridine and cyclopentaquinoline derivatives with fluorobenzoic acid moiety induce cell cycle arrest and apoptosis in lung cancer cells by activation of DNA damage signaling. Tumor Biol. 2017, 39, 1010428317695011. [Google Scholar] [CrossRef]
- Takac, P.; Kello, M.; Vilkova, M.; Vaskova, J.; Michalkova, R.; Mojzisova, G.; Mojzis, J. Antiproliferative effect of acridine chalcone is mediated by induction of oxidative stress. Biomolecules 2020, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.K.F.; Duarte, S.S.; Lisboa, T.M.; Ferreira, R.C.; Lopes, A.L.d.; Carvalho, D.; Rodrigues-Mascarenhas, S.; da Silva, P.M.; Segundo, M.A.; de Moura, R.O. Antitumor effect of a novel spiro-acridine compound is associated with up-regulation of Th1-type responses and antiangiogenic action. Molecules 2020, 25, 29. [Google Scholar] [CrossRef]
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M. Toxicity and antitumor activity of a thiophene–acridine hybrid. Molecules 2020, 25, 64. [Google Scholar] [CrossRef]
- Almeida, S.M.V.; Silva, L.P.B.G.; Lima, L.R.A.; Botelho, S.P.S.; Lima, M.d.C.; Pitta, I.d.; Beltrão, E.I.C.; Junior, L.B.C. Dimethyl-2-[(acridin-9-yl) methylidene]-malonate as fluorescent probe for histochemical analysis. Microsc. Res. Tech. 2017, 80, 608–614. [Google Scholar] [CrossRef]
- Azab, H.A.; Hussein, B.H.; El-Azab, M.F.; Gomaa, M.; El-Falouji, A.I. Bis (acridine-9-carboxylate)-nitro-europium (III) dihydrate complex a new apoptotic agent through Flk-1 down regulation, caspase-3 activation and oligonucleosomes DNA fragmentation. Bioorg. Med. Chem. 2013, 21, 223–234. [Google Scholar] [CrossRef]
- Borowa-Mazgaj, B.; Mróz, A.; Augustin, E.; Paluszkiewicz, E.; Mazerska, Z. The overexpression of CPR and P450 3A4 in pancreatic cancer cells changes the metabolic profile and increases the cytotoxicity and pro-apoptotic activity of acridine antitumor agent, C-1748. Biochem. Pharmacol. 2017, 142, 21–38. [Google Scholar] [CrossRef]
- Campbell, A.P.; Wakelin, L.P.; Denny, W.A.; Finch, A.M. Homobivalent conjugation increases the allosteric effect of 9-aminoacridine at the α1-adrenergic receptors. Mol. Pharmacol. 2017, 91, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Lopes-Nunes, J.; Lopes, A.C.; Campello, M.P.C.; Paulo, A.; Queiroz, J.A.; Cruz, C. Aptamer-guided acridine derivatives for cervical cancer. Org. Biomol. Chem. 2019, 17, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Hazawa, M.; Hosokawa, Y.; Igarashi, J.; Suga, H.; Kashiwakura, I. Novel acridine-based N-acyl-homoserine lactone analogs induce endoreduplication in the human oral squamous carcinoma cell line SAS. Biol. Pharm. Bull. 2012, 35, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, D.; Li, W.; Yin, J.; Zhang, Y.; Yuan, Z.; Gao, C.; Liu, F.; Jiang, Y. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg. Med. Chem. 2018, 26, 3958–3966. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Li, X.; Lu, X.; Zhang, L.; Li, R.; Zhang, N.; Liu, S.; Yang, X.; Wang, Y.; Zhao, Y. A novel acridine derivative, LS-1-10 inhibits autophagic degradation and triggers apoptosis in colon cancer cells. Cell Death Dis. 2017, 8, e3086. [Google Scholar] [CrossRef] [PubMed]
- Gardette, M.; Papon, J.; Bonnet, M.; Desbois, N.; Labarre, P.; Wu, T.-D.; Miot-Noirault, E.; Madelmont, J.-C.; Guerquin-Kern, J.-L.; Chezal, J.-M. Evaluation of new iodinated acridine derivatives for targeted radionuclide therapy of melanoma using 125I, an Auger electron emitter. Investig. New Drugs 2011, 29, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Gardette, M.; Viallard, C.; Paillas, S.; Guerquin-Kern, J.-L.; Papon, J.; Moins, N.; Labarre, P.; Desbois, N.; Wong-Wah-Chung, P.; Palle, S. Evaluation of two 125I-radiolabeled acridine derivatives for Auger-electron radionuclide therapy of melanoma. Investig. New Drugs 2014, 32, 587–597. [Google Scholar] [CrossRef]
- Ghosh, R.; Bhowmik, S.; Guha, D. 9-phenyl acridine exhibits antitumour activity by inducing apoptosis in A375 cells. Mol. Cell. Biochem. 2012, 361, 55–66. [Google Scholar] [CrossRef]
- Girek, M.; losiński, K.K.; Grobelski, B.; Pizzimenti, S.; Cucci, M.A.; Daga, M.; Barrera, G.; Pasieka, Z.; Czarnecka, K.; Szymański, P. Novel tetrahydroacridine derivatives with iodobenzoic moieties induce G0/G1 cell cycle arrest and apoptosis in A549 non-small lung cancer and HT-29 colorectal cancer cells. Mol. Cell. Biochem. 2019, 460, 123–150. [Google Scholar] [CrossRef]
- Haider, M.R.; Ahmad, K.; Siddiqui, N.; Ali, Z.; Akhtar, M.J.; Fuloria, N.; Fuloria, S.; Ravichandran, M.; Yar, M.S. Novel 9-(2-(1-arylethylidene) hydrazinyl) acridine derivatives: Target Topoisomerase 1 and growth inhibition of HeLa cancer cells. Bioorg. Chem. 2019, 88, 102962. [Google Scholar] [CrossRef]
- Hansda, S.; Ghosh, G.; Ghosh, R. 9-phenyl acridine photosensitizes A375 cells to UVA radiation. Heliyon 2020, 6, e04733. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J. Acridine derivatives as inhibitors of the ire1α–xbp1 pathway are cytotoxic to human multiple myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef]
- Mangueira, V.M.; Batista, T.M.; Brito, M.T.; de Sousa, T.K.G.; da Cruz, R.M.D.; de Abrantes, R.A.; Veras, R.C.; de Medeiros, I.A.; Medeiros, K.K.d.; Pereira, A.L.d. A new acridine derivative induces cell cycle arrest and antiangiogenic effect on Ehrlich ascites carcinoma model. Biomed. Pharmacother. 2017, 90, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; He, K.; Yoo, M.-H.; Chan, C.-B.; Liu, X.; Zhang, Z.; Olson, J.J.; Xiao, G.; Wang, L.; Mao, H. Acridine yellow G blocks glioblastoma growth via dual inhibition of epidermal growth factor receptor and protein kinase C kinases. J. Biol. Chem. 2012, 287, 6113–6127. [Google Scholar] [CrossRef] [PubMed]
- Paluszkiewicz, E.; Horowska, B.; Borowa-Mazgaj, B.; Peszyńska-Sularz, G.; Paradziej-Lukowicz, J.; Augustin, E.; Konopa, J.; Mazerska, Z. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020, 204, 112599. [Google Scholar] [CrossRef]
- Ramesh, K.B.; Pasha, M.A. Study on one-pot four-component synthesis of 9-aryl-hexahydro-acridine-1, 8-diones using SiO2–I as a new heterogeneous catalyst and their anticancer activity. Bioorg. Med. Chem. Lett. 2014, 24, 3907–3913. [Google Scholar] [CrossRef]
- Raza, A.; Jacobson, B.A.; Benoit, A.; Patel, M.R.; Jay-Dixon, J.; Hiasa, H.; Ferguson, D.M.; Kratzke, R.A. Novel acridine-based agents with topoisomerase II inhibitor activity suppress mesothelioma cell proliferation and induce apoptosis. Investig. New Drugs 2012, 30, 1443–1448. [Google Scholar] [CrossRef]
- Roe, S.; Gunaratnam, M.; Spiteri, C.; Sharma, P.; Alharthy, R.D.; Neidle, S.; Moses, J.E. Synthesis and biological evaluation of hybrid acridine-HSP90 ligand conjugates as telomerase inhibitors. Org. Biomol. Chem. 2015, 13, 8500–8504. [Google Scholar] [CrossRef]
- Salem, O.M.; Vilková, M.; JanoĿková, J.; Jendželovskỳ, R.; FedoroĿko, P.; Žilecká, E.; Kašpárková, J.; Brabec, V.; Imrich, J.; Kožurková, M. New spiro tria (thia) zolidine acridines as topoisomerase inhibitors, DNA binders and cytostatic compounds. Int. J. Biol. Macromol. 2016, 86, 690–700. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, A.; Sharma, A.; Kaur, G. Identification of amino acid appended acridines as potential leads to anti-cancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3854–3858. [Google Scholar] [CrossRef]
- Ungvarsky, J.; Plsikova, J.; Janovec, L.; Koval, J.; Mikes, J.; Mikesová, L.; Harvanova, D.; Fedorocko, P.; Kristian, P.; Kasparkova, J. Novel trisubstituted acridines as human telomeric quadruplex binding ligands. Bioorg. Chem. 2014, 57, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Wilks, T.R.; Pitto-Barry, A.; Kirby, N.; Stulz, E.; O’Reilly, R.K. Construction of DNA–polymer hybrids using intercalation interactions. Chem. Commun. 2014, 50, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Han, M.; Cowan, J.A. Toward the Design of a Catalytic Metallodrug: Selective Cleavage of G-Quadruplex Telomeric DNA by an Anticancer Copper–Acridine–ATCUN Complex. Angew. Chem. 2015, 127, 1921–1925. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, B.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.M.; Jacobson, B.A.; Jay-Dixon, J.; Patel, M.R.; Kratzke, R.A.; Raza, A. Targeting topoisomerase II activity in NSCLC with 9-aminoacridine derivatives. Anticancer Res. 2015, 35, 5211–5217. [Google Scholar] [PubMed]
- Sun, Y.-W.; Chen, K.-Y.; Kwon, C.-H.; Chen, K.-M. CK0403, a 9-aminoacridine, is a potent anti-cancer agent in human breast cancer cells. Mol. Med. Rep. 2016, 13, 933–938. [Google Scholar] [CrossRef][Green Version]
- Aguilera, R.F.; Artali, R.; Benoit, A.; Gómez, R.G.; Casadellà, R.E.i.; Ferguson, D.M.; Sham, Y.Y.; Mazzini, S. Structure and stability of human telomeric G-quadruplex with preclinical 9-amino acridines. PLoS ONE 2013, 8, E57701. [Google Scholar]
- Joubert, J.P.; Smit, F.J.; Plessis, L.D.; Smith, P.J.; N’Da, D.D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin–acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27. [Google Scholar] [CrossRef]
- Ju, W.; Zhang, M.; Petrus, M.; Maeda, M.; Pise-Masison, C.A.; Waldmann, T.A. Combination of 9-aminoacridine with Campath-1H provides effective therapy for a murine model of adult T-cell leukaemia. Retrovirology 2014, 11, 43. [Google Scholar] [CrossRef]
- Kava, H.W.; Murray, V. CpG methylation increases the DNA binding of 9-aminoacridine carboxamide Pt analogues. Bioorg. Med. Chem. 2016, 24, 4701–4710. [Google Scholar] [CrossRef]
- Luan, X.; Gao, C.; Zhang, N.; Chen, Y.; Sun, Q.; Tan, C.; Liu, H.; Jin, Y.; Jiang, Y. Exploration of acridine scaffold as a potentially interesting scaffold for discovering novel multi-target VEGFR-2 and Src kinase inhibitors. Bioorganic Med. Chem. 2011, 19, 3312–3319. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Murray, V. The sequence selectivity of DNA-targeted 9-aminoacridine cisplatin analogues in a telomere-containing DNA sequence. JBIC J. Biol. Inorg. Chem. 2011, 16, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.; Blake, A.J.; Benoit, A.; David, M.F.; Robert, A.K. Efficacy of substituted 9-aminoacridine derivatives in small cell lung cancer. Investig. New Drugs 2013, 31, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Sharma, S.; Silakari, O.; Singh, M.; Kaur, M. Synthesis, characterization and antitumor activity of 2-methyl-9-substituted acridines. Arab. J. Chem. 2017, 10, S956–S963. [Google Scholar] [CrossRef]
- Walunj, D.; Egarmina, K.; Tuchinsky, H.; Shpilberg, O.; Hershkovitz-Rokah, O.; Grynszpan, F.; Gellerman, G. Expedient synthesis and anticancer evaluation of dual-action 9-anilinoacridine methyl triazene chimeras. Chem. Biol. Drug Des. 2021, 97, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Li, X.; Li, L.; Zhang, B.; Gao, C.; Chen, Y.; Tan, C.; Liu, H.; Xie, W.; Yang, T. Design, synthesis and evaluation of acridine derivatives as multi-target Src and MEK kinase inhibitors for anti-tumor treatment. Bioorg. Med. Chem. 2016, 24, 261–269. [Google Scholar] [CrossRef]
- Lo, W.-L.; Chu, P.-Y.; Lee, T.-H.; Su, T.-L.; Chien, Y.; Chen, Y.-W.; Huang, P.-I.; Tseng, L.-M.; Tu, P.-H.; Kao, S.-Y. A Combined DNA-affinic molecule and N-mustard alkylating agent has an anti-cancer effect and induces autophagy in oral cancer cells. Int. J. Mol. Sci. 2012, 13, 3277–3290. [Google Scholar] [CrossRef]
- Kalirajan, R.; Pandiselvi, A.; Gowramma, B.; Balachandran, P. In-silico design, ADMET screening, MM-GBSA binding free energy of some novel isoxazole substituted 9-anilinoacridines as HER2 inhibitors targeting breast cancer. Curr. Drug Res. Rev. Former. Curr. Drug Abus. Rev. 2018, 11, 118–128. [Google Scholar] [CrossRef]
- Perez, S.A.; de Haro, C.; Vicente, C.; Donaire, A.; Zamora, A.; Zajac, J.; Kostrhunova, H.; Brabec, V.; Bautista, D.; Ruiz, J. New acridine thiourea gold (I) anticancer agents: Targeting the nucleus and inhibiting vasculogenic mimicry. ACS Chem. Biol. 2017, 12, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Bertrand, B.; Fernandez-Cestau, J.; Waller, Z.A.; O’Connell, M.A.; Searcey, M.; Bochmann, M. Acridine-decorated cyclometallated gold (III) complexes: Synthesis and anti-tumour investigations. Dalton Trans. 2018, 47, 13523–13534. [Google Scholar] [CrossRef]
- Paulíková, H.; Vantová, Z.; Hunáková, L.; Čižeková, L.; Čarná, M.; Kožurková, M.; Sabolová, D.; Kristian, P.; Hamul’aková, S.; Imrich, J. DNA binding acridine–thiazolidinone agents affecting intracellular glutathione. Bioorg. Med. Chem. 2012, 20, 7139–7148. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.W.; Silva, T.G.; Pitta, M.G.d.; Bezerra, D.P.; Costa-Lotufo, L.V.; de Moraes, M.O.; Pessoa, C.; de Moura, M.A.F.; de Abreu, F.C.; de Lima, M.d.C.A. Synthesis and cytotoxic activity of new acridine-thiazolidine derivatives. Bioorg. Med. Chem. 2012, 20, 3533–3539. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.G.; Marques, F.B.; da Fonseca, M.B.; Rogerio, K.R.; Graebin, C.S.; Andricopulo, A.D. Discovery of a series of acridinones as mechanism-based tubulin assembly inhibitors with anticancer activity. PLoS ONE 2016, 11, e0160842. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Khanaposhtani, M.; Safavi, M.; Sabourian, R.; Mahdavi, M.; Pordeli, M.; Saeedi, M.; Ardestani, S.K.; Foroumadi, A.; Shafiee, A.; Akbarzadeh, T. Design, synthesis, in vitro cytotoxic activity evaluation, and apoptosis-induction study of new 9 (10H)-acridinone-1, 2, 3-triazoles. Mol. Divers. 2015, 19, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, M.; Chu, R.; Fedejko-Kap, B.; Augustin, E.; Mazerska, Z.; Radominska-Pandya, A.; Chambers, T.C. Metabolic transformation of antitumor acridinone C-1305 but not C-1311 via selective cellular expression of UGT1A10 increases cytotoxic response: Implications for clinical use. Drug Metab. Dispos. 2013, 41, 414–421. [Google Scholar] [CrossRef]
- Chen, K.; Chu, B.; Liu, F.; Li, B.; Gao, C.; Li, L.; Sun, Q.; Shen, Z.; Jiang, Y. New benzimidazole acridine derivative induces human colon cancer cell apoptosis in vitro via the ROS-JNK signaling pathway. Acta Pharmacol. Sin. 2015, 36, 1074–1084. [Google Scholar] [CrossRef]
- Yuan, Z.; Chen, S.; Chen, C.; Chen, J.; Chen, C.; Dai, Q.; Gao, C.; Jiang, Y. Design, synthesis and biological evaluation of 4-amidobenzimidazole acridine derivatives as dual PARP and Topo inhibitors for cancer therapy. Eur. J. Med. Chem. 2017, 138, 1135–1146. [Google Scholar] [CrossRef]
- Gao, C.; Li, B.; Zhang, B.; Sun, Q.; Li, L.; Li, X.; Chen, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and biological evaluation of benzimidazole acridine derivatives as potential DNA-binding and apoptosis-inducing agents. Bioorg. Med. Chem. 2015, 23, 1800–1807. [Google Scholar] [CrossRef]
- Behbahani, F.S.; Tabeshpour, J.; Mirzaei, S.; Golmakaniyoon, S.; Tayarani-Najaran, Z.; Ghasemi, A.; Ghodsi, R. Synthesis and biological evaluation of novel benzo [c] acridine-diones as potential anticancer agents and tubulin polymerization inhibitors. Arch. Der Pharm. 2019, 352, 1800307. [Google Scholar] [CrossRef]
- Guo, Q.-L.; Su, H.-F.; Wang, N.; Liao, S.-R.; Lu, Y.-T.; Ou, T.-M.; Tan, J.-H.; Li, D.; Huang, Z.-S. Synthesis and evaluation of 7-substituted-5, 6-dihydrobenzo [c] acridine derivatives as new c-KIT promoter G-quadruplex binding ligands. Eur. J. Med. Chem. 2017, 130, 458–471. [Google Scholar] [CrossRef]
- Kumar, N.P.; Sharma, P.; Reddy, T.S.; Shankaraiah, N.; Bhargava, S.K.; Kamal, A. Microwave-assisted one-pot synthesis of new phenanthrene fused-tetrahydrodibenzo-acridinones as potential cytotoxic and apoptosis inducing agents. Eur. J. Med. Chem. 2018, 151, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Torikai, K.; Koga, R.; Liu, X.; Umehara, K.; Kitano, T.; Watanabe, K.; Oishi, T.; Noguchi, H.; Shimohigashi, Y. Design and synthesis of benzoacridines as estrogenic and anti-estrogenic agents. Bioorg. Med. Chem. 2017, 25, 5216–5237. [Google Scholar] [CrossRef] [PubMed]
- Paradziej-Lukowicz, J.; Skwarska, A.; Peszyńska-Sularz, G.; Brillowska-Dąbrowska, A.; Konopa, J. Anticancer imidazoacridinone C-1311 inhibits hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF) and angiogenesis. Cancer Biol. Ther. 2011, 12, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3, e293. [Google Scholar] [CrossRef] [PubMed]
- Augustin, E.; Paw\lowska, M.; Polewska, J.; Potega, A.; Mazerska, Z. Modulation of CYP3A4 activity and induction of apoptosis, necrosis and senescence by the anti-tumour imidazoacridinone C-1311 in human hepatoma cells. Cell Biol. Int. 2013, 37, 109–120. [Google Scholar] [CrossRef]
- Dunstan, M.S.; Barnes, J.; Humphries, M.; Whitehead, R.C.; Bryce, R.A.; Leys, D.; Stratford, I.J.; Nolan, K.A. Novel inhibitors of NRH: Quinone oxidoreductase 2 (NQO2): Crystal structures, biochemical activity, and intracellular effects of imidazoacridin-6-ones. J. Med. Chem. 2011, 54, 6597–6611. [Google Scholar] [CrossRef]
- Fedejko-Kap, B.; Bratton, S.M.; Finel, M.; Radominska-Pandya, A.; Mazerska, Z. Role of human UDP-glucuronosyltransferases in the biotransformation of the triazoloacridinone and imidazoacridinone antitumor agents C-1305 and C-1311: Highly selective substrates for UGT1A10. Drug Metab. Dispos. 2012, 40, 1736–1743. [Google Scholar] [CrossRef]
- Polewska, J.; Skwarska, A.; Augustin, E.; Konopa, J. DNA-damaging imidazoacridinone C-1311 induces autophagy followed by irreversible growth arrest and senescence in human lung cancer cells. J. Pharmacol. Exp. Ther. 2013, 346, 393–405. [Google Scholar] [CrossRef]
- Potega, A.; Dabrowska, E.; Niemira, M.; Kot-Wasik, A.; Ronseaux, S.; Henderson, C.J.; Wolf, C.R.; Mazerska, Z. The imidazoacridinone antitumor drug, C-1311, is metabolized by flavin monooxygenases but not by cytochrome P450s. Drug Metab. Dispos. 2011, 39, 1423–1432. [Google Scholar] [CrossRef]
- Skwarska, A.; Augustin, E.; Beffinger, M.; Wojtczyk, A.; Konicz, S.; Laskowska, K.; Polewska, J. Targeting of FLT3-ITD kinase contributes to high selectivity of imidazoacridinone C-1311 against FLT3-activated leukemia cells. Biochem. Pharmacol. 2015, 95, 238–252. [Google Scholar] [CrossRef]
- Wiśniewska, A.; Niemira, M.; Jagiełło, K.; Potęga, A.; Świst, M.; Henderson, C.; Skwarska, A.; Augustin, E.; Konopa, J.; Mazerska, Z. Diminished toxicity of C-1748, 4-methyl-9-hydroxyethylamino-1-nitroacridine, compared with its demethyl analog, C-857, corresponds to its resistance to metabolism in HepG2 cells. Biochem. Pharmacol. 2012, 84, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; You, C.; Zheng, C.; Gu, Y.; Gu, H.; Zhang, R.; Wu, H.; Sun, B. 3-Nitroacridine derivatives arrest cell cycle at G0/G1 phase and induce apoptosis in human breast cancer cells may act as DNA-target anticancer agents. Life Sci. 2018, 206, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wu, H.; You, C.; Gao, Z.; Sun, K.; Wang, M.; Chen, F.; Sun, B. 1,3-dimethyl-6-nitroacridine derivatives induce apoptosis in human breast cancer cells by targeting DNA. Drug Dev. Ind. Pharm. 2019, 45, 212–221. [Google Scholar] [CrossRef]
- Kalirajan, R.; Kulshrestha, V.; Sankar, S.; Jubie, S. Docking studies, synthesis, characterization of some novel oxazine substituted 9-anilinoacridine derivatives and evaluation for their antioxidant and anticancer activities as topoisomerase II inhibitors. Eur. J. Med. Chem. 2012, 56, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Kalirajan, R.; Sankar, S.; Jubie, S.; Gowramma, B. Molecular docking studies and in-silico ADMET screening of some novel oxazine substituted 9-anilinoacridines as topoisomerase II inhibitors. Indian J. Pharm. Educ. Res. 2017, 51, 110–115. [Google Scholar] [CrossRef]
- Kalirajan, R.; Kulshrestha, V.; Sankar, S. Synthesis, characterization and antitumour activity of some novel oxazine substituted 9-anilinoacridines and their 3D-QSAR studies. Indian J. Pharm. Sci. 2018, 80, 921–929. [Google Scholar] [CrossRef]
- Cheung-Ong, K.; Song, K.T.; Ma, Z.; Shabtai, D.; Lee, A.Y.; Gallo, D.; Heisler, L.E.; Brown, G.W.; Bierbach, U.; Giaever, G. Comparative chemogenomics to examine the mechanism of action of DNA-targeted platinum-acridine anticancer agents. ACS Chem. Biol. 2012, 7, 1892–1901. [Google Scholar] [CrossRef]
- Ding, S.; Qiao, X.; Kucera, G.L.; Bierbach, U. Design of a platinum–acridine–endoxifen conjugate targeted at hormone-dependent breast cancer. Chem. Commun. 2013, 49, 2415–2417. [Google Scholar] [CrossRef][Green Version]
- Fahrenholtz, C.D.; Ding, S.; Bernish, B.W.; Wright, M.L.; Zheng, Y.; Yang, M.; Yao, X.; Donati, G.L.; Gross, M.D.; Bierbach, U. Design and cellular studies of a carbon nanotube-based delivery system for a hybrid platinum-acridine anticancer agent. J. Inorg. Biochem. 2016, 165, 170–180. [Google Scholar] [CrossRef]
- Graham, L.A.; Suryadi, J.; West, T.K.; Kucera, G.L.; Bierbach, U. Synthesis, aqueous reactivity, and biological evaluation of carboxylic acid ester-functionalized platinum–acridine hybrid anticancer agents. J. Med. Chem. 2012, 55, 7817–7827. [Google Scholar] [CrossRef]
- Kostrhunova, H.; Malina, J.; Pickard, A.J.; Stepankova, J.; Vojtiskova, M.; Kasparkova, J.; Muchova, T.; Rohlfing, M.L.; Bierbach, U.; Brabec, V. Replacement of a thiourea with an amidine group in a monofunctional platinum–acridine antitumor agent. Effect on DNA interactions, DNA adduct recognition and repair. Mol. Pharm. 2011, 8, 1941–1954. [Google Scholar] [CrossRef] [PubMed]
- Pickard, A.J.; Liu, F.; Bartenstein, T.F.; Haines, L.G.; Levine, K.E.; Kucera, G.L.; Bierbach, U. Redesigning the DNA-Targeted Chromophore in Platinum–Acridine Anticancer Agents: A Structure–Activity Relationship Study. Chem.-Eur. J. 2014, 20, 16174–16187. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Zeitany, A.E.; Wright, M.W.; Essader, A.S.; Levine, K.E.; Kucera, G.L.; Bierbach, U. Analysis of the DNA damage produced by a platinum–acridine antitumor agent and its effects in NCI-H460 lung cancer cells. Metallomics 2012, 4, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Smyre, C.L.; Saluta, G.; Kute, T.E.; Kucera, G.L.; Bierbach, U. Inhibition of DNA synthesis by a platinum–acridine hybrid agent leads to potent cell kill in nonsmall cell lung cancer. ACS Med. Chem. Lett. 2011, 2, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Changchien, J.-J.; Chen, Y.-J.; Huang, C.-H.; Cheng, T.-L.; Lin, S.-R.; Chang, L.-S. Quinacrine induces apoptosis in human leukaemia K562 cells via p38 MAPK-elicited BCL2 down-regulation and suppression of ERK/c-Jun-mediated BCL2L1 expression. Toxicol. Appl. Pharmacol. 2015, 284, 33–41. [Google Scholar] [CrossRef]
- Huang, C.-H.; Lee, Y.-C.; Chen, Y.-J.; Wang, L.-J.; Shi, Y.-J.; Chang, L.-S. Quinacrine induces the apoptosis of human leukaemia U937 cells through FOXP3/miR-183/β-TrCP/SP1 axis-mediated BAX upregulation. Toxicol. Appl. Pharmacol. 2017, 334, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Tripathi, N.; Kathuria, D.; Siddharth, S.; Nayak, A.; Bharatam, P.V.; Kundu, C. Quinacrine and curcumin synergistically increased the breast cancer stem cells death by inhibiting ABCG2 and modulating DNA damage repair pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105682. [Google Scholar] [CrossRef] [PubMed]
- Siddharth, S.; Nayak, D.; Nayak, A.; Das, S.; Kundu, C.N. ABT-888 and quinacrine induced apoptosis in metastatic breast cancer stem cells by inhibiting base excision repair via adenomatous polyposis coli. DNA Repair 2016, 45, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Sheng, L.; Xu, K.; Wang, Y.; Zhu, H.; Zhang, P.; Mu, Q.; Ouyang, G. Anticancer effect of quinacrine on diffuse large B-cell lymphoma via inhibition of MSI2-NUMB signaling pathway Corrigendum in/10.3892/mmr. 2020.11813. Mol. Med. Rep. 2018, 17, 522–530. [Google Scholar]
- Solomon, V.R.; Pundir, S.; Le, H.-T.; Lee, H. Design and synthesis of novel quinacrine-[1,3]-thiazinan-4-one hybrids for their anti-breast cancer activity. Eur. J. Med. Chem. 2017, 143, 1028–1038. [Google Scholar] [CrossRef]
- Solomon, V.R.; Almnayan, D.; Lee, H. Design, synthesis and characterization of novel quinacrine analogs that preferentially kill cancer over non-cancer cells through the down-regulation of Bcl-2 and up-regulation of Bax and Bad. Eur. J. Med. Chem. 2017, 137, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Rego, M.J.d.; de Moura, R.O.; Jacob, I.T.; Lins, T.U.L.e.; Pereira, M.C.; Lima, M.d.C.A.; Galdino-Pitta, M.R.; Pitta, I.d.; Pitta, M.G.d. Synthesis and anticancer evaluation of thiazacridine derivatives reveals new selective molecules to hematopoietic neoplastic cells. Comb. Chem. High Throughput Screen. 2017, 20, 713–718. [Google Scholar]
- Barros, F.W.; Bezerra, D.P.; Ferreira, P.M.; Cavalcanti, B.C.; Silva, T.G.; Pitta, M.G.; de Lima, M.d.C.; Galdino, S.L.; Pitta, I.d.; Costa-Lotufo, L.V. Inhibition of DNA topoisomerase I activity and induction of apoptosis by thiazacridine derivatives. Toxicol. Appl. Pharmacol. 2013, 268, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chagas, M.B.O.; Cordeiro, N.C.C.; Marques, K.M.R.; Pitta, M.G.R.; Rêgo, M.; Lima, M.C.A.; Pitta, M.G.R.; Pitta, I.R. New thiazacridine agents: Synthesis, physical and chemical characterization, and in vitro anticancer evaluation. Hum. Exp. Toxicol. 2017, 36, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Lafayette, E.A.; de Almeida, V.; Mônica, S.; Pitta, M.G.d.; Beltrão, E.I.C.; da Silva, T.G.; de Moura, R.O.; Pitta, I.d.; de Carvalho, L.B.; de Lima, M.D.C.A. Synthesis, DNA binding and topoisomerase I inhibition activity of thiazacridine and imidazacridine derivatives. Molecules 2013, 18, 15035–15050. [Google Scholar] [CrossRef]
- Cui, Z.; Chen, S.; Wang, Y.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur. J. Med. Chem. 2017, 136, 372–381. [Google Scholar] [CrossRef]
- Karelou, M.; Kourafalos, V.; Tragomalou, A.P.; Marakos, P.; Pouli, N.; Tsitsilonis, O.E.; Gikas, E.; Kostakis, I.K. Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules 2020, 25, 4584. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Propyl-AcrDTU |  | Leukaemia L1210 cells | [1] |
| Tetrandrine-based receptors |  | Anti-proliferative studies | [2] |
| 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C1748) |  | Phase II enzymes—UDP-glucuronosyltransferases (UGTs) | [3] |
| - |  | Human lung adenocarcinoma cells | [4] |
| Acridine chalcone 1C ((2 E)-3-(acridin-9-yl)-1-(2,6-dimethoxyphenyl)prop-2-en-1-one) |  | Human colorectal HCT116 cells | [5] |
| Novel spiro-acridine (E)-50-oxo-10-((3,4,5-trimethoxybenzylidene)amino)-10,50-dihydro-10H-spiro[acridine-9,20-pyrrole]40-carbonitrile (AMTAC-17) |  | Anti-tumour activity of AMTAC-17 | [6] |
| 2-((6-Chloro-2-methoxy-acridin-9-yl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]-thiophene-3-carbonitrile (ACS03) |  | Anti-tumour activity | [7] |
| Dimethyl 2-[(acridin-9-yl) methylidene]-malonate (LPSF/IP-81) |  | Invasive ductal carcinoma of human breast. | [8] |
| Bis (acridine-9-carboxylate)-nitro-europium (III) dihydrate |  | Anti-angiogenic and apoptotic activity against an animal model of carcinogenesis | [9] |
| 9-amino-1-nitroacridine |  | Pancreatic cancer | [10] |
| 9-amino acridine derivatives |  | On alpha-adrenergic receptors | [11] |
| AT11-L0 |  | Anti-proliferative activity | [12] |
| Novel acridine-based N-acyl-homoserine lactone (AHL) analogs |  | Human oral squamous carcinoma | [13] |
| New series of acridine hydroxamic acid derivatives |  | Topo II inhibition activity | [14] |
| 9-chloro-2-(3-(dimethylamino) propyl) pyrrolo [2,3,4-kl] acridin-1(2H)-one (LS-1-10) |  | Colon cancer cells | [15] |
| Two iodinated acridine derivatives |  | Targeted radionuclide therapy | [16] |
| [125I] ICF01035 and [125I] ICF01040 |  | Melanoma-targeted in mice bearing B16F0 | [17] |
| 9-phenyl acridine (ACPH) |  | Anti-cancer agent | [18] |
| Tetra hydro acridine derivatives with iodobenzoic moiety |  | Human lung adenocarcinoma, human colorectal adenocarcinoma. | [19] |
| A series of 9-(2-(1-arylethylidene) hydrazinyl) acridine |  | Anti-cancer activity | [20] |
| 9-phenylacridine (ACPH) |  | Antitumour activity | [21] |
| A class of acridine derivatives |  | In vivo MM tumour growth | [22] |
| N0-(2-chloro-6-methoxy-acridin-9-yl)-2-cyano-3-(4-dimethylaminophenyl)-acrilohidrazida (ACS-AZ10). ACS-AZ10 |  | Anti-tumour activity | [23] |
| Acridine yellow G |  | Inhibits both EGFR and PKCs | [24] |
| Unsymmetrical bisacridine derivatives (UAs) |  | Cytotoxicity screening against several tumour cell lines | [25] |
| 9-aryl-hexahydro-acridine-1,8-diones |  | Anti-cancer activity against HepG2 and MCF-7 cell lines | [26] |
| Series of acridine-based catalytic inhibitors |  | Pancreatic cancer and hTopoII catalytic inhibitors | [27] |
| Four hybrid acridine-HSP90 |  | Telomerase inhibitor BRACO-19 | [28] |
| Three new diphenyl substituted spiro triazolidine- and thiazolidinone-acridines |  | Interaction with calf thymus DNA | [29] |
| Amino acid appended acridines |  | Anti-proliferative activity, arrested cells in G0/G1 phase of the cell cycle | [30] |
| A novel series of tri-substituted acridines |  | Mimicking the effects of BRACO19 | [31] |
| DNA–polymer hybrids |  | DNA–polymerisation | [32] |
| A novel DNA-cleaving agent CuGGHK-Acr |  | Targets G4 telomeric DNA | [33] |
| A series of 9-benzyl acridine derivatives |  | Anti-proliferative inhibitors | [34] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Acridine-based catalytic inhibitors |  | Anti-proliferative activity | [35] |
| CK0403 |  | Treatment of breast cancer | [36] |
| 9-amino acridines |  | Anti-cancer activity | [37] |
| 9-aminoacridine and artemisinin–acridine |  | Anti-malarial activity, anti-cancer activity | [38] |
| 9-aminoacridine (9AA) |  | Anti-leukaemic cells | [39] |
| 9-aminoacridine carboxamide |  | DNA-targeted intercalator | [40] |
| 9-aminoacridine derivatives |  | Anti-proliferative activity | [41] |
| Four acridine Pt complexes |  | Detailed DNA sequence specificity | [42] |
| Small library of substituted 9-aminoacridine derivatives |  | Ability to inhibit proliferation and induce cellular death in SCLC | [43] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| 2-methyl-9 substituted (AS 0–8) acridines |  | Lung carcinoma, breast cancer. | [44] |
| CK0403 |  | Anti-cancer activity | [45] |
| New series of 9-anilinoacridines containing phenyl-urea moieties |  | Novel dual Src and MEK inhibitors. | [46] |
| BO-1051 |  | Against oral cancer | [47] |
| Novel isoxazole substituted 9 –anilinoacridines |  | HER2 inhibitors. | [48] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Two new 1-acridin-9-yl-3-methylthiourea |  | Human ovarian carcinoma cisplatin-sensitive A2780 cell line, breast cancer cell lines | [49] |
| Seven new cyclometalated Au (III) complexes |  | Cancer cells including leukaemia, lung and breast cancer cells | [50] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Three new acridine–thiazolidinone derivatives |  | Leukemic cells, human epithelial ovarian cancer cell lines. | [51] |
| Series of novel hybrid 5-acridin-9-ylmethylene-3-benzyl-thiazolidine-2,4-diones |  | DNA interaction assays were performed | [52] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Aseries of acridinones inspired by the structure of podophyllo toxin |  | Triple-negative breast cancer cell line MDA-MB-231 | [53] |
| A new series of 9(10H)-acridinone-1,2,3 triazole derivatives |  | Against human breast cancer cell lines. | [54] |
| C-1305 C-1311 |  | Anti-tumour agents | [55] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| New benzimidazole acridine derivative |  | Human colon cancer cell lines | [56] |
| A series of 4-amidobenzimidazole acridines |  | Inhibitory activities against Topo and PARP-1 | [57] |
| Novel benzimidazole acridine derivatives |  | New DNA-targeted compounds | [58] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Tubulin polymerisation inhibitors |  | Eight cancer cell lines including MCF-7, A2780, HeLa, HepG2, DU145, A549, PC3, and LNCAP | [59] |
| New 7-substituted-5, 6-dihydrobenzo[c]acridine derivatives |  | Induce apoptosis through activation of the caspase-3 cascade pathway | [60] |
| Phenanthrene fused-tetra hydrodibenzo-acridinones. |  | Cervical (HeLa), prostate (PC-3), fibrosarcoma (HT-1080), ovarian (SKOV-3) cancer cells | [61] |
| 12-arylbenzoacridines |  | Selective estrogen-receptor modulators (SERMs). | [62] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Imidazoacridinone C-1311 |  | Anti-cancer activity | [63] |
| A novel photo activation-based pharmacological Trojan horse approach |  | MDR tumour cells | [64] |
| C-1311 |  | Anti-tumour agent C-1311in hepatoma cells | [65] |
| Crystal structures of NQO2 containing two of the imidazoacridin-6-ones |  | Inhibits the enzymatic function of NQO2 in cells. | [66] |
| Imidazoacridinone and triazoloacridinone |  | Liver cancer | [67] |
| C-1311 |  | Human non-small-cell lung cancer (NSCLC) cell lines, A549 and H460. In A549 cells | [68] |
| C-1311 and C-1330 |  | Human live microsomal enzymes | [69] |
| Imidazoacridinone C-1311 |  | Against FLT3-ITD kinase | [70] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| C-1748, 9-(20-hydroxyethylamino)-4-methyl-1-nitroacridine |  | Anti-tumour activity | [71] |
| 3-nitroacridine derivatives |  | DNA-binding affinity and good inhibitory effect of tumour cell proliferation | [72] |
| New series of 1,3-dimethyl-6-nitroacridine derivatives |  | Anti-cancer activity against four cancer cells | [73] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| A series of 9-anilinoacridines substituted with oxazine derivatives |  | Antioxidant and anti-cancer activity | [74] |
| Novel oxazine substituted 9-anilinoacridines |  | Topoisomerase II | [75] |
| A series of oxazine substituted 9-anilinoacridines |  | Anti-tumour activity on Dalton’s lymphoma ascites cells | [76] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Non-classical platinum-acridine hybrid agents |  | Distinct modules of DNA repair | [77] |
| A novel pharmacophore comprising a DNA-targeted platinum–acridine hybrid agent |  | Breast cancer cell lines | [78] |
| P3A1 |  | Cancer cells using MWCNTs as a drug carrier | [79] |
| [Pt(en)(L/L0)](NO3)2 [PtCl(en)(LH/L0H)](NO3)2 |  | Inhibits H460 lung cancer cell proliferation | [80] |
| Platinum−acridine hybrid agents |  | Anti-cancer activity | [81] |
| Platinum acridine hybrid agent [PtCl(en)(L)](NO3)2 |  | Delineate mechanistic differences between the platinum acridine hybrid agent | [82] |
| 7-aminobenz[c] acridine |  | Tested in five NSCLC cell lines | [83] |
| Platinum–acridine agent |  | Structurally characterised | [84] |
| Platinum-acridine anti-cancer agent [PtCl(en)(LH)](NO3)2 |  | Chemo resistant non-small-cell lung cancer (NSCLC) cell lines | [85] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Quinacrine |  | Human leukaemia K562cells | [86] |
| Quinacrine |  | Human leukaemia U937 cells. | [87] |
| curcumin (Cur) and quinacrine (QC) |  | Induced breast epithelial transformed (MCF-10A-Tr) generated metastatic cells | [88] |
| Quinacrine |  | Inhibition of PARP-1PARylation | [89] |
| Quinacrine |  | Cytotoxic effect of QC on DLBCL cells | [90] |
| Quinacrine |  | Against different cancer cell types | [91,92] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Three new thiazacridine derivatives |  | Induced neoplastic cell death primarily by apoptosis | [93] |
| An acridine and thiazolidine nucleus |  | Tested in human colon carcinoma HCT-8 cells | [94] |
| Series of new thiazacridine agents |  | HepG2 cells | [95] |
| Thiazacridine and imidazacridine derivatives |  | Tumours suppressors in some cancer cell lines | [96] |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| A new series of azaacridine derivatives |  | Potent EGFR and Src dual inhibitors | [97] |
| A series of new aza-acridine analogues |  | Anti-tumour agent | [97] |
| Potent Compound | Cancer Cell Lines | Control | IC50 (µM) | Reference |
|---|---|---|---|---|
| Propyl-AcrDTU | HL-60 | - | 7.2–8 | [1] |
| MAnT | HeLa cervical cancer cell line | Tetrandrine | 2.74 | [2] |
| C-1748 | HT29 Cell | Sorafenib | 39.7 | [3] |
| Cmpd-1 | A549 Cells | Etoposide | 15 | [4] |
| Cmpd-2 | A549 Cells | “ | 10 | |
| Cmpd-3 | A549 Cells | “ | 15 | |
| Cmpd-4 | A549 Cells | “ | 10 | |
| ACSO3 | HCT-116 Cells | Doxorubicin | 23.11 ± 1.03 | [7] |
| HeLa | “ | ˃50 | ||
| MCF-7 | “ | ˃50 | ||
| K562 | “ | ˃50 | ||
| HL-60 | “ | ˃50 | ||
| HaCat | “ | 62.18 ± 1.15 | ||
| PBMC | “ | 115.2 ± 5.82 | ||
| Cisplatin | MCF-7 | - | 12.7 ± 2.2 | [9] |
| 9-Acridine carboxylic acid | “ | - | 21.5 ± 3.1 | |
| Eu(III)-complex | “ | - | 14.3 ± 2.6 | |
| C-1748 | MiaPaCa-2 | - | 0.015 | [10] |
| AsPC-1 | - | 0.075 | ||
| B1 | SAS Cells | - | 1.5 | [13] |
| B2 | “ | - | 11.7 | |
| B3 | “ | - | 5.3 | |
| B4 | “ | - | 18.0 | |
| B5 | “ | - | 9.4 | |
| 80c | U937 HCT-116 | m-AMSA | 0.90 | [14] |
| LS-1-10 | DLD1 Cells | Amsacrine | 1.31 | [15] |
| ACPH | A375 | Camptothecin | 20.74 | [18] |
| 1i | A549 HT-29 | Etoposide | 59.12–14.87 17.32–5.90 | [19] |
| 4b,4d,4e | HeLa, HepG2, HEK 239 | Camptothecin | 18.89–52.64 18.7–108.34 31.38–277.13 | [20] |
| C2 | malignant glioma and other human cancers. | erlotinib | 0.75–5 | [24] |
| C-1311 | DU-145 HCT-116 MBA-MB-231 | Gemcitabine Erlotinib | 0.01–0.03 0.01–0.03 0.01–0.03 | [25] |
| 4b 4f 4g 4i 4j | HepG2 MCF-7 | Doxorubicin | 1.4 ± 0.11 4.7 ± 0.09 2.2 ± 0.09 5.3 ± 0.16 4.8 ± 0.12 4.4 ± 0.10 2.6 ± 0.11 5.9 ± 0.15 1.6 ± 0.14 5.0 ± 0.18 | [26] |
| SR374 SR375 SR361 SR362 | MCF7 A549 GIST48 WI38 | - | 1.6, 0.4, 2.9, <0.1 0.3, 0.1, 0.5, 0.1 1.5, 1.6, 2.8, 2.3 7.4, 4.5, 10.3, 0.7 | [28] |
| Cmpd 5 | HL-60 | Acridine | 25.87 32.16 | [29] |
| Cmpd 13 Cmpd 15 Cmpd 16 Cmpd 17 Cmpd 18 | HT-29 | BRACO19 | 13.17–12.55 41.12–27.38 69.99–22.12 48.80–23.25 44.89–33.23 | [31] |
| CuGGHK-Acr GGHK-Acr Ho-Acr | HuH-7 MCF-7 Caco2 | - | 9.8 ± 2.3 26.6 ± 2.2 16.7 ± 1.6 | [33] |
| 8p | A549 Cells | Doxorubicin Etoposide Amsacrine | 0.61 ± 0.06 | [34] |
| 9AmAcPtCl2 | HeLa Cells | Cisplatin | 0.4 | [40] |
| 7r | HepG-2 MCF-7 | Colchicin | 4.2 23.8 | [41] |
| 7b | H1299 WM264 HCT116 | Cisplatin | 2.9 0.8 15.4 | [45] |
| 8m | K562 HepG-2 | Imatinib | 4.08 ± 0.14 9.41 ± 1.09 | [46] |
| BO-1051 | SAS Cells OECM1 | - | 2.39 1.97 | [47] |
| Cmpd 2 | A2780 MDA-MB-231 SK-BR-3 MCF-7 | CDDP AuC1PPh3 | 0.88 ± 0.20 2.75 ± 0.40 3.62 ± 0.14 5.32 ± 0.65 | [50] |
| Complex 11 | A549 MCF-7 HL60 | Cisplatin | 7.6 ± 0.6 1.5 ± 0.1 1.1 ± 0.1 | [50] |
| 2C | HL-60 L1210 A2780 | Cisplatin | 1.3 ± 0.2 3.1 ± 0.4 7.7 ± 0.5 | [51] |
| Cmpd-9 | PC-3 COLO-205 | Amsacrine | 5.5–9.5 8.6–42.3 | [52] |
| Cmpd-6 Cmpd-7 Cmpd-9 Cmpd-10 | MDA-MB-231 DU-145 | Colchicine | 3 ± 1, 3.2 ± 0.7 0.190 ± 0.007 1.1 ± 0.2 1.0 ± 0.3 0.11 ± 0.02 0.12 ± 0.02 | [53] |
| 8c | MCF-7 T-47D MDA-MB-231 | etoposide | 11 ± 4.8 14.5 ± 5.2 16.6 ± 5.9 | [54] |
| C-1305 C-1311 | KB-3 | - | 0.263 ± 0.016 0.106 ± 0.014 | [55] |
| 8m | SW480 HCT116 | - | 6.77 ± 0.19 3.33 ± 0.02 | [56] |
| 11L | PARP-1 MCF-7 | Olaparib m-AMSA | 0.45 ± 0.03 2.14 ± 0.92 | [57] |
| 8I | K562 HepG-2 | Colchicine Imatinib | 2.68 8.11 | [58] |
| 4g | HUVEC LNCAP | Β-Lapachone | 49.19 ± 2.3 18.54 ± 2.11 | [59] |
| 2b | HeLa K562 A549 | - | 3.2 9.2 5.7 | [60] |
| 8m | SKOV-3 | Cisplatin | 0.24 ± 0.05 | [61] |
| Cmpd-1 | MCF-7 | Tamoxifen citrate | 0.951 | [62] |
| 6a1 | HCT116 | NQO2 | 14 ± 4 | [66] |
| C-1311 | MV4-11 MOLM13 | - | 0.03 ± 0.01 0.04 ± 0.02 | [70] |
| Cmpd-2 | MCF-7 MDA-MB-231 SGC7901 MGC803 | - | 6.79 ± 2.01 6.46 ± 2.19 17.25 ± 3.84 10.94 ± 2.26 | [72] |
| Cmpd-1 Cmpd-6 | MCF-7 MDA-MB-231 SGC7901 MGC803 | - | 8.83 ± 0.98 10.02 ± 0.78 41.47 ± 3.24 23.96 ± 1.54 | [73] |
| 5a | DLA Cells | ascorbic acid | 20.03 ± 0.2583 | [74] |
| 8 | MCF-7 MDA-MB-231 | Tamoxifen | 1.6 ± 0.4 13.2 ± 0.1 | [78] |
| Cmpd-3 | NCI-H460 OVCAR-3 PANC-1 | Cisplatin | 0.008 ± 0.002 1.1 ± 0.1 0.09 ± 0.01 | [80] |
| 3a 3b | H460 | - | 12 ± 2 2.8 ± 0.3 | [81] |
| P1-A1 P1-B1 P1-B2 | NCI-H460 | - | 0.0052 ± 0.0001 0.24 ± 0.01 2.4 ± 0.5 | [83] |
| Cmpd-1 | NCI-H460 NCI-H522 | Cisplatin | 8 ± 2 18 ± 2 | [85] |
| QC | OCI-Ly01 SU-DHL-8 | Quinacrine | 1.8 2 | [90] |
| Cmpd-25 | MDA-MB-468 MDA-MB-231 MCF-7 184B5 | Cisplatin Quinacrine | 1.73 ± 0.80 2.80 ± 1.30 0.69 ± 0.41 4.96 ± 0.24 | [91] |
| Cmpd-11 | MDA-MB-468 MDA-MB-231 MCF-7 184B5 | Cisplatin Quinacrine | 2.40 ± 1.01 1.92 ± 0.20 1.24 ± 0.51 16.16 ± 0.81 | [92] |
| LPSF/AC-119 | Raji Jurkat | Amsacrine | 0.6 1.53 | [93] |
| AC-4 AC-7 AC-10 AC-23 | HCT-8 | Amsacrine | 3.1 5.3 3.6 2.3 | [94] |
| 7a 7b | HepG2 | Amsacrine | 68.03 48.63 | [95] |
| 13b | K562 A549 | Imatinib | 0.22 ± 0.03 0.253 ± 0.16 | [97] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varakumar, P.; Rajagopal, K.; Aparna, B.; Raman, K.; Byran, G.; Gonçalves Lima, C.M.; Rashid, S.; Nafady, M.H.; Emran, T.B.; Wybraniec, S. Acridine as an Anti-Tumour Agent: A Critical Review. Molecules 2023, 28, 193. https://doi.org/10.3390/molecules28010193
Varakumar P, Rajagopal K, Aparna B, Raman K, Byran G, Gonçalves Lima CM, Rashid S, Nafady MH, Emran TB, Wybraniec S. Acridine as an Anti-Tumour Agent: A Critical Review. Molecules. 2023; 28(1):193. https://doi.org/10.3390/molecules28010193
Chicago/Turabian StyleVarakumar, Potlapati, Kalirajan Rajagopal, Baliwada Aparna, Kannan Raman, Gowramma Byran, Clara Mariana Gonçalves Lima, Salma Rashid, Mohammed H. Nafady, Talha Bin Emran, and Sławomir Wybraniec. 2023. "Acridine as an Anti-Tumour Agent: A Critical Review" Molecules 28, no. 1: 193. https://doi.org/10.3390/molecules28010193
APA StyleVarakumar, P., Rajagopal, K., Aparna, B., Raman, K., Byran, G., Gonçalves Lima, C. M., Rashid, S., Nafady, M. H., Emran, T. B., & Wybraniec, S. (2023). Acridine as an Anti-Tumour Agent: A Critical Review. Molecules, 28(1), 193. https://doi.org/10.3390/molecules28010193