
From the Streets to the Judicial Evidence: Determination of Traditional Illicit Substances in Drug Seizures by a Rapid and Sensitive UHPLC-MS/MS-Based Platform

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Development of the UHPLC-MS/MS Method
2.2. Method Validation
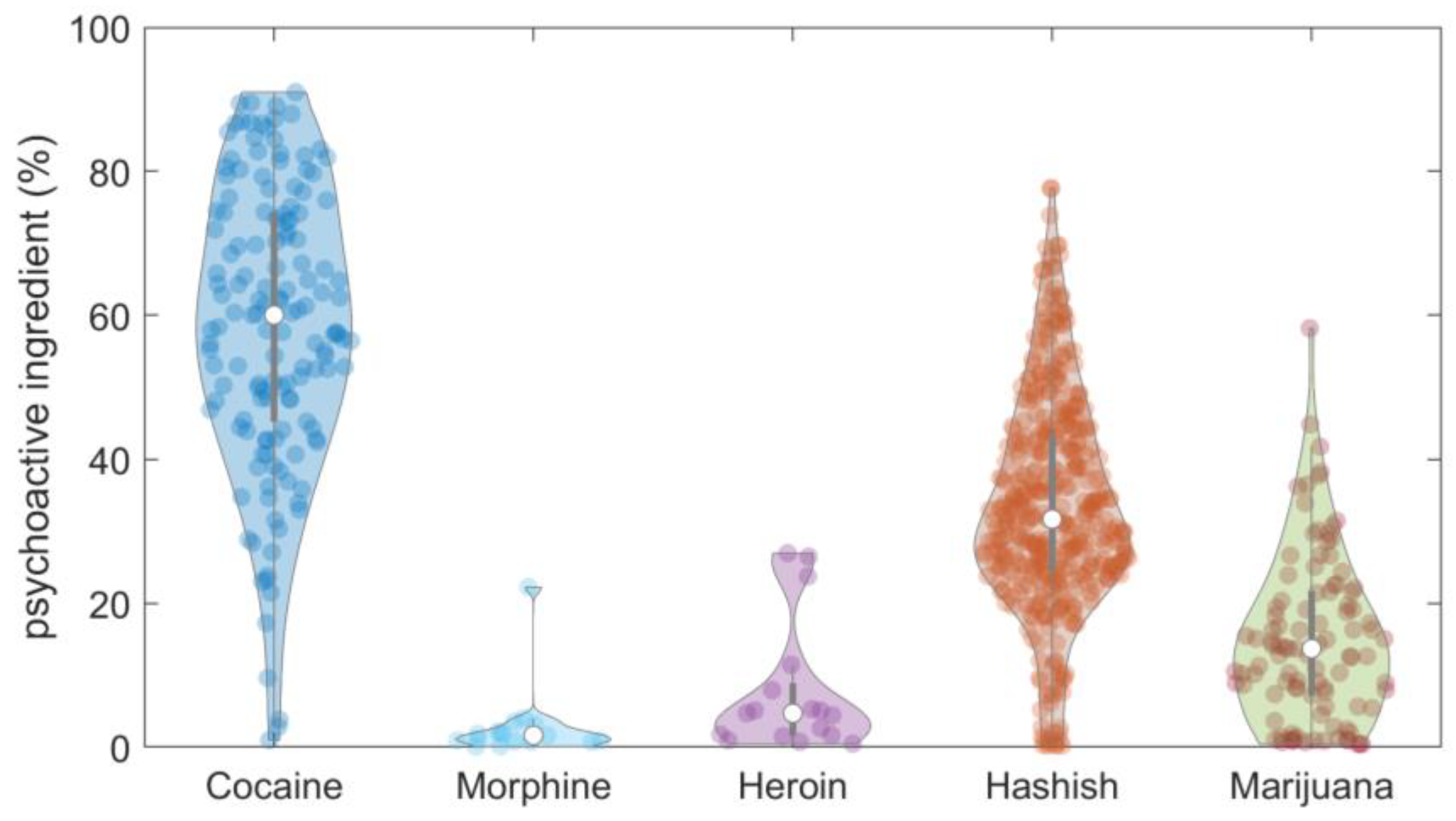
3. Analytical Results of Street Seizures
4. Materials and Methods
4.1. From Seizures Registration to Final Report Generation
4.2. Street Seizure Registration and Sample Collection
- -
- Registration. A dedicated custom-made software developed at the NPS Laboratory (Figure 6a) allows for easy registration of all the meaningful information about seized materials. To every single seizure, a unique alphanumeric ID is assigned, and a detailed descriptive technical sheet is generated. After seizure coding, the NPS workers register visual and morphological descriptions of the seizure-relevant aspects, e.g., type and shape of wrapping materials, color, odor, forms, marks, total number of similar items of each type of supposed substance, their gross and net weights, and any other characteristic that can be considered essential. The gross and net weights of each single item are measured and registered up to a maximum of 20 similar items. For more than 20 similar items, the total gross and net weights of the seizure are estimated based on the average weight of the sampled items and the total number of items in the seizure. One or more digital images of the seizures are taken before and after unpacking. The photographic appendix is a valuable part of the final report, especially for the prosecutor’s data handling and decision. Any other relevant visual aspect considered to be of interest to the prosecutor’s findings that could emerge during the laboratory processing of the seizure is recorded as well.
4.3. Chemical and Reagents
4.4. Sample Preparation Procedure
4.5. UHPLC-MS/MS Conditions
4.6. Final Report Generation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Drug Report. 2021. Available online: //www.unodc.org/unodc/en/data-and-analysis/wdr2021.html (accessed on 26 November 2022).
- Bertol, E.; Bigagli, L.; D’Errico, S.; Mari, F.; Palumbo, D.; Pascali, J.P.; Vaiano, F. Analysis of Illicit Drugs Seized in the Province of Florence from 2006 to 2016. Forensic Sci. Int. 2018, 284, 194–203. [Google Scholar] [CrossRef] [PubMed]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2021: Trends and Developments; Publications Office: Luxembourg, 2021. [Google Scholar]
- Statistical Bulletin. 2022. Available online: https://www.emcdda.europa.eu/data/stats2022_en (accessed on 30 November 2022).
- European Monitoring Centre for Drugs and Drug Addiction. Relazione Europea Sulla Droga 2021: Tendenze e Sviluppi; Publications Office: Luxembourg, 2021. [Google Scholar]
- Gazzetta Ufficiale. Available online: https://www.gazzettaufficiale.it/eli/id/1990/10/31/090G0363/sg (accessed on 27 November 2022).
- Gazzetta Ufficiale. Available online: https://www.gazzettaufficiale.it/atto/serie_generale/caricaDettaglioAtto/originario?atto.dataPubblicazioneGazzetta=2006-04-24&atto.codiceRedazionale=06A04031&elenco30giorni=false (accessed on 27 November 2022).
- Classification of Controlled Drugs—Topic Overview. Available online: https://www.emcdda.europa.eu/publications/topic-overviews/classification-of-controlled-drugs/html_en (accessed on 30 November 2022).
- Ministero Della Salute Testo Unico Sugli Stupefacenti, le Ultime Modifiche. Available online: https://www.salute.gov.it/portale/medicinaliStupefacenti/dettaglioNotizieMedicinaliStupefacenti.jsp?lingua=italiano&menu=notizie&p=dalministero&id=1608 (accessed on 1 December 2022).
- Zamengo, L.; Bettin, C.; Frison, G.; Gregio, M.; Sciarrone, R. Drugs WorkBook (DWB): A Tool for the Analysis of Illicit Drugs in Seized Materials. Sci. Justice 2013, 53, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, A.; Basilicata, P.; Coraggio, L.; Guadagni, R.; Simonelli, A.; Pieri, M. Illicit Drugs Seizures in 2013–2018 and Characteristics of the Illicit Market within the Neapolitan Area. Forensic Sci. Int. 2021, 321, 110738. [Google Scholar] [CrossRef] [PubMed]
- Elkins, K.M.; Weghorst, A.C.; Quinn, A.A.; Acharya, S. Colour Quantitation for Chemical Spot Tests for a Controlled Substances Presumptive Test Database: Colour Quantitation for Presumptive Drug Test Database. Drug Test. Anal. 2017, 9, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Philp, M.; Fu, S. A Review of Chemical ‘Spot’ Tests: A Presumptive Illicit Drug Identification Technique. Drug Test. Anal. 2018, 10, 95–108. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, D.; Heffron, J.J.A. Rapid Analysis of Illicit Drugs by Mass Spectrometry: Results from Seizures in Ireland. Analyst 2000, 125, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Fiorentin, T.R.; Fogarty, M.; Limberger, R.P.; Logan, B.K. Determination of Cutting Agents in Seized Cocaine Samples Using GC–MS, GC–TMS and LC–MS/MS. Forensic Sci. Int. 2019, 295, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Yüksel, B.; Şen, N. Development and Validation of a GC-FID Method for Determination of Cocaine in Illicit Drug Samples. Marmara Pharm. J. 2018, 22, 511–518. [Google Scholar] [CrossRef]
- Fiorentin, T.R.; Logan, B.K.; Martin, D.M.; Browne, T.; Rieders, E.F. Assessment of a Portable Quadrupole-Based Gas Chromatography Mass Spectrometry for Seized Drug Analysis. Forensic Sci. Int. 2020, 313, 110342. [Google Scholar] [CrossRef] [PubMed]
- Brettell, T.A.; Lum, B.J. Analysis of Drugs of Abuse by Gas Chromatography–Mass Spectrometry (GC-MS). In Analysis of Drugs of Abuse; Methods in Molecular Biology; Musah, R.A., Ed.; Springer: New York, NY, USA, 2018; pp. 29–42. ISBN 978-1-4939-8579-1. [Google Scholar]
- Merone, G.M.; Tartaglia, A.; Rossi, S.; Santavenere, F.; Bassotti, E.; D’Ovidio, C.; Bonelli, M.; Rosato, E.; de Grazia, U.; Locatelli, M.; et al. Fast Quantitative LC-MS/MS Determination of Illicit Substances in Solid and Liquid Unknown Seized Samples. Anal. Chem. 2021, 93, 16308–16313. [Google Scholar] [CrossRef] [PubMed]
- Chiuminatto, U.; Gosetti, F.; Dossetto, P.; Mazzucco, E.; Zampieri, D.; Robotti, E.; Gennaro, M.C.; Marengo, E. Automated Online Solid Phase Extraction Ultra High Performance Liquid Chromatography Method Coupled with Tandem Mass Spectrometry for Determination of Forty-Two Therapeutic Drugs and Drugs of Abuse in Human Urine. Anal. Chem. 2010, 82, 5636–5645. [Google Scholar] [CrossRef] [PubMed]
- Jovanov, P.; Petrin-Miličević, M.; Radosavljević-Stevanović, N.; Vraneš, M.; Belić, S.; Sakač, M.; Nikolov, J.; Gadžurić, S. Rapid Determination of the Primary Alkaloids in Illicit Heroin by High-Performance Liquid Chromatography with Tandem Mass Spectrometry (HPLC–MS/MS). Anal. Lett. 2021, 54, 1224–1232. [Google Scholar] [CrossRef]
- Wimmer, K.; Schneider, S. Screening for Illicit Drugs on Euro Banknotes by LC–MS/MS. Forensic Sci. Int. 2011, 206, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Stempfer, M.; Reinstadler, V.; Lang, A.; Oberacher, H. Analysis of Cannabis Seizures by Non-Targeted Liquid Chromatography-Tandem Mass Spectrometry. J. Pharm. Biomed. Anal. 2021, 205, 114313. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-H.; Lee, H.-H.; Chen, B.-H.; Lin, Y.-C.; Chao, Y.-Y.; Huang, Y.-L. Using Ambient Mass Spectrometry and LC–MS/MS for the Rapid Detection and Identification of Multiple Illicit Street Drugs. J. Food Drug Anal. 2019, 27, 439–450. [Google Scholar] [CrossRef] [PubMed]
- European Union. Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the Performance of Analytical Methods for Residues of Pharmacologically Active Substances Used in Food-Producing Animals and on the Interpretation of Results as Well as on the Methods to Be Used for Sampling and Repealing Decisions 2002/657/EC and 98/179/EC (Text with EEA Relevance); European Union: Luxembourg, 2021; Volume 180. [Google Scholar]
- EMA, International Council on Harmonisation of Technical Requirements for Registration Pharmaceuticals Human Use (ICH). Available online: https://www.ema.europa.eu/en/partners-networks/international-activities/multilateral-coalitions-initiatives/international-council-harmonisation-technical-requirements-registration-pharmaceuticals-human-use (accessed on 28 November 2022).
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, A.; Famiglini, G.; Palma, P.; Pierini, E.; Termopoli, V.; Trufelli, H. Overcoming Matrix Effects in Liquid Chromatography−Mass Spectrometry. Anal. Chem. 2008, 80, 9343–9348. [Google Scholar] [CrossRef] [PubMed]
- SWGDRUG Home Page. Available online: https://www.swgdrug.org/ (accessed on 28 November 2022).
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal Suppression/Enhancement in High-Performance Liquid Chromatography Tandem Mass Spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, B.; Fletcher, P.; Seamusholden; Gorur-Shandilya, S. Bastibe/Violinplot-Matlab: A good starting point (v. 0.1). Zenodo 2021. [Google Scholar] [CrossRef]
- Conlan, X.A.; Theakstone, A.G. CHAPTER 1. Detection Strategies for Traditional Illicit Substances. In Detection Science; Dennany, L., Ed.; Royal Society of Chemistry: Cambridge, UK, 2021; pp. 1–40. ISBN 978-1-83916-022-6. [Google Scholar]
- Dujourdy, L.; Csesztregi, T.; Bovens, M.; Franc, A.; Nagy, J. Sampling of Illicit Drugs for Quantitative Analysis. Part I: Heterogeneity Study of Illicit Drugs in Europe. Forensic Sci. Int. 2013, 231, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Best Practice Manuals|ENFSI 2016. Available online: https://enfsi.eu/about-enfsi/structure/working-groups/documents-page/documents/best-practice-manuals/ (accessed on 26 November 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | LOD (ng mL−1) | LOQ (ng mL−1) | R2 | RSD% Intra-Day (n = 5) | RSD% Inter-Day (n = 35) | Recovery (%) |
|---|---|---|---|---|---|---|
| 6-MAM | 0.01 | 0.04 | 0.9965 | 0.9 | 11.3 | 105.3 ± 0.7 |
| Amphetamine | 0.12 | 0.41 | 0.9998 | 0.1 | 10.4 | 101.1 ± 0.8 |
| Cocaine | 0.01 | 0.04 | 0.9989 | 4.1 | 10.7 | 106.1 ± 1.3 |
| Heroin | 0.01 | 0.02 | 0.9991 | 0.4 | 8.7 | 96.5 ± 0.9 |
| GBL | 1.02 | 3.02 | 0.9978 | 2.6 | 8.1 | 109.1 ± 0.7 |
| GHB | 0.11 | 0.32 | 0.9994 | 0.8 | 10.3 | 109.1 ± 0.7 |
| Ketamine | 0.01 | 0.05 | 0.9945 | 0.1 | 9.7 | 105.5 ± 0.8 |
| LSD | 0.01 | 0.03 | 0.9992 | 2.7 | 8.1 | 94.5 ± 0.7 |
| MDMA | 0.04 | 0.12 | 0.9963 | 0.7 | 12.6 | 105.5 ± 0.8 |
| Methamphetamine | 0.04 | 0.13 | 0.9992 | 3.4 | 11.6 | 105.7 ± 0.3 |
| Morphine | 0.09 | 0.30 | 0.9991 | 1.3 | 2.3 | 96.1 ± 0.1 |
| Trans-∆9-THC | 0.04 | 0.47 | 0.9968 | 2.6 | 8.5 | 94.3 ± 0.5 a 94.3 ± 0.3 b |
| THCA | 0.09 | 0.31 | 0.9983 | 2.1 | 8.0 | 94.3 ± 0.5 |
| Compound | Street Seizures | Extraction Solvent (Volume) | Instrumentation | Run Time (min) | LODs | LOQs | Ref. |
|---|---|---|---|---|---|---|---|
| Trans-∆9-THC | Herbal and resin cannabis | Cyclohexane (9 mL) | GC-FID | 12 | 3 ng µL−1 | 4.2 ng µL−1 | [11] |
| Heroin | Powder | ACN (Not declared) | 4.5 ng µL−1 | 5.5 ng µL−1 | |||
| Cocaine | Powder | ACN (Not declared) | 3.5 ng µL−1 | 4.5 ng µL−1 | |||
| MDMA (and other 6 compounds) | Tablets | Absolute ethanol (20 mL) | GC-MS | 33 | 32 ng µL−1 | Not declared | [14] |
| Cocaine | Powder | Methanol/ Chloroform (Not declared) | GC-FID | 13 | 1.8 ng µL−1 | 5.6 ng µL−1 | [16] |
| Cocaine | Powder | Methanol (10 mL) | Portable GC-MS | 15 | From 40 ng µL−1 to 100 ng µL−1 | Not declared | [17] |
| Heroin | |||||||
| Methamphetamine (and other 21 compounds) | |||||||
| Cocaine | Solid and liquid materials | ACN (5 mL) | LC-MS/MS | 15 | 1.67 ng mL−1 | 5 ng mL−1 | [19] |
| MDMA | |||||||
| Trans-∆9-THC | |||||||
| Heroin | |||||||
| Ketamine | |||||||
| 6-MAM (and other 31 compounds) | |||||||
| Heroin (and other 3 compounds) | Powder | Not declared | LC-MS/MS | 6 | 0.4 mg L−1 | 1 mg L−1 | [21] |
| Cocaine | Solid and liquid materials | Methanol (10 mL) | LC-MS/MS | 8 | From 0.01 ng mL−1 to 1.02 ng mL−1 | From 0.04 ng/mL to 3.04 ng/mL | This validated method |
| Heroin | |||||||
| 6-MAM | |||||||
| Amphetamine | |||||||
| GHB | |||||||
| GBL | |||||||
| Ketamine | |||||||
| LSD | |||||||
| MDMA | |||||||
| Methamphetamine | |||||||
| Morphine | |||||||
| Trans-∆9-THC | |||||||
| THCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gosetti, F.; Consonni, V.; Ballabio, D.; Orlandi, M.E.; Amodio, A.; Valeria Picci, M.; Visentin, M.; Termopoli, V. From the Streets to the Judicial Evidence: Determination of Traditional Illicit Substances in Drug Seizures by a Rapid and Sensitive UHPLC-MS/MS-Based Platform. Molecules 2023, 28, 164. https://doi.org/10.3390/molecules28010164
Gosetti F, Consonni V, Ballabio D, Orlandi ME, Amodio A, Valeria Picci M, Visentin M, Termopoli V. From the Streets to the Judicial Evidence: Determination of Traditional Illicit Substances in Drug Seizures by a Rapid and Sensitive UHPLC-MS/MS-Based Platform. Molecules. 2023; 28(1):164. https://doi.org/10.3390/molecules28010164
Chicago/Turabian StyleGosetti, Fabio, Viviana Consonni, Davide Ballabio, Marco Emilio Orlandi, Angelo Amodio, Maria Valeria Picci, Marco Visentin, and Veronica Termopoli. 2023. "From the Streets to the Judicial Evidence: Determination of Traditional Illicit Substances in Drug Seizures by a Rapid and Sensitive UHPLC-MS/MS-Based Platform" Molecules 28, no. 1: 164. https://doi.org/10.3390/molecules28010164
APA StyleGosetti, F., Consonni, V., Ballabio, D., Orlandi, M. E., Amodio, A., Valeria Picci, M., Visentin, M., & Termopoli, V. (2023). From the Streets to the Judicial Evidence: Determination of Traditional Illicit Substances in Drug Seizures by a Rapid and Sensitive UHPLC-MS/MS-Based Platform. Molecules, 28(1), 164. https://doi.org/10.3390/molecules28010164