
Allosteric Binding of MDMA to the Human Serotonin Transporter (hSERT) via Ensemble Binding Space Analysis with ΔG Calculations, Induced Fit Docking and Monte Carlo Simulations



Abstract
:1. Introduction
2. Results and Discussion
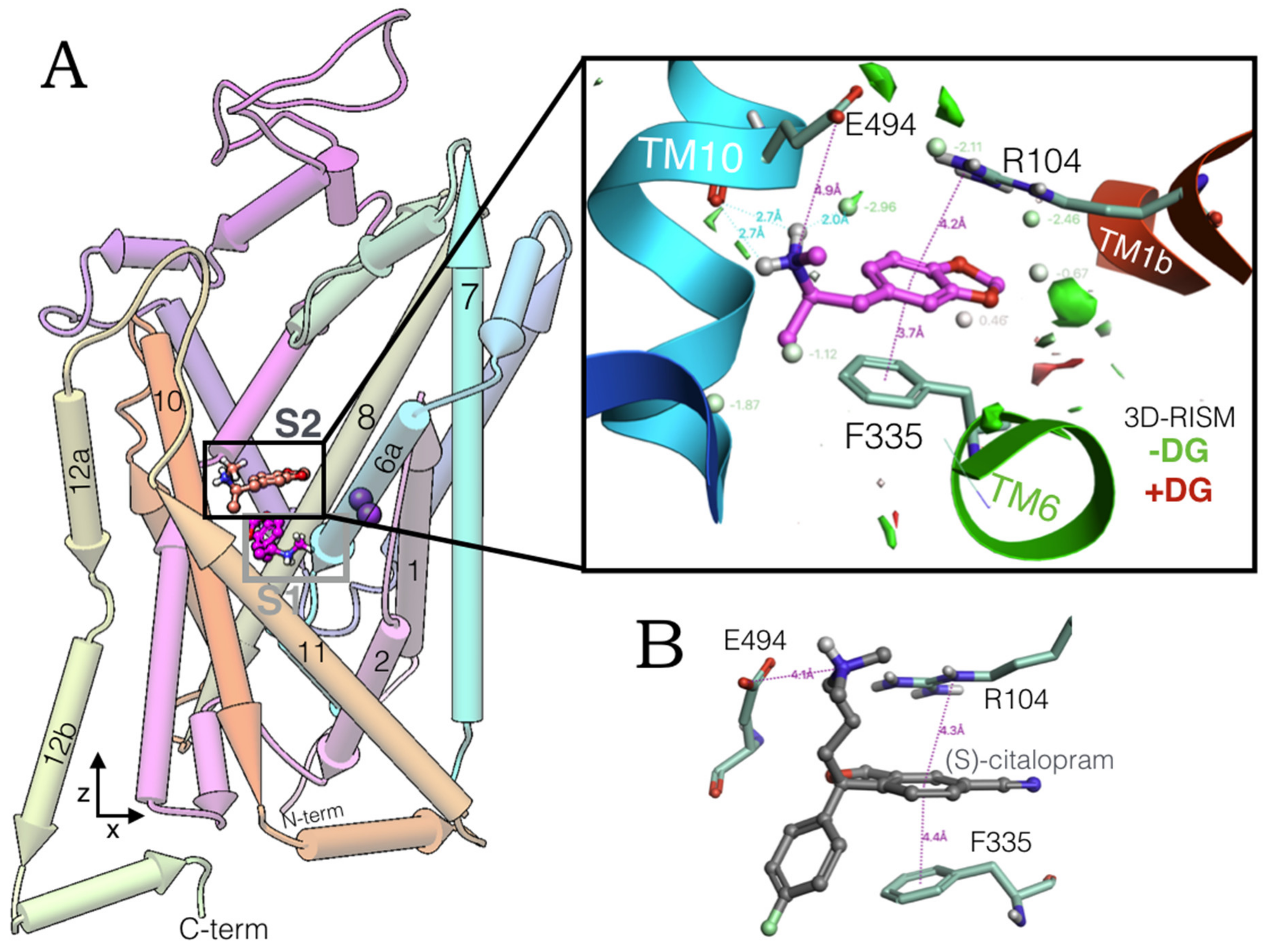
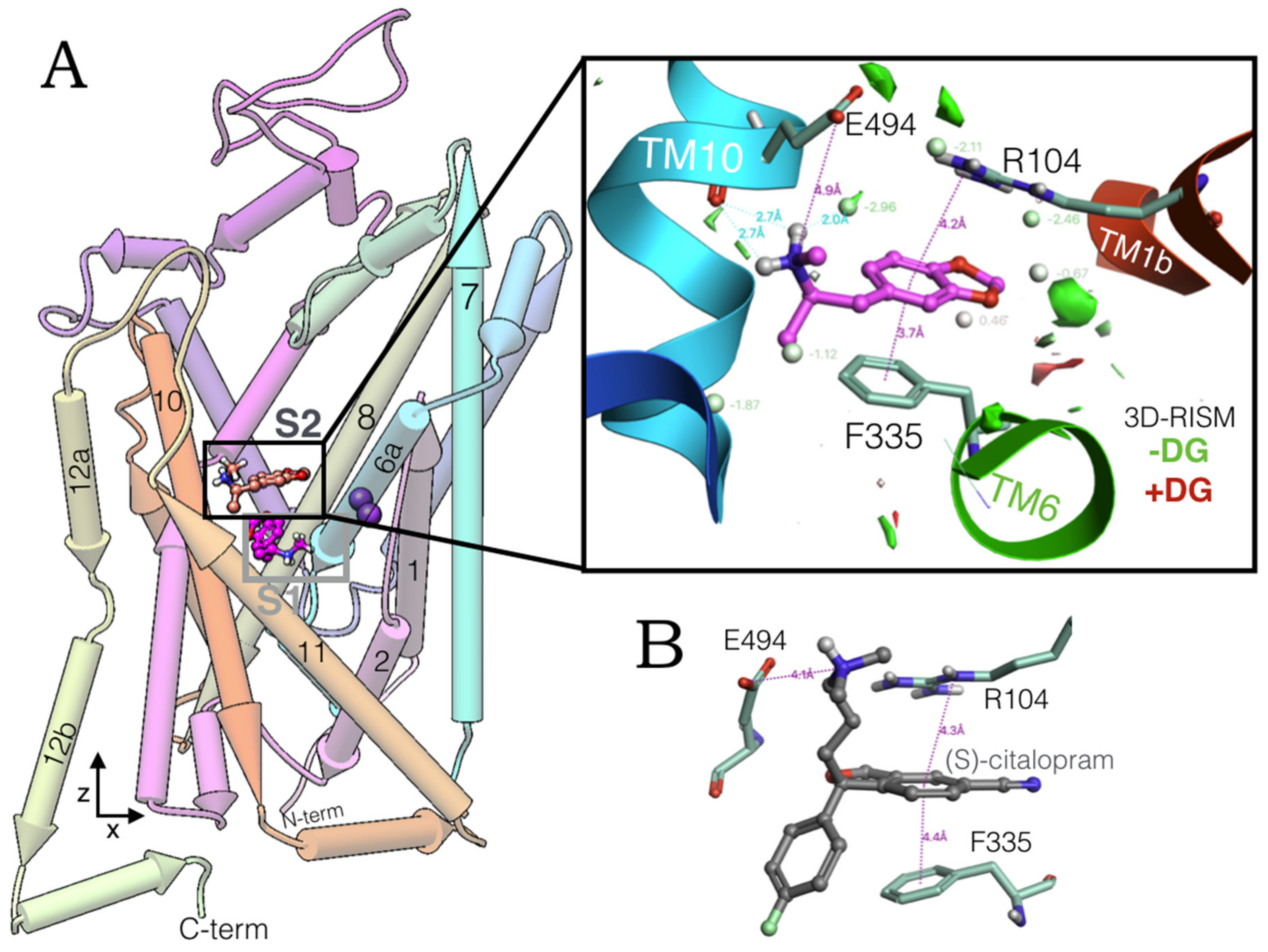
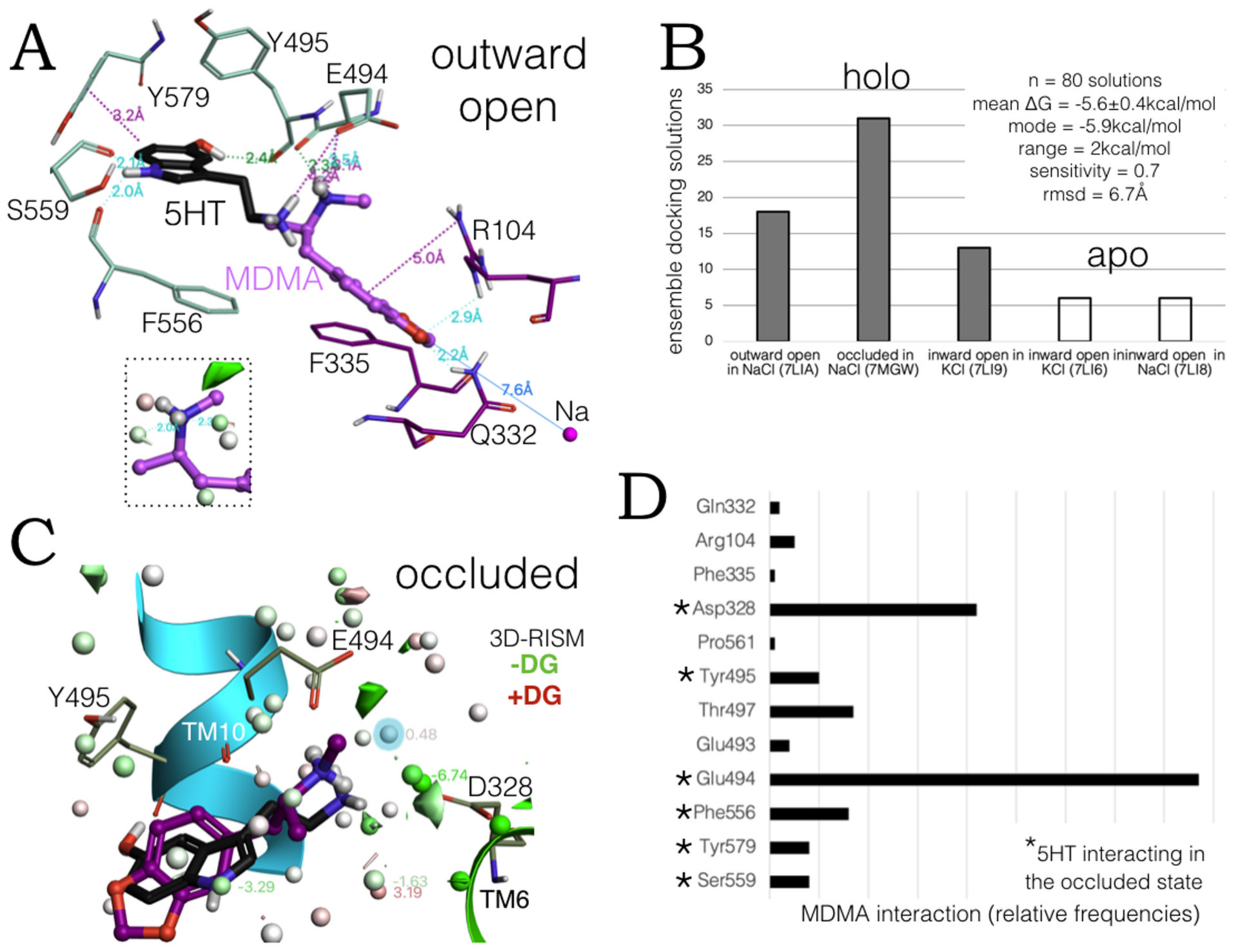
2.1. MDMA Allosteric Occupation of the hSERT Overlaps the Binding Site of Escitalopram
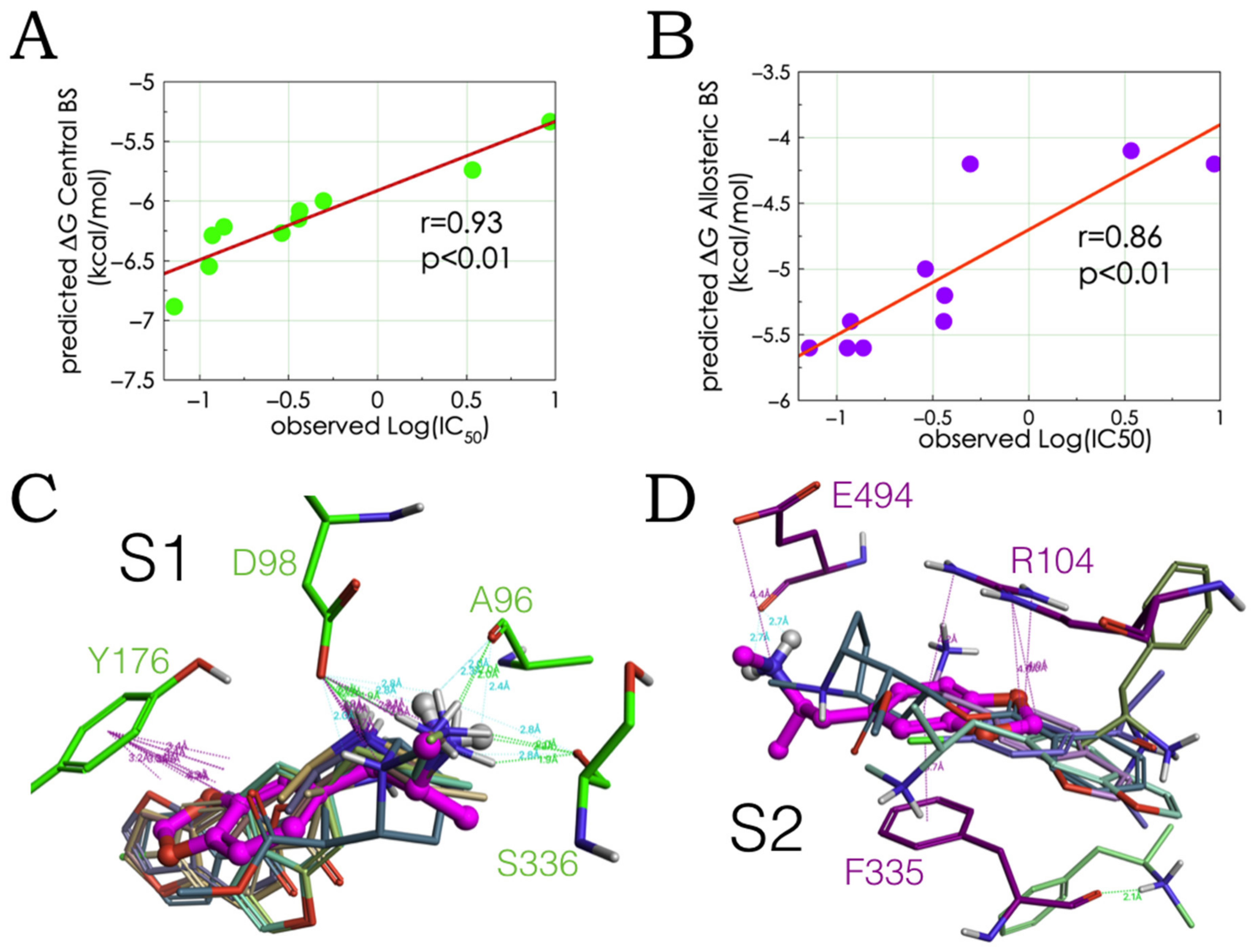
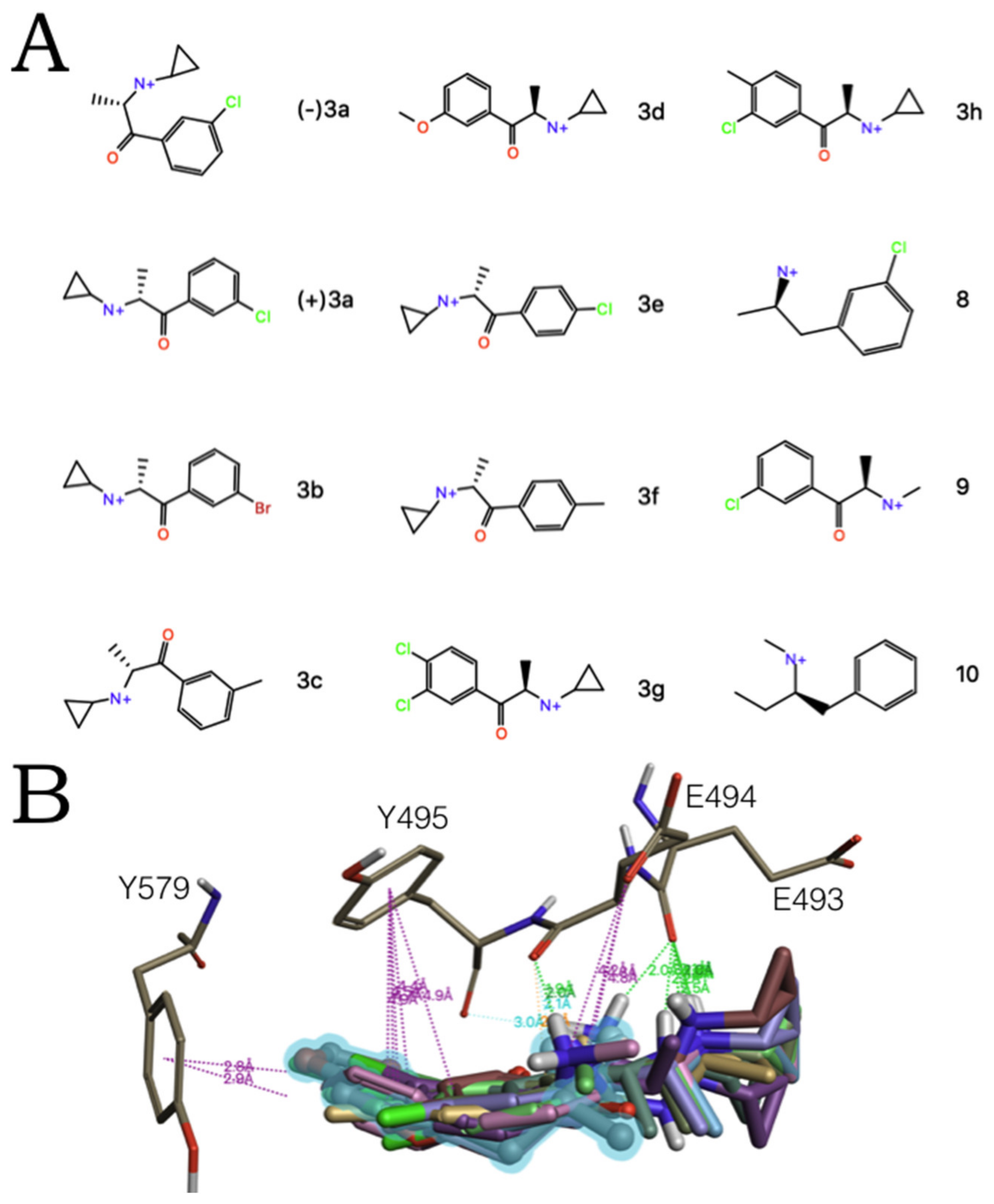
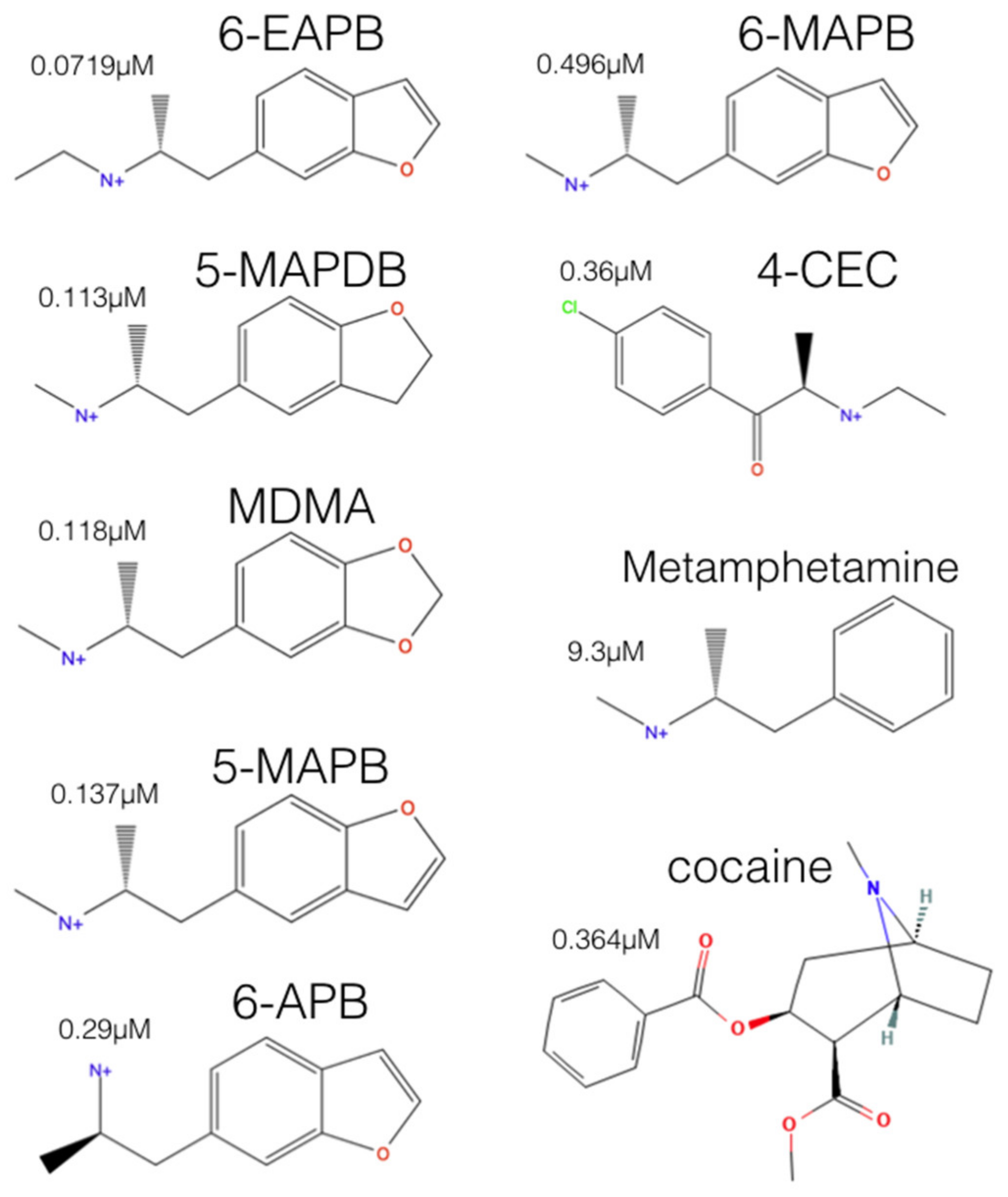
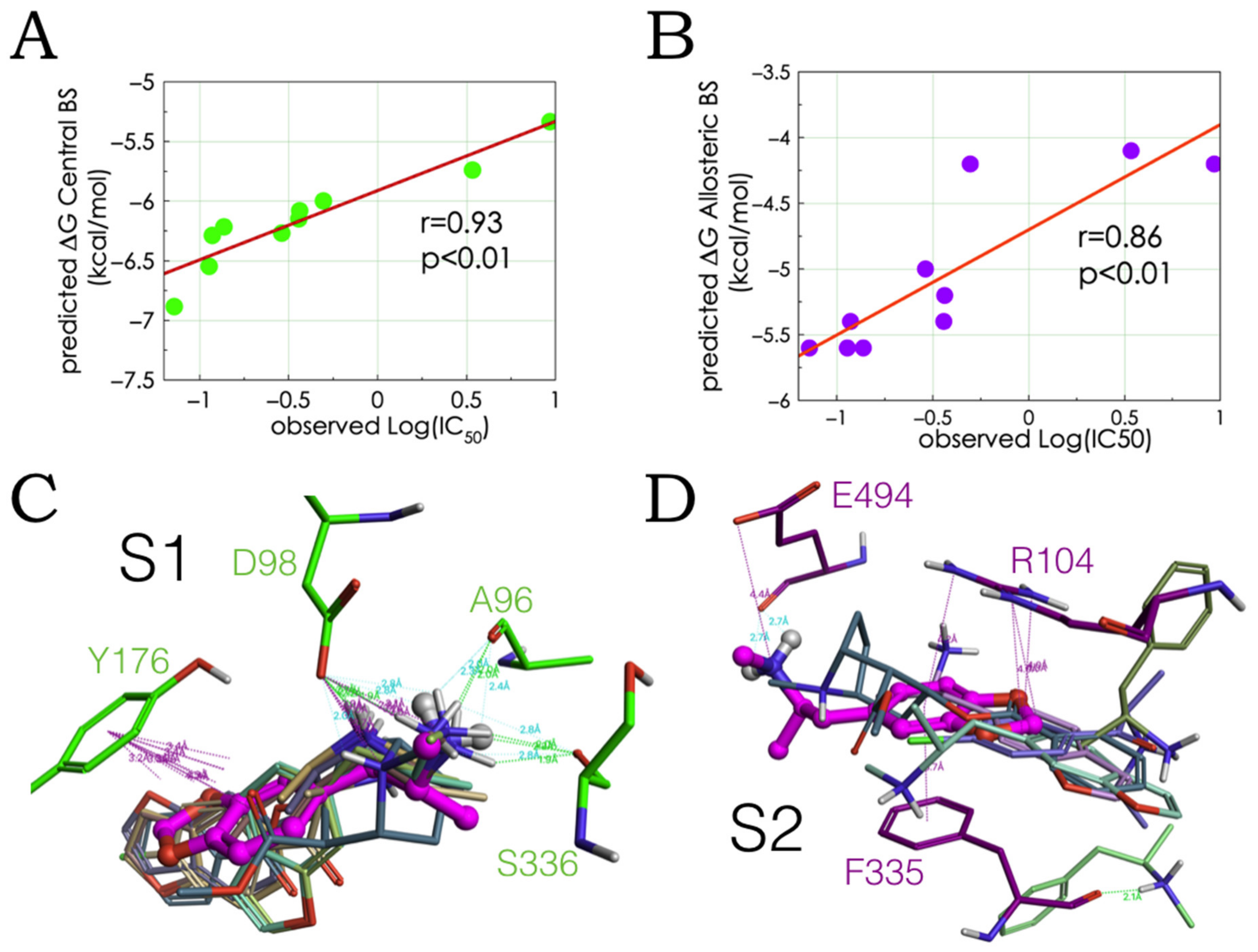
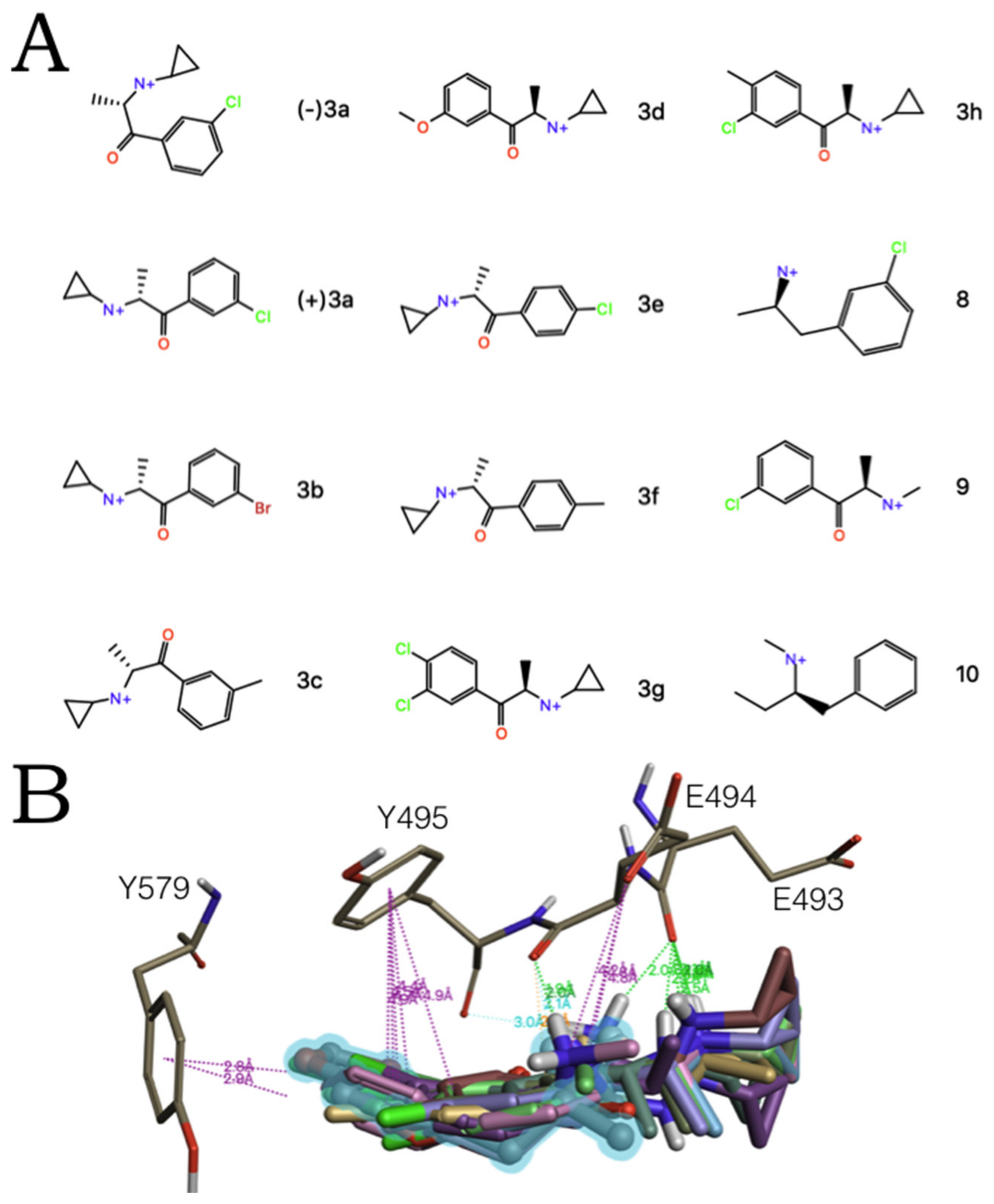
2.2. Experimental-Computational Quantitative Correlation of 5-HT Reuptake Inhibitors to S1 and S2 of the hSERT
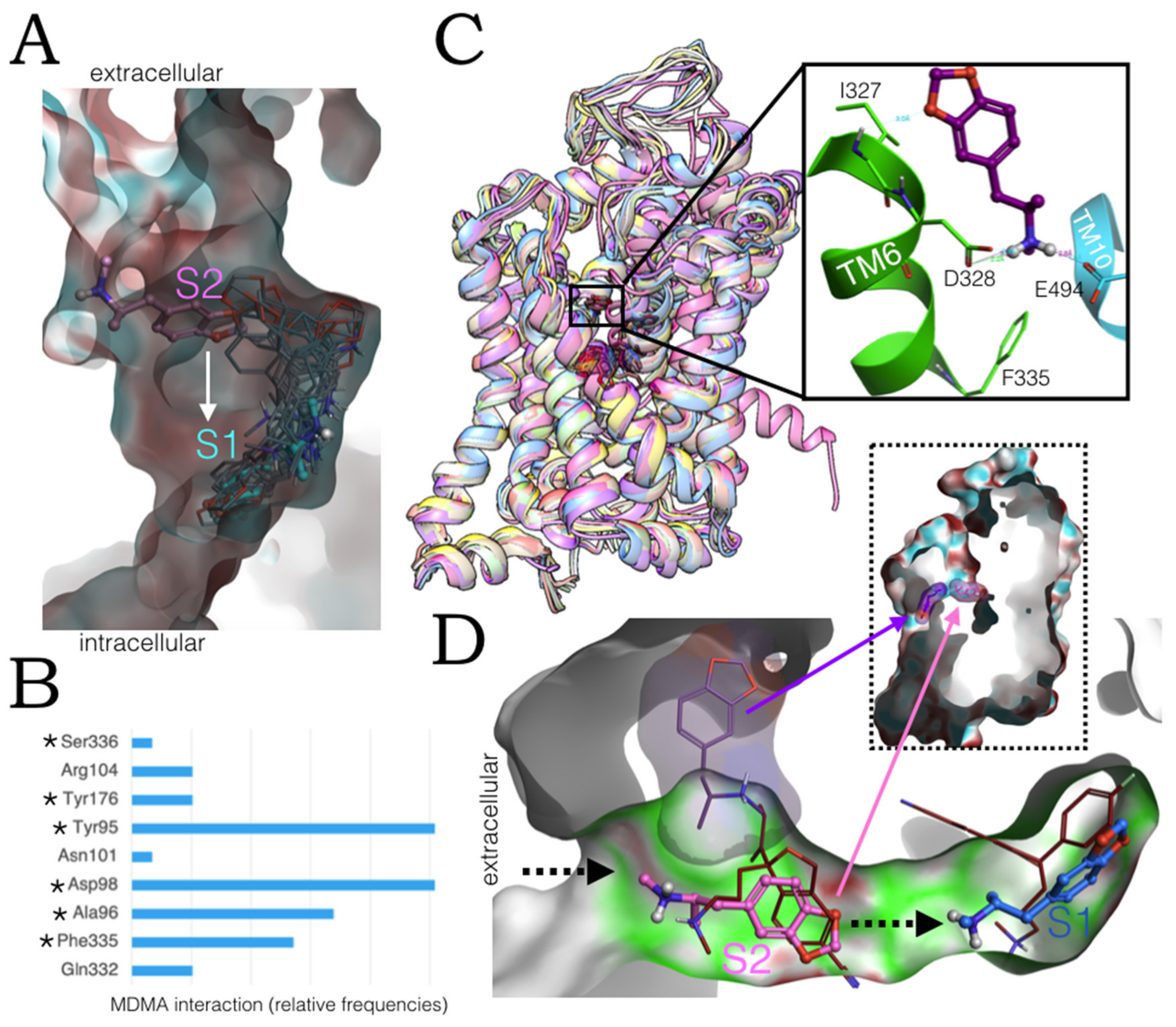
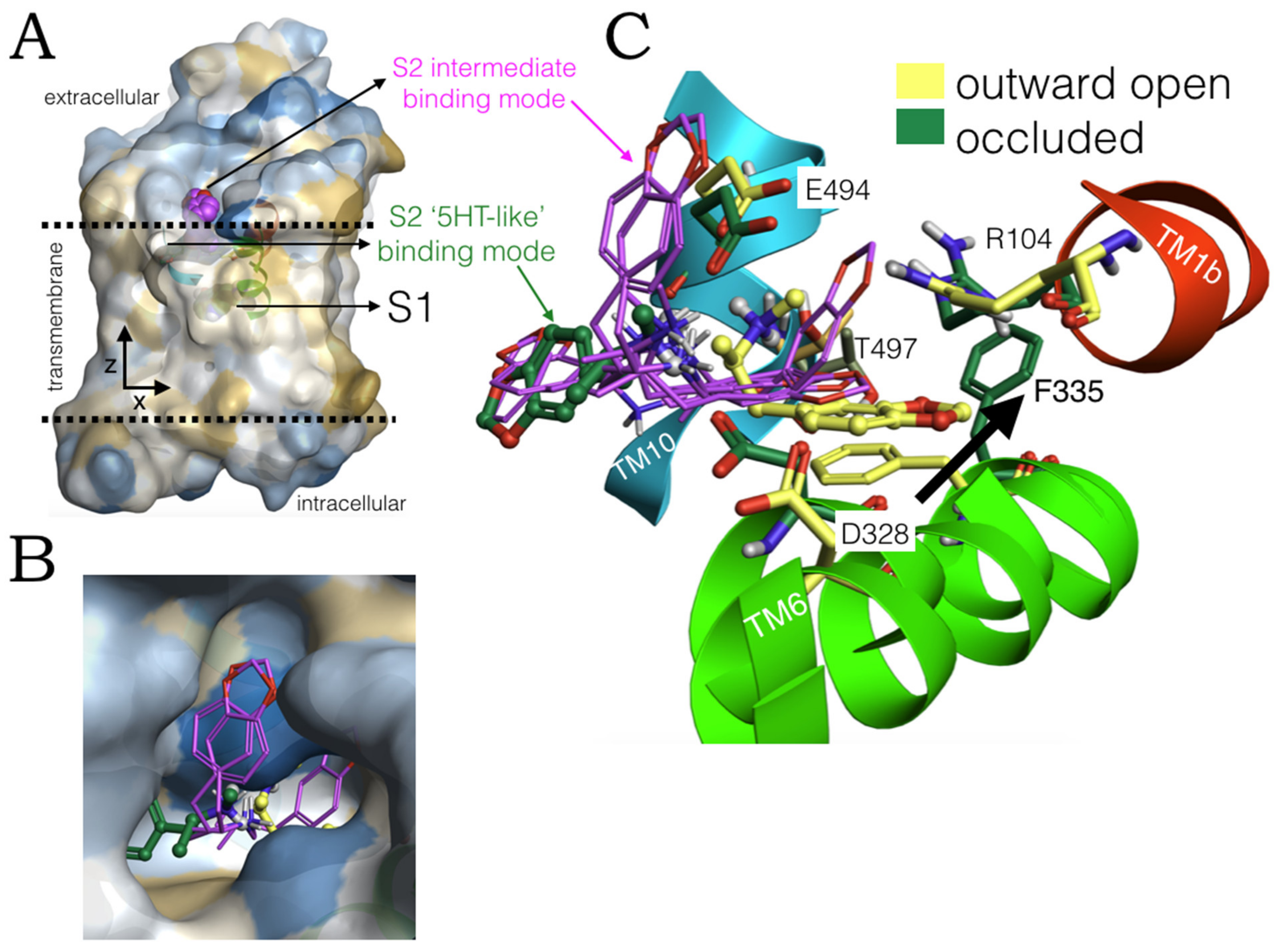
2.3. Ensemble Binding Space Analysis of MDMA on the Allosteric Site of the hSERT: 5-HT and Escitalopram Analogous Binding Modes
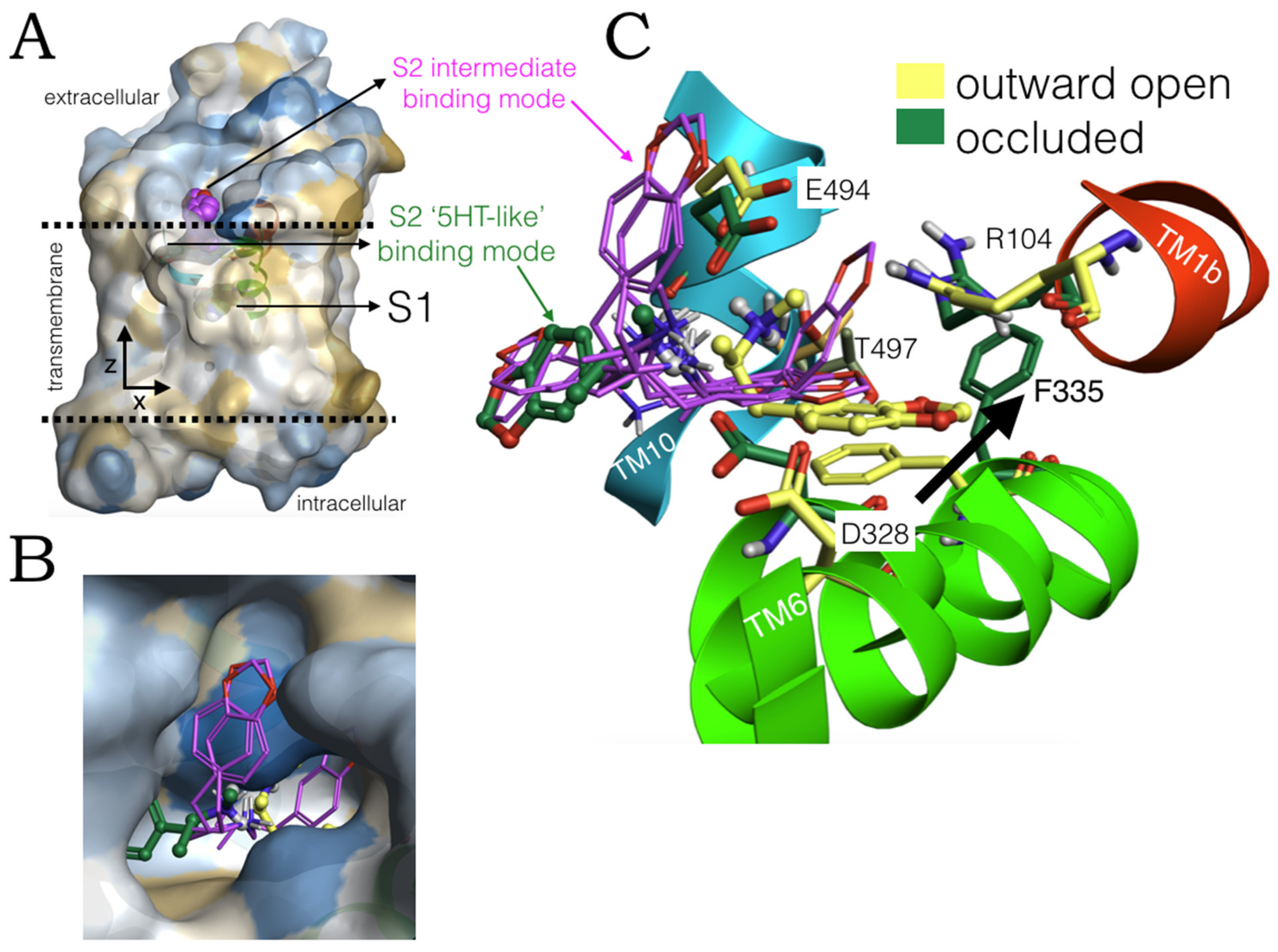
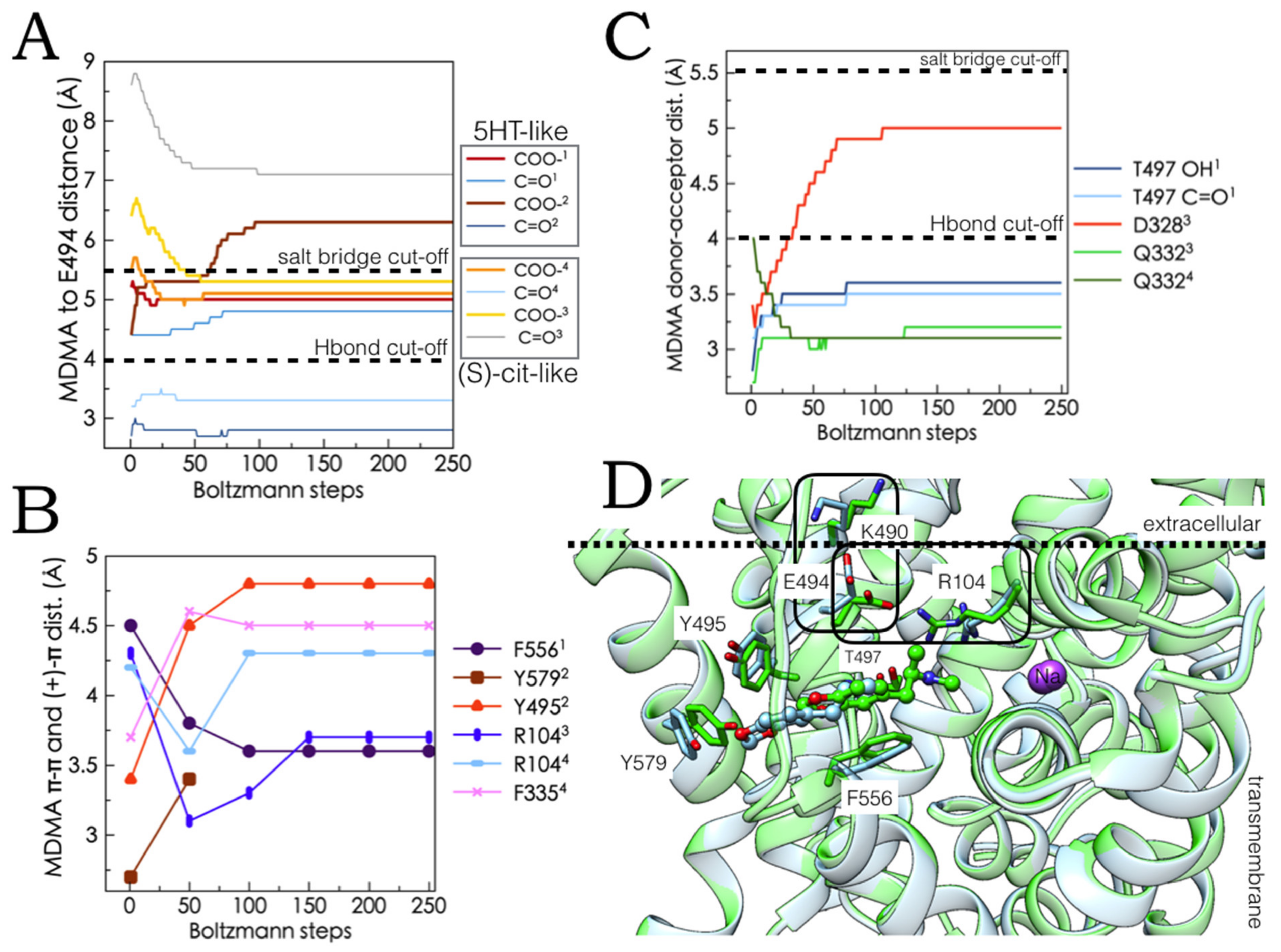
2.4. Intermediate Poses between the ‘5-HT-like’and the ‘Escitalopram-like’ Binding Modes of MDMA
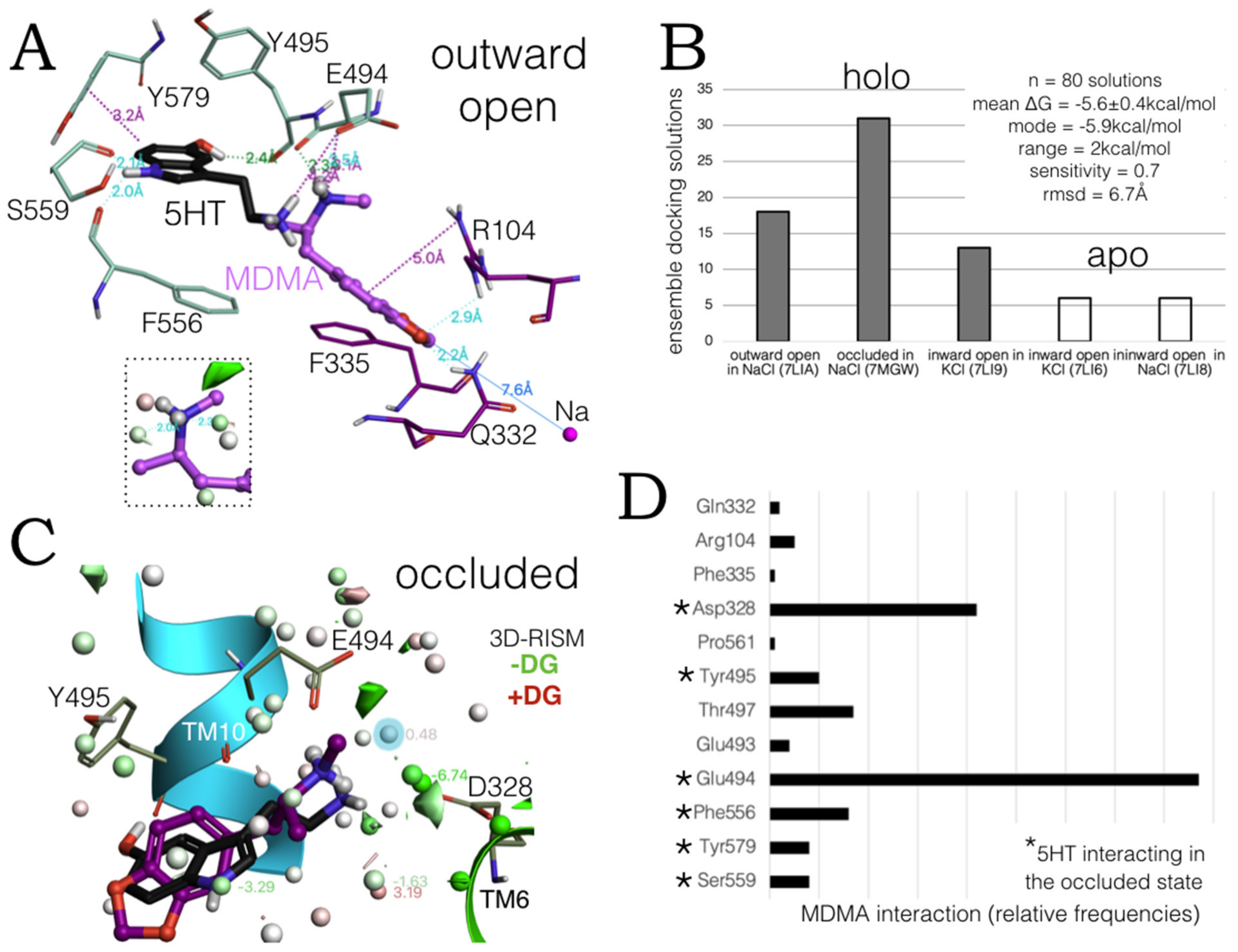
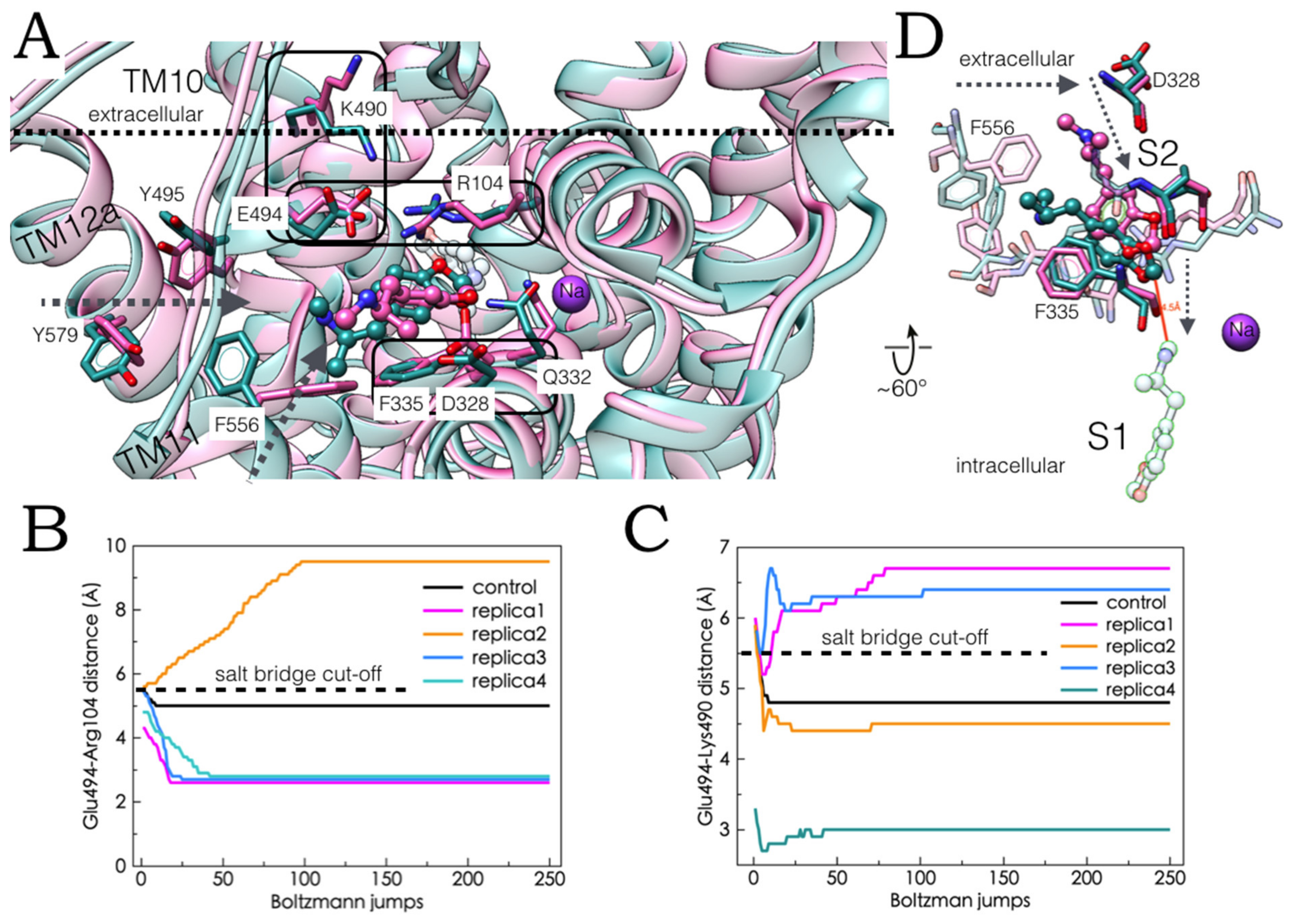
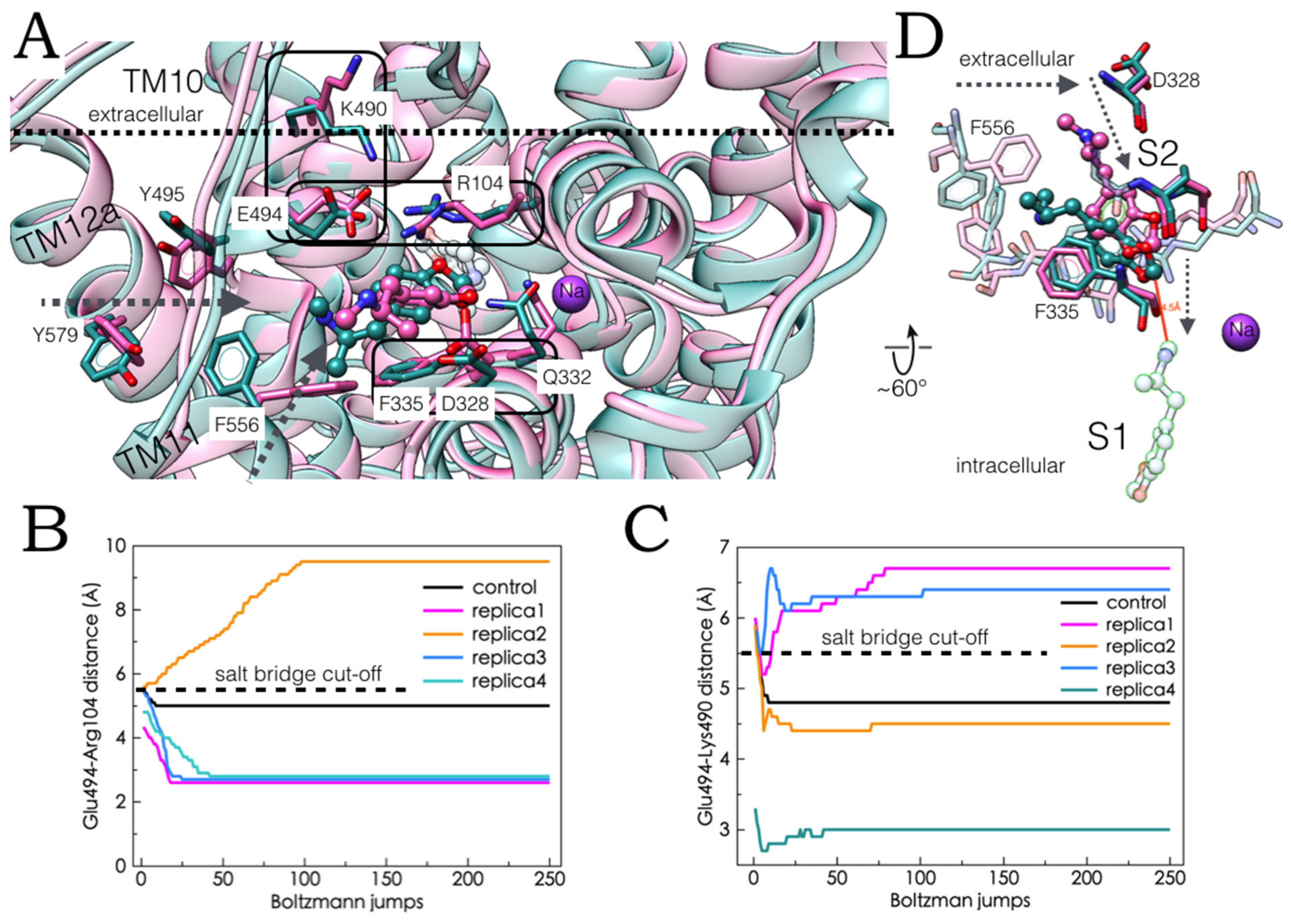
2.5. Monte Carlo (MC) Simulations on Double-Bound MDMA/hSERT Models to Identify Allosteric Determinants
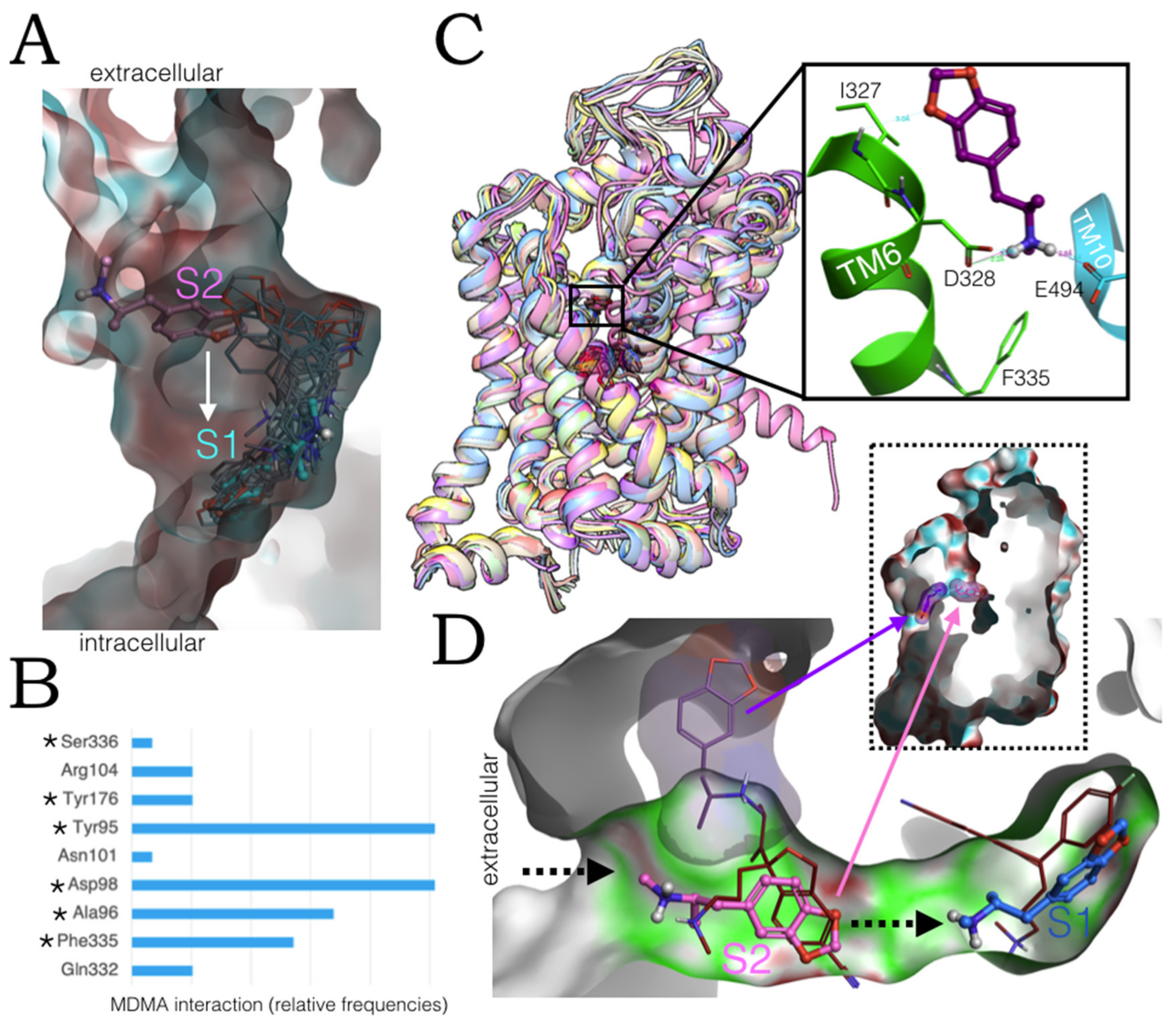
2.6. Ensemble Binding Space Analysis and Electrostatic Complementarity of the Pathway of MDMA from S2 to S1
3. Methods
3.1. Induced Fit and Ensemble Docking
3.2. Solvation Thermodynamics and Gibbs Free Energies Calculations
3.3. Monte Carlo (MC) Energy Perturbations and Electrostatic Complementarity (EC)
3.4. Ensemble Binding Space Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bauer, M.R.; Mackey, M.D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62, 3036–3050. [Google Scholar] [CrossRef]
- Berger, U.V.; Gu, X.F.; Azmitia, E.C. The substituted amphetamines 3,4-methylenedioxymethamphetamine, methamphetamine, p-chloroamphetamine and fenfluramine induce 5-hydroxytryptamine release via a common mechanism blocked by fluoxe-tine and cocaine. Eur. J. Pharmacol. 1992, 215, 153–160. [Google Scholar] [CrossRef]
- Blough, B.E.; Landavazo, A.; Partilla, J.S.; Baumann, M.H.; Decker, A.M.; Page, K.M.; Rothman, R.B. Hybrid Dopamine Uptake Blocker–Serotonin Releaser Ligands: A New Twist on Transporter-Focused Therapeutics. ACS Med. Chem. Lett. 2014, 5, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.G.; Sachpatzidis, A.; Rudnick, G. The third transmembrane domain of the serotonin transporter contains residues as-sociated with substrate and cocaine binding. J. Biol. Chem. 1997, 272, 28321–28327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, C.G. Out of the night-time and into the day: Ketamine and MDMA as therapies for mental disorders. Aust. N. Z. J. Psychiatry 2021, 55, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, L.E.; Andrews, A.M.; Olson, D.E. Dark Classics in Chemical Neuroscience: 3,4-Methylenedioxymethamphetamine. ACS Chem. Neurosci. 2018, 9, 2408–2427. [Google Scholar] [CrossRef]
- Erol, I.; Aksoydan, B.; Kantarcioglu, I.; Salmas, R.E.; Durdagi, S. Identification of novel serotonin reuptake inhibitors targeting central and allosteric binding sites: A virtual screening and molecular dynamics simulations study. J. Mol. Graph. Model. 2017, 74, 193–202. [Google Scholar] [CrossRef]
- Eshleman, A.J.; Nagarajan, S.; Wolfrum, K.M.; Reed, J.F.; Swanson, T.L.; Nilsen, A.; Janowsky, A. Structure-activity relationships of bath salt components: Substituted cathinones and benzofurans at biogenic amine transporters. Psychopharmacology 2018, 236, 939–952. [Google Scholar] [CrossRef]
- Heifets, B.D.; Malenka, R.C. Better living through chemistry: MDMA’s prosocial mechanism as a starting point for improved therapeutics. Neuropsychopharmacology 2020, 46, 261. [Google Scholar] [CrossRef]
- Islas, A.; Moreno, L.G.; Scior, T. Induced fit, ensemble binding space docking and Monte Carlo simulations of MDMA ‘ecstasy’ and 3D pharmacophore design of MDMA derivatives on the human serotonin transporter (hSERT). Heliyon 2021, 7, e07784. [Google Scholar] [CrossRef]
- Luethi, D.; Liechti, M.E. Designer drugs: Mechanism of action and adverse effects. Arch. Toxicol. 2020, 94, 1085–1133. [Google Scholar] [CrossRef] [Green Version]
- Maples-Keller, J.L.; Norrholm, S.D.; Burton, M.; Reiff, C.; Coghlan, C.; Jovanovic, T.; Yasinski, C.; Jarboe, K.; Rakofsky, J.; Rauch, S.; et al. A randomized controlled trial of 3,4-methylenedioxymethamphetamine (MDMA) and fear extinction retention in healthy adults. J. Psychopharmacol. 2022, 36, 368–377. [Google Scholar] [CrossRef]
- Marseille, E.; Mitchell, J.M.; Kahn, J.G. Updated cost-effectiveness of MDMA-assisted therapy for the treatment of posttraumatic stress disorder in the United States: Findings from a phase 3 trial. PLoS ONE 2022, 17, e0263252. [Google Scholar] [CrossRef]
- Möller, I.R.; Slivacka, M.; Nielsen, A.K.; Rasmussen, S.G.F.; Gether, U.; Loland, C.J.; Rand, K.D. Conformational dynamics of the human serotonin transporter during substrate and drug binding. Nat. Commun. 2019, 10, 1687. [Google Scholar] [CrossRef]
- Navratna, V.; Gouaux, E. Insights into the mechanism and pharmacology of neurotransmitter sodium symporters. Curr. Opin. Struct. Biol. 2019, 54, 161–170. [Google Scholar] [CrossRef]
- Niello, M.; Cintulova, D.; Hellsberg, E.; Jäntsch, K.; Holy, M.; Ayatollahi, L.H.; Cozzi, N.V.; Freissmuth, M.; Sandtner, W.; Ecker, G.F.; et al. para-Trifluoromethyl-methcathinone is an allosteric modulator of the serotonin transporter. Neuropharmacology 2019, 161, 107615. [Google Scholar] [CrossRef]
- Ortore, G.; Orlandini, E.; Betti, L.; Giannaccini, G.; Mazzoni, M.R.; Camodeca, C.; Nencetti, S. Focus on Human Monoamine Trans-porter Selectivity. New Human DAT and NET Models, Experimental Validation, and SERT Affinity Exploration. ACS Chem. Neurosci. 2020, 11, 3214–3232. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA suite of programs: An versatile platform for cheminformatics and drug design projects. Bioinformatics 2020, 37, 1174–1175. [Google Scholar] [CrossRef]
- Plenge, P.; Shi, L.; Beuming, T.; Te, J.; Newman, A.H.; Weinstein, H.; Gether, U.; Loland, C.J. Steric Hindrance Mutagenesis in the Conserved Extracellular Vestibule Impedes Allosteric Binding of Antidepressants to the Serotonin Transporter. J. Biol. Chem. 2012, 287, 39316–39326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plenge, P.; Abramyan, A.M.; Sørensen, G.; Mørk, A.; Weikop, P.; Gether, U.; Bang-Andersen, B.; Shi, L.; Loland, C.J. The mechanism of a high-affinity allosteric inhibitor of the serotonin transporter. Nat. Commun. 2020, 11, 1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plenge, P.; Yang, D.; Salomon, K.; Laursen, L.; Kalenderoglou, I.E.; Newman, A.H.; Gouaux, E.; Coleman, J.A.; Loland, C.J. The antide-pressant drug vilazodone is an allosteric inhibitor of the serotonin transporter. Nat. Commun. 2021, 12, 5063. [Google Scholar] [CrossRef] [PubMed]
- Sandtner, W.; Stockner, T.; Hasenhuetl, P.; Partilla, J.S.; Seddik, A.; Zhang, Y.-W.; Cao, J.; Holy, M.; Steinkellner, T.; Rudnick, G.; et al. Binding Mode Selection Determines the Action of Ecstasy Homologs at Monoamine Transporters. Mol. Pharmacol. 2015, 89, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.W.; Sicignano, D.J.; Hernandez, A.V.; White, C.M. MDMA-assisted psychotherapy for treatment of post-traumatic stress disorder: A systematic review with meta-analysis. J. Clin. Pharmacol. 2021, 62, 463–471. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Zeifman, A.A.; Stroylov, V.S.; Chilov, G.G. TSAR, a new graph-theoretical approach to computational modeling of protein side-chain flexibility: Modeling of ionization properties of proteins. Proteins Struct. Funct. Bioinform. 2011, 79, 2693–2710. [Google Scholar] [CrossRef]
- Tancer, M.; Johanson, C.-E. The effects of fluoxetine on the subjective and physiological effects of 3,4-methylenedioxymethamphetamine (MDMA) in humans. Psychopharmacologia 2006, 189, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.J.; Christoffel, D.J.; Heifets, B.D.; Ben-Dor, G.A.; Selimbeyoglu, A.; Hung, L.W.; Deisseroth, K.; Malenka, R.C. 5-HT release in nucleus accumbens rescues social deficits in mouse autism model. Nature 2018, 560, 589–594. [Google Scholar] [CrossRef]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Fu, T.; Deng, S.; Yang, F.; Yang, J.; Zhu, F. Molecular Mechanism for the Allosteric Inhibition of the Human Serotonin Transporter by Antidepressant escitalopram. ACS Chem. Neurosci. 2022, 13, 340–351. [Google Scholar] [CrossRef]
- Yang, D.; Gouaux, E. Illumination of serotonin transporter mechanism and role of the allosteric site. Sci. Adv. 2021, 7, eabl3857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Rudnick, G. The Cytoplasmic Substrate Permeation Pathway of Serotonin Transporter. J. Biol. Chem. 2006, 281, 36213–36220. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5-HT-Like Orientation | Escitalopram Orientation | p-Value |
|---|---|---|
| −7.1 ± 0.9 kcal/mol * | −6.3 ± 0.6 kcal/mol | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islas, Á.A.; Scior, T. Allosteric Binding of MDMA to the Human Serotonin Transporter (hSERT) via Ensemble Binding Space Analysis with ΔG Calculations, Induced Fit Docking and Monte Carlo Simulations. Molecules 2022, 27, 2977. https://doi.org/10.3390/molecules27092977
Islas ÁA, Scior T. Allosteric Binding of MDMA to the Human Serotonin Transporter (hSERT) via Ensemble Binding Space Analysis with ΔG Calculations, Induced Fit Docking and Monte Carlo Simulations. Molecules. 2022; 27(9):2977. https://doi.org/10.3390/molecules27092977
Chicago/Turabian StyleIslas, Ángel A., and Thomas Scior. 2022. "Allosteric Binding of MDMA to the Human Serotonin Transporter (hSERT) via Ensemble Binding Space Analysis with ΔG Calculations, Induced Fit Docking and Monte Carlo Simulations" Molecules 27, no. 9: 2977. https://doi.org/10.3390/molecules27092977
APA StyleIslas, Á. A., & Scior, T. (2022). Allosteric Binding of MDMA to the Human Serotonin Transporter (hSERT) via Ensemble Binding Space Analysis with ΔG Calculations, Induced Fit Docking and Monte Carlo Simulations. Molecules, 27(9), 2977. https://doi.org/10.3390/molecules27092977