
Characterizing the ZrC(111)/c-ZrO2(111) Hetero-Ceramic Interface: First Principles DFT and Atomistic Thermodynamic Modeling

 ,
,
Abstract
1. Introduction
2. Structural Models and Calculation Schemes
2.1. General Computational Details
2.2. Finite Temperature Molecular Dynamics (MD)
2.3. Bulk ZrC and c-ZrO2 Phases
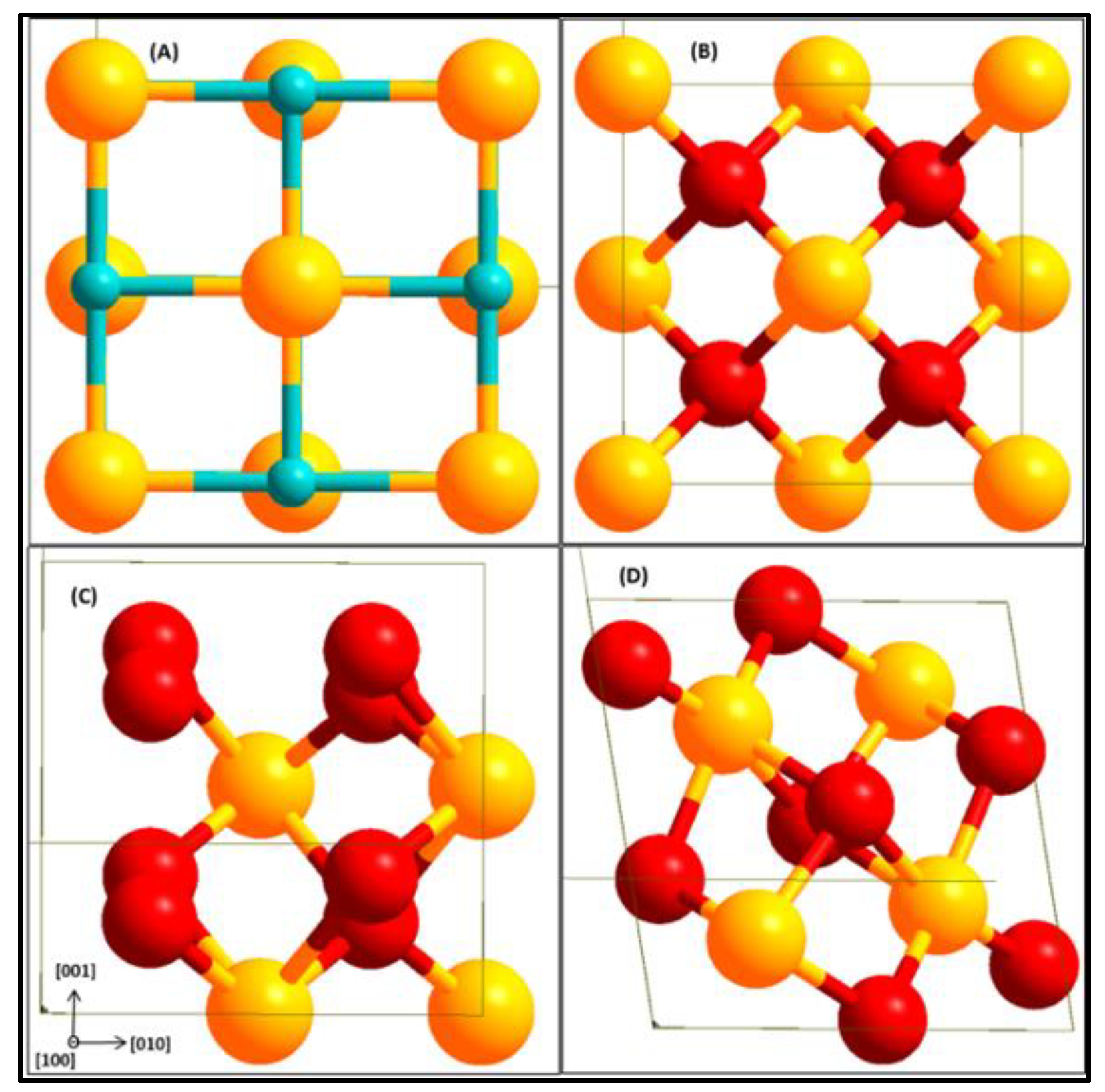
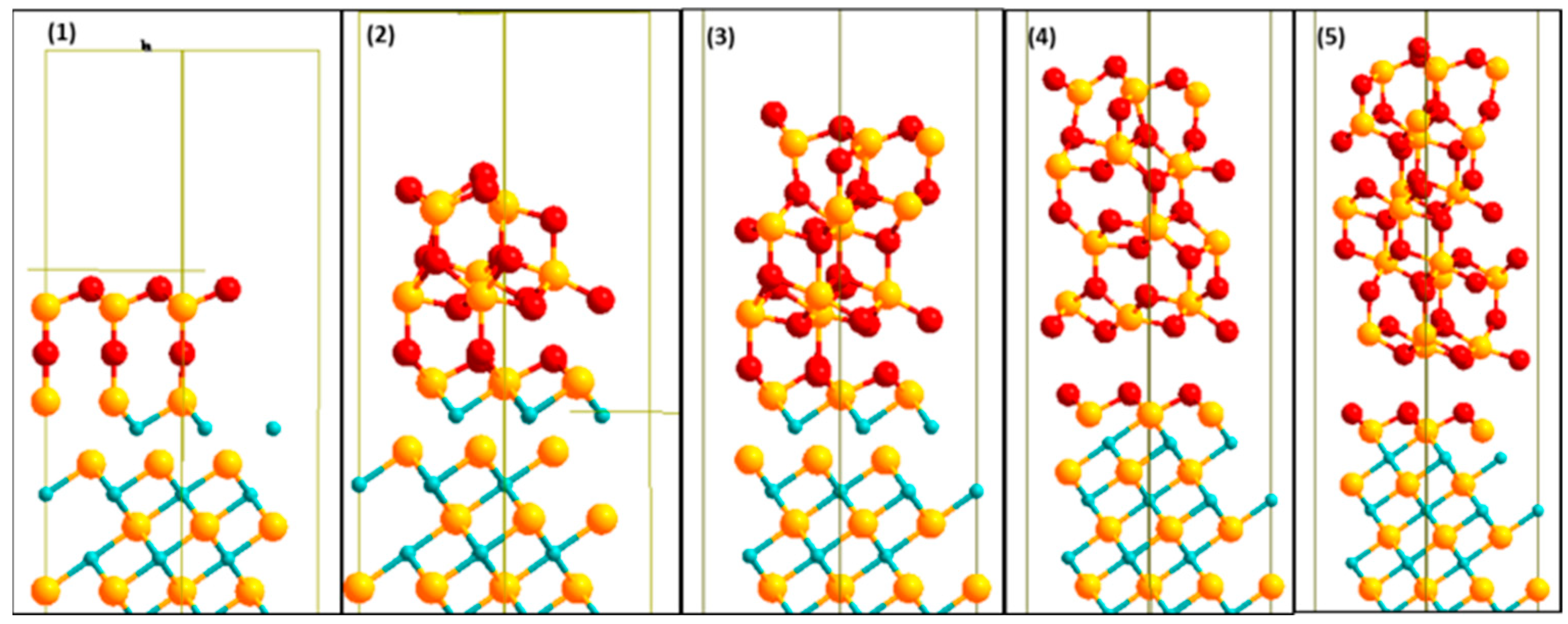
2.4. Interface Model Construction
2.4.1. Commensurate Phases and Surface Structures
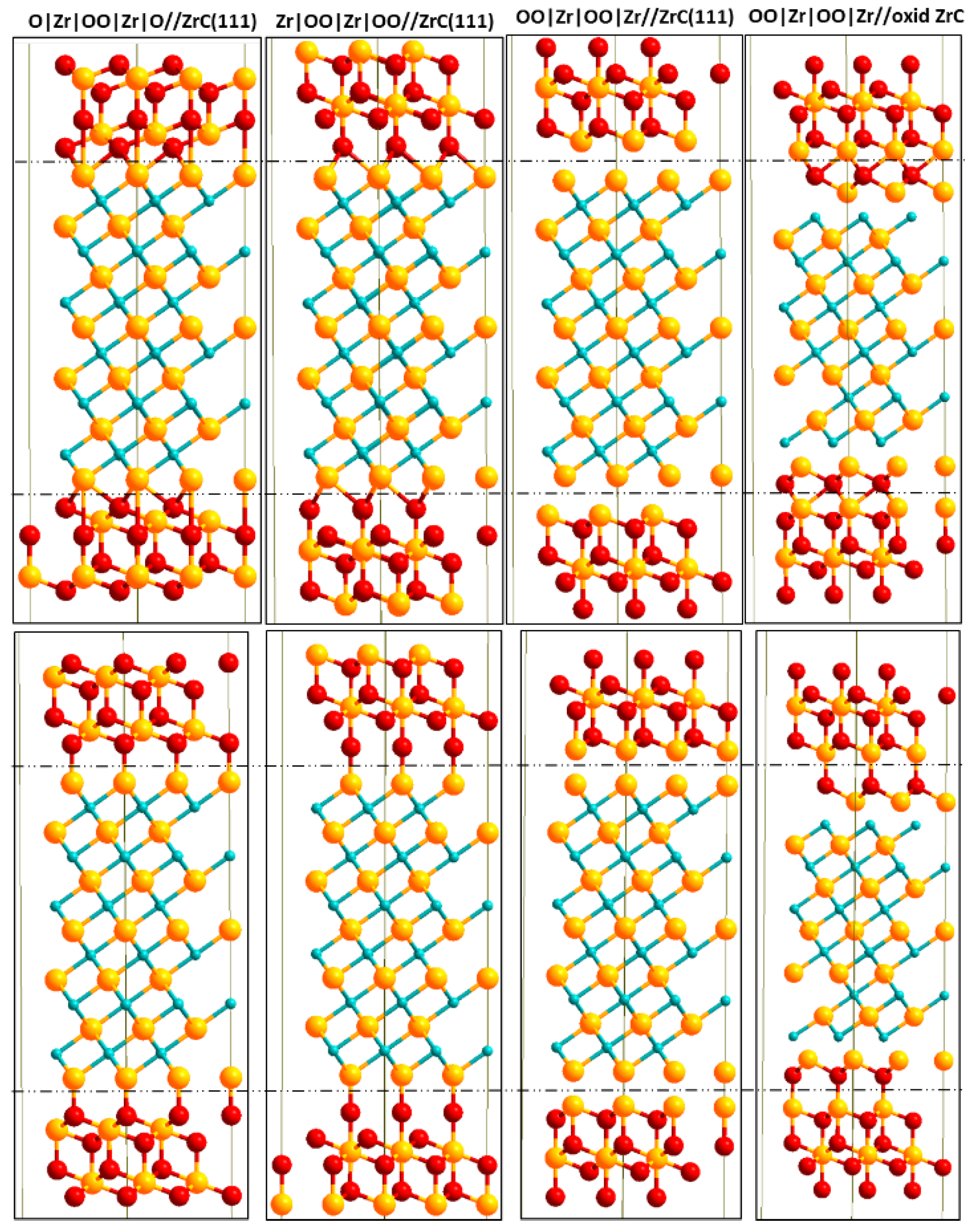
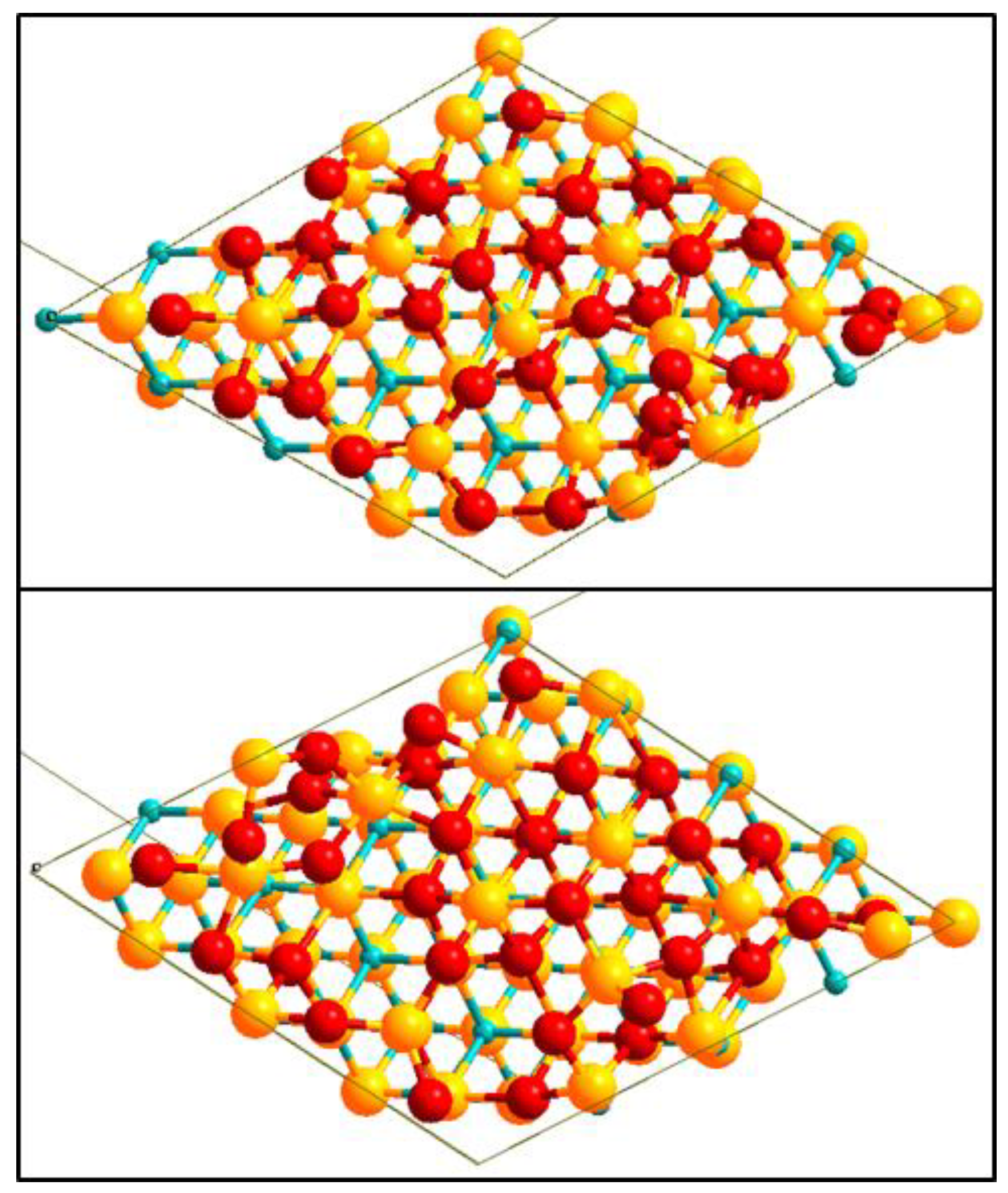
2.4.2. Geometrical Models for Interface
2.4.3. Mechanics and Cohesion at the Interface
2.5. Interfacial Thermodynamics
- -
- For , the upper boundary is and lower boundary is where are the formation energies of ZrC and c-ZrO2, respectively. Thus, the Zr chemical potential range is defined as:
- -
- For O chemical potential, the upper limit is the lower boundary is and the range of chemical potentials for O is:
3. Results and Discussion
3.1. ZrC-ZrO2: From Experiments to Theoretical Studies
3.2. Finite Temperature Molecular Dynamic Simulation
3.3. Surface and Bulk Properties of ZrC and ZrO2
3.4. Interface Cohesion and Structure
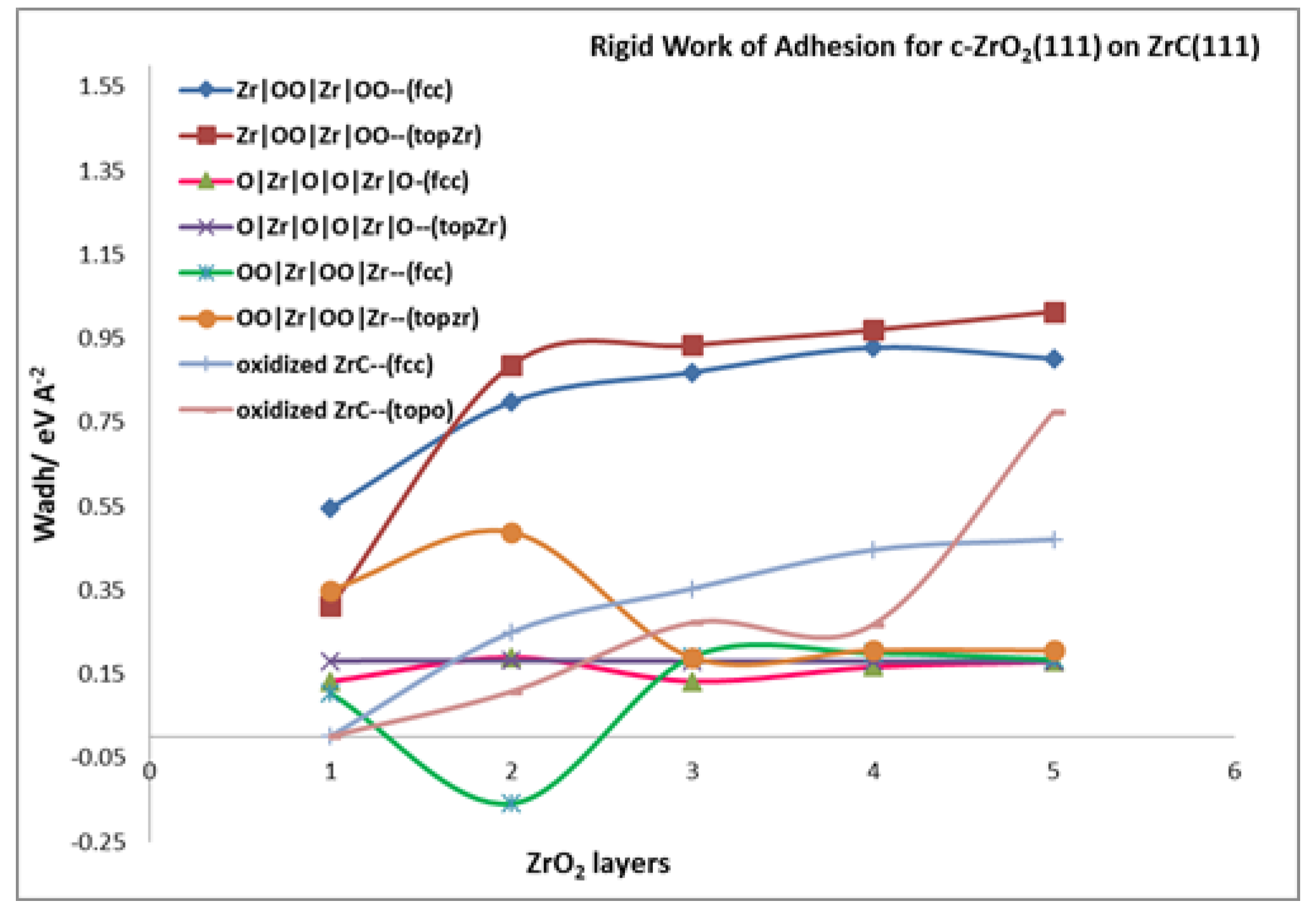
3.4.1. Rigid Work of Adhesion
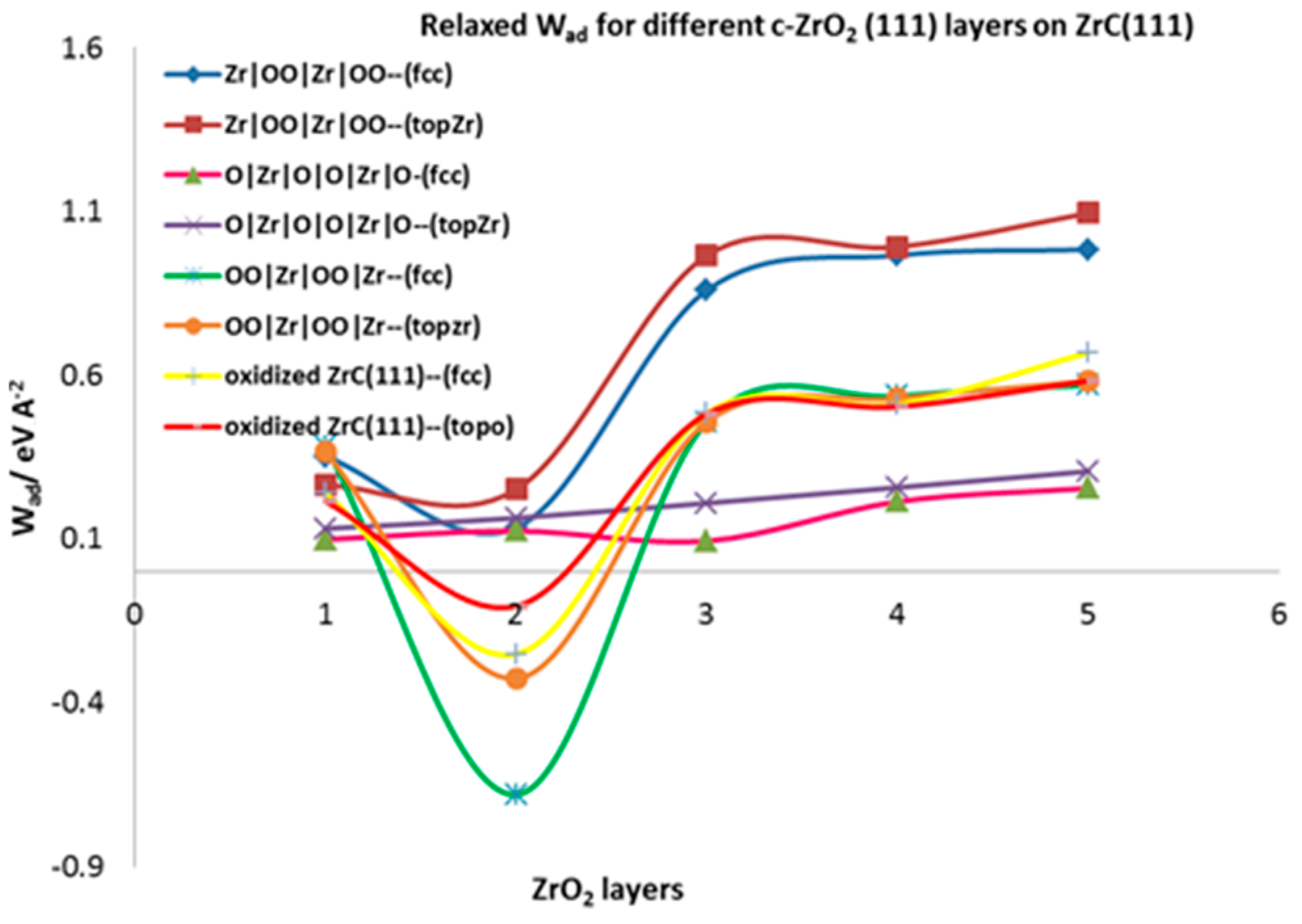
3.4.2. Relaxed Work of Adhesion
3.4.3. Relaxed Work of Adhesion
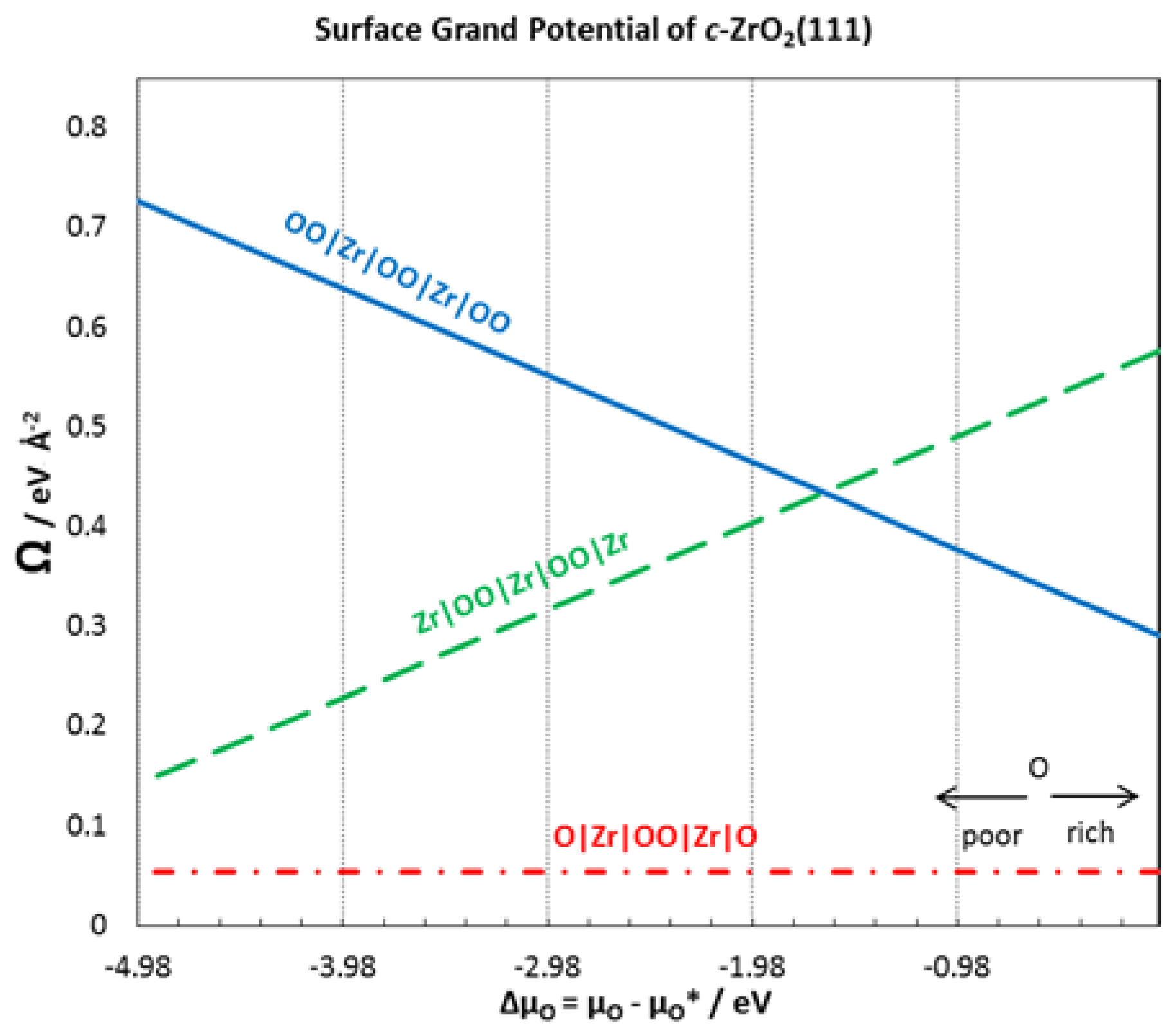
3.5. Thermodynamic Stability of the Interface
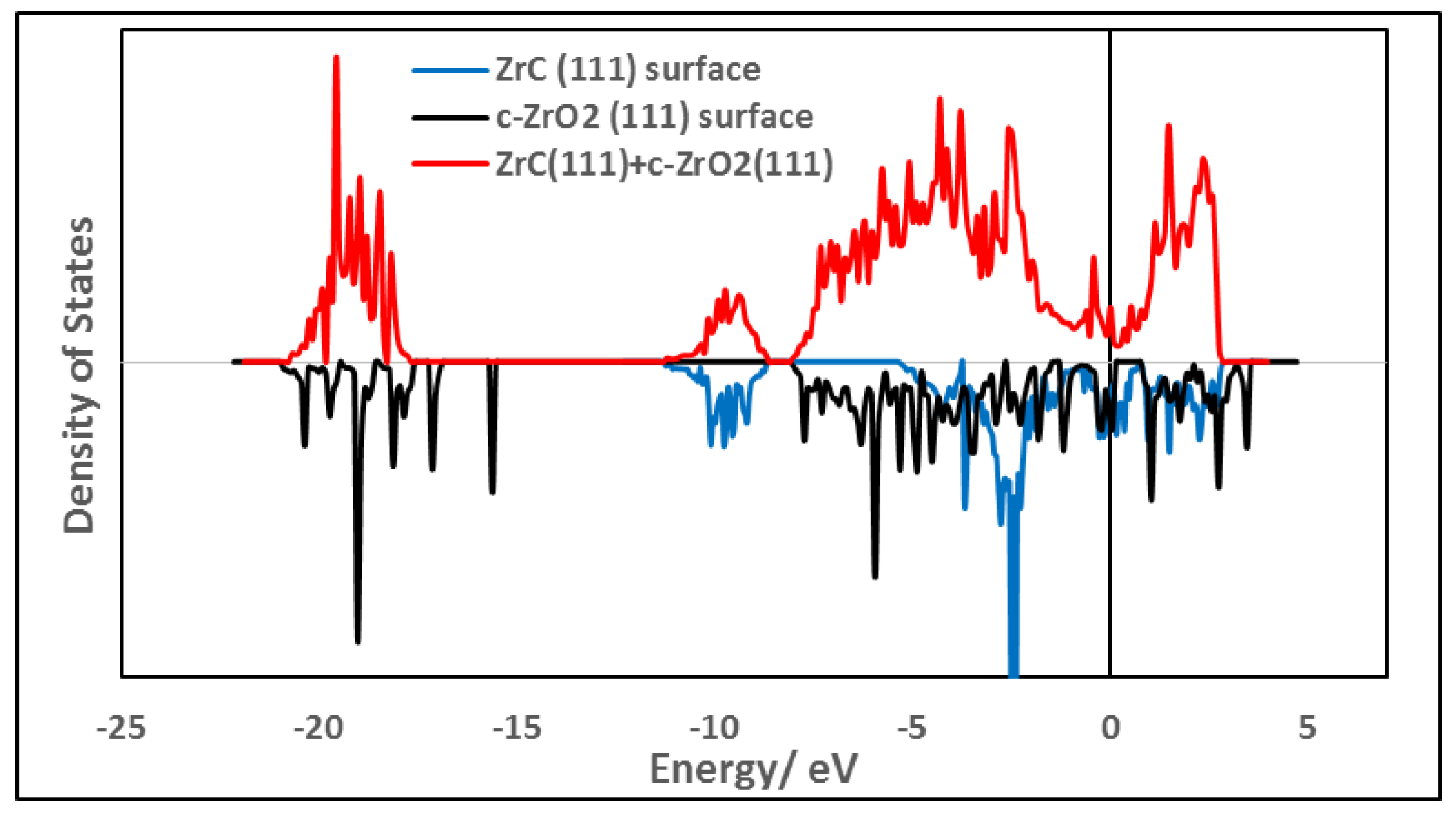
3.6. Interfacial Electronic Properties
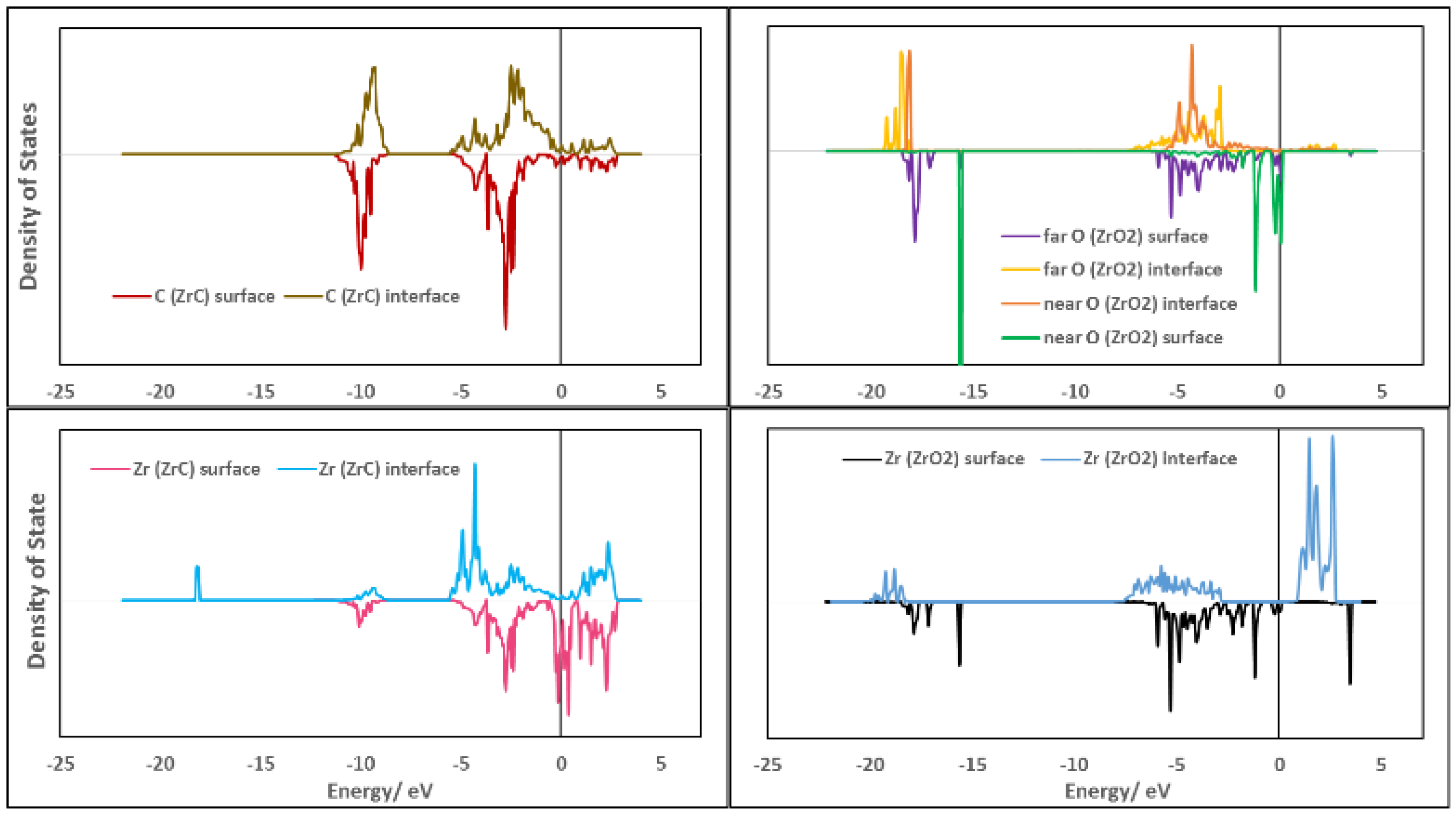
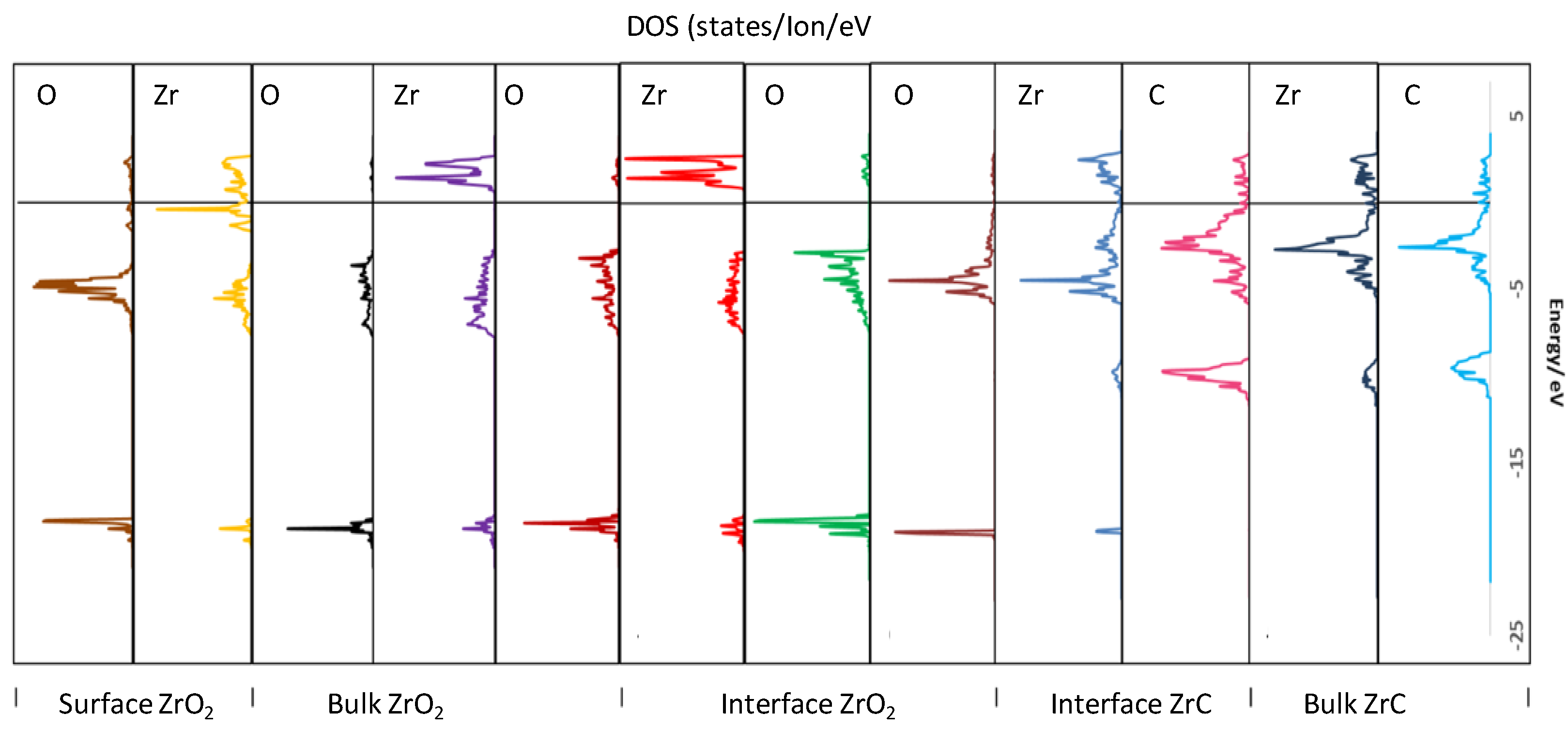
3.6.1. Density of States
3.6.2. Charge Transfer Analysis
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gosset, D.; Dollé, M.; Simeone, D.; Baldinozzi, G.; Thomé, L. Structural Behaviour of Nearly Stoichiometric ZrC under Ion Irradiation. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2008, 266, 2801–2805. [Google Scholar] [CrossRef]
- Gosset, D.; Dollé, M.; Simeone, D.; Baldinozzi, G.; Thomé, L. Structural Evolution of Zirconium Carbide under Ion Irradiation. J. Nucl. Mater. 2008, 373, 123–129. [Google Scholar] [CrossRef]
- Li, H.; Zhang, L.; Cheng, L.; Wang, Y. Oxidation Analysis of 2D C/ZrC–SiC Composites with Different Coating Structures in CH4 Combustion Gas Environment. Ceram. Int. 2009, 35, 2277–2282. [Google Scholar] [CrossRef]
- Osei-Agyemang, E.; Paul, J.-F.; Lucas, R.; Foucaud, S.; Cristol, S. Stability, Equilibrium Morphology and Hydration of ZrC(111) and (110) Surfaces with H2O: A Combined Periodic DFT and Atomistic Thermodynamic Study. Phys. Chem. Chem. Phys. 2015, 17, 21401–21413. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.; Carter, E.A. Structure, Bonding, and Adhesion at the ZrC(1 0 0)/Fe(1 1 0) Interface from First Principles. Surf. Sci. 2004, 560, 103–120. [Google Scholar] [CrossRef]
- Kato, S.; Ozawa, K.; Edamoto, K.; Otani, S. Photoelectron Spectroscopy Study of the Oxidation of ZrC(100). Jpn. J. Appl. Phys. 2000, 39, 5217–5222. [Google Scholar] [CrossRef]
- Viñes, F.; Sousa, C.; Illas, F.; Liu, P.; Rodriguez, J.A. Density Functional Study of the Adsorption of Atomic Oxygen on the (001) Surface of Early Transition-Metal Carbides. J. Phys. Chem. C 2007, 111, 1307–1314. [Google Scholar] [CrossRef]
- Kitaoka, H.; Ozawa, K.; Edamoto, K.; Otani, S. The Interaction of Water with Oxygen-Modified ZrC(100) Surfaces. Solid State Commun. 2001, 118, 23–26. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Gomes, J.; Nakamura, K.; Viñes, F.; Sousa, C.; Illas, F. Interaction of Oxygen with ZrC(001) and VC(001): Photoemission and First-Principles Studies. Phys. Rev. B 2005, 72, 075427. [Google Scholar] [CrossRef]
- Osei-Agyemang, E.; Paul, J.-F.; Lucas, R.; Foucaud, S.; Cristol, S. Oxidation and Equilibrium Morphology of Zirconium Carbide Low Index Surfaces Using DFT and Atomistic Thermodynamic Modeling. J. Phys. Chem. C 2016, 120, 8759–8771. [Google Scholar] [CrossRef]
- Shimada, S.; Yoshimatsu, M.; Inagaki, M.; Otani, S. Formation and Characterization of Carbon at the ZrCZrO2 Interface by Oxidation of ZrC Single Crystals. Carbon 1998, 36, 1125–1131. [Google Scholar] [CrossRef]
- Shimada, S.; Yoshimatsu, M.; Yunazar, F.; Otani, S. Deposition and Characterization of Carbon at the Interface by Oxidation of Single Crystals of Carbides (HfC, ZrC, TiC). Tanso 1999, 1999, 223–228. [Google Scholar] [CrossRef][Green Version]
- Noda, T.; Yamazaki, M.; Ozawa, K.; Edamoto, K.; Otani, S. Oxygen Adsorption on a ZrC(111) Surface: Angle-Resolved Photoemission Study. Surf. Sci. 2000, 450, 27–33. [Google Scholar] [CrossRef]
- Noda, T.; Nakane, T.; Ozawa, K.; Edamoto, K.; Tanaka, S.; Otani, S. Photoemission Study of the Oxidation of ZrC(111). Solid State Commun. 1998, 107, 145–148. [Google Scholar] [CrossRef]
- Ozawa, K.; Yoshii, T.; Noda, T.; Edamoto, K.; Tanaka, S. Coadsorption of Oxygen and Cesium on ZrC(1 1 1). Surf. Sci. 2002, 511, 421–434. [Google Scholar] [CrossRef]
- Tokumitsu, S.; Anazawa, T.; Tanabe, A.; Sekine, R.; Miyazaki, E.; Edamoto, K.; Kato, H.; Otani, S. Interaction of Hydrogen with ZrC(111) Surface: Angle-Resolved Photoemission Study. Surf. Sci. 1996, 351, 165–171. [Google Scholar] [CrossRef]
- Vojvodic, A.; Ruberto, C.; Lundqvist, B.I. Atomic and Molecular Adsorption on Transition-Metal Carbide (111) Surfaces from Density-Functional Theory: A Trend Study of Surface Electronic Factors. J. Phys. Condens. Matter Inst. Phys. J. 2010, 22, 375504. [Google Scholar] [CrossRef]
- Aono, M.; Oshima, C.; Zaima, S.; Otani, S.; Ishizawa, Y. Quantitative Surface Atomic Geometry and Two-Dimensional Surface Electron Distribution Analysis by a New Technique in Low-Energy Ion Scattering. Jpn. J. Appl. Phys. 1981, 20, L829–L832. [Google Scholar] [CrossRef]
- Hayami, W.; Souda, R.; Aizawa, T.; Otani, S.; Ishizawa, Y. Analysis of the HfC(111) Surface Structure by Impact Collision Ion Scattering Spectroscopy (ICISS). Surf. Sci. 1992, 276, 299–307. [Google Scholar] [CrossRef]
- Hayami, W.; Souda, R.; Aizawa, T.; Otani, S.; Ishizawa, Y. Analysis of the NbC(111)-(√3 × √3 )R30°-Al Surface Structure by Impact-Collision Ion-Scattering Spectroscopy. Phys. Rev. B 1993, 47, 13752–13758. [Google Scholar] [CrossRef]
- Christensen, A.; Carter, E.A. First-Principles Characterization of a Heteroceramic Interface: ZrO2(001) Deposited on an Alpha-Al2O3(1102) Substrate. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 62, 16968–16983. [Google Scholar] [CrossRef]
- Christensen, A.; Carter, E.A. Adhesion of Ultrathin ZrO2(111) Films on Ni(111) from First Principles. J. Chem. Phys. 2001, 114, 5816–5831. [Google Scholar] [CrossRef]
- Lucas, R.; Pizon, D.; Laborde, E.; Trolliard, G.; Foucaud, S.; Maître, A. A simple route for organic covalent grafting onto zirconium carbide particles. Appl. Surf. Sci. 2013, 287, 411–414. [Google Scholar] [CrossRef]
- Dasan, A.; Lucas, R.; Laborde, E.; Piriou, C.; Foucaud, S. Towards a surface functionalisation and grafting of a polycarbosilane onto zirconium carbide particles for the development of hybrid core-shell structures. Appl. Surf. Sci. 2019, 495, 143409. [Google Scholar] [CrossRef]
- Osei-Agyemang, E.; Dasan, A.; Lucas, R.; Foucaud, S.; Paul, J.F.; Cristol, S.; Laborde, E. Grafting of organic molecular precursors onto ZrC(100)//t-ZrO2(001) surfaces: When experimental and theoretical studies meet. Appl. Surf. Sci. 2022, 576, 151622. [Google Scholar] [CrossRef]
- Hafner, J. Ab-Initio Simulations of Materials Using VASP: Density-Functional Theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Mermin, N.D. Thermal Properties of the Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1441–A1443. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-Precision Sampling for Brillouin-Zone Integration in Metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Martin, L.; Vallverdu, G.; Martinez, H.; Cras, F.L.; Baraille, I. First Principles Calculations of Solid–solid Interfaces: An Application to Conversion Materials for Lithium-Ion Batteries. J. Mater. Chem. 2012, 22, 22063–22071. [Google Scholar] [CrossRef]
- Christensen, A.; Carter, E.A. First-Principles Study of the Surfaces of Zirconia. Phys. Rev. B 1998, 58, 8050–8064. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Fan, X.-L.; Liu, C.; Ran, R.-X. First Principles Study of Structural and Electronic Properties of Cubic Phase of ZrO2 and HfO2. Phys. B Condens. Matter 2014, 434, 7–13. [Google Scholar] [CrossRef]
- Liu, L.M.; Wang, S.Q.; Ye, H.Q. First-Principles Study of Metal/nitride Polar Interfaces: Ti/TiN. Surf. Interface Anal. 2003, 35, 835–841. [Google Scholar] [CrossRef]
- Wang, X.-G.; Smith, J.R. Si/Cu Interface Structure and Adhesion. Phys. Rev. Lett. 2005, 95, 156102. [Google Scholar] [CrossRef]
- Hashibon, A.; Elsässer, C.; Mishin, Y.; Gumbsch, P. First-Principles Study of Thermodynamical and Mechanical Stabilities of Thin Copper Film on Tantalum. Phys. Rev. B 2007, 76, 245434. [Google Scholar] [CrossRef]
- Hashibon, A.; Elsässer, C. Approaches to Atomistic Triple-Line Properties from First-Principles. Scr. Mater. 2010, 62, 939–944. [Google Scholar] [CrossRef]
- Dalverny, A.-L.; Filhol, J.-S.; Doublet, M.-L. Interface Electrochemistry in Conversion Materials for Li-Ion Batteries. J. Mater. Chem. 2011, 21, 10134–10142. [Google Scholar] [CrossRef]
- Malyi, O.I.; Chen, Z.; Shu, G.G.; Wu, P. Effect of Sulfur Impurity on the Stability of Cubic Zirconia and Its Interfaces with Metals. J. Mater. Chem. 2011, 21, 12363–12368. [Google Scholar] [CrossRef]
- Dai, H.; Du, J.; Wang, L.; Peng, C.; Liu, X. First-Principle Study of the AlP/Si Interfacial Adhesion. Phys. B Condens. Matter 2010, 405, 573–578. [Google Scholar] [CrossRef]
- Radican, K.; Berdunov, N.; Manai, G.; Shvets, I.V. Epitaxial Molybdenum Oxide Grown on Mo(110): LEED, STM, and Density Functional Theory Calculations. Phys. Rev. B 2007, 75, 155434. [Google Scholar] [CrossRef]
- Hashibon, A.; Elsässer, C.; Rühle, M. Structure at Abrupt Copper–alumina Interfaces: An Ab Initio Study. Acta Mater. 2005, 53, 5323–5332. [Google Scholar] [CrossRef]
- Finnis, M.W. The Theory of Metal—Ceramic Interfaces. J. Phys. Condens. Matter 1996, 8, 5811. [Google Scholar] [CrossRef]
- Liu, M.W.; Liu, X. Ultrathin Metal Films; Springer: Berlin, Germany, 2004. [Google Scholar]
- Suresh, L.B.F.; Suresh, S. Thin Film Materials: Stress, Defect Formation and Surface Evolution; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Osei-Agyemang, E.; Paul, J.F.; Lucas, R.; Foucaud, S.; Cristol, S. Periodic DFT and Atomistic Thermodynamic Modeling of Reactivity of H2, O2, and H2O Molecules on Bare and Oxygen Modified ZrC(100) Surface. J. Phys. Chem. C 2014, 118, 12952–12961. [Google Scholar] [CrossRef]
- Bogicevic, A.; Jennison, D.R. Variations in the Nature of Metal Adsorption on Ultrathin Al2O3 Films. Phys. Rev. Lett. 1999, 82, 4050–4053. [Google Scholar] [CrossRef]
- Musolino, V.; Selloni, A.; Car, R. Atomic and Electronic Structure of Cu Clusters on MgO. Surf. Sci. 1998, 402–404, 413–417. [Google Scholar] [CrossRef]
- Pacchioni, G.; Rösch, N. Supported Nickel and Copper Clusters on MgO(100): A First-principles Calculation on the Metal/oxide Interface. J. Chem. Phys. 1996, 104, 7329–7337. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZrC | c-ZrO2 | Overlap Area(S1–2)/Å2 | Misfit (ϒ) |
|---|---|---|---|
| (111) | (001) | 44.874 | 0.169 |
| (111) | (110) | 44.874 | 0.157 |
| (111) | (111) | 44.874 | 0.072 |
| Number of Layers | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Ωi/eV Å−2 | 0.068 | 0.053 | 0.055 | 0.055 | 0.054 | 0.054 |
| Interface Model | OO|Zr|OO|Zr—ZrC(111) | Zr|OO|Zr|OO—ZrC(111) | O|Zr|OO|Zr|O—ZrC(111) | OO|Zr|OO|Zr—4O-ZrC(111) | ||||
|---|---|---|---|---|---|---|---|---|
| c-ZrO2(111) Layers | fcc | top | fcc | top | fcc | top | fcc | top |
| Rigid work of Adhesion, | ||||||||
| 1 | 0.104 | 0.350 | 0.545 | 0.312 | 0.132 | 0.182 | 0.002 | 0.002 |
| 2 | −0.158 | 0.488 | 0.798 | 0.886 | 0.190 | 0.182 | 0.250 | 0.108 |
| 3 | 0.191 | 0.190 | 0.869 | 0.934 | 0.133 | 0.181 | 0.353 | 0.273 |
| 4 | 0.200 | 0.207 | 0.928 | 0.970 | 0.67 | 0.180 | 0.446 | 0.268 |
| 5 | 0.185 | 0.206 | 0.902 | 1.014 | 0.180 | 0.181 | 0.471 | 0.776 |
| Relaxed work of Adhesion, | ||||||||
| 1 | 0.383 | 0.366 | 0.351 | 0.264 | 0.098 | 0.131 | 0.240 | 0.215 |
| 2 | −0.682 | −0.327 | 0.134 | 0.251 | 0.124 | 0.164 | −0.252 | −0.106 |
| 3 | 0.458 | 0.457 | 0.859 | 0.965 | 0.093 | 0.209 | 0.484 | 0.481 |
| 4 | 0.536 | 0.529 | 0.966 | 0.991 | 0.214 | 0.257 | 0.511 | 0.503 |
| 5 | 0.570 | 0.582 | 0.983 | 1.095 | 0.254 | 0.306 | 0.669 | 0.583 |
| Model Site | /eV Å−2 |
|---|---|
| O|Zr|OO|Zr|O-ZrC(111) | |
| fcc | −0.562 |
| top | −0.605 |
| Zr|OO|Zr|OO-ZrC(111) | |
| fcc | −1.555 |
| top | −0.451 |
| OO|Zr|OO|Zr-ZrC(111) | |
| fcc | −0.257 |
| top | −0.251 |
| Zr|OO|Zr-oxidized ZrC(111) | |
| fcc | −0.444 |
| Ion Type | ZrO2 Layers on ZrC | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |||
| ZrO2 layer at Interface | Zr | 0.05 | 0.01 | −0.09 | −0.07 | −0.08 | |
| O | 0.04 | 0.42 | 0.21 | 0.25 | 0.23 | ||
| O | 0.38 | 0.73 | 0.67 | 0.75 | 0.75 | ||
| Interface Plane | |||||||
| ZrC layer at Interface | Zr | −0.49 | −0.89 | −0.94 | −0.92 | −0.93 | |
| C | 0.00 | −0.04 | −0.05 | −0.07 | −0.07 | ||
| Layer in Slab | 4 ZrO2 (Surface) | 3 ZrO2 (Bulk) | 2 ZrO2 (Bulk) | 1 ZrO2 (Interface) | 1 ZrC (Interface) | 2 ZrC (Bulk) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ion Type | O | Zr | O | Zr | O | O | Zr | O | O | Zr | O | O | Zr | C | Zr | C |
| Absolute Q | 7.14 | 2.63 | 7.18 | 1.79 | 7.17 | 7.11 | 1.72 | 7.09 | 7.11 | 1.72 | 7.13 | 7.17 | 2.00 | 5.74 | 2.28 | 5.71 |
| ∆Qs (vs. surface) | −0.04 | 0.07 | 0.01 | 0.04 | 0.09 | −0.08 | −0.01 | −0.02 | −0.07 | −0.07 | 0.25 | 0.75 | −0.92 | −0.07 | −0.07 | −0.03 |
| ∆Qb (vs. bulk) | −0.01 | 0.94 | 0.03 | 0.10 | 0.02 | −0.04 | 0.03 | −0.06 | −0.04 | 0.03 | −0.02 | 0.02 | −0.34 | 0.08 | −0.06 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osei-Agyemang, E.; Paul, J.-F.; Lucas, R.; Foucaud, S.; Cristol, S.; Mamede, A.-S.; Nuns, N.; Addad, A. Characterizing the ZrC(111)/c-ZrO2(111) Hetero-Ceramic Interface: First Principles DFT and Atomistic Thermodynamic Modeling. Molecules 2022, 27, 2954. https://doi.org/10.3390/molecules27092954
Osei-Agyemang E, Paul J-F, Lucas R, Foucaud S, Cristol S, Mamede A-S, Nuns N, Addad A. Characterizing the ZrC(111)/c-ZrO2(111) Hetero-Ceramic Interface: First Principles DFT and Atomistic Thermodynamic Modeling. Molecules. 2022; 27(9):2954. https://doi.org/10.3390/molecules27092954
Chicago/Turabian StyleOsei-Agyemang, Eric, Jean-François Paul, Romain Lucas, Sylvie Foucaud, Sylvain Cristol, Anne-Sophie Mamede, Nicolas Nuns, and Ahmed Addad. 2022. "Characterizing the ZrC(111)/c-ZrO2(111) Hetero-Ceramic Interface: First Principles DFT and Atomistic Thermodynamic Modeling" Molecules 27, no. 9: 2954. https://doi.org/10.3390/molecules27092954
APA StyleOsei-Agyemang, E., Paul, J.-F., Lucas, R., Foucaud, S., Cristol, S., Mamede, A.-S., Nuns, N., & Addad, A. (2022). Characterizing the ZrC(111)/c-ZrO2(111) Hetero-Ceramic Interface: First Principles DFT and Atomistic Thermodynamic Modeling. Molecules, 27(9), 2954. https://doi.org/10.3390/molecules27092954