
Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex

Abstract
:1. Introduction
2. NSP14 Is Essential to RNA Replication and Transcription
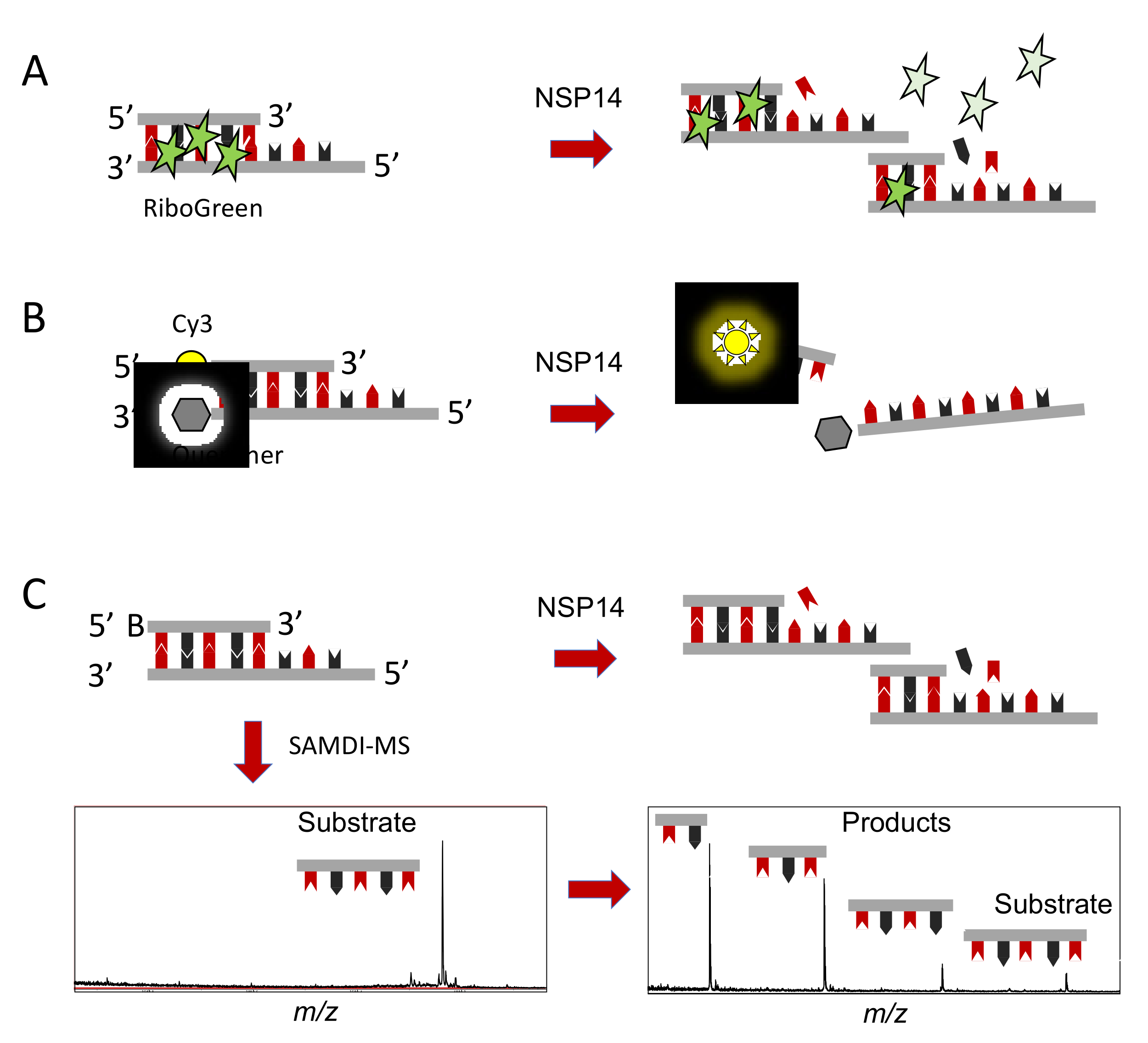
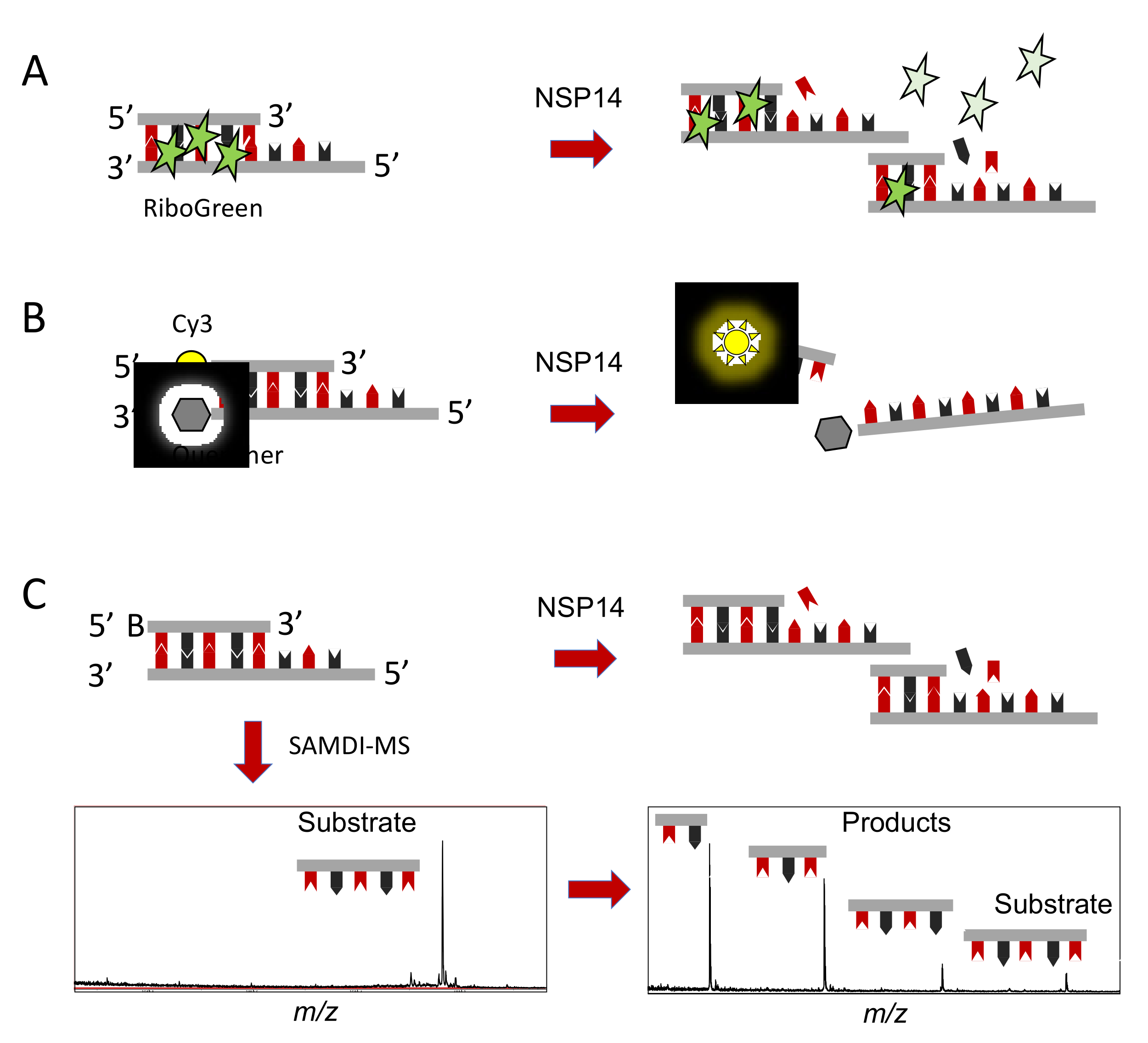
3. In Vitro Assays to Identify and Characterize NSP14 Proofreading Inhibitors
4. Known Small Molecules That Interfere with the Proofreading Activity of NSP14
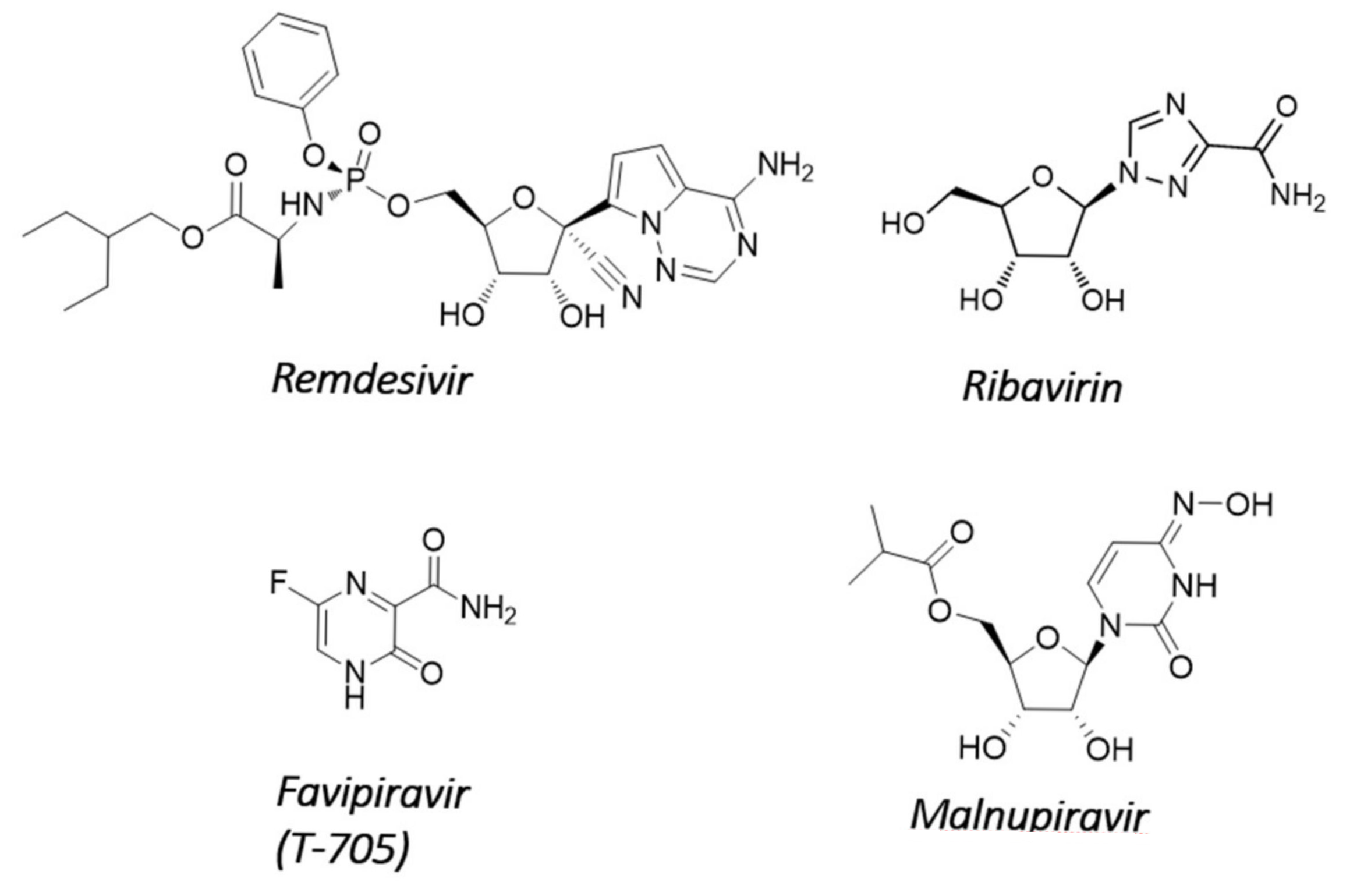
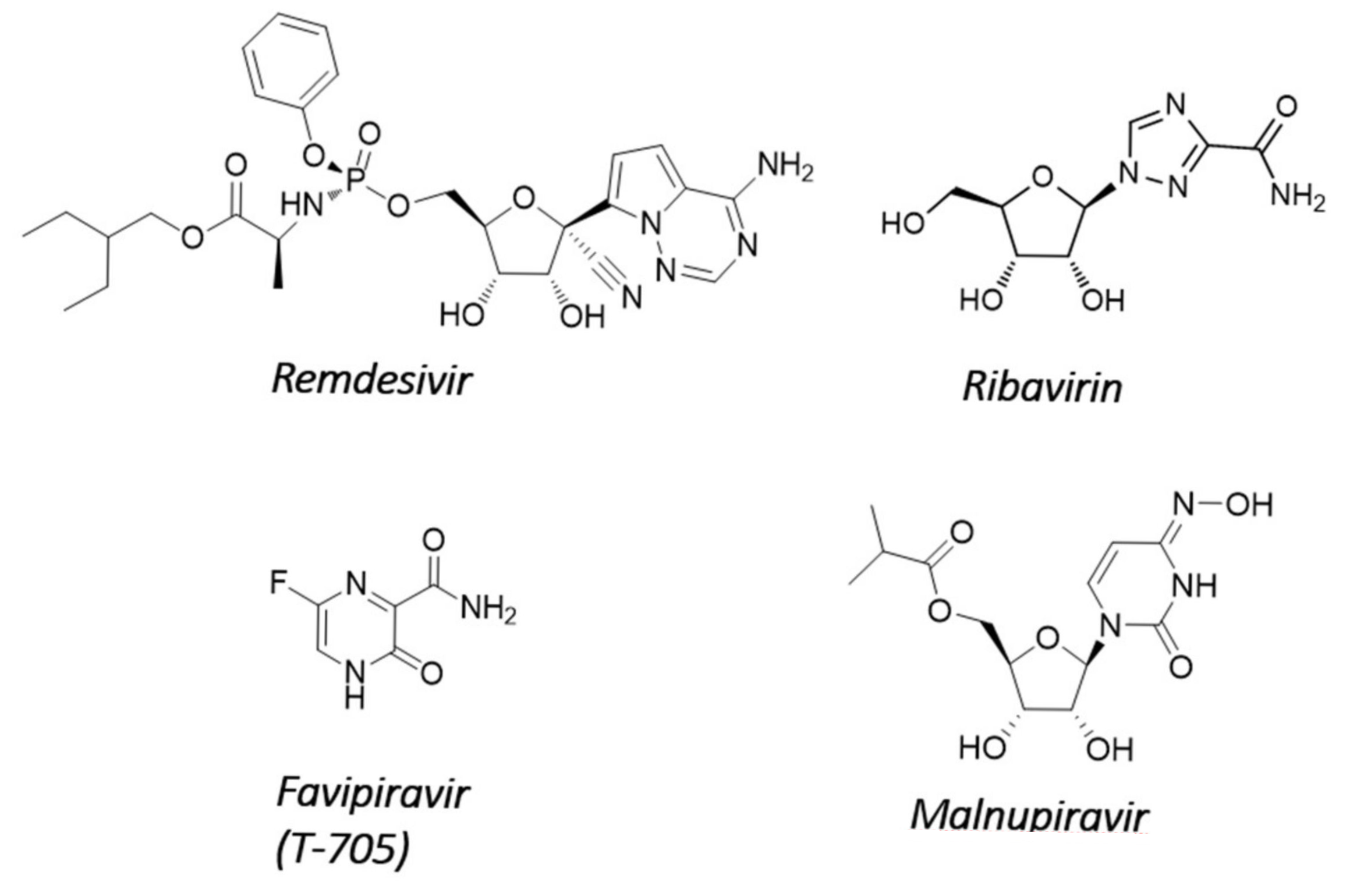
4.1. Nucleosides Causing Delayed Chain Termination or Error Catastrophe
4.2. Non-Nucleoside Inhibitors
5. Conclusions: Is There a Case for NSP14 Proofreading Inhibitors?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Group, C.S. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.; Ruben, M.; Overkleeft, H.S.; van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef] [Green Version]
- Shannon, A.; Fattorini, V.; Sama, B.; Selisko, B.; Feracci, M.; Falcou, C.; Gauffre, P.; El Kazzi, P.; Delpal, A.; Decroly, E.; et al. A dual mechanism of action of AT-527 against SARS-CoV-2 polymerase. Nat. Commun. 2022, 13, 621. [Google Scholar] [CrossRef]
- Sweis, R.F.; Pliushchev, M.; Brown, P.J.; Guo, J.; Li, F.; Maag, D.; Petros, A.M.; Soni, N.B.; Tse, C.; Vedadi, M.; et al. Discovery and development of potent and selective inhibitors of histone methyltransferase g9a. ACS Med. Chem. Lett. 2014, 5, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Devkota, K.; Schapira, M.; Perveen, S.; Khalili Yazdi, A.; Li, F.; Chau, I.; Ghiabi, P.; Hajian, T.; Loppnau, P.; Bolotokova, A.; et al. Probing the SAM Binding Site of SARS-CoV-2 Nsp14 In Vitro Using SAM Competitive Inhibitors Guides Developing Selective Bisubstrate Inhibitors. SLAS Discov. 2021, 26, 1200–1211. [Google Scholar] [CrossRef]
- Ogando, N.S.; El Kazzi, P.; Zevenhoven-Dobbe, J.C.; Bontes, B.W.; Decombe, A.; Posthuma, C.C.; Thiel, V.; Canard, B.; Ferron, F.; Decroly, E.; et al. Structure-function analysis of the nsp14 N7-guanine methyltransferase reveals an essential role in Betacoronavirus replication. Proc. Natl. Acad. Sci. USA 2021, 118, e2108709118. [Google Scholar] [CrossRef]
- Selvaraj, C.; Dinesh, D.C.; Panwar, U.; Abhirami, R.; Boura, E.; Singh, S.K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 4582–4593. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, R.; Sutto-Ortiz, P.; Guiraud, M.; Canard, B.; Vasseur, J.J.; Decroly, E.; Debart, F. Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur. J. Med. Chem. 2020, 201, 112557. [Google Scholar] [CrossRef] [PubMed]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3′->5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beese, L.S.; Steitz, T.A. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism. EMBO J. 1991, 10, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Jiang, M.; Hu, T.; Liu, Q.; Chen, X.S.; Guo, D. Biochemical characterization of exoribonuclease encoded by SARS coronavirus. J. BioChem. Mol. Biol. 2007, 40, 649–655. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [Green Version]
- Ogando, N.S.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Bredenbeek, P.J.; Posthuma, C.C.; Snijder, E.J. The Enzymatic Activity of the nsp14 Exoribonuclease Is Critical for Replication of MERS-CoV and SARS-CoV-2. J. Virol. 2020, 94, e01246-20. [Google Scholar] [CrossRef]
- Pan, J.; Peng, X.; Gao, Y.; Li, Z.; Lu, X.; Chen, Y.; Ishaq, M.; Liu, D.; Dediego, M.L.; Enjuanes, L.; et al. Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PLoS ONE 2008, 3, e3299. [Google Scholar] [CrossRef]
- Bouvet, M.; Imbert, I.; Subissi, L.; Gluais, L.; Canard, B.; Decroly, E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [Google Scholar] [CrossRef] [Green Version]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, e3900-9. [Google Scholar] [CrossRef] [Green Version]
- Ferron, F.; Subissi, L.; Silveira De Morais, A.T.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.K.; Appleby, T.C.; Bilello, J.P.; Feng, J.Y.; Schmitz, U.; Campbell, E.A. An atomistic model of the coronavirus replication-transcription complex as a hexamer assembled around nsp15. J. Biol. Chem. 2021, 297, 101218. [Google Scholar] [CrossRef] [PubMed]
- Moeller, N.H.; Shi, K.; Demir, O.; Belica, C.; Banerjee, S.; Yin, L.; Durfee, C.; Amaro, R.E.; Aihara, H. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc. Natl. Acad. Sci. USA 2022, 119, e2106379119. [Google Scholar] [CrossRef]
- Yan, L.; Yang, Y.; Li, M.; Zhang, Y.; Zheng, L.; Ge, J.; Huang, Y.C.; Liu, Z.; Wang, T.; Gao, S.; et al. Coupling of N7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell 2021, 184, 3474–3485.e11. [Google Scholar] [CrossRef]
- Liu, C.; Shi, W.; Becker, S.T.; Schatz, D.G.; Liu, B.; Yang, Y. Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science 2021, 373, 1142–1146. [Google Scholar] [CrossRef]
- Ogando, N.S.; Ferron, F.; Decroly, E.; Canard, B.; Posthuma, C.C.; Snijder, E.J. The Curious Case of the Nidovirus Exoribonuclease: Its Role in RNA Synthesis and Replication Fidelity. Front. Microbiol. 2019, 10, 1813. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef] [Green Version]
- Case, J.B.; Li, Y.; Elliott, R.; Lu, X.; Graepel, K.W.; Sexton, N.R.; Smith, E.C.; Weiss, S.R.; Denison, M.R. Murine Hepatitis Virus nsp14 Exoribonuclease Activity Is Required for Resistance to Innate Immunity. J. Virol. 2018, 92, e01531-17. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [Green Version]
- Sevajol, M.; Subissi, L.; Decroly, E.; Canard, B.; Imbert, I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014, 194, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [PubMed]
- Kikkert, M. Innate Immune Evasion by Human Respiratory RNA Viruses. J. Innate. Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Shokri, S.; Mahmoudvand, S.; Taherkhani, R.; Farshadpour, F. Modulation of the immune response by Middle East respiratory syndrome coronavirus. J. Cell Physiol. 2019, 234, 2143–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, J.B.; Ashbrook, A.W.; Dermody, T.S.; Denison, M.R. Mutagenesis of S-Adenosyl-l-Methionine-Binding Residues in Coronavirus nsp14 N7-Methyltransferase Demonstrates Differing Requirements for Genome Translation and Resistance to Innate Immunity. J. Virol. 2016, 90, 7248–7256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canal, B.; McClure, A.W.; Curran, J.F.; Wu, M.; Ulferts, R.; Weissmann, F.; Zeng, J.; Bertolin, A.P.; Milligan, J.C.; Basu, S.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp14/nsp10 exoribonuclease. BioChem. J. 2021, 478, 2445–2464. [Google Scholar] [CrossRef]
- Scholle, M.D.; Liu, C.; Deval, J.; Gurard-Levin, Z.A. Label-Free Screening of SARS-CoV-2 NSP14 Exonuclease Activity Using SAMDI Mass Spectrometry. SLAS Discov. 2021, 26, 766–774. [Google Scholar] [CrossRef]
- Liu, C.; Boland, S.; Scholle, M.D.; Bardiot, D.; Marchand, A.; Chaltin, P.; Blatt, L.M.; Beigelman, L.; Symons, J.A.; Raboisson, P.; et al. Dual inhibition of SARS-CoV-2 and human rhinovirus with protease inhibitors in clinical development. Antiviral. Res. 2021, 187, 105020. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci 2020, 253, 117592. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.S.; Nguyen, L.P.; Lee, G.H.; Lee, S.G.; Lyoo, K.S.; Kim, B.; Hwang, S.B. Asunaprevir, a Potent Hepatitis C Virus Protease Inhibitor, Blocks SARS-CoV-2 Propagation. Mol. Cells 2021, 44, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.S.; Hui, K.P.Y.; Lai, H.M.; He, X.; Khan, K.S.; Kaur, S.; Huang, J.; Li, Z.; Chan, A.K.N.; Cheung, H.H.; et al. Simeprevir Potently Suppresses SARS-CoV-2 Replication and Synergizes with Remdesivir. ACS Cent. Sci. 2021, 7, 792–802. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schafer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., 3rd; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428.e13. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [Green Version]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Hobartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef]
- Bravo, J.P.K.; Dangerfield, T.L.; Taylor, D.W.; Johnson, K.A. Remdesivir is a delayed translocation inhibitor of SARS-CoV-2 replication. Mol. Cell 2021, 81, 1548–1552.e4. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Huffman, J.H.; Khare, G.P.; Allen, L.B.; Witkowski, J.T.; Robins, R.K. Broad-spectrum antiviral activity of Virazole: 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B. Metabolism and antiviral activity of ribavirin. Virus Res. 2005, 107, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Gordon, S.C.; Schiff, E.R.; Shiffman, M.L.; Lee, W.M.; Rustgi, V.K.; Goodman, Z.D.; Ling, M.H.; Cort, S.; Albrecht, J.K. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med. 1998, 339, 1485–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.B.; Walsh, E.E.; Hruska, J.F.; Betts, R.F.; Hall, W.J. Ribavirin treatment of experimental respiratory syncytial viral infection. A controlled double-blind study in young adults. JAMA 1983, 249, 2666–2670. [Google Scholar] [CrossRef]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef]
- Stockman, L.J.; Bellamy, R.; Garner, P. SARS: Systematic review of treatment effects. PLoS Med. 2006, 3, e343. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chan, K.H.; Jiang, Y.; Kao, R.Y.; Lu, H.T.; Fan, K.W.; Cheng, V.C.; Tsui, W.H.; Hung, I.F.; Lee, T.S.; et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 2004, 31, 69–75. [Google Scholar] [CrossRef]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003, 361, 2045–2046. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.C.; Blanc, H.; Surdel, M.C.; Vignuzzi, M.; Denison, M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, A.; Selisko, B.; Le, N.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Hill, C.S.; Sarkar, S.; Tse, L.V.; Woodburn, B.M.D.; Schinazi, R.F.; Sheahan, T.P.; Baric, R.S.; Heise, M.T.; Swanstrom, R. beta-d-N4-hydroxycytidine Inhibits SARS-CoV-2 Through Lethal Mutagenesis But Is Also Mutagenic To Mammalian Cells. J. Infect. Dis. 2021, 224, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Ghasemnejad-Berenji, M.; Pashapour, S. Favipiravir and COVID-19: A Simplified Summary. Drug Res. 2021, 71, 166–170. [Google Scholar] [CrossRef]
- Painter, G.R.; Natchus, M.G.; Cohen, O.; Holman, W.; Painter, W.P. Developing a direct acting, orally available antiviral agent in a pandemic: The evolution of molnupiravir as a potential treatment for COVID-19. Curr. Opin. Virol. 2021, 50, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.L.; Pruijssers, A.J.; Chappell, J.D.; Gribble, J.; Lu, X.; Andres, E.L.; Bluemling, G.R.; Lockwood, M.A.; Sheahan, T.P.; Sims, A.C.; et al. Small-Molecule Antiviral beta-d-N (4)-Hydroxycytidine Inhibits a Proofreading-Intact Coronavirus with a High Genetic Barrier to Resistance. J. Virol. 2019, 93, e01348-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schafer, A.; Dinnon, K.H., 3rd; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef]
- Malone, B.; Campbell, E.A. Molnupiravir: Coding for catastrophe. Nat. Struct. Mol. Biol. 2021, 28, 706–708. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rona, G.; Zeke, A.; Miwatani-Minter, B.; de Vries, M.; Kaur, R.; Schinlever, A.; Garcia, S.F.; Goldberg, H.V.; Wang, H.; Hinds, T.R.; et al. The NSP14/NSP10 RNA repair complex as a Pan-coronavirus therapeutic target. Cell Death Differ. 2022, 29, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sacramento, C.Q.; Jockusch, S.; Chaves, O.A.; Tao, C.; Fintelman-Rodrigues, N.; Chien, M.; Temerozo, J.R.; Li, X.; Kumar, S.; et al. Combination of antiviral drugs inhibits SARS-CoV-2 polymerase and exonuclease and demonstrates COVID-19 therapeutic potential in viral cell culture. Commun. Biol. 2022, 5, 154. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, N.; Nair, D.T. Ritonavir may inhibit exoribonuclease activity of nsp14 from the SARS-CoV-2 virus and potentiate the activity of chain terminating drugs. Int. J. Biol. Macromol. 2021, 168, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Gorgulla, C.; Padmanabha Das, K.M.; Leigh, K.E.; Cespugli, M.; Fischer, P.D.; Wang, Z.F.; Tesseyre, G.; Pandita, S.; Shnapir, A.; Calderaio, A.; et al. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. iScience 2021, 24, 102021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deval, J.; Gurard-Levin, Z.A. Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex. Molecules 2022, 27, 2918. https://doi.org/10.3390/molecules27092918
Deval J, Gurard-Levin ZA. Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex. Molecules. 2022; 27(9):2918. https://doi.org/10.3390/molecules27092918
Chicago/Turabian StyleDeval, Jerome, and Zachary A. Gurard-Levin. 2022. "Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex" Molecules 27, no. 9: 2918. https://doi.org/10.3390/molecules27092918
APA StyleDeval, J., & Gurard-Levin, Z. A. (2022). Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex. Molecules, 27(9), 2918. https://doi.org/10.3390/molecules27092918