
An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review

Abstract
1. Introduction
2. Macrocyclic Compounds for the Treatment of Malignant Tumors
2.1. Anaplastic Lymphoma Kinase (ALK) and C-Ros Oncogene 1 (ROS1) Macrocyclic Inhibitors
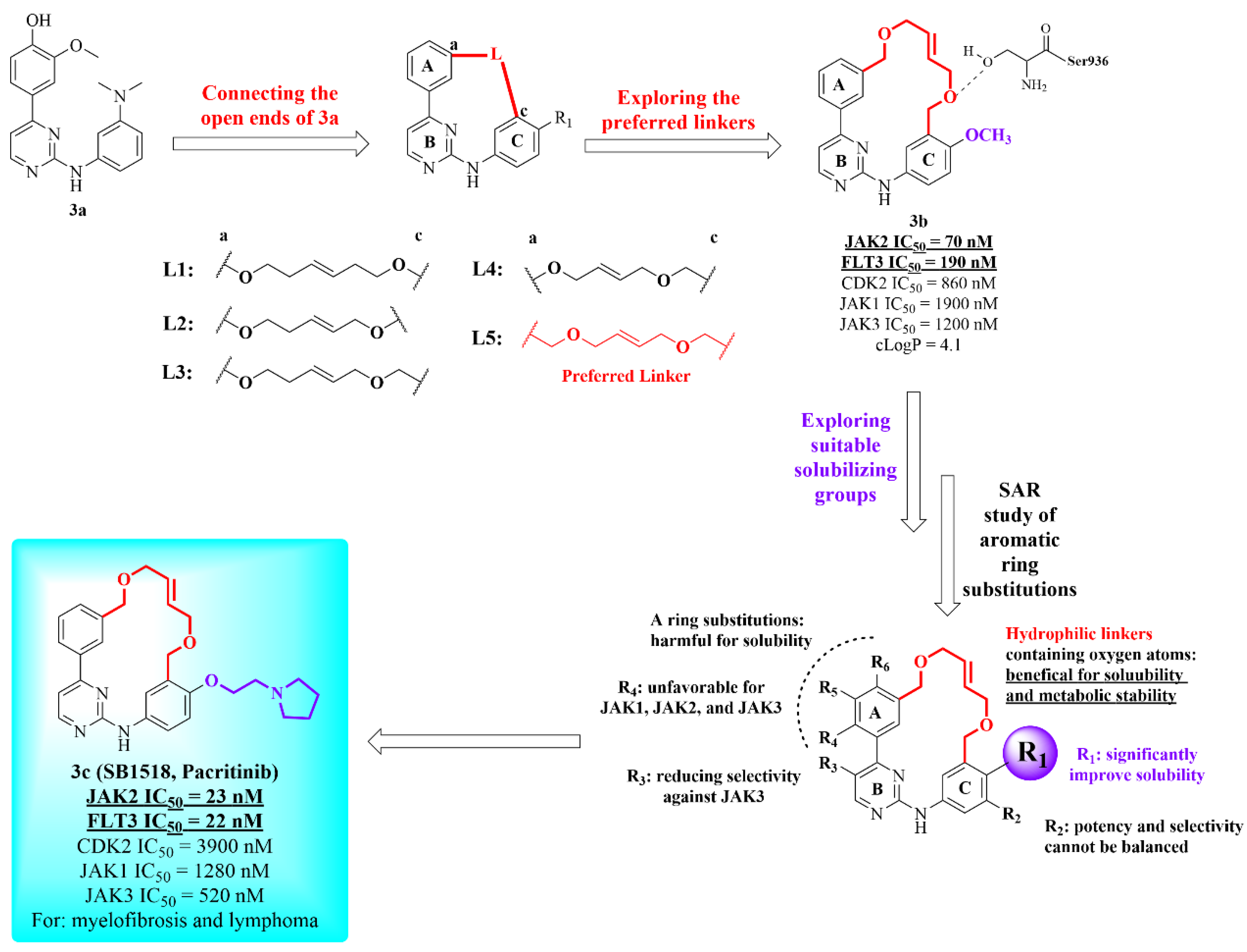
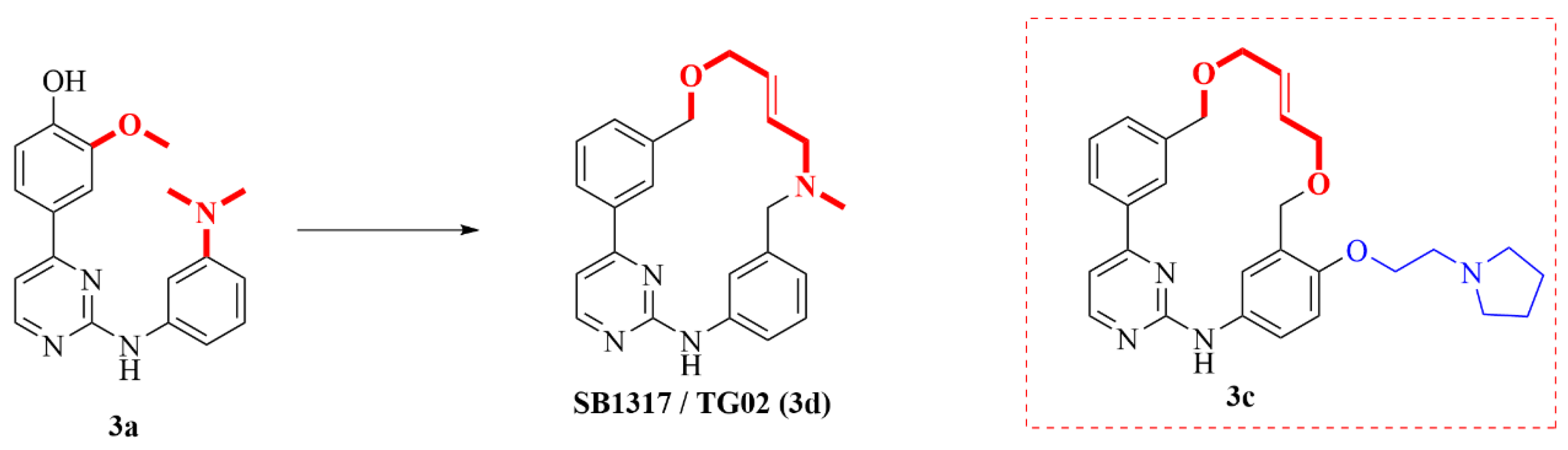
2.2. Janus Kinase 2 (JAK2) Macrocyclic Inhibitors
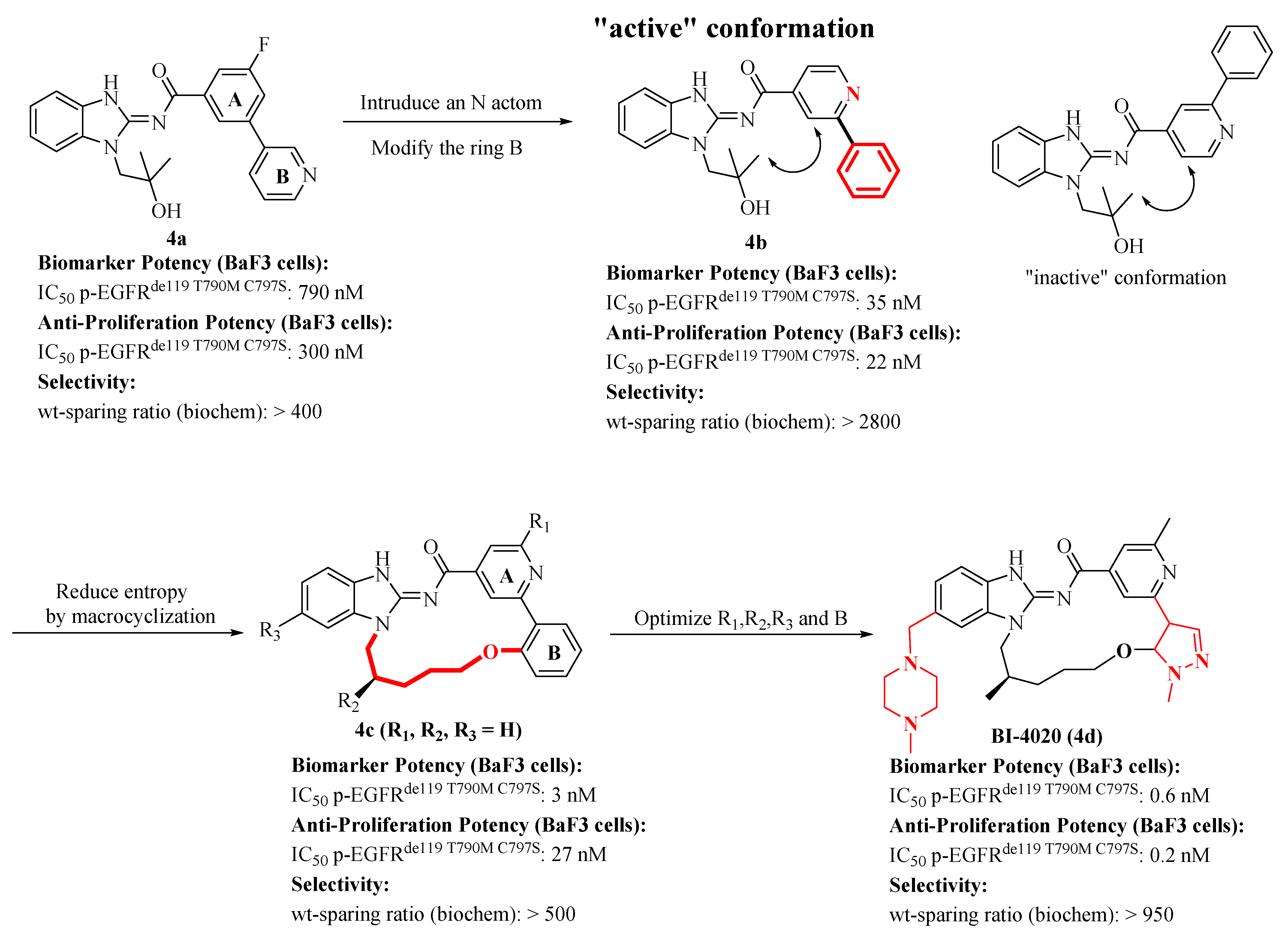
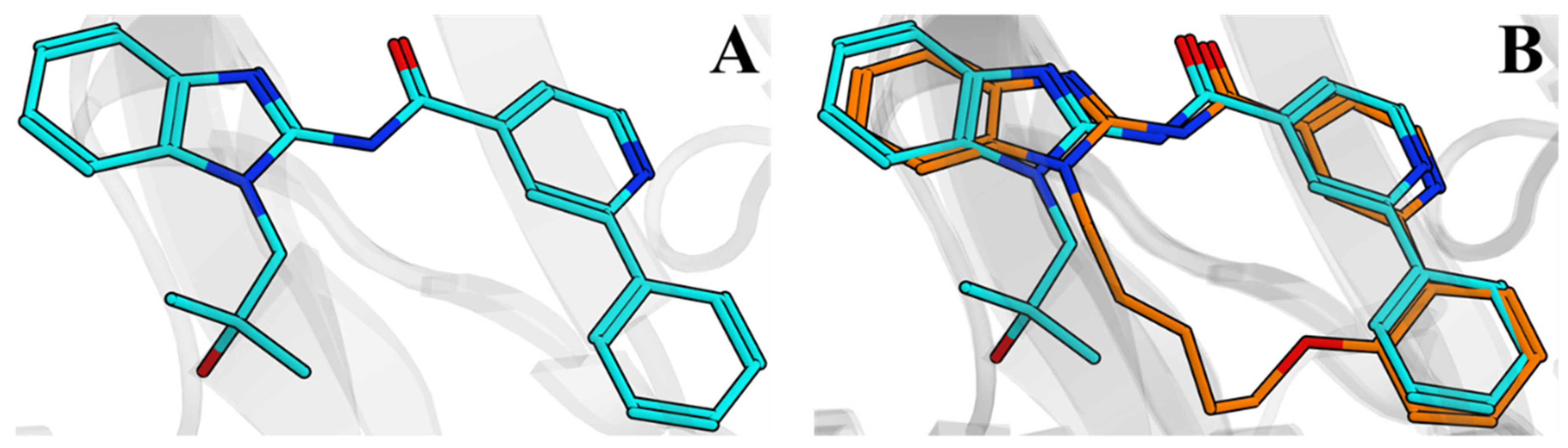
2.3. Discovery of a Next Generation of EGFR Tyrosine Kinase Inhibitors
2.4. Discovery of Novel Pyridones and Pyridone Macrocycles as Potent Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitors
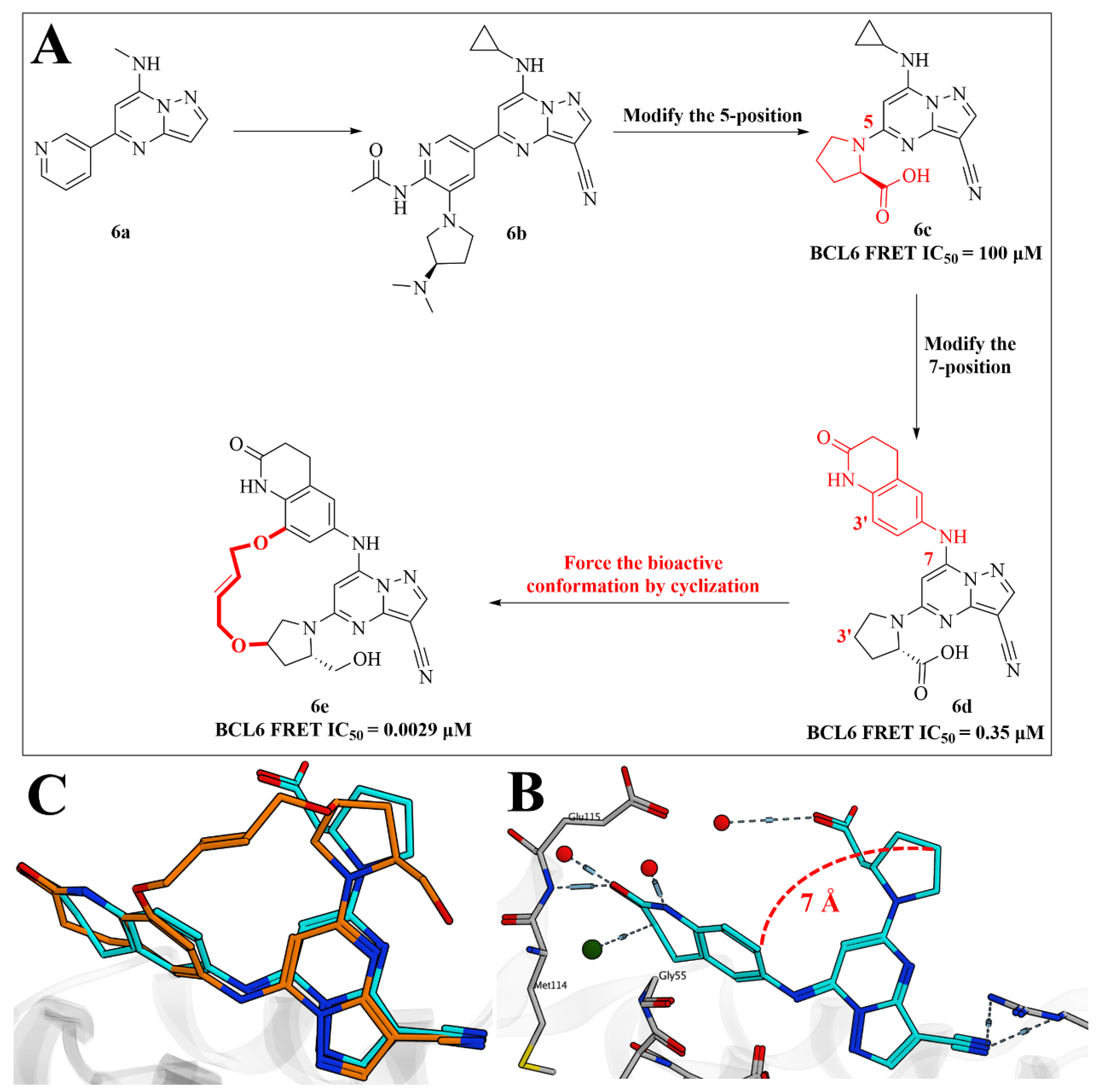
2.5. Discovery of Pyrazolo [1,5-a]pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors
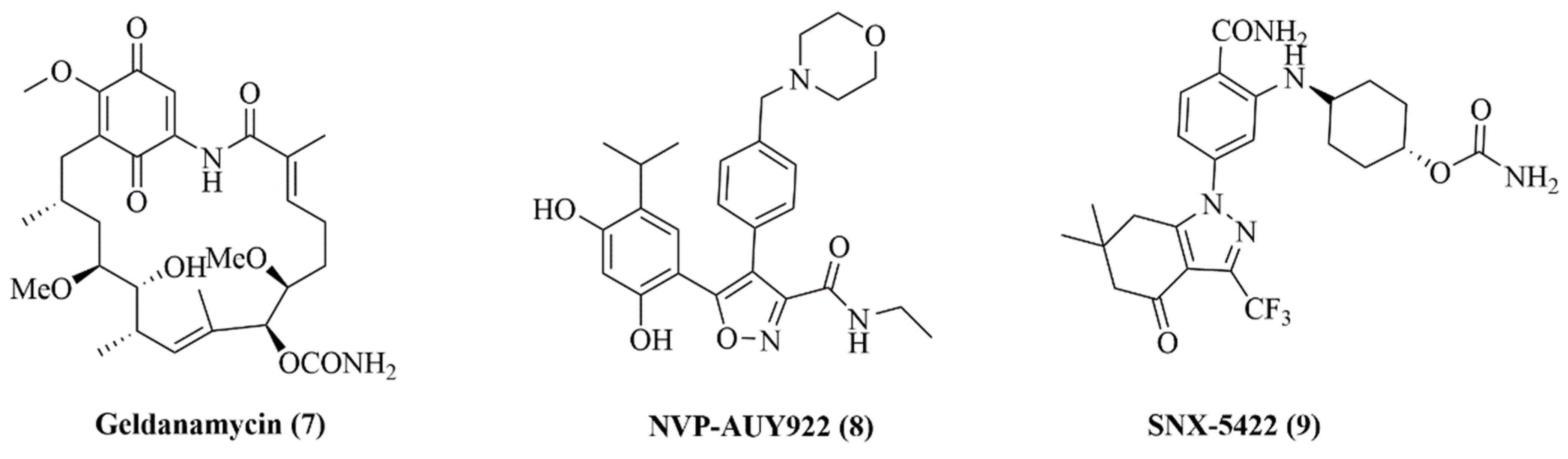
2.6. Design of Macrocyclic Hsp90 Inhibitors with Increased Metabolic Stability and Potent Cell-Proliferation Activity
2.7. Structure-Based Design, Synthesis, and Biological Evaluation of Potent and Selective Macrocyclic Checkpoint Kinase 1 Inhibitors
2.8. Non-Natural Macrocyclic Inhibitors of Histone Deacetylases (HDACs): Design, Synthesis, and Activity
2.9. Structure-Based Design and Synthesis of Novel Macrocyclic Pyrazolo[1,5-a][1,3,5]triazine Compounds as Potent Inhibitors of Protein Kinase CK2
2.10. Structure-Based Drug Design of a Highly Potent CDK1, 2, 4, 6 Inhibitor with Novel Macrocyclic Quinoxalin-2-One Structure
2.11. Macrocyclic Pyrrolobenzodiazepine Dimers as Antibody-Drug Conjugate Payloads
2.12. Design and Synthesis of a New, Conformationally Constrained, Macrocyclic Small-Molecule Inhibitor of STAT3 via ‘Click Chemistry’
2.13. Structure-Based Design of a Macrocyclic PROTAC
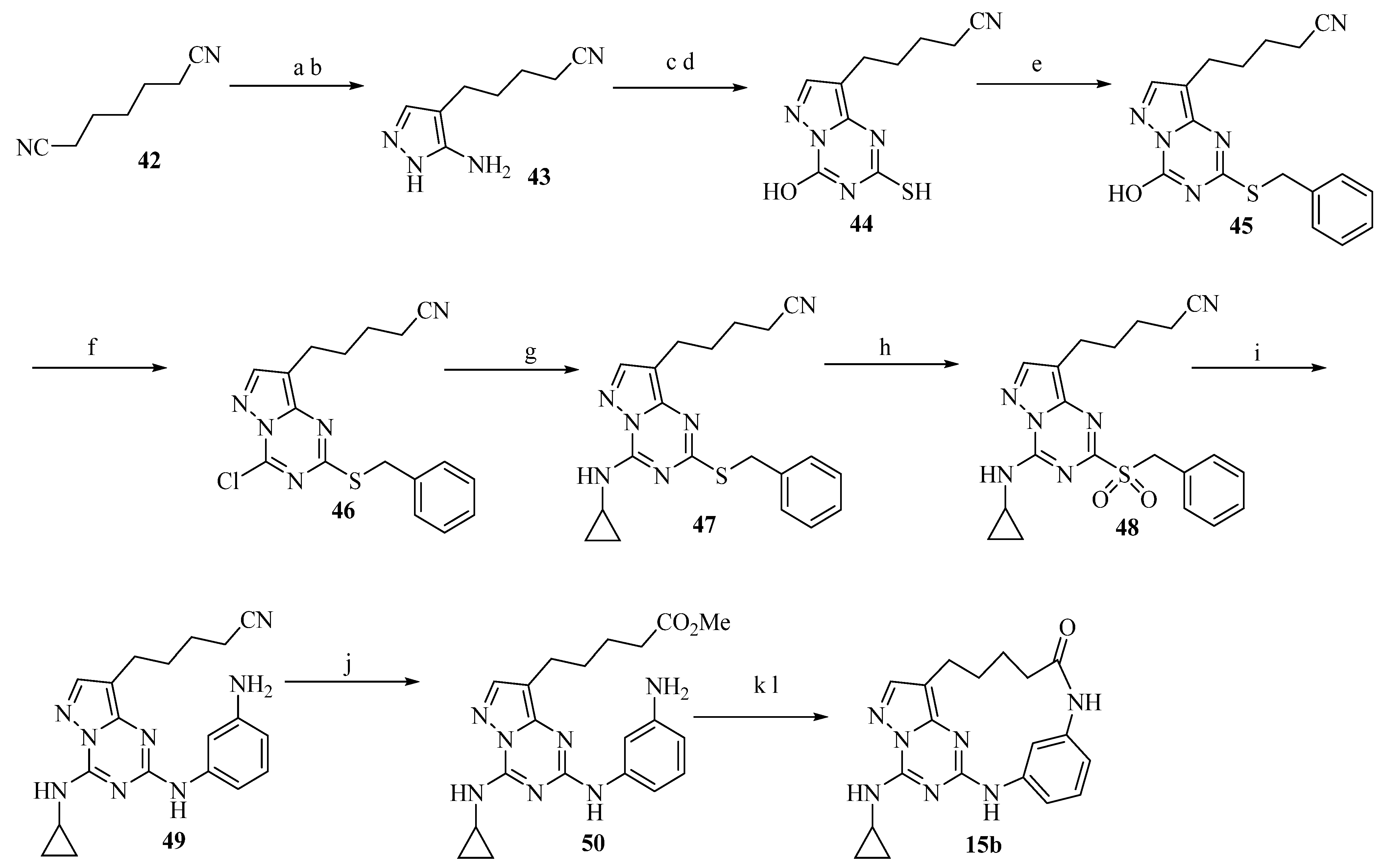
2.14. Structure-Based Design of GLS1 Macrocyclic Inhibitors Targeting Allosteric Binding Site
2.15. Omipalisib Inspired Macrocycles As Dual PI3K/mTOR Inhibitors and Triple PI3K/mTOR/PIM-1 Inhibitors
3. Synthesis of Macrocyclic Compounds
3.1. Synthesis of Macrocyclic Compounds Using Coupling Reaction
3.1.1. Synthesis of HSP90 Macrocyclic Inhibitors Using Buchwald-Hartwig Coupling Reaction
3.1.2. Synthesis of BET Macrocyclic Inhibitors using Sonogashira Coupling Reaction
3.2. Synthesis of CDK Macrocyclic Inhibitors Using Nucleophilic Substitution Reaction
3.3. Synthesis of CK2 Macrocyclic Inhibitors using a Condensation Reaction
3.4. Synthesis of Chk1 and HDAC Macrocyclic Inhibitors Using Ring-Closing Metathesis (RCM) Reaction
3.5. Synthesis of STAT3 Macrocyclic Inhibitors Using “Click Chemistry” Cycloaddition Reaction
4. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Surade, S.; Blundell, T.L. Structural biology and drug discovery of difficult targets: The limits of ligandability. Chem. Biol. 2012, 19, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Mallinson, J.; Collins, I. Macrocycles in new drug discovery. Future Med. Chem. 2012, 4, 1409–1438. [Google Scholar] [CrossRef] [PubMed]
- DeLorbe, J.E.; Clements, J.H.; Whiddon, B.B.; Martin, S.F. Thermodynamic and structural effects of macrocyclic constraints in protein-ligand interactions. ACS Med. Chem. Lett. 2010, 1, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.R.; Davies, N.L.; James, K. Comparison of diffusion coefficients for matched pairs of macrocyclic and linear molecules over a drug-like molecular weight range. Org. Biomol. Chem. 2011, 9, 7727–7733. [Google Scholar] [CrossRef] [PubMed]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery an underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Marsault, E.; Peterson, M.L. Macrocycles are great cycles: Applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 2011, 54, 1961–2004. [Google Scholar] [CrossRef]
- Schreiber, S.L. Molecular diversity by design. Nature 2009, 457, 153–154. [Google Scholar] [CrossRef]
- Joanne, K. Bringing macrocycles full circle. SciBX 2012, 5, 1176. [Google Scholar]
- Giordanetto, F.; Kihlberg, J. Macrocyclic Drugs and Clinical Candidates: What Can Medicinal Chemists Learn from Their Properties? J. Med. Chem. 2014, 57, 278–295. [Google Scholar] [CrossRef]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.L.; Dinh, D.; et al. Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar]
- William, A.D.; Lee, A.C.; Blanchard, S. Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma. J. Med. Chem. 2011, 54, 4638–4658. [Google Scholar] [PubMed]
- Poulsen, A.; William, A.; Blanchard, S.; Nagaraj, H.; Williams, M.; Wang, H.; Lee, A.; Sun, E.; Teo, E.L.; Tan, E.K.; et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J. Mol. Model. 2013, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Harald, E.; Dietrich, B.; Scharn, P.M.; Bader, D.; Baum, G.; Bergner, A.; Chong, A.; Doebel, E.; Egger, S.; Engelhardt, G.C.; et al. Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 2019, 62, 10272–10293. [Google Scholar]
- Wang, L.; Pratt, J.K.; Soltwedel, T.; Sheppard, G.S.; Fidanze, S.D.; Liu, D.; Hasvold, L.A.; Mantei, R.A.; Holms, J.H.; McClellan, W.J.; et al. Fragment-Based, Structure-Enabled Discovery of Novel Pyridones and Pyridone Macrocycles as Potent Bromodomain and Extra Terminal Domain (BET) Family Bromodomain Inhibitors. J. Med. Chem. 2017, 60, 3828–3850. [Google Scholar] [CrossRef]
- Mccoull, W.; Abrams, R.D.; Anderson, E.; Blades, K.; Barton, P.; Box, M.; Burgess, J.; Byth, K.; Cao, Q.; Chuaqui, C.; et al. Discovery of Pyrazolo [1,5-a] pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors. J. Med. Chem. 2017, 60, 4386–4402. [Google Scholar] [CrossRef]
- Suda, A.; Koyano, H.; Hayase, T.; Hada, K.; Kawasaki, K.I.; Komiyama, S.; Hasegawa, K.; Fukami, T.A.; Sato, S.; Miura, T.; et al. Design and synthesis of novel macrocyclic 2-amino-6-arylpyrimidine Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1136–1141. [Google Scholar] [CrossRef]
- Messaoudi, S.; Peyrat, J.F.; Brion, J.D.; Alami, M. Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agent Med. Chem. 2008, 8, 761–782. [Google Scholar] [CrossRef]
- Whitesell, L.; Mimnaugh, E.G.; Costa, B.D.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.J.; Collins, I.; Davies, N.G.M.; Drysdale, M.J.; et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef]
- Zapf, C.W.; Bloom, J.D.; McBean, J.L.; Dushin, R.G.; Nittoli, T.; Ingalls, C.; Sutherland, A.G.; Sonye, J.P.; Eid, C.N.; Golas, J.; et al. Design and SAR of macrocyclic Hsp90 inhibitors with increased metabolic stability and potent cell-proliferation activity. Bioorg. Med. Chem. Lett. 2011, 21, 2278–2282. [Google Scholar] [CrossRef]
- Biamonte, M.A.; Water, R.V.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.C. Heat Shock Protein 90: Inhibitors in Clinical Trials. J. Med. Chem. 2010, 53, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Barta, T.E.; Veal, J.M.; Rice, J.W.; Partridge, J.M.; Fadden, R.P.; Ma, W.; Jenks, M.; Geng, L.F.; Hanson, G.J.; Huang, K.H.; et al. Discovery of Benzamide Tetrahydro-4H-carbazol-4-ones as Novel Small Molecule Inhibitors of Hsp90. Bioorg. Med. Chem. Lett. 2008, 18, 3517–3521. [Google Scholar] [CrossRef]
- Zapf, C.W.; Bloom, J.D.; McBean, J.L.; Dushin, R.G.; Nittoli, T.; Otteng, M.; Ingalls, C.; Golas, J.M.; Liu, H.; Lucas, J.; et al. Macrocyclic lactams as potent Hsp90 inhibitors with excellent tumor exposure and extended biomarker activity. Bioorg. Med. Chem. Lett. 2011, 21, 3411–3416. [Google Scholar] [CrossRef] [PubMed]
- Walworth, N.; Davey, S.; Beach, D. Fission yeast Chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 1993, 363, 368–371. [Google Scholar] [CrossRef]
- Chen, Y.H.; Sanchez, Y. Chk1 in the DNA damage response: Conserved roles from yeasts to mammals. DNA Repair. 2004, 3, 1025–1032. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Rivera, S.C.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.S.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–255. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Prudhomme, M. Combining DNA damaging agents and checkpoint 1 inhibitors. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 435–438. [Google Scholar] [CrossRef]
- Kong, N.; Fotouhi, N.; Wovkulich, P.M.; Roberts, J. Cell cycle inhibitors for the treatment of cancer. Drug Future 2003, 28, 881–896. [Google Scholar] [CrossRef]
- Kawabe, T. G2 checkpoint abrogators as anticancer drugs. Mol. Cancer Ther. 2004, 3, 513–519. [Google Scholar] [PubMed]
- Tao, Z.F.; Lin, N.H. Chk1 inhibitors for novel cancer treatment. Anti-Cancer Agents Med. Chem. 2006, 6, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.F.; Wang, L.; Stewart, K.D.; Chen, Z.H.; Gu, W.; Bui, M.H.; Merta, P.; Zhang, H.Y.; Kovar, P.; Johnson, E.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of Potent and Selective Macrocyclic Checkpoint Kinase 1 Inhibitors. J. Med. Chem. 2007, 50, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Xiao, Z.; Gu, W.Z.; Xue, J.; Bui, M.H.; Kovar, P.; Li, G.Q.; Wang, G.; Tao, Z.F.; Tong, Y.S.; et al. Selective Chk1 inhibitors differentially sensitize a broad range of p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, 119, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.T.; Li, G.Q.; Mantei, R.A.; Chen, Z.H.; Kovar, P.; Gu, W.; Xiao, Z.; Zhang, H.Y.; Sham, H.L.; Sowin, T.; et al. 1-(5-Chloro-2-alkoxyphenyl)-3-(5-cyano-pyrazi-2-yl)ureas as potent and selective inhibitors of Chk1 kinase: Synthesis, preliminary SAR, and biological activities. J. Med. Chem. 2005, 48, 3118–3121. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, H.; Xu, W.F. Strategies in developing promising histone deacetylase inhibitors. Med. Res. Rev. 2010, 30, 585–602. [Google Scholar] [CrossRef]
- Paris, M.; Porcelloni, M.; Binaschi, M.; Fattori, D. Histone Deacetylase Inhibitors: From Bench to Clinic. J. Med. Chem. 2008, 51, 1505–1529. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Hanessian, S.; Auzzas, L.; Giannini, G.; Marzi, M.; Cabri, W.; Barbarino, M.; Vesci, L.; Pisano, C. ω-Alkoxy Analogues of SAHA (Vorinostat) as Inhibitors of HDAC: A Study of Chain-Length and Stereochemical Dependence. Bioorg. Med. Chem. Lett. 2007, 17, 6261–6265. [Google Scholar] [CrossRef]
- Hanessian, S.; Auzzas, L.; Larsson, A.; Zhang, J.B.; Giannini, G.; Gallo, G.; Ciacci, A.; Cabri, W. Vorinostat-like Molecules as Structural, Stereochemical and Pharmacological Tools. ACS Med. Chem. Lett. 2010, 1, 70–74. [Google Scholar] [CrossRef][Green Version]
- Auzzas, L.; Larsson, A.; Matera, R.; Baraldi, A.; Simard, B.D.; Giannini, G.; Cabri, W.; Battistuzzi, G.; Gallo, G.; Ciacci, A.; et al. Non-Natural Macrocyclic Inhibitors of Histone Deacetylases: Design, Synthesis, and Activity. J. Med. Chem. 2010, 53, 8387–8399. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Lu, J.; Averill, A.; Almassy, R.; Chu, S. Structure-based design and synthesis of novel macrocyclic pyrazolo[1,5-a] [1,3,5]triazine compounds as potent inhibitors of protein kinase CK2 and their anticancer activities. Bioorg. Med. Chem. Lett. 2008, 18, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, N.; Sugimoto, T.; Shibata, J.; Nakamura, K.; Masutani, K.; Ikuta, M.; Hirai, H. Structure-based drug design of a highly potent CDK1,2,4,6 inhibitor with novel macrocyclic quinoxalin-2-one structure. Bioorg. Med. Chem. Lett. 2006, 16, 5122–5126. [Google Scholar] [CrossRef]
- Hirai, H.; Hirose, M.; Kamijyo, K.; Kawanishi, N.; Masutani, K.; Shibata, J.; Sugimoto, T. Novel Quinoxalinone Derivatives. International Patent WO 2004039809, 18 April 2022. [Google Scholar]
- Donnell, A.F.; Zhang, Y.; Stang, E.M.; Donna, D.W.; Andrew, J.T.; Heidi, L.P.; Gretchen, M.S.; Chin, P.; Chetana, R.; Robert, M.B.; et al. Macrocyclic pyrrolobenzodiazepine dimers as antibody- drug conjugate payloads. Bioorg. Med. Chem. Lett. 2017, 27, 5267–5271. [Google Scholar] [CrossRef]
- Chen, J.Y.; Coleska, Z.N.; Yang, C.Y.; Gomez, C.; Gao, W.; Krajewski, K.; Jiang, S.; Roller, P.; Wang, S.M. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via ‘click chemistry’. Bioorg. Med. Chem. Lett. 2007, 17, 3939–3942. [Google Scholar] [CrossRef]
- Valeur, E.; Guéret, S.M.; Adihou, H.; Gopalakrishnan, R.; Lemurell, M.; Waldmann, H.; Grossmann, T.N.; Plowright, A.T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem. Int. Ed. Engl. 2017, 56, 10294–10323. [Google Scholar] [CrossRef] [PubMed]
- Alihodžić, S.; Bukvić, M.; Elenkov, I.J.; Hutinec, A.; Koštrun, S.; Pešić, D.; Saxty, G.; Tomašković, L.; Žiher, D. Current Trends in Macrocyclic Drug Discovery and beyond-Ro5. Prog. Med. Chem. 2018, 57, 113–233. [Google Scholar]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.Z.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Roy, M.J.; Winkler, S.; Hughes, S.J.; Whitworth, C.; Galant, M.; Farnaby, W.; Rumpel, K.; Ciulli, A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. [Google Scholar] [CrossRef]
- Natalya, N.P.; Craig, B.T. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar]
- Brian, J.A.; Zachary, E.S.; Chi, V.D. From krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar]
- Sarah, C.; Zimmermann, B.D.; Takashi, T. Recent progress in the discovery of allosteric inhibitors of kidney type glutaminase. J. Med. Chem. 2019, 62, 46–59. [Google Scholar]
- Xu, X.; Wang, J.B.; Wang, M.; Yuan, X.Y.; Li, L.; Zhang, C.; Huang, H.D.; Jing, T.; Wang, C.C.; Tong, C.; et al. Structure-Enabled Discovery of Novel Macrocyclic Inhibitors Targeting Glutaminase 1 Allosteric Binding Site. J. Med. Chem. 2021, 64, 4588–4611. [Google Scholar] [CrossRef]
- Alvarez, R.M.; García, A.B.; Riesco-Fagundo, C.; Martín, J.I.; Varela, C.; Hergueta, A.R.; Cantalapiedra, E.G.; Oyarzabal, J.; Geronimo, B.D.; Lorenzo, M.; et al. Omipalisib inspired macrocycles as dual PI3K/mTOR inhibitors. Eur. J. Med. Chem. 2021, 211, 113109. [Google Scholar] [CrossRef]
- Martínez-González, S.; Alvarez, R.M.; Martín, J.I.; García, A.B.; Riesco-Fagundo, C.; Varela, C.; Hergueta, A.R.; Cantalapiedra, E.G.; Albarrán, M.I.; Gómez-Casero, E.; et al. Macrocyclization as a Source of Desired Polypharmacology. Discovery of Triple PI3K/mTOR/PIM Inhibitors. ACS Med. Chem. Lett. 2021, 12, 1794–1801. [Google Scholar] [CrossRef]
- Miller, S.J.; Blackwell, H.E.; Grubbs, R.H. Application of Ring-Closing Metathesis to the Synthesis of Rigidified Amino Acids and Peptides. J. Am. Chem. Soc. 1996, 118, 9606–9614. [Google Scholar] [CrossRef]
- Tsantrizos, Y.S. TMC-435, an NS3/4A protease inhibitor for the treatment of HCV infection. Curr. Opin. Investig. Drugs. 2009, 10, 871–881. [Google Scholar]
- Weghe, P.V.; Eustache, J. The application of olefin metathesis to the synthesis of biologically active macrocyclic agents. Curr. Top. Med. Chem. 2005, 5, 1495–1519. [Google Scholar] [CrossRef]
- Pirali, T.; Tron, G.C.; Zhu, J.P. One-pot synthesis of macrocycles by a tandem three-component reaction and intramolecular [3 + 2] cycloaddition. Org. Lett. 2006, 8, 4145–4148. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.; Egyed, M.; Szoke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus Best Available Therapy for the Treatment of Myelofibrosis Irrespective of Baseline Cytopenias (PERSIST-1): An International, Randomised, Phase 3 Trial. Lancet Haematol. 2017, 4, e225. [Google Scholar] [CrossRef]
- Available online: https://www.onclive.com/view/fda-approves-pacritinib-for-intermediate-or-high-risk-myelofibrosis (accessed on 18 April 2022).
- Pallis, M.; Abdul-Aziz, A.; Burrows, F.; Seedhouse, C.; Grundy, M.; Russell, N. The Multi-Kinase Inhibitor TG02 Overcomes Signalling Activation by Survival Factors to Deplete MCL1 and XIAP and Induce Cell Death in Primary Acute Myeloid Leukaemia Cells. Br. J. Haematol. 2012, 159, 191–203. [Google Scholar] [CrossRef]
- Malizzia, L.J.; Hsu, A. Temsirolimus, an MTOR Inhibitor for Treatment of Patients with Advanced Renal Cell Carcinoma. Clin. J. Oncol. Nurs. 2008, 12, 639–646. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 Trial of Everolimus for Metastatic Renal Cell Carcinoma: Final Results and Analysis of Prognostic Factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef]
- MacDonald, A.S.; Group, R.G.S. A Worldwide, Phase III, Randomized, Controlled, Safety and Efficacy Study of a Sirolimus/Cyclosporine Regimen for Prevention of Acute Rejection in Recipients of Primary Mismatched Renal Allografts. Transplantation 2001, 71, 271–280. [Google Scholar] [CrossRef]
- Zhang, W.; Borthakur, G.; Gao, C.; Chen, Y.; Mu, H.; Ruvolo, V.R.; Nomoto, K.; Zhao, N.; Konopleva, M.; Andreeff, M. The Dual MEK/FLT3 Inhibitor E6201 Exerts Cytotoxic Activity against Acute Myeloid Leukemia Cells Harboring Resistance-Conferring FLT3 Mutations. Cancer Res. 2016, 76, 1528–1537. [Google Scholar] [CrossRef]
- Babiker, H.M.; Byron, S.A.; Hendricks, W.P.D.; Elmquist, W.F.; Gampa, G.; Vondrak, J.; Aldrich, J.; Cuyugan, L.; Adkins, J.; De Luca, V.; et al. E6201, an Intravenous MEK1 Inhibitor, Achieves an Exceptional Response in BRAF V600E-Mutated Metastatic Malignant Melanoma with Brain Metastases. Investig. New Drugs 2019, 37, 636–645. [Google Scholar] [CrossRef]
- Hanke, T.; Merk, D.; Steinhilber, D.; Geisslinger, G.; SchubertZsilavecz, M. Small Molecules with Anti-Inflammatory Properties in Clinical Development. Pharmacol. Ther. 2016, 157, 163–187. [Google Scholar] [CrossRef]
- Drilon, A. TRK Inhibitors in TRK Fusion-Positive Cancers. Ann. Oncol. 2019, 30, VIII23–VIII30. [Google Scholar] [CrossRef] [PubMed]
- Janssen Pharmaceutica, N.V. MTKI Quinazoline Derivatives. International Patent WO 2006061417, 13 July 2006. [Google Scholar]
- Konings, I.R.H.M.; De Jonge, M.J.A.; Burger, H.; Van Der Gaast, A.; Van Beijsterveldt, L.E.C.; Winkler, H.; Verweij, J.; Yuan, Z.; Hellemans, P.; Eskens, F.A.L.M. Phase 1 and Pharmacological Study of the Broad-Spectrum Tyrosine Kinase Inhibitor JNJ 26483327 in Patients with Advanced Solid Tumours. Br. J. Cancer 2010, 103, 987–992. [Google Scholar] [CrossRef] [PubMed][Green Version]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | ALK Ki (nM) ALK-L1996M Ki (nM) | pALK Cell IC50 (nM) pALK-L1196M Cell IC50 (nM) | LipE | MDR BA/AB (Ratio) | TrB Ki (nM) (Selectivity) |
|---|---|---|---|---|---|
| 1 (Crizotinib) | 0.74 8.2 | 80 843 | 4.1 | 12.5/0.28 (44.5) | -- |
| 2a | 4.0 38 | 95 3200 | 3.1 | 16.3/20.0 (0.82) | -- |
| 2b | <0.1 0.57 | 1.0 20 | 4.4 | -- | -- |
| 2c | <0.2 0.29 | 0.7 14 | 5.7 | 28.3/8.1 (4.2) | -- |
| 2d (Lorlatinib) | <0.07 0.70 | 1.3 21 | 5.4 | 28.0/19.3 (1.5) | 23 (38×) |
| Compd. |  | MTS EC50, µM | |
|---|---|---|---|
| Compd. Alone | Compd. + Dox | ||
| 11e | 4-H, 5′-H | >59 | 37.8 |
| 11f | 4-H, 5′-CN | 57.6 | 3.04 |
| 11g | 4-NH2, 5′-CN | >59 | 0.43 |
| Comp. | HDAC IC50 (nM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Class I | Class IIa | Class IIb | Class IV | ||||||||
| 1 | 2 | 3 | 8 | 4 | 5 | 7 | 9 | 6 | 10 | 11 | |
| 12 | 258 | 921 | 350 | 243 | 493 | 378 | 344 | 316 | 28 | 456 | 362 |
| 13a | 53 | 254 | 131 | 331 | 648 | 134 | 432 | 247 | 20 | 179 | 197 |
| 13b | 31 | 158 | 55 | 198 | 79 | 62 | 27 | 60 | 0.84 | 62 | 22 |
| 13c | 28 | 171 | 16 | 39 | 52 | 24 | 15 | 22 | 0.75 | 46 | 34 |
| 13d | 41 | 128 | 63 | 45 | 64 | 65 | 34 | 47 | 0.40 | 65 | 45 |
| Comp. | H460 (Lung) IC50/μM | HCT-116 (Colon) IC50/μM |
|---|---|---|
| 12 | 3.40 | 1.20 |
| 13a | 0.52 | 0.22 |
| 13b | 1.05 | 0.69 |
| 13c | 0.51 | 0.40 |
| 13d | 6.80 | 3.74 |
| Compound | R | Ki (nM) | HCT116 IC50 (µM) | PC3 IC50 (µM) |
|---|---|---|---|---|
 | ||||
| 14c |  | 1.3 | 0.25 | 0.5 |
| 14d |  | 1.4 | 0.19 | 0.23 |
| 14e |  | 5.2 | 0.24 | 0.38 |
| 14f |  | 3.6 | 0.33 | 0.60 |
| 14g |  | 7.4 | 0.27 | 0.33 |
| 14h |  | 4.0 | 0.13 | 0.21 |
| 14i |  | 2.0 | 0.083 | 0.12 |
| 14j |  | 2.2 | 0.13 | 0.14 |
| Compounds | Structures | Targets | Clinical Trial |
|---|---|---|---|
| 2d (Lorlatinib) |  | ALK ROS1 | approved |
| 3c (Pacritinib) |  | JAK2 FLT3 | approved |
| 3d (Zotiraciclib) |  | JAK2 FLT3 CDK2 | Phase 1/2 NCT02942264 |
| 66 (Sirolimus) |  | mTOR | approved |
| 67 (Temsirolimus) |  | mTOR | approved |
| 68 (Everolimus) |  | mTOR | approved |
| 69 (E6201) |  | MEK1 FLT3 | Phase 2 NCT00539929 NCT01268527 |
| 70 (Repotrecitinib) |  | TRKA TRKB TRKC ROS1 ALK | Phase 1/2 NCT03093116 |
| 71 (Selitrecitinib, LOXO-195) |  | TRKA TRKB TRKC | Phase 1/2 NCT03215511 |
| 72 (JNJ-26483327) |  | EGFR RET VEGFR-3 Her4 Lyn Fyn Yes | Phase 1 NCT00676299 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Fang, R.; Rao, Q. An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review. Molecules 2022, 27, 2837. https://doi.org/10.3390/molecules27092837
Liang Y, Fang R, Rao Q. An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review. Molecules. 2022; 27(9):2837. https://doi.org/10.3390/molecules27092837
Chicago/Turabian StyleLiang, Yan, Ru Fang, and Qiu Rao. 2022. "An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review" Molecules 27, no. 9: 2837. https://doi.org/10.3390/molecules27092837
APA StyleLiang, Y., Fang, R., & Rao, Q. (2022). An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review. Molecules, 27(9), 2837. https://doi.org/10.3390/molecules27092837