
Structural Analysis of the Michael-Michael Ring Closure (MIMIRC) Reaction Products

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
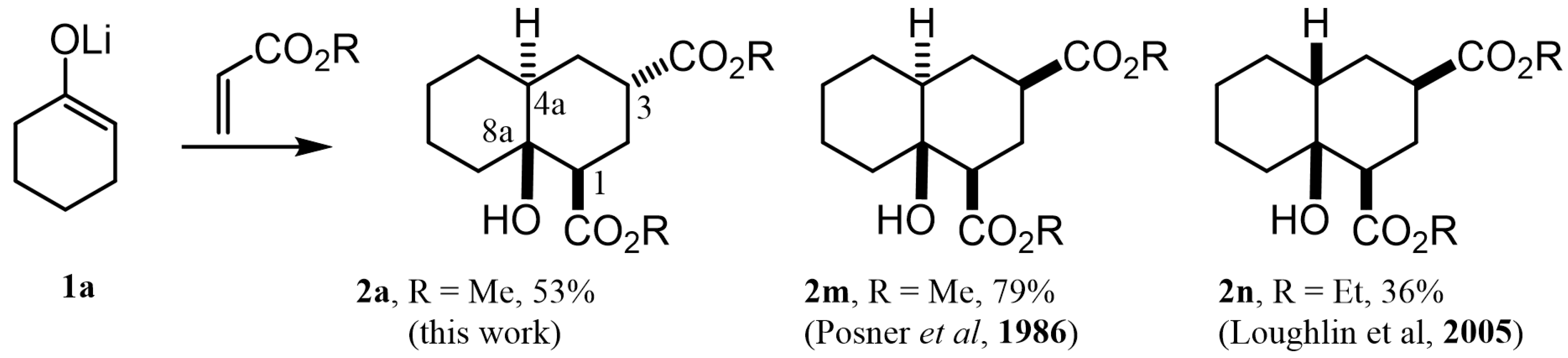
2.1. Preparation of Decalin and Hydrindane Derivatives
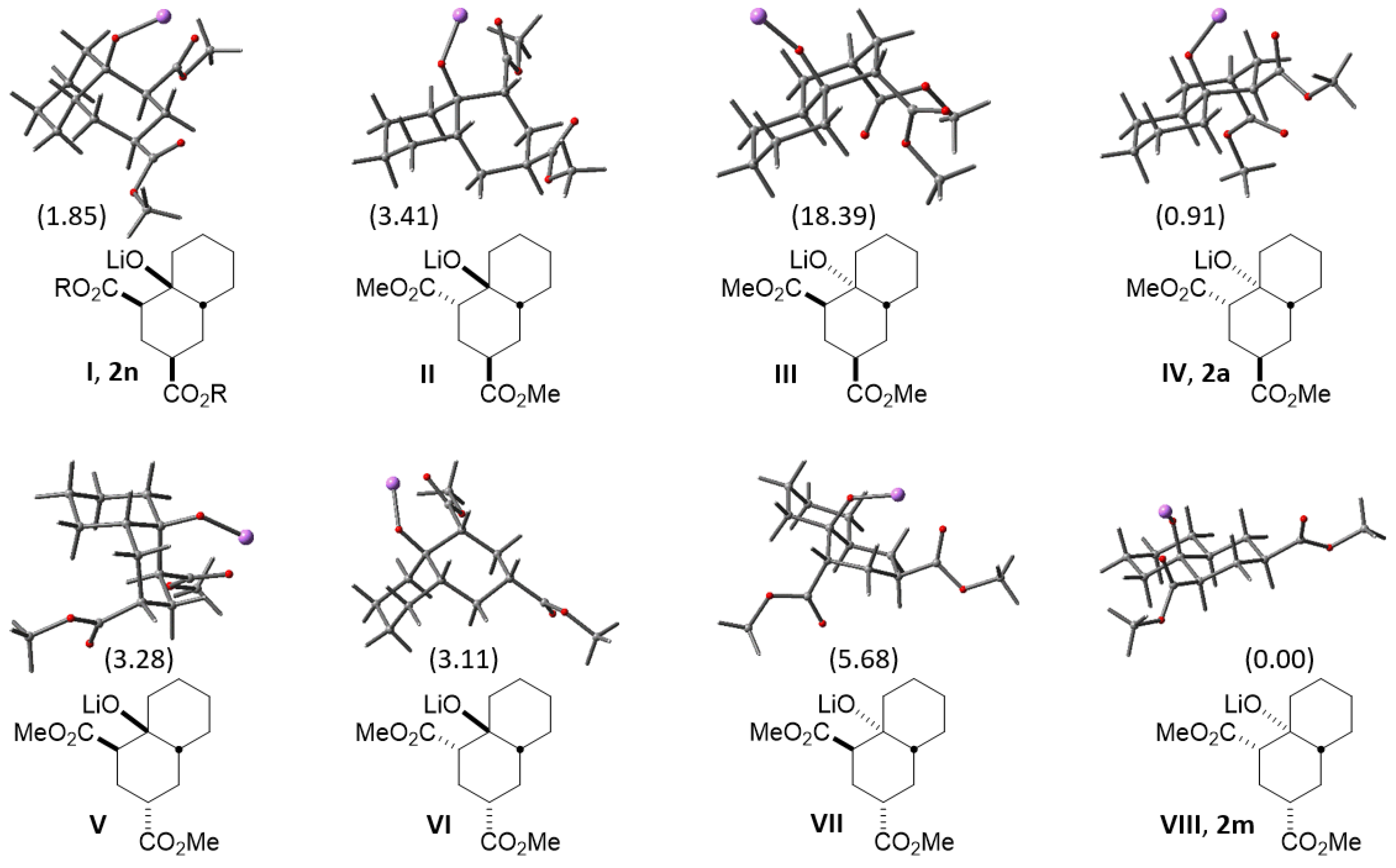
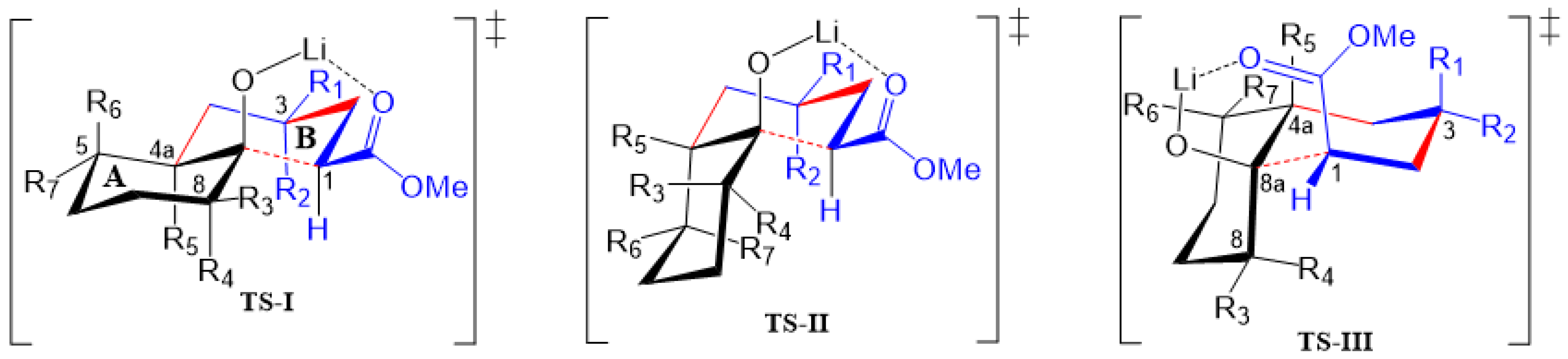
2.2. A Theoretical Structural Analysis
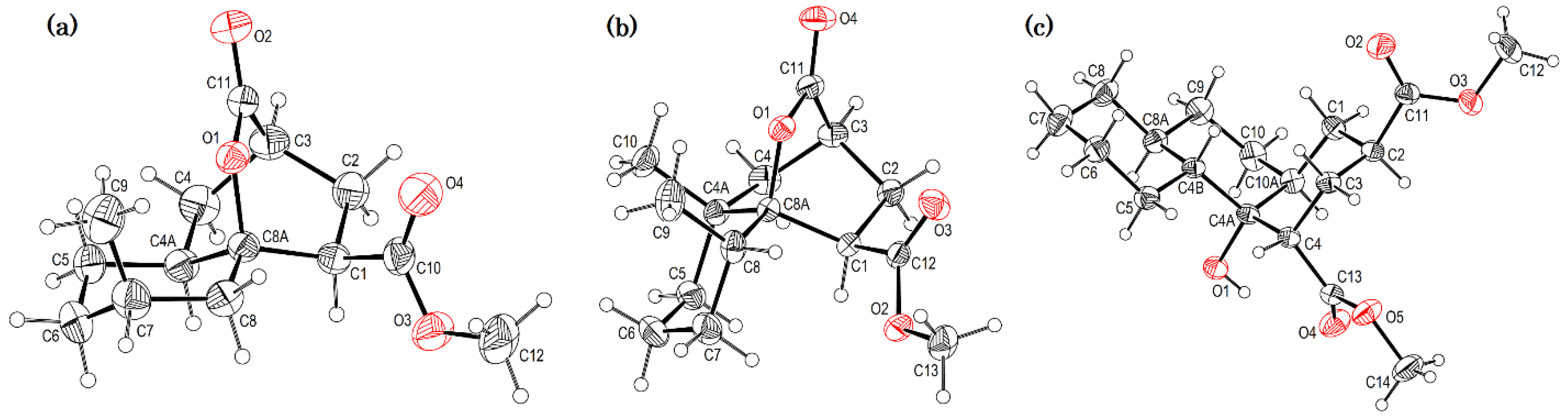
2.3. A 1H NMR-Based Structural Analysis of Decalin and Hydrindane Derivatives
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Theoretical Calculations
4.3. General Procedure for the Preparation of Decalin Derivatives 2a–j and Hydrindane 2k,l by Reacting Cycloalkanone Enolates (1a–k) with Methyl Acrylate
4.3.1. Dimethyl 8a-Hydroxydecahydronaphthalene-1,3-Dicarboxylate (2a)
4.3.2. Dimethyl 8a-Hydroxy-5-Methyl-8-(Propan-2-Ylidene) Decahydronaphthalene-1,3-Dicarboxylate (2b)
4.3.3. Methyl 7-Methyl-10-Oxooctahydro-2H-4a,2-(Epoxymethano)Naphthalene-4-CARBOXYLATE (2c)
4.3.4. Methyl 5,8a-Dimethyl-10-Oxooctahydro-2H-4a,2-(Epoxymethano)Naphthalene-4-Carboxylate (2d)
4.3.5. 4a-Hydroxy-4-(Methoxycarbonyl)-1,2,3,4,4a,7,8,8a-Octahydronaphthalene-2-Carboxylic Acid (2e)
4.3.6. Dimethyl 4a-Hydroxyoctahydro-1H-Spiro [Naphthalene-2,2′-[1,3]Dioxolane]-5,7-Dicarboxylate (2f and 2f′)
4.3.7. Methyl 5-Isopropyl-8-Methyl-10-Oxooctahydro-2H-4a,2-(Epoxyme†Hano)naph†Halene-4-Carboxylate (Mixture of 2g, 2g′ and 2g″)
4.3.8. Methyl 8-Methyl-10-Oxo-5-(Propan-2-Ylidene)Octahydro-2H-4a,2-(Epoxymethano)Naphthalene-4-Carboxylate (2h)
4.3.9. Methyl-8-Methyl-10-Oxo-5-(Prop-1-En-2-yl)1,3,4,5,6,8a-Hexahydro-2H-4a,2-(Epoxymethano)Naphthalene-4-Carboxylate (2i)
4.3.10. Dimethyl 4a-Hydroxytetradecahydrophenanthrene-2,4-Dicarboxylate (2j)
4.3.11. Dimethyl 3a-Hydroxyoctahydro-1H-Indene-4-6-Carboxylate (2k)
4.3.12. Dimethyl 3a-Hydroxy-3-Methyloctahydro-1H-Indene-4,6-Dicarboxylate (2l)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Li, G.; Kusari, S.; Spiteller, M. Natural products containing ‘decalin’ motif in microorganisms. Nat. Prod. Rep. 2014, 31, 1175–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuye, C.; Jing, Z.; Shaoping, L.; Jing, X. Total synthesis of sesterterpenoids. Nat. Prod. Rep. 2019, 36, 263–288. [Google Scholar]
- Lin, X.; Yuan, S.; Chen, S.; Chen, B.; Xu, H.; Liu, L.; Li, H.; Gao, Z. Heterologous Expression of Ilicicolin H Biosynthetic Gene Cluster and Production of a New Potent Antifungal Reagent, Ilicicolin J. Molecules 2019, 24, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Liu, C.; Li, L.; Zhou, H.; Liu, L.; Bao, L.; Chen, Q.; Song, F.; Zhang, L.; Li, E.; et al. Decalin-Containing Tetramic Acids and 4-Hydroxy-2-pyridones with Antimicrobial and Cytotoxic Activity from the Fungus Coniochaeta cephalothecoides Collected in Tibetan Plateau (Medog). J. Org. Chem. 2017, 82, 11474–11486. [Google Scholar] [CrossRef]
- Sharma, V.; Sharma, T.; Kaul, S.; Kapoor, K.K.; Dhar, M.K. Anticancer potential of labdane diterpenoid lactone “andrographolide” and its derivatives: A semi-synthetic approach. Phytochem. Rev. 2017, 16, 513–526. [Google Scholar] [CrossRef]
- Dhakal, D.; Han, J.M.; Mishra, R.; Pandey, R.P.; Kim, T.-S.; Rayamajhi, V.; Jung, H.J.; Yamaguchi, T.; Sohng, J.K. Characterization of Tailoring Steps of Nargenicin A1 Biosynthesis Reveals a Novel Analogue with Anticancer Activities. ACS Chem. Biol. 2020, 15, 1370–1380. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, J.G.; Li, T.; Zhu, G.Y.; Han, Q.B.; Hsiao, W.L.; Liu, L.; Jiang, Z.H. Immunosuppressive decalin derivatives from red yeast rice. J. Nat. Prod. 2012, 75, 567–571. [Google Scholar] [CrossRef]
- Zhang, W.; Kaplan, A.; Davison, E.; Freeman, J.; Brimble, M.; Wuest, W. Building trans-bicyclo[4.4.0]decanes/decenes in complex multifunctional frameworks: The case for antibiotic development. Nat. Prod. Rep. 2021, 38, 880–889. [Google Scholar] [CrossRef]
- Dhambri, S.; Mohammad, S.; Nguyen Van Buu, O.; Galvani, G.; Meyer, Y.; Lannou, M.-I.; Sorin, G.; Ardisson, J. Recent Advances in the Synthesis of Natural Multifunctionalized Decalins. Nat. Prod. Rep. 2015, 32, 841–864. [Google Scholar] [CrossRef]
- Posner, G.H.; Mallamo, J.P.; Black, A.Y. Tandem Michael-Michael-Ring-Closure (MIMIRC) reactions. Tetrahedron 1981, 23, 3921–3926. [Google Scholar] [CrossRef]
- Posner, G.H.; Shu-Bin, L. An extremely efficient method for one-pot, three-component, 2 + 2 + 2 construction of functionalized cyclohexenes. J. Am. Chem. Soc. 1985, 107, 1424–1426. [Google Scholar] [CrossRef]
- Xin, B.; Wenbo, X.; Yanmin, Y.; Lili, Z.; Guangxin, L. Total Syntheses of a Family of Cadinane Sesquiterpenes. J. Org. Chem. 2018, 83, 5825–5828. [Google Scholar]
- Thomas, W.P.; Pronin, S.V. New Methods and Strategies in the Synthesis of Terpenoid Natural Products. Acc. Chem. Res. 2021, 54, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Kotha, S.; Chavan, A.S.; Dipak, M.K. Synthetic approach to cis and trans-decalins via Diels—Alder reaction and ring-closing metathesis as key steps: Further extension to dioxapropellane derivative by ring-closing metathesis. Tetrahedron 2011, 67, 501–504. [Google Scholar] [CrossRef]
- Myers, J.T.; Wilde, J.H.; Sabat, M.; Dickie, D.A.; Harman, D.W. Molybdenum-Promoted Synthesis of Isoquinuclidines with Bridgehead CF3 Groups. Organometallics 2020, 39, 1404–1412. [Google Scholar] [CrossRef]
- Parasuraman, K.; Chennaiah, A.; Dubbu, S.; Sheriff, A.K.; Vankar, Y.D. Stereoselective synthesis of substituted 1,2-annulated sugars by domino double-Michael addition reaction. Carbohyd. Res. 2019, 477, 26–31. [Google Scholar] [CrossRef]
- Posner, G.H.; Shu-Bin, L.; Asirvatham, E.; Silversmith, E.F.; Shulman, E.M. Sequential Michael-Michael-Aldol Additions for Easy, One-Pot, 2 + 2 + 2 Construction of Polyfunctionalized Cyclohexanols. J. Am. Chem. Soc. 1986, 108, 511–512. [Google Scholar] [CrossRef]
- Bin, Y.; Li-Xin, Q.; Yi-Bing, Z.; Yu-Lin, W. Tandem Syntheses of Cyclohexane Derivatives via Sequential Michael-Michael-Aldol Reaction. Tetrahedron 1994, 50, 9061–9066. [Google Scholar]
- Jaafar, A.; Alilou, E.H.; Réglier, M.; Waegell, B. Unexpected [2 + 2 + 2] MIMIRC annulation between a lithium dienolate and methyl acrylate. Tetrahedron Lett. 1991, 32, 5531–5534. [Google Scholar] [CrossRef]
- Loughlin, D.W.A.; Rowen, C.C.; Healy, P.C. Diversion from Bicyclo[4.2.0]octanol Formation Through the Use of Vinyl Electrophiles. Synthesis 2005, 13, 2220–2226. [Google Scholar] [CrossRef]
- PC Spartan Pro; Wavefunction Inc.: Irvine, CA, USA, 2000.
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, H.E.; Traxler, M.D. The Stereochemistry of the Ivanov and Reformatsky Reactions. I. J. Am. Chem. Soc. 1957, 79, 1920–1923. [Google Scholar] [CrossRef]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; De Leeuw, F.A.A.M.; Altona, C. The relationship between Proton-Proton NMR Coupling Constants and Substituent Electronegativities—I: An empirical generalization of the Karplus equation. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Flores-Sandoval, C.A.; Cerda-García-Rojas, C.M.; Joseph-Nathan, P. Conformational study of the tricyclic sesquiterpene β-panasinsene aided by ab initio calculations and simulated 1H NMR parameters. Mag. Res. Chem. 2001, 39, 173–178. [Google Scholar] [CrossRef]
- Cerda-García-Rojas, C.M.; Zepeda, L.G.; Joseph-Nathan, P. A PC program for calculation of dihedral angles from 1H-NMR data. Tetrahedron Comp. Method. 1990, 3, 113–118. [Google Scholar] [CrossRef]
- Navarro-Vázquez, A.; Cobas, J.C.; Sardina, F.J.; Casanueva, J.; Díez, E. A Graphical Tool for the Prediction of Vicinal Proton−Proton 3JHH Coupling Constants. J. Chem. Inf. Comput. Sci. 2004, 44, 1680–1685. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Ketone | Product | Yield (%) b | Entry a | Ketone | Product | Yield (%) b |
|---|---|---|---|---|---|---|---|
| 1 | 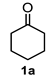 | 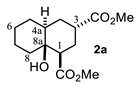 | 43 | 7 | 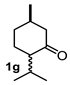 | 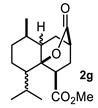 | 11 |
| 2 | 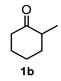 | 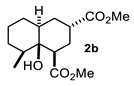 | 42 | 8 | 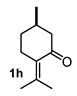 | 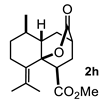 | 51 |
| 3 | 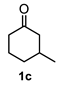 | 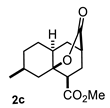 | 77 | 9 | 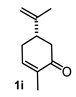 | 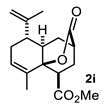 | 37 |
| 4 | 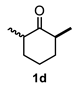 | 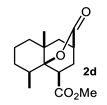 | 52 | 10 |  | 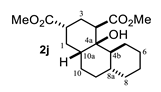 | 58 |
| 5 | 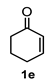 |  | 31 | 11 | 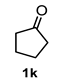 | 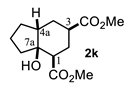 | 75 |
| 6 | 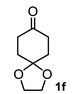 | 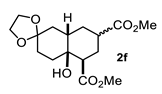 | 51 | 12 | 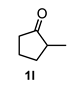 | 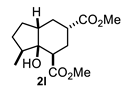 | 91 |
| Conformer | I b | II b | III | IV | V b | VI b | VII | VIII |
|---|---|---|---|---|---|---|---|---|
| a | 1.85 | 3.41 | 18.39 | 0.91 | 3.28 | 3.11 | 5.68 | 0.00 |
| b | 2.77 | 4.52 | 18.48 | 1.39 | 4.28 | 3.87 | 7.43 | 0.97 |
| c | 9.31 | 10.00 | 18.51 | 9.22 | 11.38 | 10.01 | 11.85 | 7.51 |
| d | 10.89 | 11.16 | 19.97 | 9.42 | 12.11 | 10.06 | 13.23 | 8.94 |
| e | 10.33 | |||||||
| Geometry | ΔG (kcal/mol) | Population (%) | Diast. Ratio (%) a |
|---|---|---|---|
| VIIIa | 0.00 | 64.1 | 76.6 (VIII, 2m) |
| VIIIb | 0.97 | 12.5 | |
| IVa | 0.91 | 13.9 | 20.0 (IV, 2a) |
| IVb | 1.39 | 6.1 | |
| Ia | 1.85 | 2.8 | 3.4 (I, 2n) |
| Ib | 2.77 | 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montenegro-Sustaita, M.M.; Jiménez-Vázquez, H.A.; Vargas-Díaz, E.; Herbert-Pucheta, J.E.; Zepeda-Vallejo, L.G. Structural Analysis of the Michael-Michael Ring Closure (MIMIRC) Reaction Products. Molecules 2022, 27, 2810. https://doi.org/10.3390/molecules27092810
Montenegro-Sustaita MM, Jiménez-Vázquez HA, Vargas-Díaz E, Herbert-Pucheta JE, Zepeda-Vallejo LG. Structural Analysis of the Michael-Michael Ring Closure (MIMIRC) Reaction Products. Molecules. 2022; 27(9):2810. https://doi.org/10.3390/molecules27092810
Chicago/Turabian StyleMontenegro-Sustaita, Mabel M., Hugo A. Jiménez-Vázquez, Elena Vargas-Díaz, J. Enrique Herbert-Pucheta, and L. Gerardo Zepeda-Vallejo. 2022. "Structural Analysis of the Michael-Michael Ring Closure (MIMIRC) Reaction Products" Molecules 27, no. 9: 2810. https://doi.org/10.3390/molecules27092810
APA StyleMontenegro-Sustaita, M. M., Jiménez-Vázquez, H. A., Vargas-Díaz, E., Herbert-Pucheta, J. E., & Zepeda-Vallejo, L. G. (2022). Structural Analysis of the Michael-Michael Ring Closure (MIMIRC) Reaction Products. Molecules, 27(9), 2810. https://doi.org/10.3390/molecules27092810