
Photoinduced Electron Transfer in Organized Assemblies—Case Studies

 ,
,
Abstract
:
1. Introduction
2. Fundaments of Electron Transfer in D-B-A Supramolecular Systems
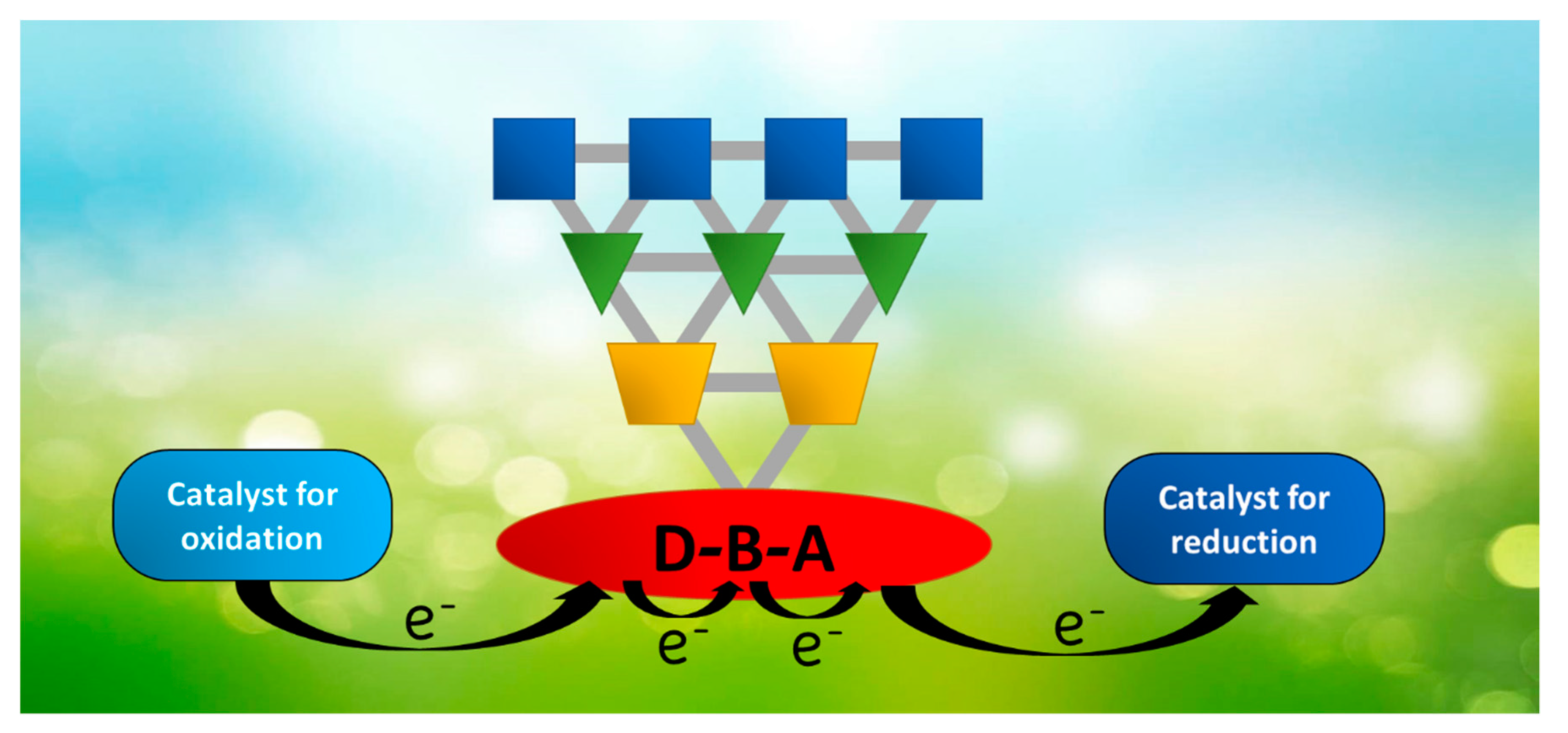
2.1. Multicomponent Systems: Supramolecular vs. Molecular Entities
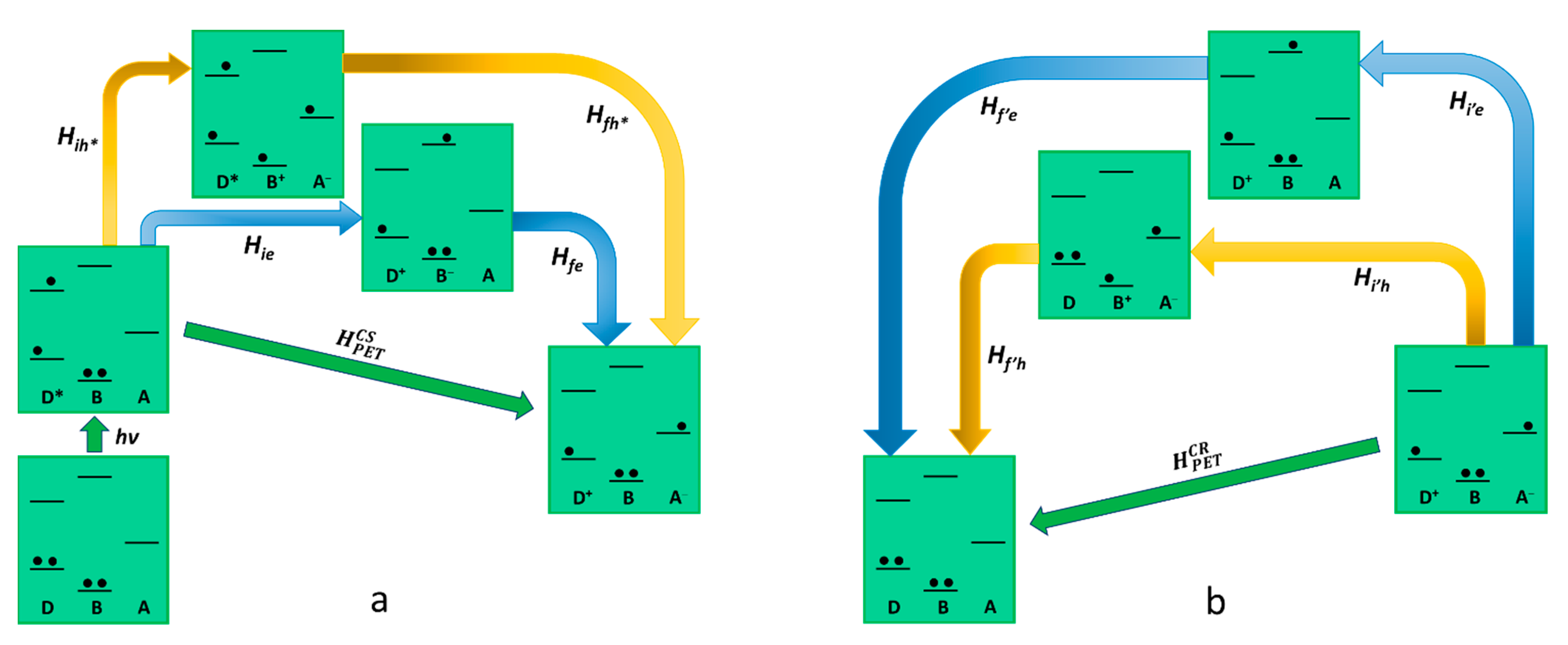
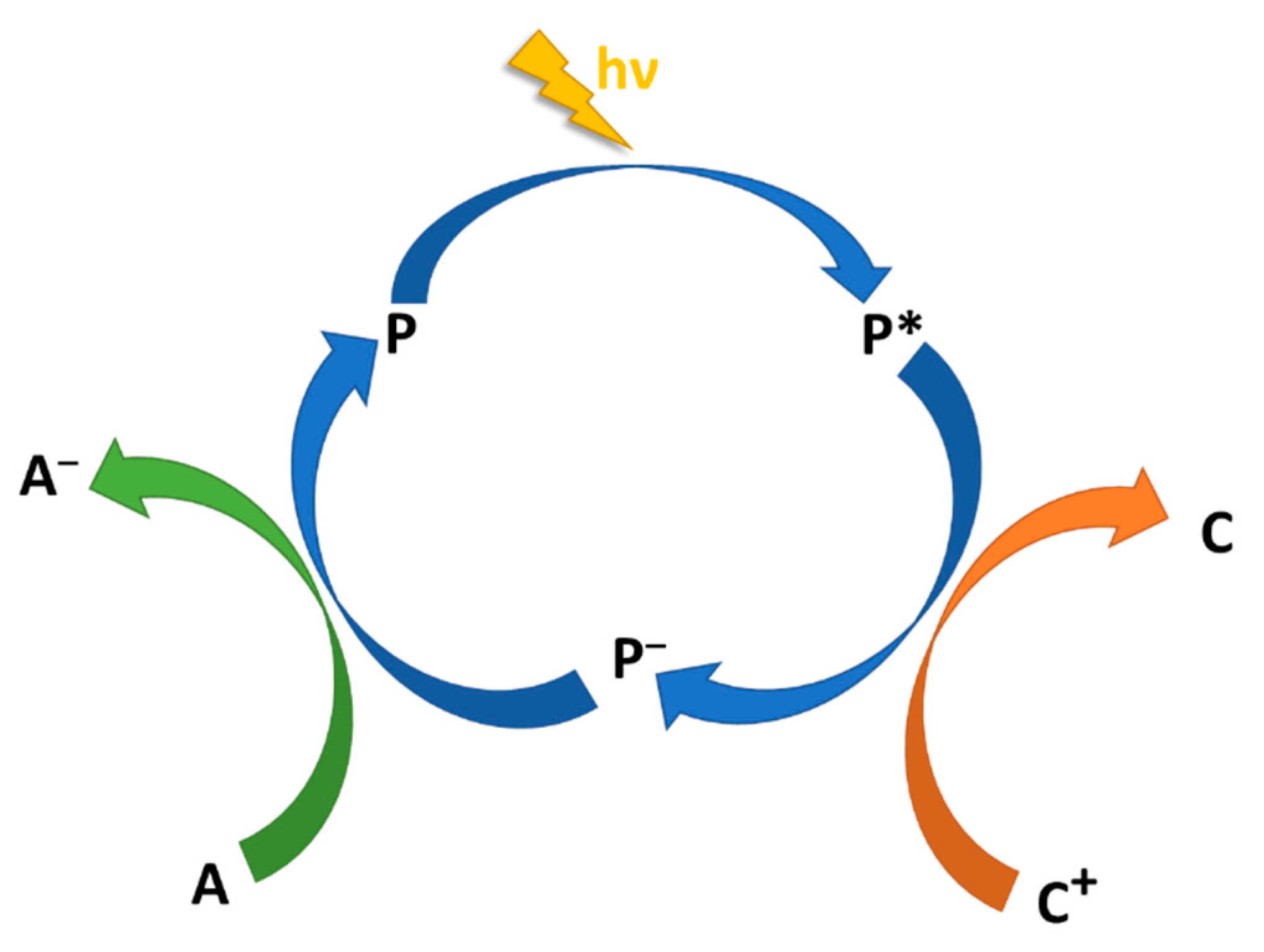
2.2. Photoinduced Charge Separation, Charge Recombination, and the Superexchange Mechanism
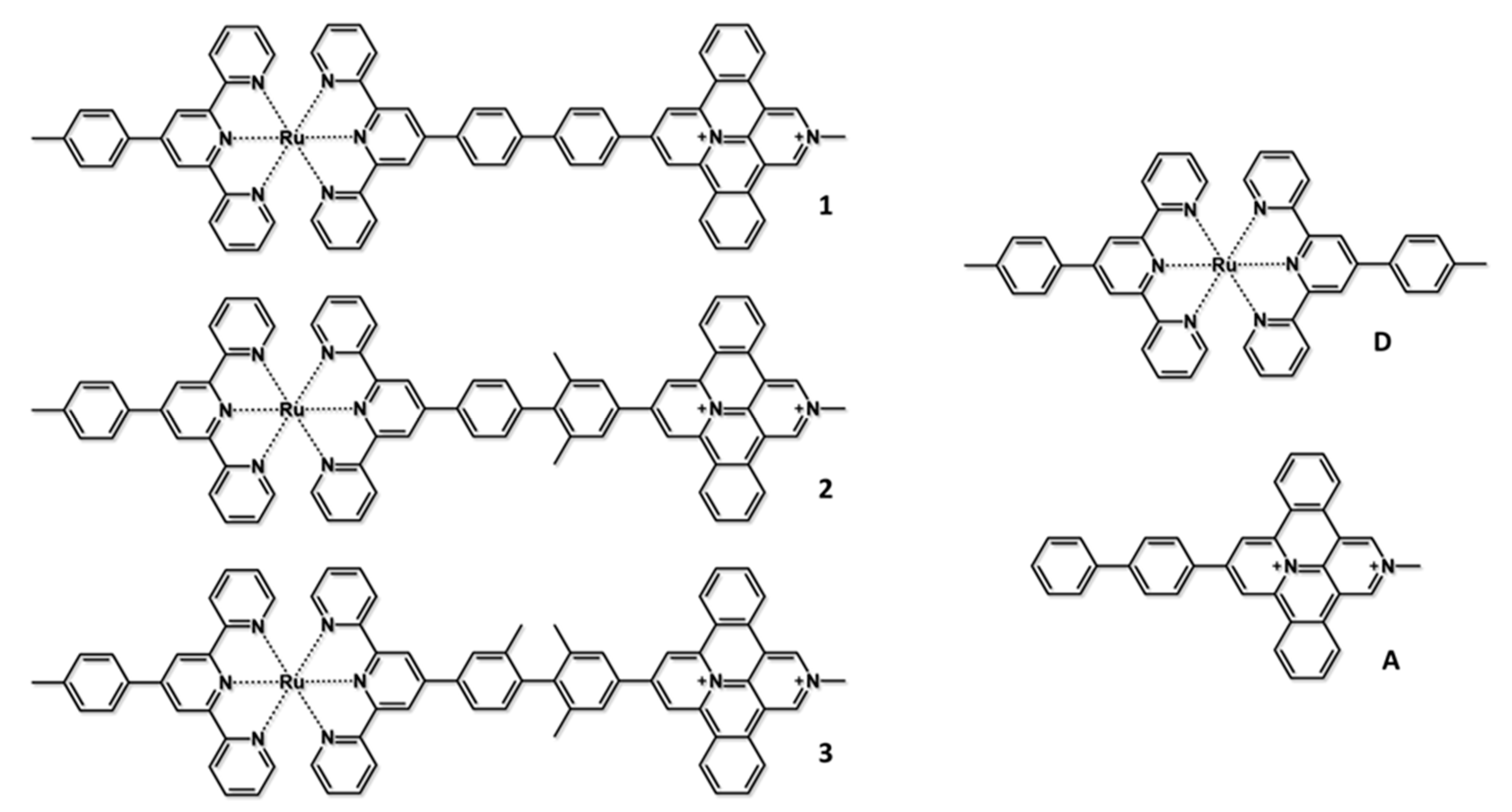
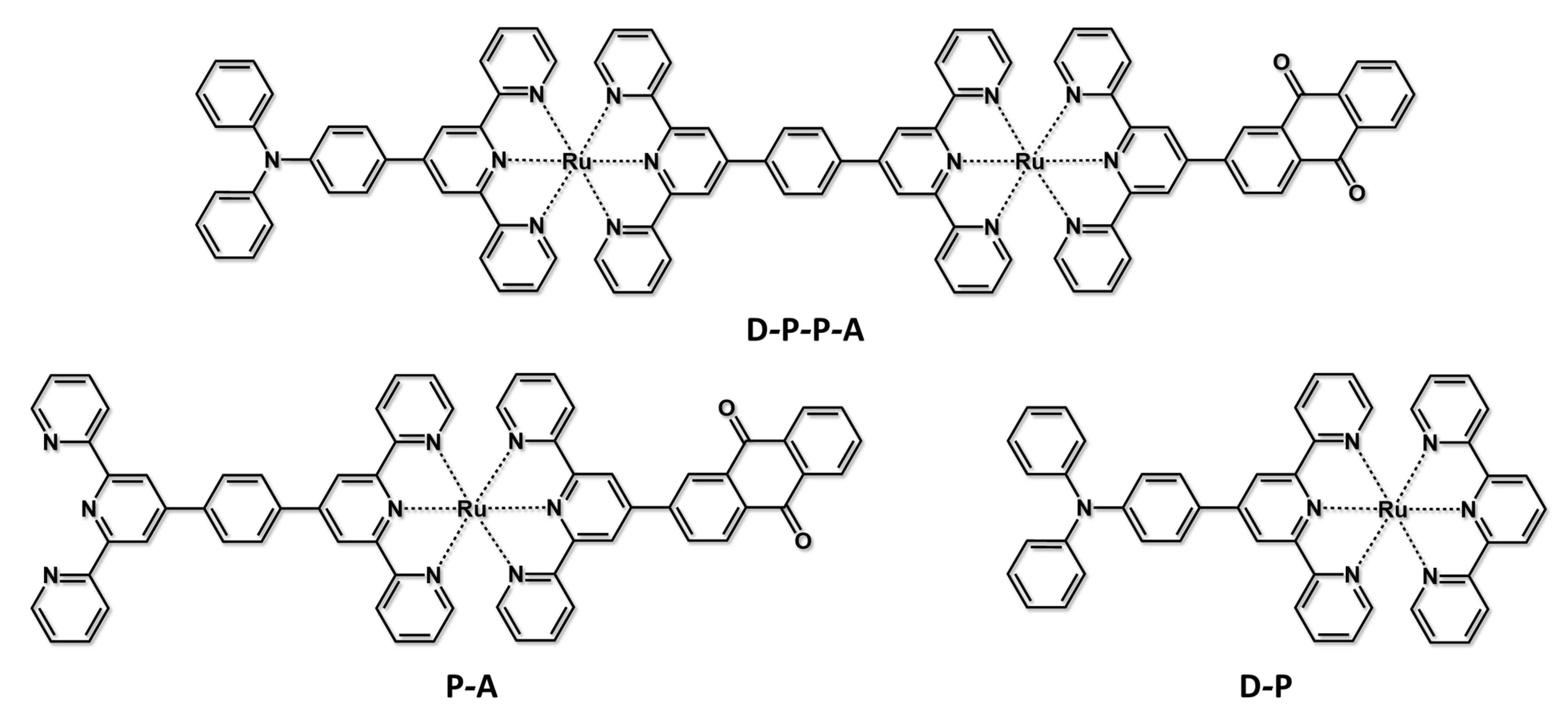
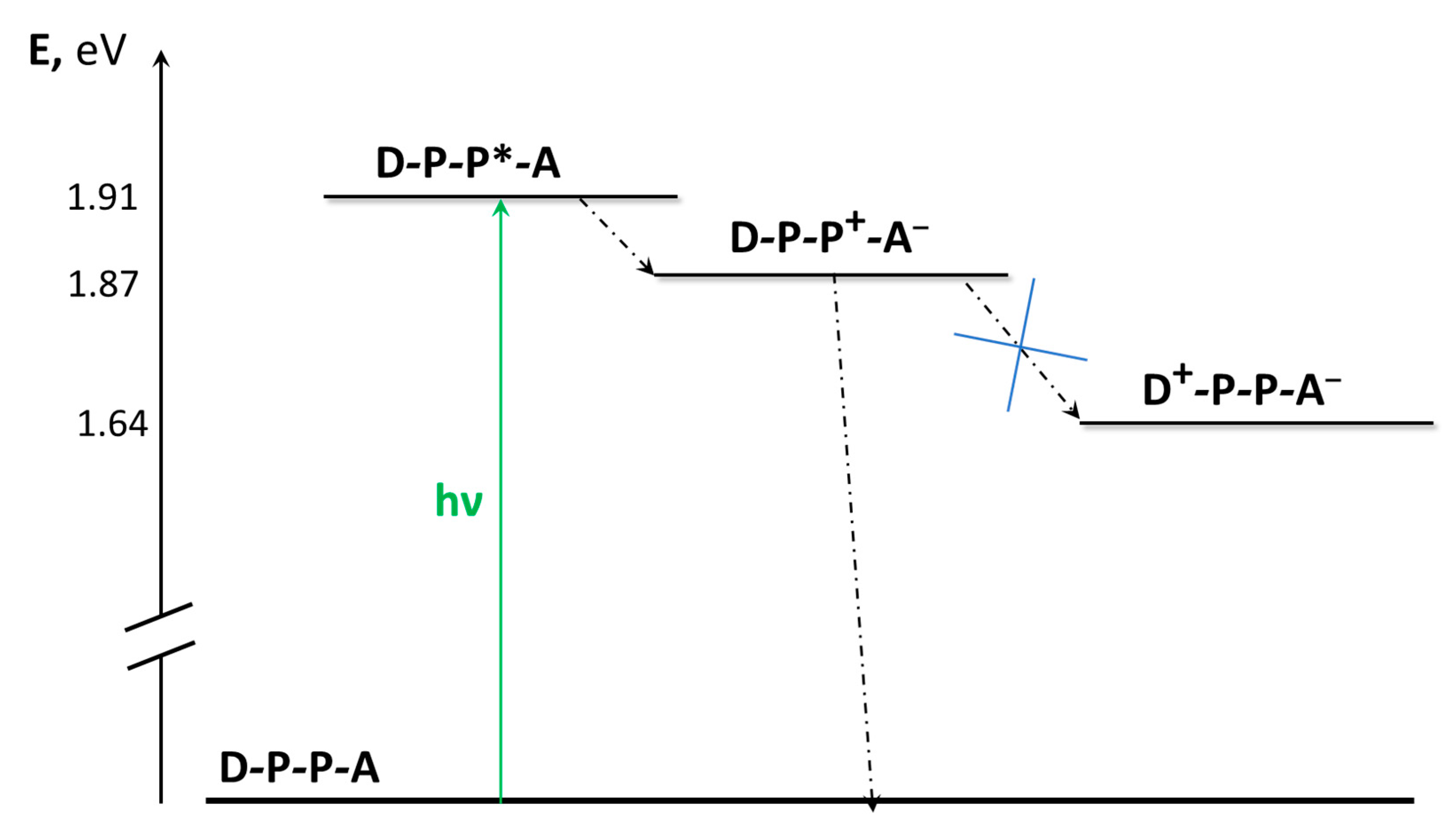
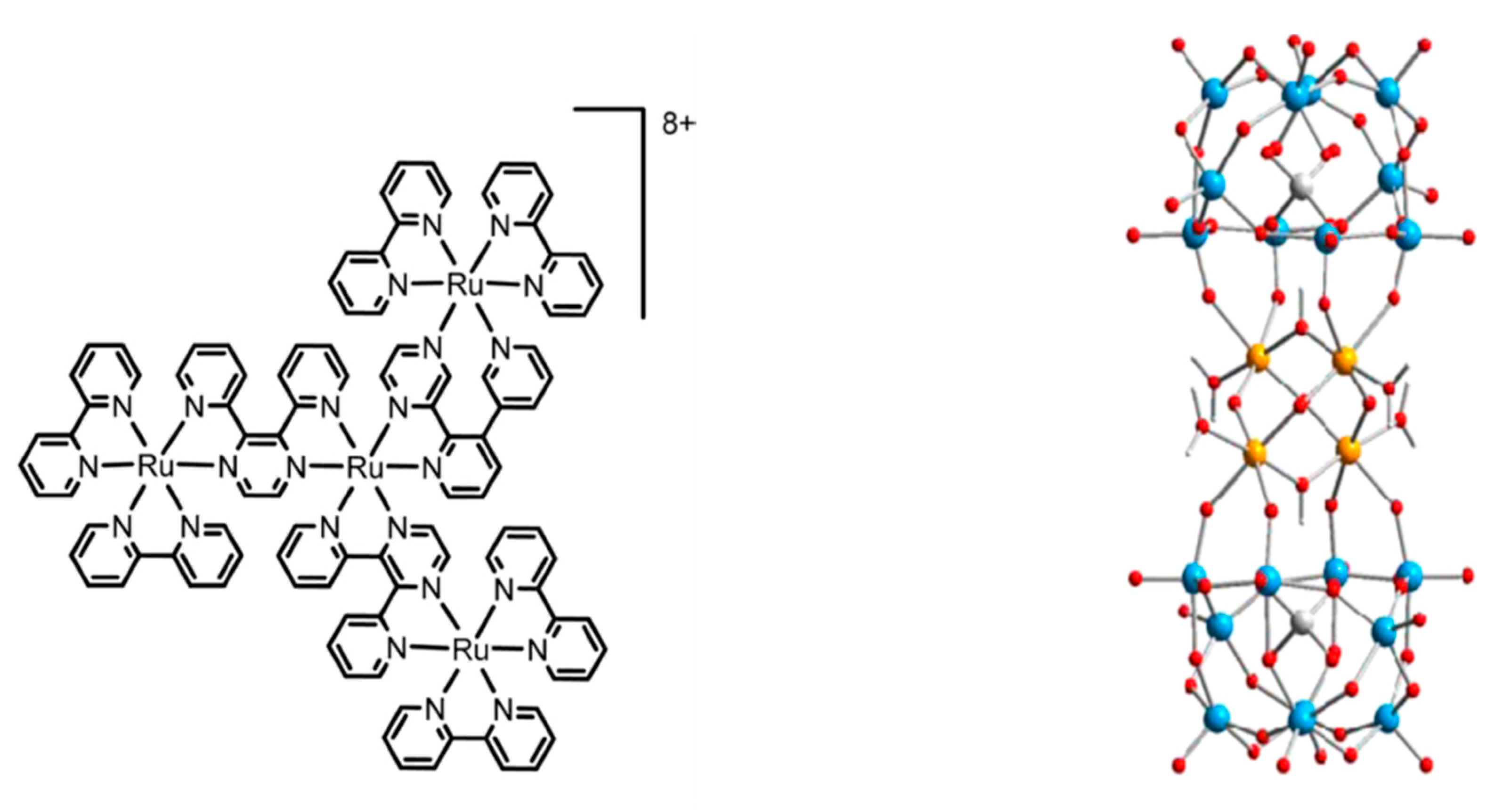
3. Examples of Photoinduced Electron Transfer Processes Recently Studied in the Messina Team
3.1. Covalently-Linked Multicomponent Species
3.2. Self-Assembled Systems
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Balzani, V. Electron Transfer in Chemistry; Balzani, V., Ed.; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Mancuso, A.; Barattucci, A.; Bonaccorsi, P.; Giannetto, A.; La Ganga, G.; Musarra-Pizzo, M.; Salerno, T.M.G.; Santoro, A.; Sciortino, M.T.; Puntoriero, F.; et al. Carbohydrates and Charges on Oligo(phenylenethynylenes): Towards the Design of Cancer Bullets. Chem. Eur. J. 2018, 24, 16972–16976. [Google Scholar] [CrossRef] [PubMed]
- Sjulstok, E.; Olsen, J.M.H.; Solov’yov, I.A. Quantifying electron transfer reactions in biological systems: What interactions play the major role? Sci. Rep. 2015, 5, 18446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S. Light-driven enzymatic catalysis of DNA repair: A review of recent biophysical studies on photolyase. Biochim. Biophys. Acta—Bioenerg. 2005, 1707, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barattucci, A.; Campagna, S.; Papalia, T.; Galletta, M.; Santoro, A.; Puntoriero, F.; Bonaccorsi, P. BODIPY on Board of Sugars: A Short Enlightened Journey up to the Cells. ChemPhotoChem 2020, 4, 647–658. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Electron flow through metalloproteins. Biochim. Biophys. Acta—Bioenerg. 2010, 1797, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Grätzel, M. Dye-sensitized solar cells. J. Photochem. Photobiol. C Photochem. Rev. 2003, 4, 145–153. [Google Scholar] [CrossRef]
- Farnum, B.H.; Wee, K.-R.; Meyer, T.J. Self-assembled molecular p/n junctions for applications in dye-sensitized solar energy conversion. Nat. Chem. 2016, 8, 845–852. [Google Scholar] [CrossRef]
- Fazio, E.; Spadaro, S.; Corsaro, C.; Neri, G.; Leonardi, S.G.; Neri, F.; Lavanya, N.; Sekar, C.; Donato, N.; Neri, G. Metal-Oxide Based Nanomaterials: Synthesis, Characterization and Their Applications in Electrical and Electrochemical Sensors. Sensors 2021, 21, 2494. [Google Scholar] [CrossRef]
- Natali, M.; Campagna, S.; Scandola, F. Photoinduced electron transfer across molecular bridges: Electron- and hole-transfer superexchange pathways. Chem. Soc. Rev. 2014, 43, 4005–4018. [Google Scholar] [CrossRef]
- Albinsson, B.; Mårtensson, J. Long-range electron and excitation energy transfer in donor–bridge–acceptor systems. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 138–155. [Google Scholar] [CrossRef]
- Barbara, P.F.; Meyer, T.J.; Ratner, M.A. Contemporary Issues in Electron Transfer Research. J. Phys. Chem. 1996, 100, 13148–13168. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Mimicking Photosynthetic Solar Energy Transduction. Acc. Chem. Res. 2001, 34, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Paddon-Row, M.N. Covalently Linked Systems Based on Organic Components in. Electron Transf. Chem. 2001, 3, 178–271. [Google Scholar]
- Arrigo, A.; Nastasi, F.; La Ganga, G.; Puntoriero, F.; Zappalà, G.; Licciardello, A.; Cavazzini, M.; Quici, S.; Campagna, S. Solvent-control of photoinduced electron transfer via hydrogen bonding in a molecular triad made of a dinuclear chromophore subunit. Chem. Phys. Lett. 2017, 683, 96–104. [Google Scholar] [CrossRef]
- Arrigo, A.; Santoro, A.; Puntoriero, F.; Lainé, P.P.; Campagna, S. Photoinduced electron transfer in donor–bridge–acceptor assemblies: The case of Os(II)-bis(terpyridine)-(bi)pyridinium dyads. Coord. Chem. Rev. 2015, 304–305, 109–116. [Google Scholar] [CrossRef]
- Puntoriero, F.; Arrigo, A.; Santoro, A.; Ganga, G.L.; Tuyèras, F.; Campagna, S.; Dupeyre, G.; Lainé, P.P. Photoinduced Intercomponent Processes in Selectively Addressable Bichromophoric Dyads Made of Linearly Arranged Ru(II) Terpyridine and Expanded Pyridinium Components. Inorg. Chem. 2019, 58, 5807–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrigo, A.; Santoro, A.; Indelli, M.T.; Natali, M.; Scandola, F.; Campagna, S. On the effect of the nature of the bridge on oxidative or reductive photoinduced electron transfer in donor–bridge–acceptor systems. PCCP 2014, 16, 818–826. [Google Scholar] [CrossRef]
- Lewis, N.S. Toward Cost-Effective Solar Energy Use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.J. The art of splitting water. Nature 2008, 451, 778–779. [Google Scholar] [CrossRef]
- Kohler, L.; Kaveevivitchai, N.; Zong, R.; Thummel, R.P. Component Analysis of Dyads Designed for Light-Driven Water Oxidation. Inorg. Chem. 2014, 53, 912–921. [Google Scholar] [CrossRef]
- Liu, F.; Concepcion, J.J.; Jurss, J.W.; Cardolaccia, T.; Templeton, J.L.; Meyer, T.J. Mechanisms of Water Oxidation from the Blue Dimer to Photosystem II. Inorg. Chem. 2008, 47, 1727–1752. [Google Scholar] [CrossRef] [PubMed]
- Jurss, J.W.; Concepcion, J.C.; Norris, M.R.; Templeton, J.L.; Meyer, T.J. Surface Catalysis of Water Oxidation by the Blue Ruthenium Dimer. Inorg. Chem. 2010, 49, 3980–3982. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Lanzafame, P.; Perathoner, S.; Centi, G.; Cozza, D.; Giorgianni, G.; Migliori, M.; Giordano, G. High performance of Au/ZTC based catalysts for the selective oxidation of bio-derivative furfural to 2-furoic acid11This paper in honor of Professor James G. Goodwin, Jr., in the occasion of his 75th birthday, to celebrate his outstanding contribution to catalysis sciences and technology. Catal. Commun. 2021, 149, 106234. [Google Scholar]
- Papanikolaou, G.; Lanzafame, P.; Giorgianni, G.; Abate, S.; Perathoner, S.; Centi, G. Highly selective bifunctional Ni zeo-type catalysts for hydroprocessing of methyl palmitate to green diesel. Catal. Today 2020, 345, 14–21. [Google Scholar] [CrossRef]
- Teets, T.S.; Nocera, D.G. Photocatalytic hydrogen production. Chem. Commun. 2011, 47, 9268–9274. [Google Scholar] [CrossRef]
- Crabtree, R.H. Energy Production and Storage: Inorganic Chemical Strategies for a Warming World; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Wang, F.; Wang, W.-G.; Wang, H.-Y.; Si, G.; Tung, C.-H.; Wu, L.-Z. Artificial photosynthetic systems based on [FeFe]-hydrogenase mimics: The road to high efficiency for light-driven hydrogen evolution. ACS Catal 2012, 2, 407–416. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Realizing artificial photosynthesis. Faraday Discuss. 2012, 155, 9–26. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Satake, A.; Kobuke, Y. Light-Harvesting Macroring Accommodating a Tetrapodal Ligand Based on Complementary and Cooperative Coordinations. J. Am. Chem. Soc. 2004, 126, 8668–8669. [Google Scholar] [CrossRef]
- Keijer, T.; Bouwens, T.; Hessels, J.; Reek, J.N.H. Supramolecular strategies in artificial photosynthesis. Chem. Sci. 2021, 12, 50–70. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Saito, K.; Ohkubo, K.; Khoury, T.; Kashiwagi, Y.; Absalom, M.A.; Gadde, S.; D’Souza, F.; Araki, Y.; Ito, O.; et al. Multiple photosynthetic reaction centres composed of supramolecular assemblies of zinc porphyrin dendrimers with a fullerene acceptor. Chem. Commun. 2011, 47, 7980–7982. [Google Scholar] [CrossRef]
- Sugou, K.; Sasaki, K.; Kitajima, K.; Iwaki, T.; Kuroda, Y. Light-Harvesting Heptadecameric Porphyrin Assemblies. J. Am. Chem. Soc. 2002, 124, 1182–1183. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Sugou, K.; Sasaki, K. Nonameric Porphyrin Assembly: Antenna Effect on Energy Transfer. J. Am. Chem. Soc. 2000, 122, 7833–7834. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Self-Processes—Programmed Supramolecular Systems. Supramol. Chem. 1995, 139–197.
- Paddon-Row, M.N. Investigating long-range electron-transfer processes with rigid, covalently linked donor-(norbornylogous bridge)-acceptor systems. Acc. Chem. Res. 1994, 27, 18–25. [Google Scholar] [CrossRef]
- Bonin, J.; Costentin, C.; Robert, M.; Routier, M.; Savéant, J.-M. Proton-Coupled Electron Transfers: pH-Dependent Driving Forces? Fundamentals and Artifacts. J. Am. Chem. Soc. 2013, 135, 14359–14366. [Google Scholar] [CrossRef]
- Sutin, N.; Brunschwig, B.S. Electron Transfer in Weakly Interacting Systems. Mech. Asp. Inorg. React. 1982, 198, 105–135. [Google Scholar]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) polypyridine complexes: Photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Sauvage, J.P.; Collin, J.P.; Chambron, J.C.; Guillerez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; De Cola, L.; Flamigni, L. Ruthenium(II) and Osmium(II) Bis(terpyridine) Complexes in Covalently-Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties. Chem. Rev. 1994, 94, 993–1019. [Google Scholar] [CrossRef]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and Photophysics of Coordination Compounds: Ruthenium. In Photochemistry and Photophysics of Coordination Compounds I; Balzani, V., Campagna, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 117–214. [Google Scholar]
- Kestner, N.R.; Logan, J.; Jortner, J. Thermal electron transfer reactions in polar solvents. J. Phys. Chem. 1974, 78, 2148–2166. [Google Scholar] [CrossRef]
- Jortner, J. Temperature dependent activation energy for electron transfer between biological molecules. J. Chem. Phys. 1976, 64, 4860–4867. [Google Scholar] [CrossRef]
- Ulstrup, J.; Jortner, J. The effect of intramolecular quantum modes on free energy relationships for electron transfer reactions. J. Chem. Phys. 1975, 63, 4358–4368. [Google Scholar] [CrossRef] [Green Version]
- Brunschwig, B.S.; Logan, J.; Newton, M.D.; Sutin, N. A semiclassical treatment of electron-exchange reactions. Application to the hexaaquoiron (II)-hexaaquoiron (III) system. J. Am. Chem. Soc. 1980, 102, 5798–5809. [Google Scholar] [CrossRef]
- Siders, P.; Marcus, R. Quantum effects in electron-transfer reactions. J. Am. Chem. Soc. 1981, 103, 741–747. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta—Bioenerg 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Miller, J.R.; Beitz, J.V.; Huddleston, R.K. Effect of free energy on rates of electron transfer between molecules. J. Am. Chem. Soc. 1984, 106, 5057–5068. [Google Scholar] [CrossRef]
- McConnell, H.M. Intramolecular charge transfer in aromatic free radicals. J. Chem. Phys. 1961, 35, 508–515. [Google Scholar] [CrossRef]
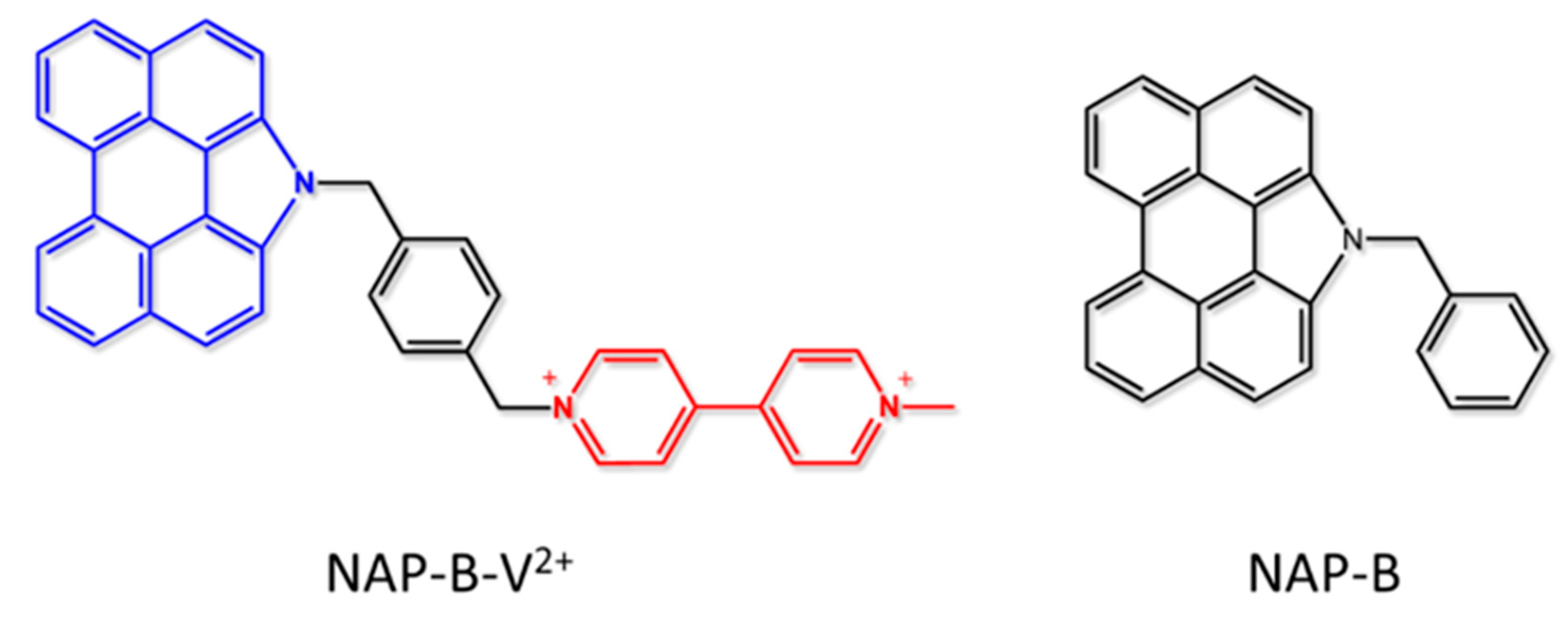
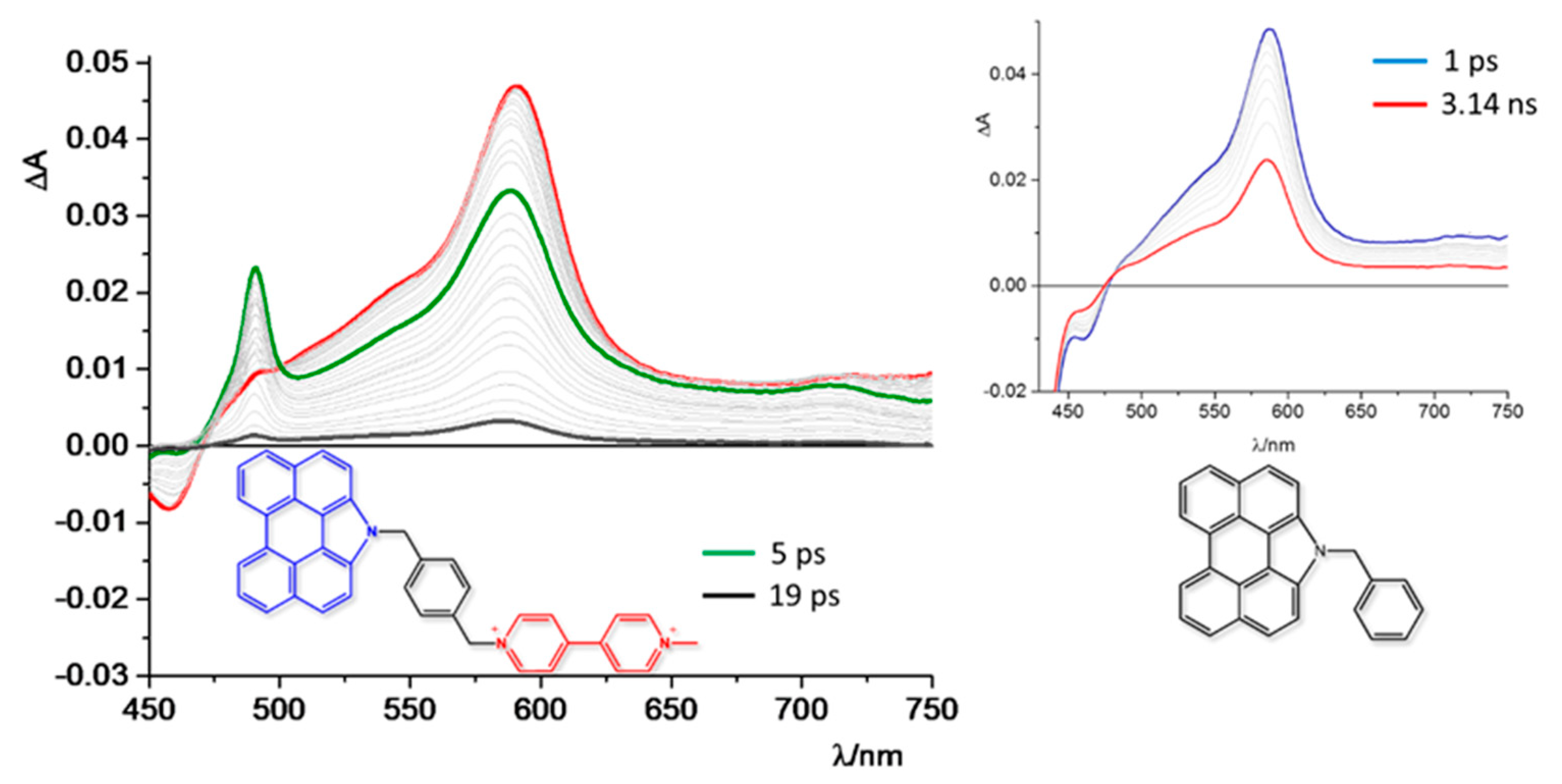
- Santoni, M.-P.; Santoro, A.; Salerno, T.M.G.; Puntoriero, F.; Nastasi, F.; Di Pietro, M.L.; Galletta, M.; Campagna, S. Photoinduced Charge Separation in a Donor–Spacer–Acceptor Dyad with N-Annulated Perylene Donor and Methylviologen Acceptor. ChemPhysChem 2015, 16, 3147–3150. [Google Scholar] [CrossRef]
- Orazietti, M.; Kuss-Petermann, M.; Hamm, P.; Wenger, O.S. Light-Driven Electron Accumulation in a Molecular Pentad. Angew. Chem. Int. Ed. 2016, 55, 9407–9410. [Google Scholar] [CrossRef]
- Bonn, A.G.; Yushchenko, O.; Vauthey, E.; Wenger, O.S. Photoinduced Electron Transfer in an Anthraquinone–[Ru(bpy)3]2+–Oligotriarylamine–[Ru(bpy)3]2+–Anthraquinone Pentad. Inorg. Chem. 2016, 55, 2894–2899. [Google Scholar] [CrossRef]
- Hankache, J.; Niemi, M.; Lemmetyinen, H.; Wenger, O.S. Hydrogen-Bonding Effects on the Formation and Lifetimes of Charge-Separated States in Molecular Triads. J. Chem. Phys. A 2012, 116, 8159–8168. [Google Scholar] [CrossRef] [PubMed]
- Kuss-Petermann, M.; Wenger, O.S. Pump-Pump-Probe Spectroscopy of a Molecular Triad Monitoring Detrimental Processes for Photoinduced Charge Accumulation. Helv. Chim. Acta 2017, 100, e1600283. [Google Scholar] [CrossRef] [Green Version]
- Gust, D.; Moore, T.A.; Moore, A.L.; Macpherson, A.N.; Lopez, A.; DeGraziano, J.M.; Gouni, I.; Bittersmann, E.; Seely, G.R. Photoinduced electron and energy transfer in molecular pentads. J. Am. Chem. Soc. 1993, 115, 11141–11152. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L.; Lee, S.-J.; Bittersmann, E.; Luttrull, D.K.; Rehms, A.A.; DeGraziano, J.M.; Ma, X.C.; Gao, F.; et al. Efficient Multistep Photoinitiated Electron Transfer in a Molecular Pentad. Science 1990, 248, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Ballardini, R.; Balzani, V.; Clemente-León, M.; Credi, A.; Gandolfi, M.T.; Ishow, E.; Perkins, J.; Stoddart, J.F.; Tseng, H.-R.; Wenger, S. Photoinduced Electron Transfer in a Triad That Can Be Assembled/Disassembled by Two Different External Inputs. Toward Molecular-Level Electrical Extension Cables. J. Am. Chem. Soc. 2002, 124, 12786–12795. [Google Scholar] [CrossRef]
- Davis, C.M.; Ohkubo, K.; Lammer, A.D.; Kim, D.S.; Kawashima, Y.; Sessler, J.L.; Fukuzumi, S. Photoinduced electron transfer in a supramolecular triad produced by porphyrin anion-induced electron transfer from tetrathiafulvalene calix[4]pyrrole to Li+@C60. Chem. Commun. 2015, 51, 9789–9792. [Google Scholar] [CrossRef] [Green Version]
- Megiatto, J.D.; Antoniuk-Pablant, A.; Sherman, B.D.; Kodis, G.; Gervaldo, M.; Moore, T.A.; Moore, A.L.; Gust, D. Mimicking the electron transfer chain in photosystem II with a molecular triad thermodynamically capable of water oxidation. Proc. Natl. Acad. Sci. USA 2012, 109, 15578–15583. [Google Scholar] [CrossRef] [Green Version]
- Cancelliere, A.M.; Puntoriero, F.; Serroni, S.; Campagna, S.; Tamaki, Y.; Saito, D.; Ishitani, O. Efficient trinuclear Ru(ii)–Re(i) supramolecular photocatalysts for CO2 reduction based on a new tris-chelating bridging ligand built around a central aromatic ring. Chem. Sci. 2020, 11, 1556–1563. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Ishitani, O. Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord. Chem. Rev. 2010, 254, 346–354. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 2018, 373, 333–356. [Google Scholar] [CrossRef]
- Natali, M.; Amati, A.; Merchiori, S.; Ventura, B.; Iengo, E. Photoinduced Proton-Coupled Electron Transfer in Supramolecular SnIV Di(l-tyrosinato) Porphyrin Conjugates. J. Phys. Chem. C 2020, 124, 8514–8525. [Google Scholar] [CrossRef]
- Holub, J.; Santoro, A.; Lehn, J.-M. Electronic absorption and emission properties of bishydrazone [2 × 2] metallosupramolecular grid-type architectures. Inorg. Chim. Acta 2019, 494, 223–231. [Google Scholar] [CrossRef]
- Wasielewski, M.R. Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems. Acc. Chem. Res. 2009, 42, 1910–1921. [Google Scholar] [CrossRef] [PubMed]
- Puntoriero, F.; Serroni, S.; La Ganga, G.; Santoro, A.; Galletta, M.; Nastasi, F.; La Mazza, E.; Cancelliere, A.M.; Campagna, S. Photo- and Redox-Active Metal Dendrimers: A Journey from Molecular Design to Applications and Self-Aggregated Systems. Eur. J. Inorg. Chem. 2018, 2018, 3887–3899. [Google Scholar] [CrossRef] [Green Version]
- Bottari, G.; Trukhina, O.; Ince, M.; Torres, T. Towards artificial photosynthesis: Supramolecular, donor–acceptor, porphyrin- and phthalocyanine/carbon nanostructure ensembles. Coord. Chem. Rev. 2012, 256, 2453–2477. [Google Scholar] [CrossRef]
- Natali, M.; Puntoriero, F.; Chiorboli, C.; La Ganga, G.; Sartorel, A.; Bonchio, M.; Campagna, S.; Scandola, F. Working the Other Way Around: Photocatalytic Water Oxidation Triggered by Reductive Quenching of the Photoexcited Chromophore. J. Phys. Chem. C 2015, 119, 2371–2379. [Google Scholar] [CrossRef]
- Puntoriero, F.; La Ganga, G.; Sartorel, A.; Carraro, M.; Scorrano, G.; Bonchio, M.; Campagna, S. Photo-induced water oxidation with tetra-nuclear ruthenium sensitizer and catalyst: A unique 4 × 4 ruthenium interplay triggering high efficiency with low-energy visible light. Chem. Commun. 2010, 46, 4725–4727. [Google Scholar] [CrossRef]
- Ronconi, F.; Santoni, M.-P.; Nastasi, F.; Bruno, G.; Argazzi, R.; Berardi, S.; Caramori, S.; Bignozzi, C.A.; Campagna, S. Charge injection into nanostructured TiO2 electrodes from the photogenerated reduced form of a new Ru(ii) polypyridine compound: The “anti-biomimetic” mechanism at work. Dalton Trans. 2016, 45, 14109–14123. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorption | Luminescence | Redox Data (V vs. SCE) | ||||
|---|---|---|---|---|---|---|
| λ [nm] (ε[M−1cm−1]) | λ [nm] | τ [ns] | φ | E1/2(red) | E1/2(ox) | |
| NAP-B-V2+ | 422 (35,450) | - | - | - | −0.38 | +0.89 |
| NAP-B | 421 (36,000) | 435 | 5.0 | 0.9 | - | 0.88 |
| D | A | 1 | 2 | 3 | |
|---|---|---|---|---|---|
| E1/2(red) (V vs. SCE) | −1.27 | −0.42 | −0.42 | −0.43 | −0.45 |
| E1/2(ox) (V vs. SCE) | +1.22 | - | +1.23 | +1.23 | +1.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoro, A.; Bella, G.; Cancelliere, A.M.; Serroni, S.; Lazzaro, G.; Campagna, S. Photoinduced Electron Transfer in Organized Assemblies—Case Studies. Molecules 2022, 27, 2713. https://doi.org/10.3390/molecules27092713
Santoro A, Bella G, Cancelliere AM, Serroni S, Lazzaro G, Campagna S. Photoinduced Electron Transfer in Organized Assemblies—Case Studies. Molecules. 2022; 27(9):2713. https://doi.org/10.3390/molecules27092713
Chicago/Turabian StyleSantoro, Antonio, Giovanni Bella, Ambra M. Cancelliere, Scolastica Serroni, Giuliana Lazzaro, and Sebastiano Campagna. 2022. "Photoinduced Electron Transfer in Organized Assemblies—Case Studies" Molecules 27, no. 9: 2713. https://doi.org/10.3390/molecules27092713
APA StyleSantoro, A., Bella, G., Cancelliere, A. M., Serroni, S., Lazzaro, G., & Campagna, S. (2022). Photoinduced Electron Transfer in Organized Assemblies—Case Studies. Molecules, 27(9), 2713. https://doi.org/10.3390/molecules27092713