
In Silico and In Vitro Antimalarial Screening and Validation Targeting Plasmodium falciparum Plasmepsin V

 and
and
Abstract
:
1. Introduction
2. Results
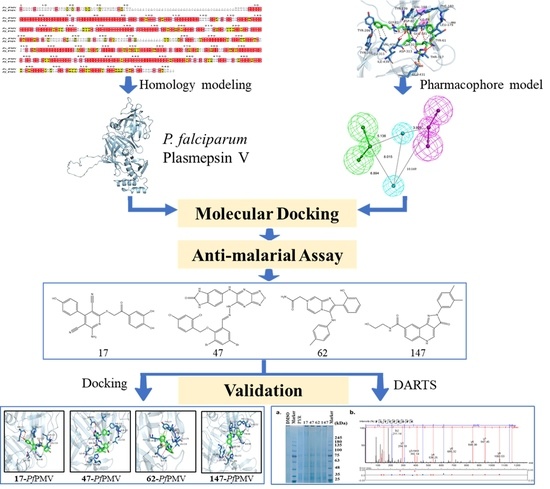
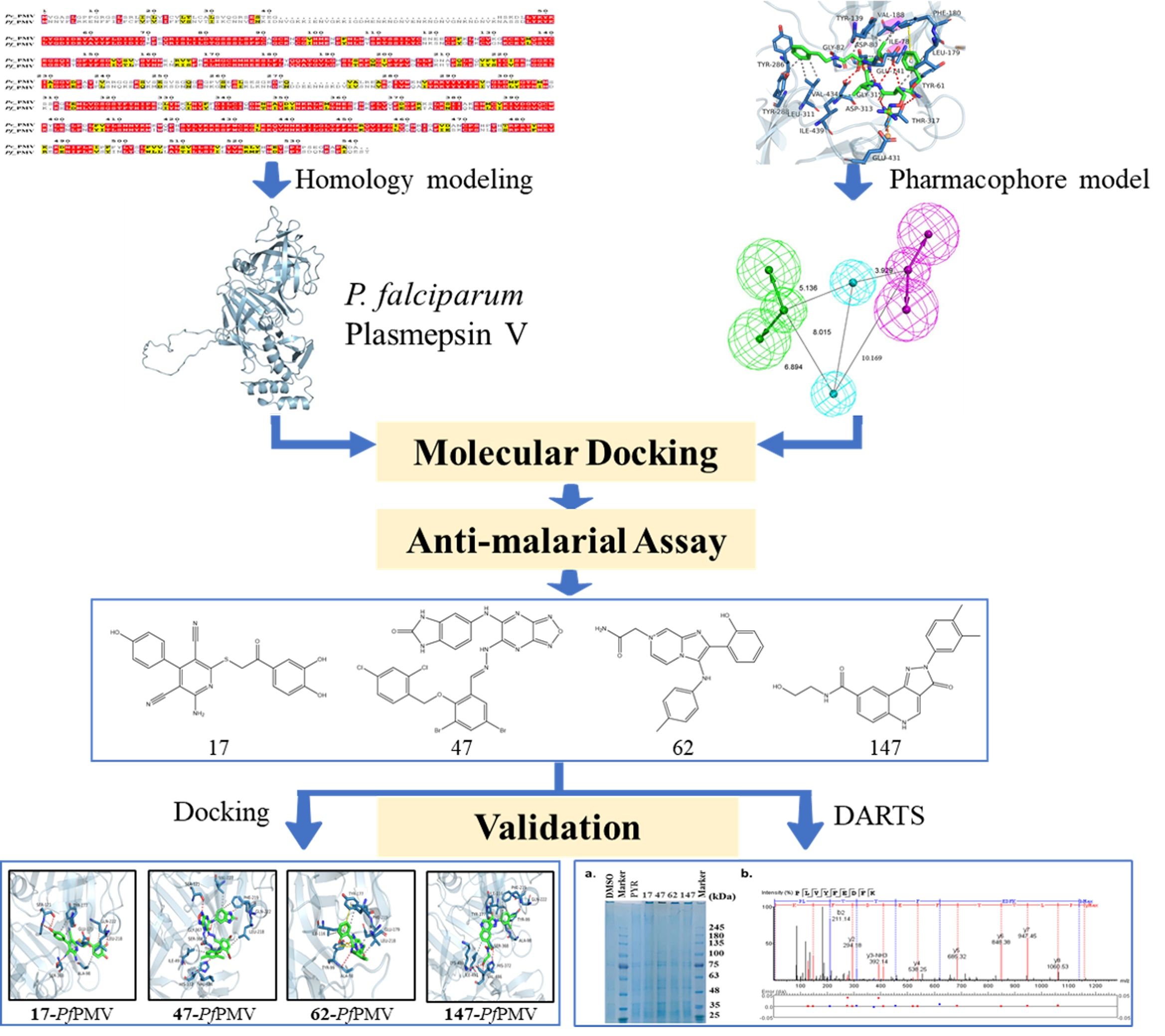
2.1. Homology Modeling of PfPMV and Virtual Screening
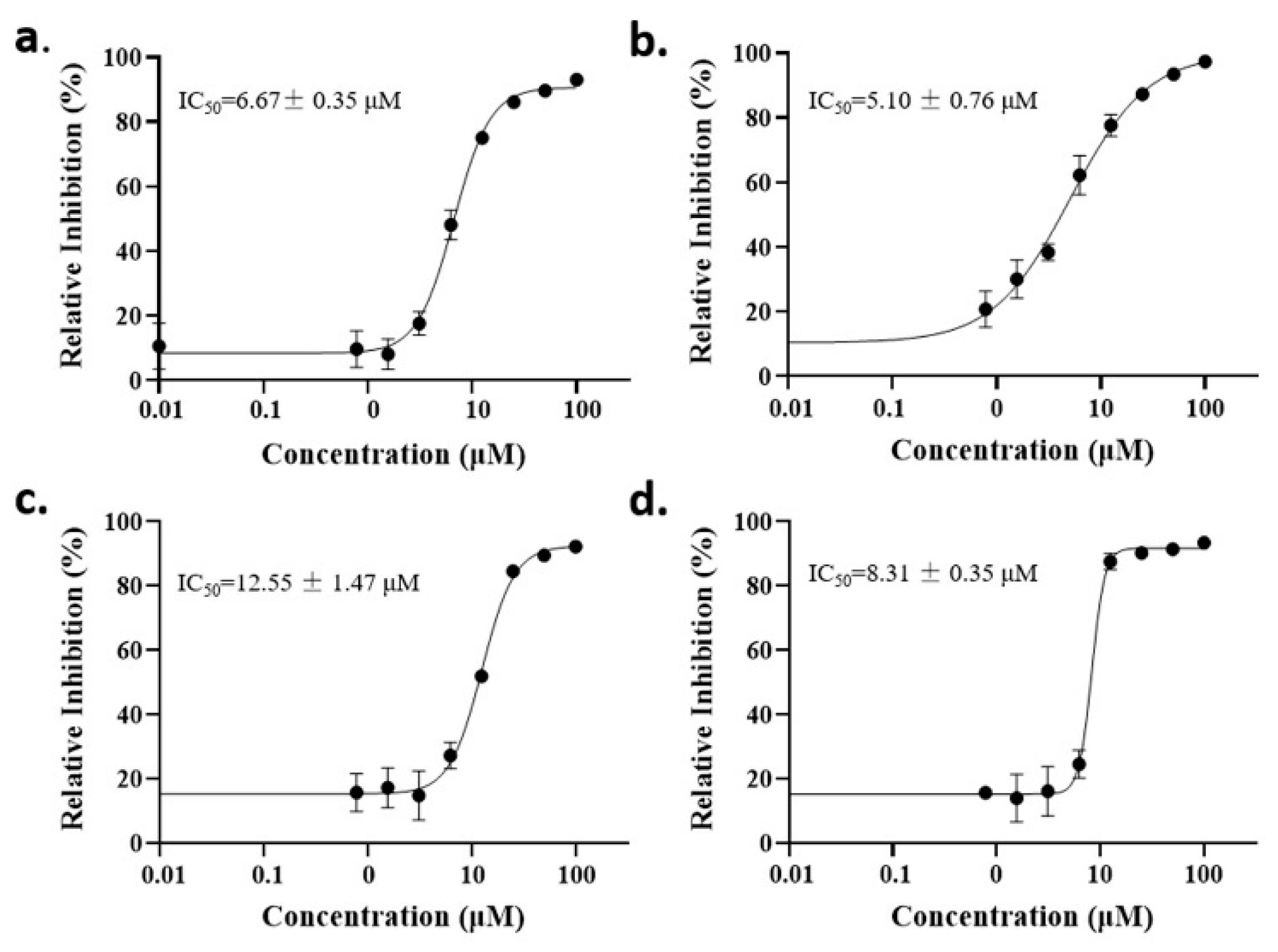
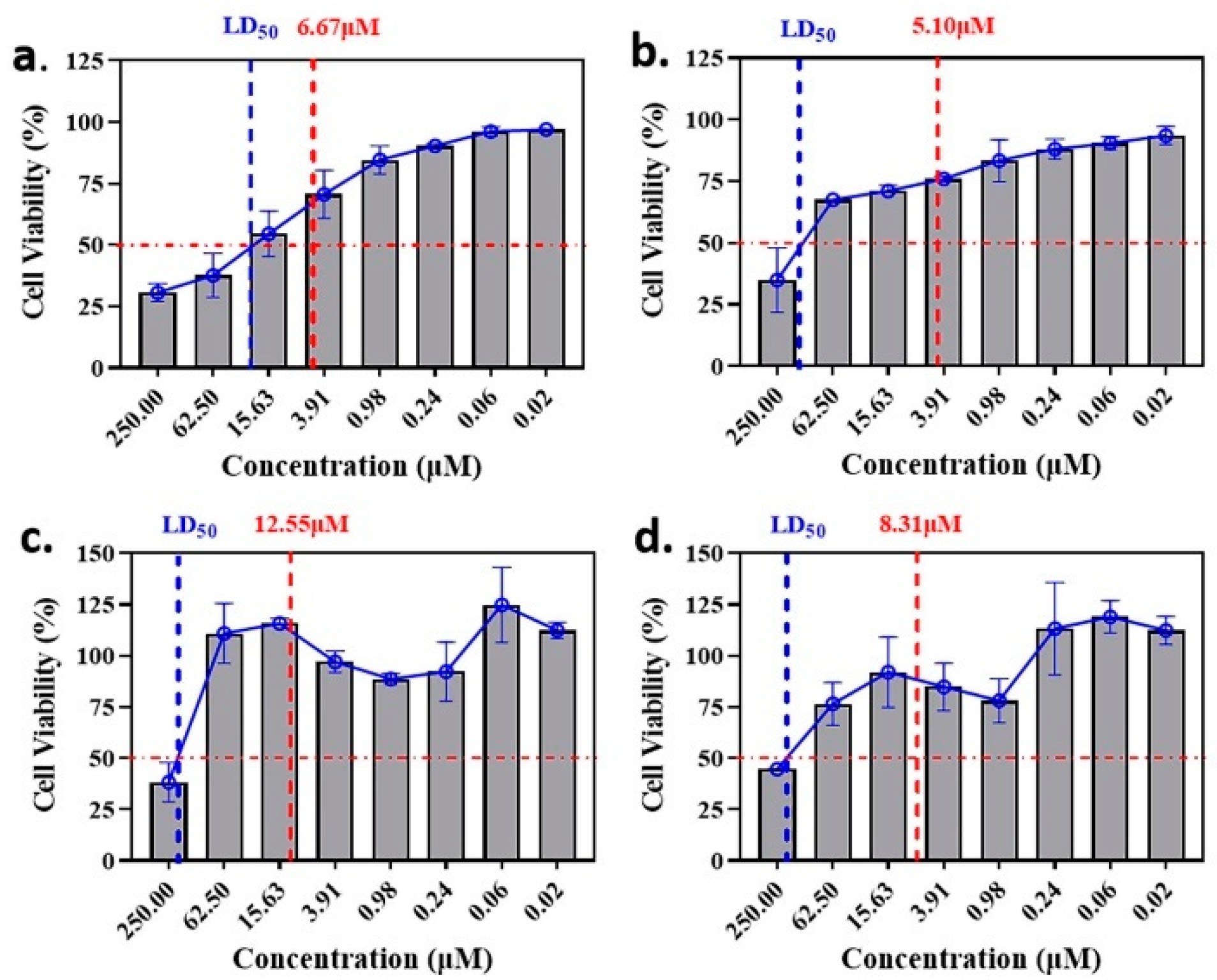
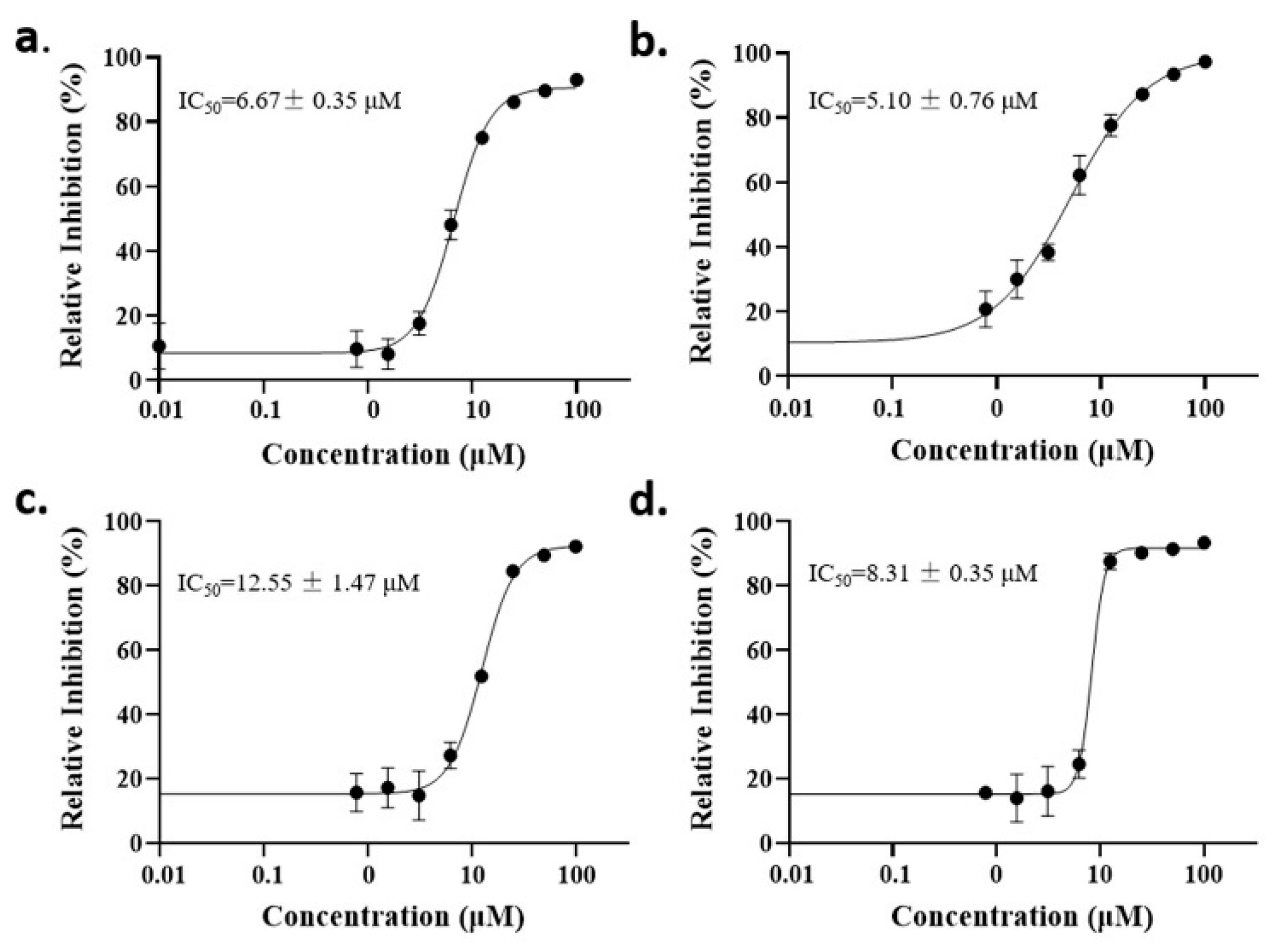
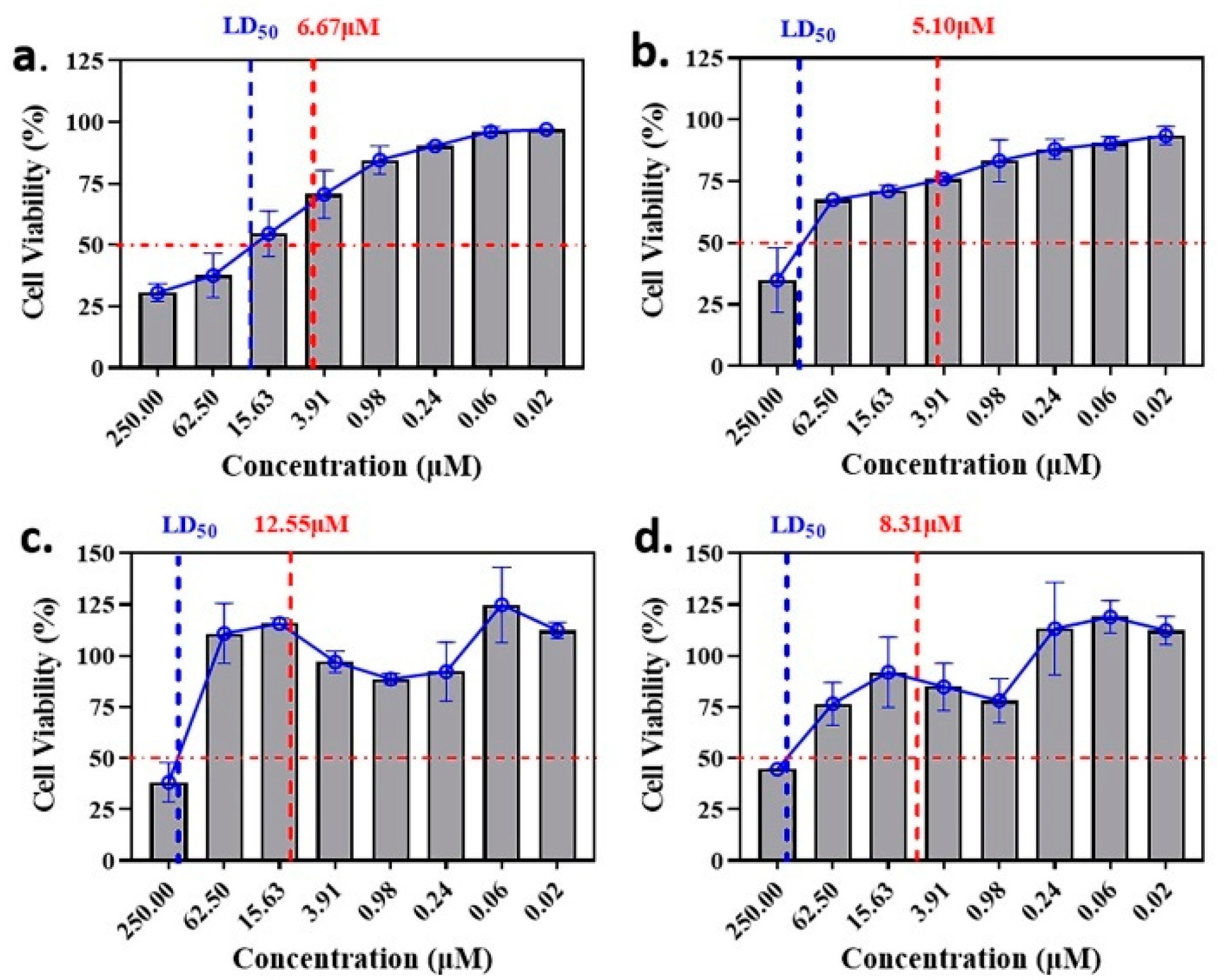
2.2. Anti-Malaria Activity and Cytotoxicity of Candidate Compounds
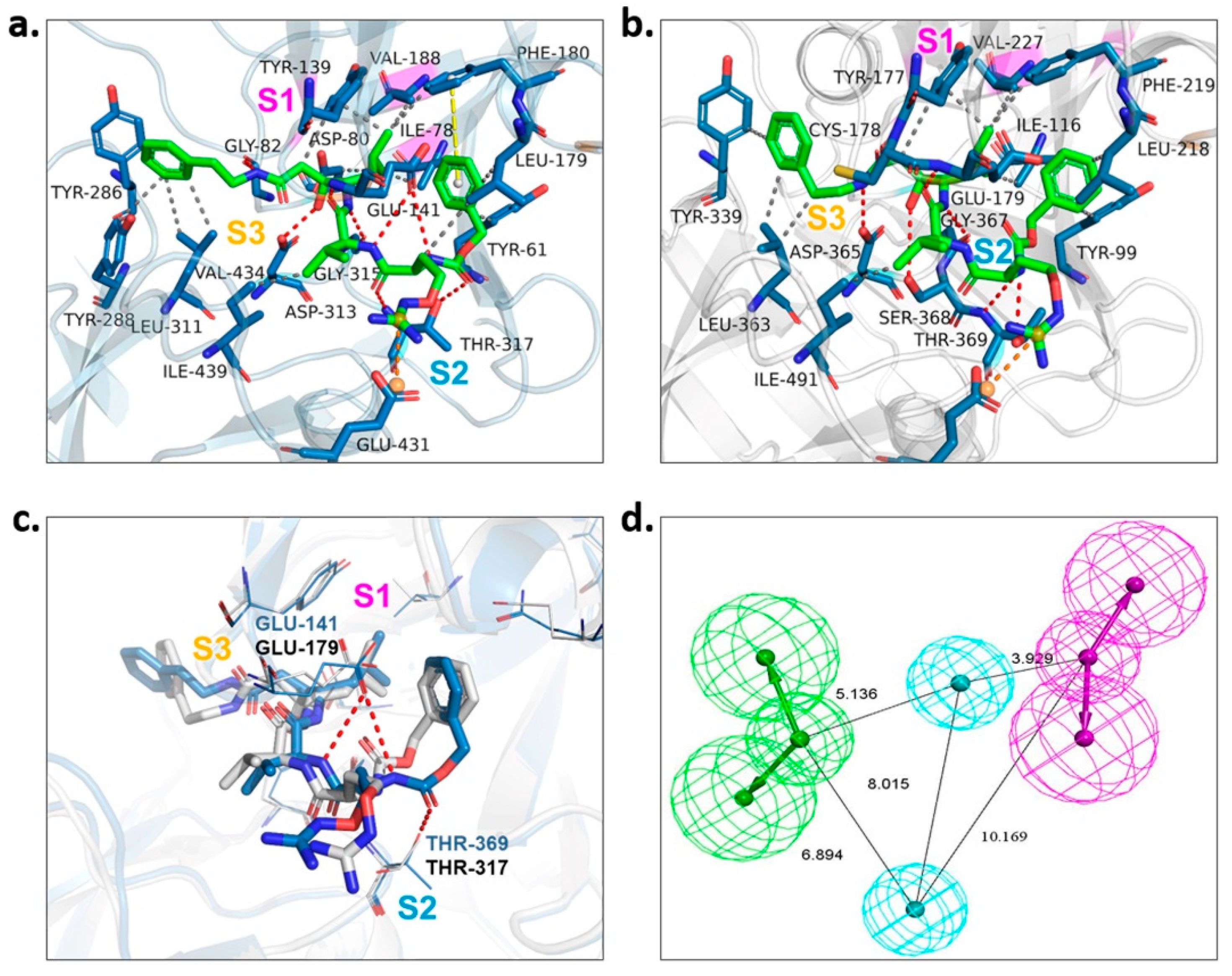
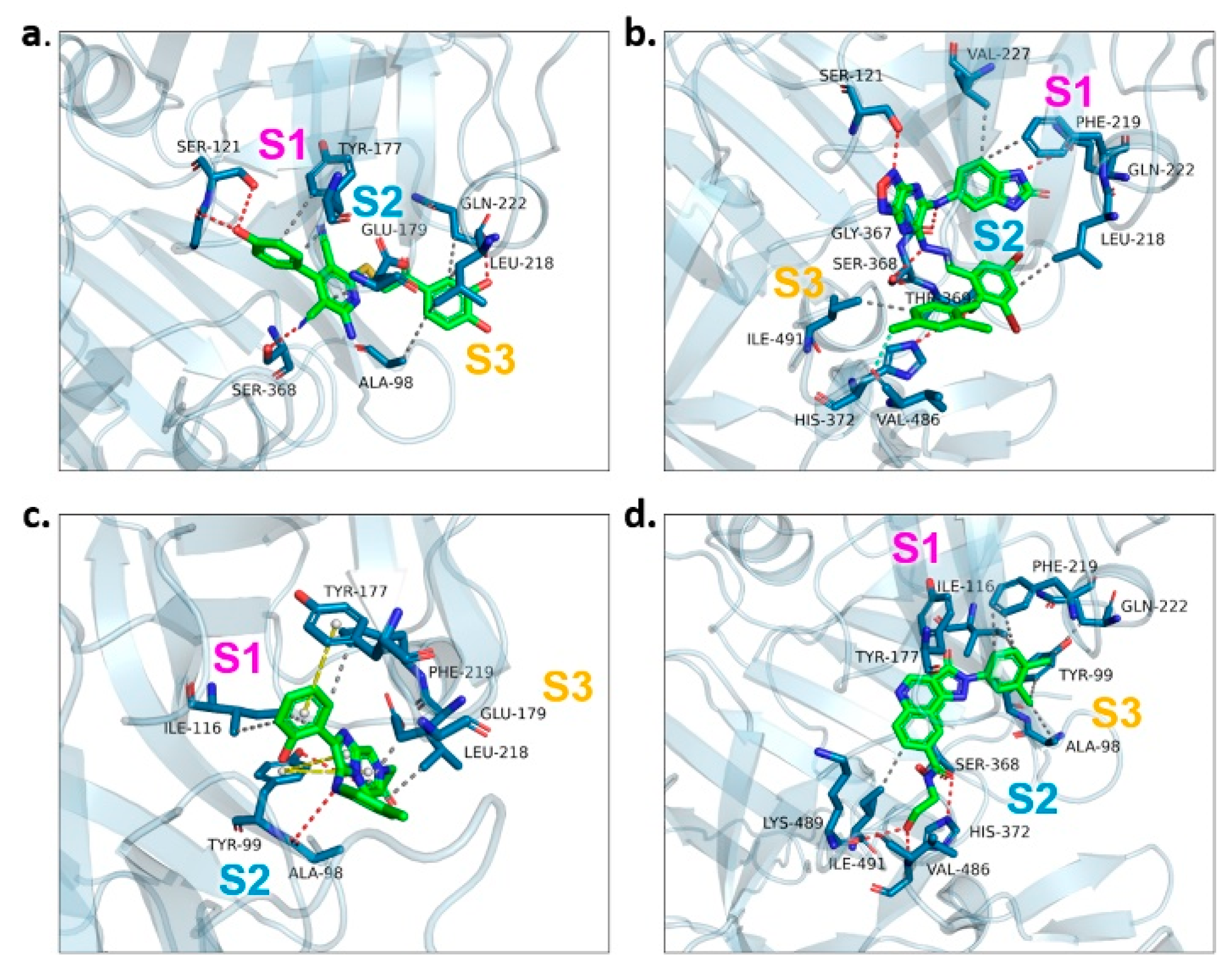
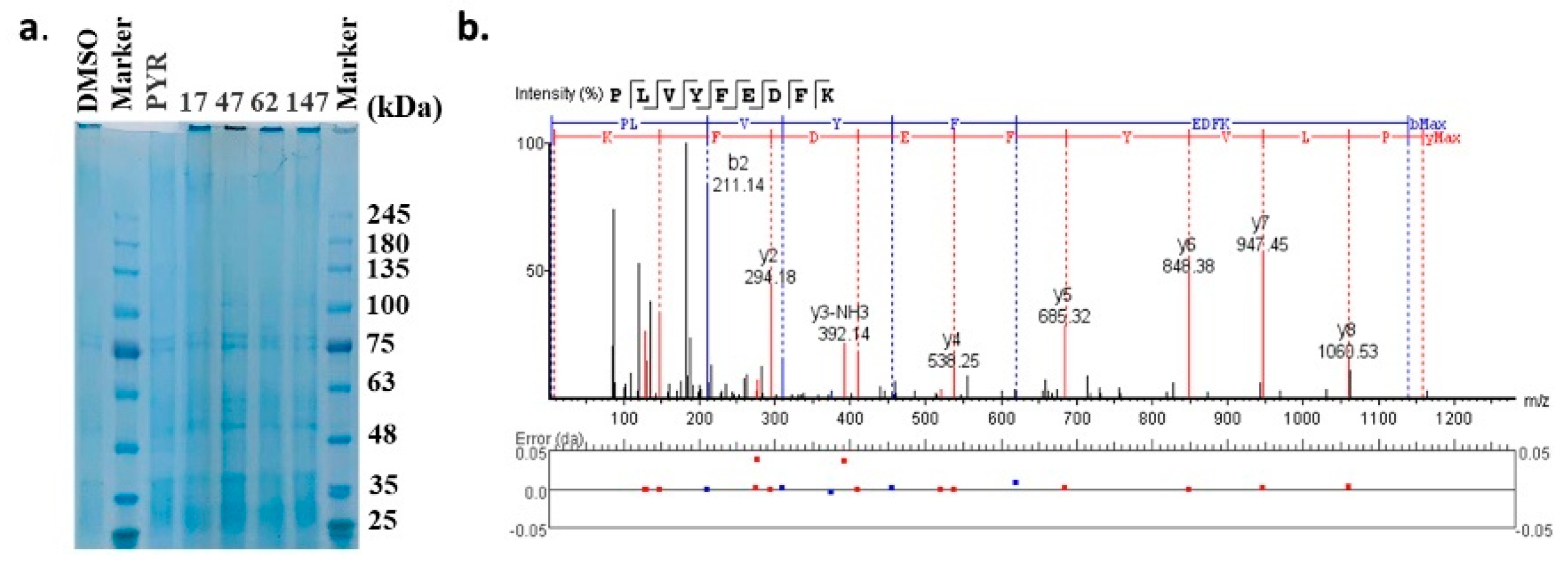
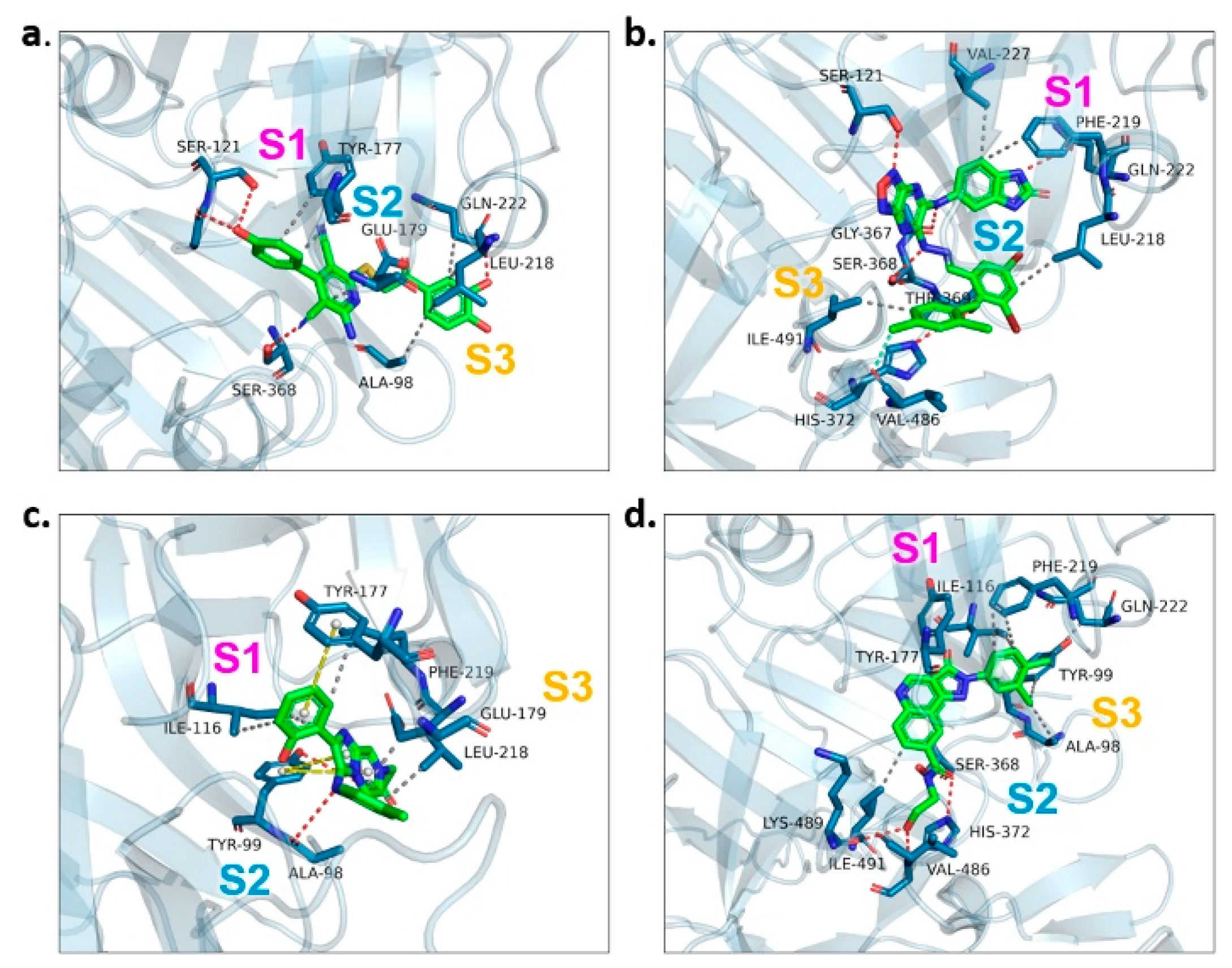
2.3. Binding Target Investigation of Candidate Compounds
3. Discussion
4. Materials and Methods
4.1. Homology Modeling of PfPMV 3D Structure
4.2. Virtual Screening through Molecular Docking
4.3. Virtual Screening by Pharmacophore Model Analysis
4.4. In Vitro P. falciparum Parasite Culture and Susceptibility Assay
4.5. Culture of L929 Cell Line and Cell Viability Assessment
4.6. Molecular Docking of Candidate Compounds to PfPMV Active Sites
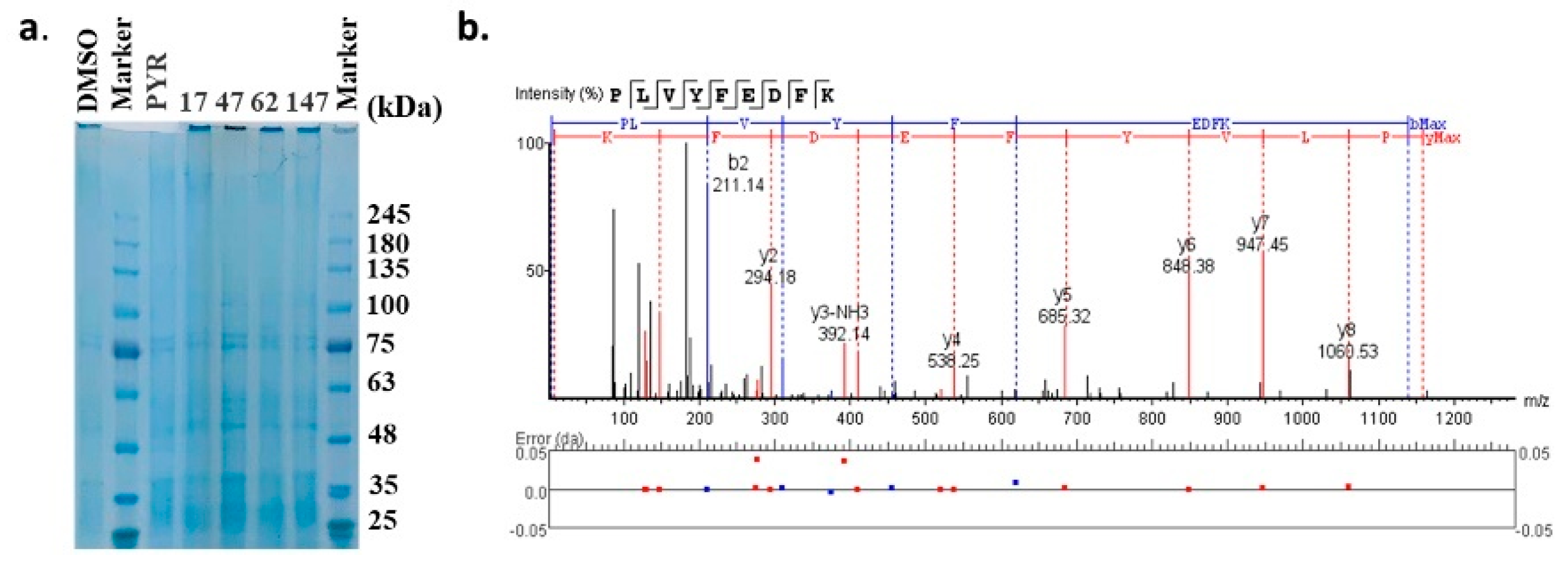
4.7. DARTS Experiment for Target Investigation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| ARTs | Artemisinin and its derivatives |
| ACTs | Artemisinin-based combination therapies |
| PMV | Plasmepsin V |
| PfPMV | Plasmodium falciparum Plasmepsin V |
| PvPMV | Plasmodium vivax Plasmepsin V |
| PEXEL | Plasmodium EXport ELement |
| IC50 | Half maximal inhibitory concentration |
| LD50 | Lethal dose of 50% |
| DARTS | Drug Affinity Responsive Target Stability |
| DMSO | Dimethyl sulfoxide |
References
- WHO. World Malaria Report. 2021. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2020/ (accessed on 10 December 2021).
- Chu, C.S.; White, N.J. Management of relapsing Plasmodium vivax malaria. Expert Rev. Anti-Infect. Ther. 2016, 14, 885–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilley, L.; Straimer, J.; Gnädig, N.F.; Ralph, S.A.; Fidock, D.A. Artemisinin action and resistance in Plasmodium falciparum. Trends Parasitol. 2016, 32, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noedl, H.; Se, Y.; Schaecher, K.; Smith, B.L.; Socheat, D.; Fukuda, M.M. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [Google Scholar] [CrossRef] [PubMed]
- Roper, C.; Pearce, R.; Nair, S.; Sharp, B.; Nosten, F.; Anderson, T. Intercontinental spread of pyrimethamine-resistant malaria. Science 2004, 305, 1124. [Google Scholar] [CrossRef] [PubMed]
- Thriemer, K.; Hong, N.V.; Rosanas-Urgell, A.; Phuc, B.Q.; Ha, D.M.; Pockele, E.; Guetens, P.; Van, N.V.; Duong, T.T.; Amambua-Ngwa, A. Delayed parasite clearance after treatment with dihydroartemisinin-piperaquine in Plasmodium falciparum malaria patients in central Vietnam. Antimicrob. Agents Chemother. 2014, 58, 7049–7055. [Google Scholar] [CrossRef] [Green Version]
- Imwong, M.; Suwannasin, K.; Kunasol, C.; Sutawong, K.; Mayxay, M.; Rekol, H.; Smithuis, F.M.; Hlaing, T.M.; Tun, K.M.; van der Pluijm, R.W. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: A molecular epidemiology observational study. Lancet Infect. Dis. 2017, 17, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L. Drug-resistant malaria advances in Mekong. Am. Assoc. Adv. Sci. 2017, 358, 155–156. [Google Scholar] [CrossRef]
- Cheuka, P.M.; Dziwornu, G.; Okombo, J.; Chibale, K. Plasmepsin Inhibitors in Antimalarial Drug Discovery: Medicinal Chemistry and Target Validation (2000 to Present). J. Med. Chem. 2020, 63, 4445–4467. [Google Scholar] [CrossRef]
- Ponsuwanna, P.; Kochakarn, T.; Bunditvorapoom, D.; Kumpornsin, K.; Otto, T.D.; Ridenour, C.; Chotivanich, K.; Wilairat, P.; White, N.J.; Miotto, O.; et al. Comparative genome-wide analysis and evolutionary history of haemoglobin-processing and haem detoxification enzymes in malarial parasites. Malaria J. 2016, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Sleebs, B.E.; Lopaticki, S.; Marapana, D.S.; O’Neill, M.T.; Rajasekaran, P.; Gazdik, M.; Gunther, S.; Whitehead, L.W.; Lowes, K.N.; Barfod, L.; et al. Inhibition of Plasmepsin V Activity Demonstrates Its Essential Role in Protein Export, PfEMP1 Display, and Survival of Malaria Parasites. PLoS Biol. 2014, 12, e1001897. [Google Scholar] [CrossRef] [Green Version]
- Marti, M.; Good, R.T.; Rug, M.; Knuepfer, E.; Cowman, A.F. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 2004, 306, 1930–1933. [Google Scholar] [CrossRef] [PubMed]
- Hiller, N.L.; Bhattacharjee, S.; van Ooij, C.; Liolios, K.; Harrison, T.; Lopez-Estrano, C.; Haldar, K. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 2004, 306, 1934–1937. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Falick, A.M.; Carlton, P.M.; Sedat, J.W.; DeRisi, J.L.; Marletta, M.A. N-terminal processing of proteins exported by malaria parasites. Mol. Biochem. Parasitol. 2008, 160, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, J.; Kwiatkowski, M.; Borner, J.; Schlüter, H.; Bruchhaus, I.; Burmester, T.; Spielmann, T.; Pick, C. The P lasmodium falciparum exportome contains non-canonical PEXEL/HT proteins. Mol. Microbiol. 2015, 97, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Babbitt, S.; Muralidharan, V.; Butler, T.; Oksman, A.; Goldberg, D.E. Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature 2010, 463, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Hodder, A.N.; Sleebs, B.E.; Czabotar, P.E.; Gazdik, M.; Xu, Y.; O’Neill, M.T.; Lopaticki, S.; Nebl, T.; Triglia, T.; Smith, B.J.; et al. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat. Struct. Mol. Biol. 2015, 22, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, W.; Hodder, A.N.; de Lezongard, R.B.; Czabotar, P.E.; Jarman, K.E.; O’Neill, M.T.; Thompson, J.K.; Sabroux, H.J.; Cowman, A.F.; Boddey, J.A.; et al. Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P-2 position of PEXEL peptidomimetics. Eur. J. Med. Chem. 2018, 154, 182–198. [Google Scholar] [CrossRef]
- Gambini, L.; Rizzi, L.; Pedretti, A.; Taglialatela-Scafati, O.; Carucci, M.; Pancotti, A.; Galli, C.; Read, M.; Giurisato, E.; Romeo, S.; et al. Picomolar Inhibition of Plasmepsin V, an Essential Malaria Protease, Achieved Exploiting the Prime Region. PLoS ONE 2015, 10, e0142509. [Google Scholar] [CrossRef] [Green Version]
- International Organization for Standardization. Biological Evaluation of Medical Devices—Part 5: In Vitro Cytotoxicity Testing; International Organization for Standardization: Geneva, Switzerland, 2009. [Google Scholar]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Bray, P.G.; Martin, R.E.; Tilley, L.; Ward, S.A.; Kirk, K.; Fidock, D.A. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol. Microbiol. 2005, 56, 323–333. [Google Scholar] [CrossRef]
- Ersmark, K.; Samuelsson, B.; Hallberg, A. Plasmepsins as potential targets for new antimalarial therapy. Med. Res. Rev. 2006, 26, 626–666. [Google Scholar] [CrossRef] [PubMed]
- Sittikul, P.; Songtawee, N.; Kongkathip, N.; Boonyalai, N. In vitro and in silico studies of naphthoquinones and peptidomimetics toward Plasmodium falciparum plasmepsin V. Biochimie 2018, 152, 159–173. [Google Scholar] [CrossRef]
- Sharma, P.P.; Kumar, S.; Kaushik, K.; Singh, A.; Singh, I.K.; Grishina, M.; Pandey, K.C.; Singh, P.; Potemkin, V.; Poonam; et al. In silico validation of novel inhibitors of malarial aspartyl protease, plasmepsin V and antimalarial efficacy prediction. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–D543. [Google Scholar] [CrossRef] [Green Version]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Wang, Z.L.; Cabrera, M.; Yang, J.Y.; Yuan, L.L.; Gupta, B.; Liang, X.Y.; Kemirembe, K.; Shrestha, S.; Brashear, A.; Li, X.L.; et al. Genome-wide association analysis identifies genetic loci associated with resistance to multiple antimalarials in Plasmodium falciparum from China-Myanmar border. Sci. Rep. 2016, 6, 33891. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Wang, C.Q.; Lucky, A.B.; Liang, X.Y.; Min, H.; Adapa, S.R.; Jiang, R.; Kim, K.; Cui, L.W. A unique GCN5 histone acetyltransferase complex controls erythrocyte invasion and virulence in the malaria parasite Plasmodium falciparum. PLoS Pathog. 2021, 17, e1009351. [Google Scholar] [CrossRef]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol. Biol. 2015, 1263, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate Compound | Molecule | H-Bonds | Hydrophobic Interactions | Halogen Bond | π-π Interaction | Docking Score |
|---|---|---|---|---|---|---|
| 17 |  | Ser-121, Gln-222, Ser-368 | Tyr-177, Glu-179, Ala-98, Leu-218, Gln-222 | - | - | −9.3 |
| 47 | 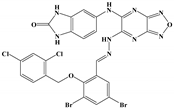 | Ser-121, Ser-38, His-372, Gly-367, Gln-222 | Ile-491, Leu-218, Phe-219, Val-227 | Val-486 | - | −10.9 |
| 62 |  | Tyr-99, Ala-98 | Ike-116, Leu-218, Glu-179, Phe-219 | - | Tyr-177 | −9.3 |
| 147 | 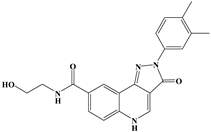 | Lys-489, Val-486, His-372 | Ile-491, Tyr-177, Ile-116, Phe-219, Gln-222, Tyr-99, Ala-98 | - | - | −10.4 |
| Identified Protein | Peptide Maximum Area | |||
|---|---|---|---|---|
| 17 | 47 | 62 | 147 | |
| Ornithine aminotransferase | 6.08 × 108 | 7.88 × 108 | 7.36 × 108 | 6.20 × 108 |
| Phosphoglycerate kinase | 4.89 × 108 | 4.53 × 108 | 3.94 × 108 | 4.16 × 108 |
| Plasmepsin Ⅲ | 2.73 × 108 | 1.72 × 108 | 2.39 × 108 | 2.29 × 108 |
| Plasmepsin Ⅳ | 2.13 × 108 | 2.08 × 108 | 1.96 × 108 | 2.02 × 108 |
| Plasmepsin Ⅱ | 1.76 × 108 | 2.08 × 108 | 1.96 × 108 | 2.02 × 108 |
| Adenosine deaminase | 1.67 × 108 | 1.89 × 108 | 1.31 × 108 | 1.43 × 108 |
| Adenosylhomocysteinase | 1.14 × 108 | 1.23 × 108 | 1.03 × 108 | 7.73 × 107 |
| Pyridoxine biosynthesis protein PDX1 | 6.94 × 107 | 9.19 × 107 | 9.71 × 107 | 3.15 × 107 |
| Plasmepsin Ⅰ | 8.21 × 107 | 9.14 × 107 | 8.67 × 107 | 4.36 × 107 |
| Serine repeat antigen 5 | 3.22 × 107 | 4.73 × 107 | 3.49 × 107 | 7.73 × 107 |
| Proliferating cell nuclear antigen | 3.74 × 107 | 7.45 × 107 | 3.75 × 107 | 4.39 × 107 |
| Surface protein P113 | 4.09 × 107 | 3.57 × 107 | 3.55 × 107 | 4.30 × 107 |
| 6-cysteine protein PF92 | 3.21 × 107 | 2.27 × 107 | 2.88 × 107 | 2.88 × 107 |
| Serine repeat antigen 6 | 2.33 × 107 | 1.94 × 107 | 1.77 × 107 | 2.28 × 107 |
| Bifunctional dihydrofolate reductase-thymidylate synthase | 1.58 × 107 | 1.85 × 107 | 1.56 × 107 | 1.62 × 107 |
| DNA repair protein RAD50 | 1.10 × 107 | 1.55 × 107 | ND | 1.74 × 107 |
| Chloroquine resistance transporter | 1.08 × 107 | 1.38 × 107 | 6.74 × 106 | 1.13 × 107 |
| Prefoldin subunit 6 | 1.37 × 107 | ND | 1.16 × 107 | 1.20 × 107 |
| Myosin A | 7.16 × 106 | 7.70 × 106 | 7.10 × 106 | 6.57 × 106 |
| Plasmepsin V | ND | 6.21 × 106 | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, X.; Wang, Z.; Chen, Q.; Li, J.; Wang, H.; Wang, Z.; Yang, L. In Silico and In Vitro Antimalarial Screening and Validation Targeting Plasmodium falciparum Plasmepsin V. Molecules 2022, 27, 2670. https://doi.org/10.3390/molecules27092670
Ji X, Wang Z, Chen Q, Li J, Wang H, Wang Z, Yang L. In Silico and In Vitro Antimalarial Screening and Validation Targeting Plasmodium falciparum Plasmepsin V. Molecules. 2022; 27(9):2670. https://doi.org/10.3390/molecules27092670
Chicago/Turabian StyleJi, Xin, Zhensheng Wang, Qianqian Chen, Jingzhong Li, Heng Wang, Zenglei Wang, and Lan Yang. 2022. "In Silico and In Vitro Antimalarial Screening and Validation Targeting Plasmodium falciparum Plasmepsin V" Molecules 27, no. 9: 2670. https://doi.org/10.3390/molecules27092670
APA StyleJi, X., Wang, Z., Chen, Q., Li, J., Wang, H., Wang, Z., & Yang, L. (2022). In Silico and In Vitro Antimalarial Screening and Validation Targeting Plasmodium falciparum Plasmepsin V. Molecules, 27(9), 2670. https://doi.org/10.3390/molecules27092670