
Thermodynamic Signatures of Structural Transitions and Dissociation of Charged Colloidal Clusters: A Parallel Tempering Monte Carlo Study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
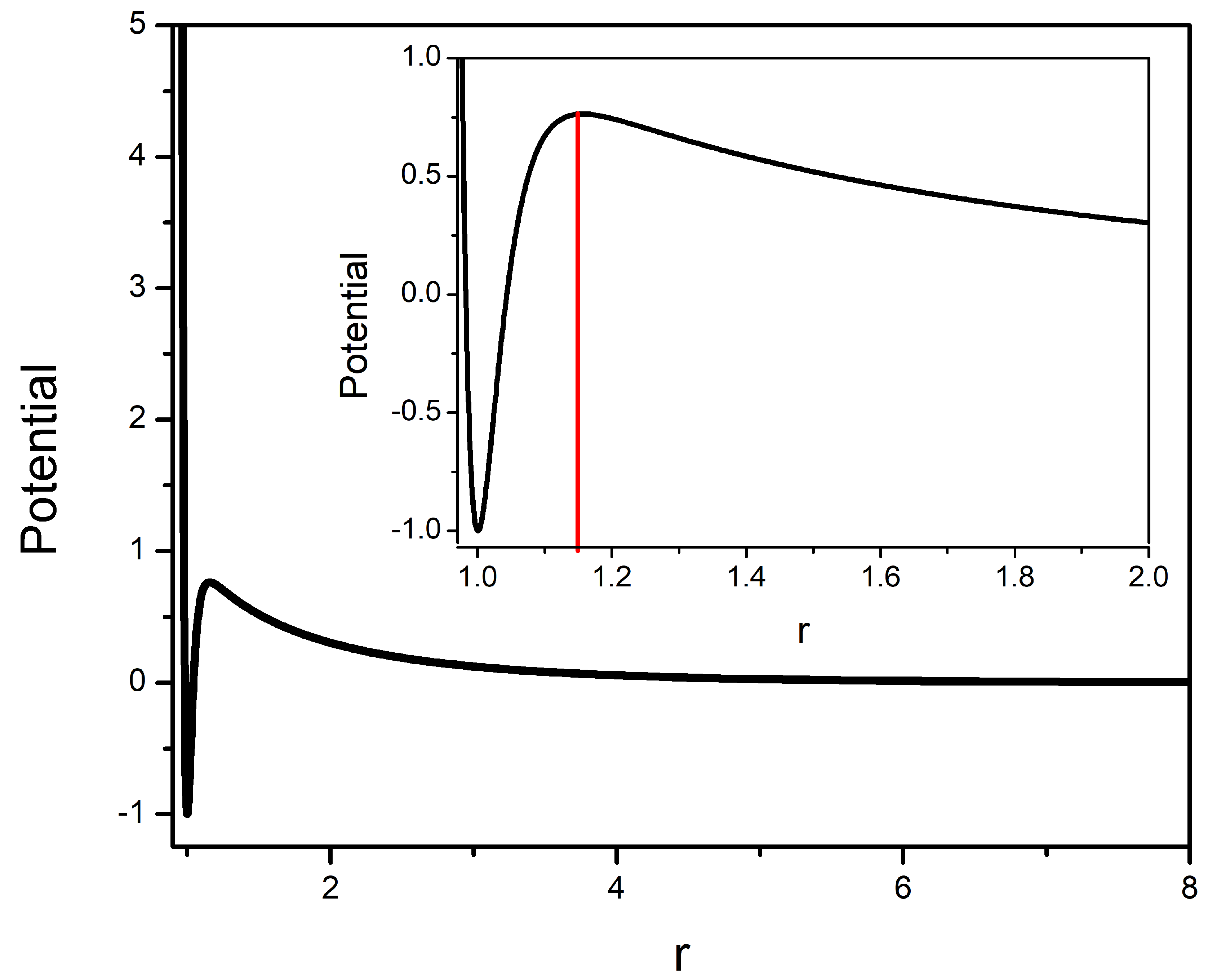
2.1. Interaction Potential
2.2. Parallel Tempering Monte Carlo Method
3. Results and Discussion
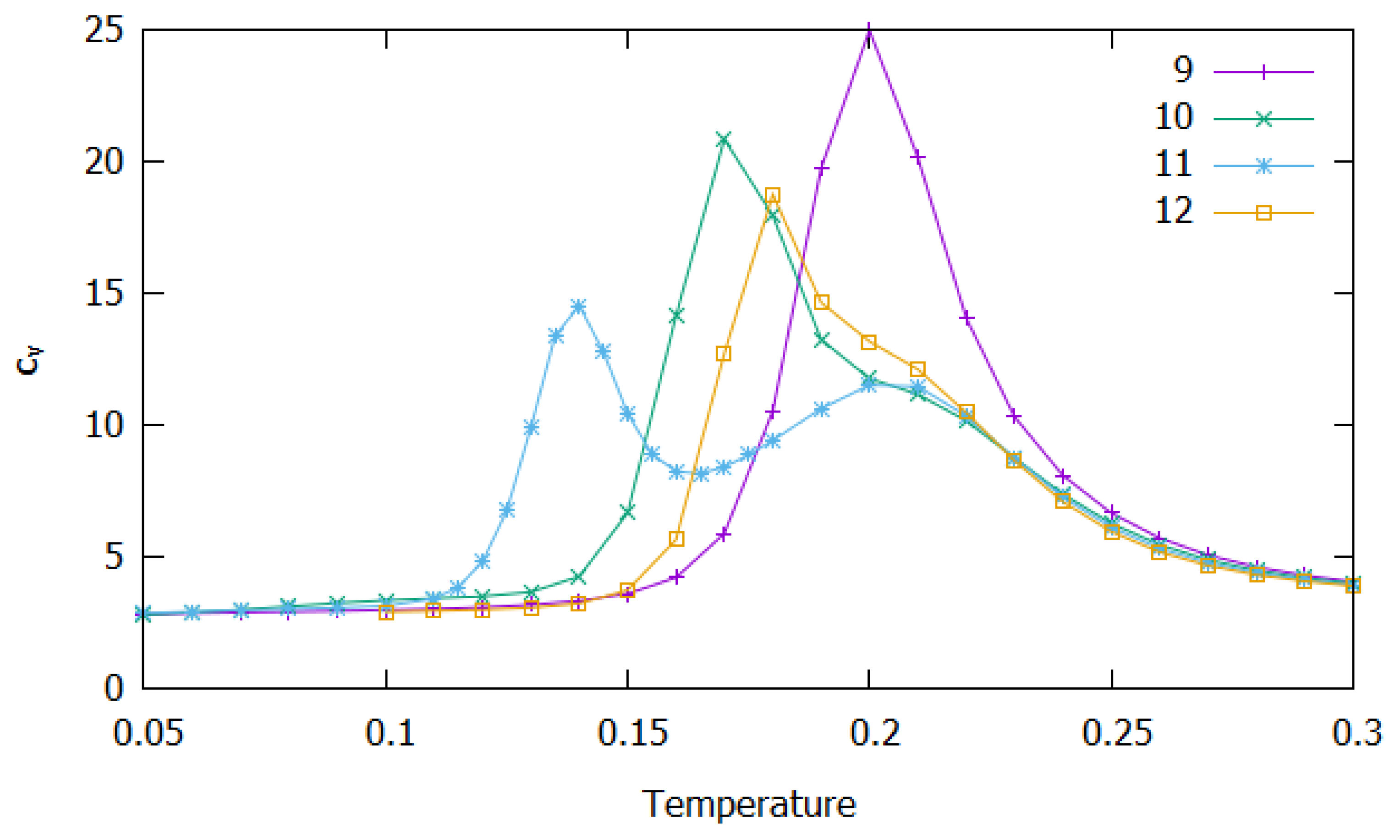
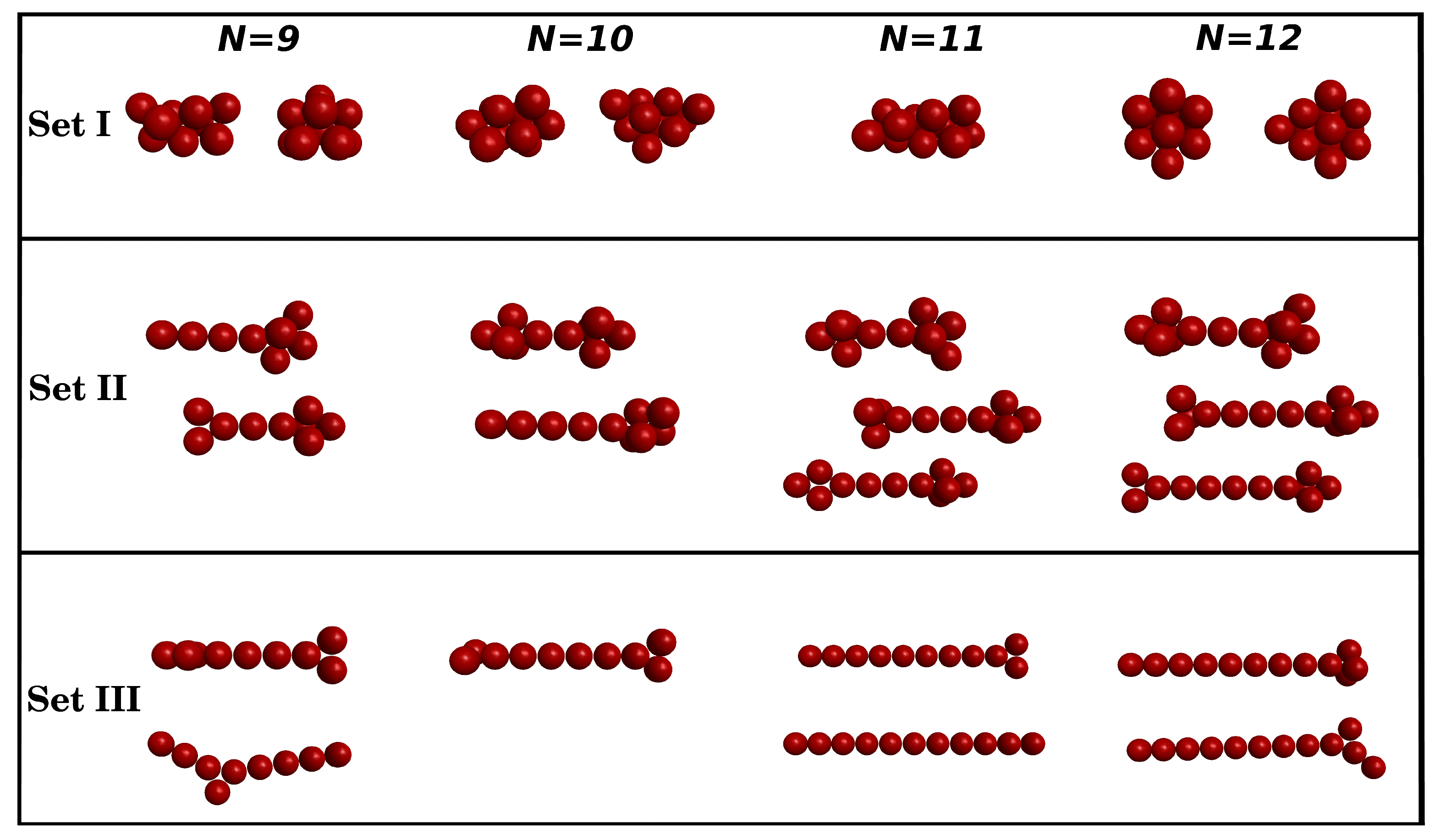
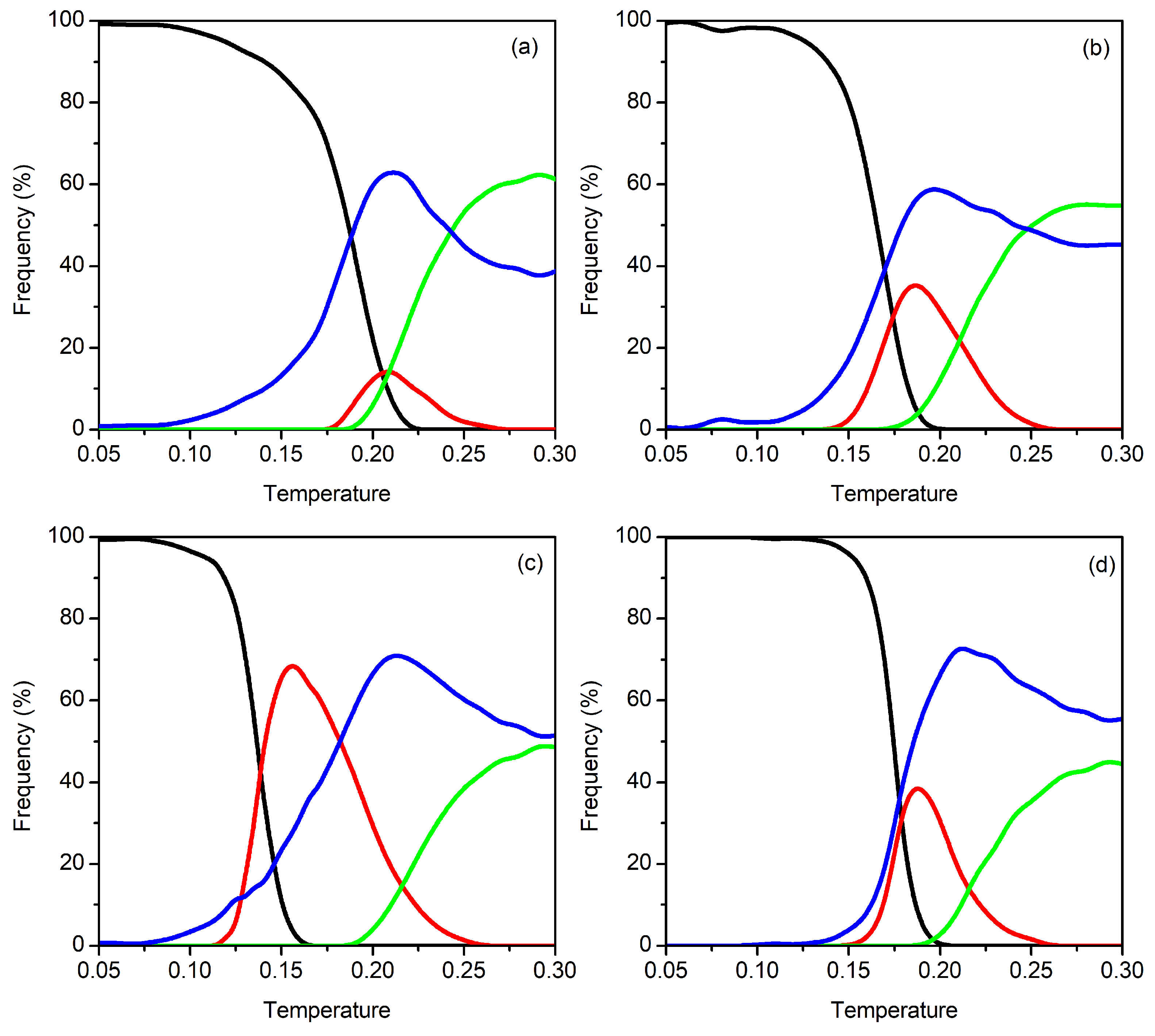
3.1. Clusters with N = 9–12 Particles
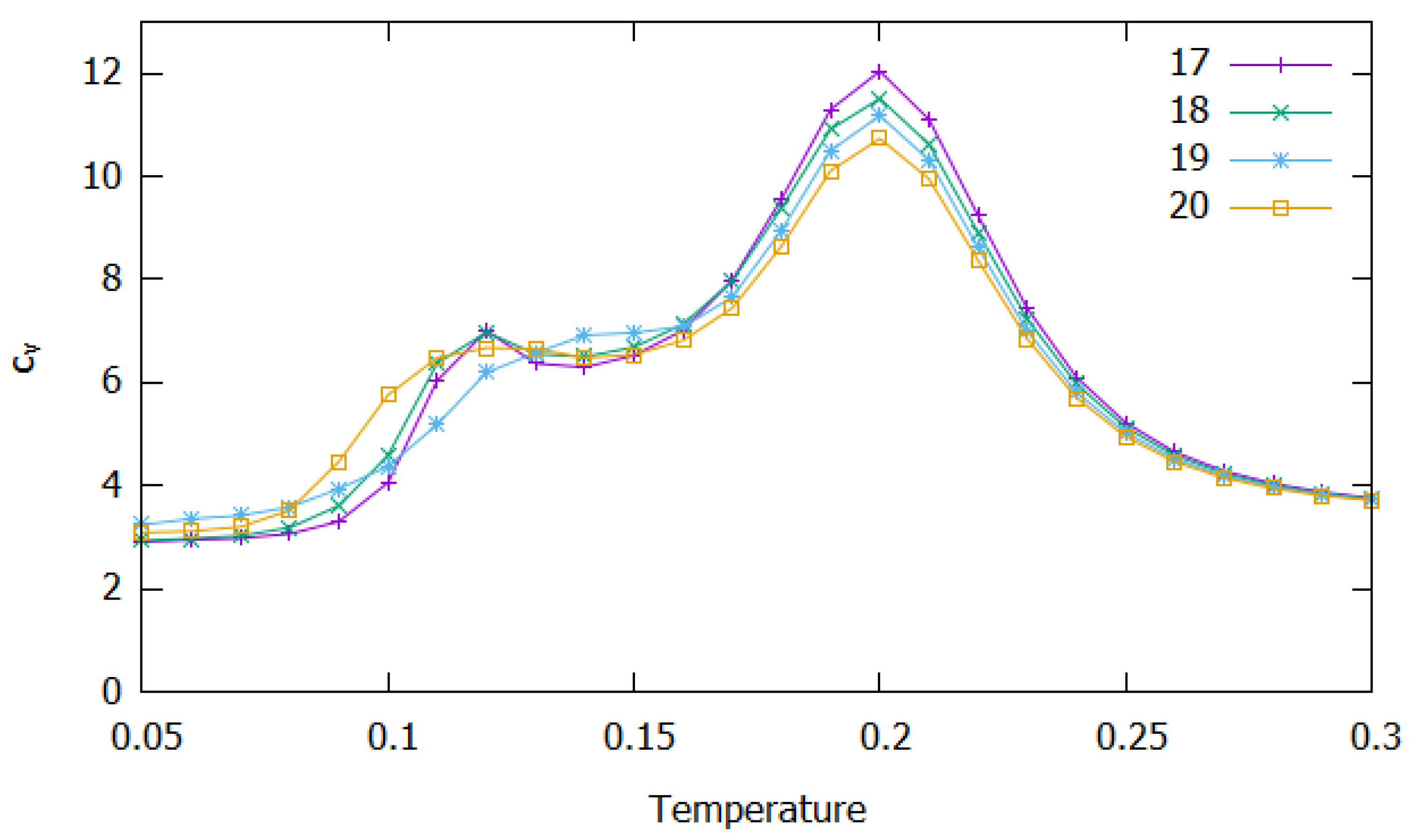
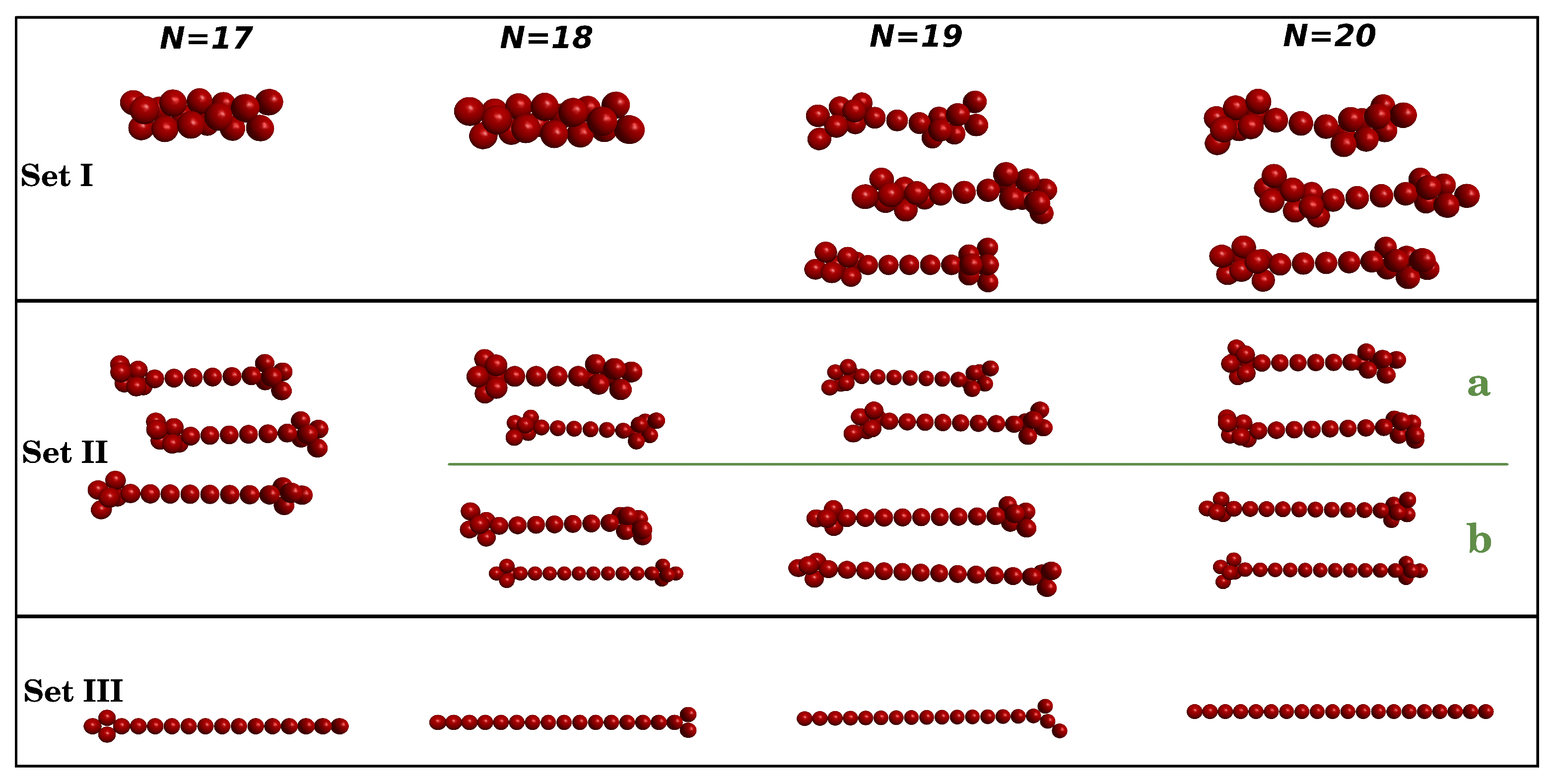
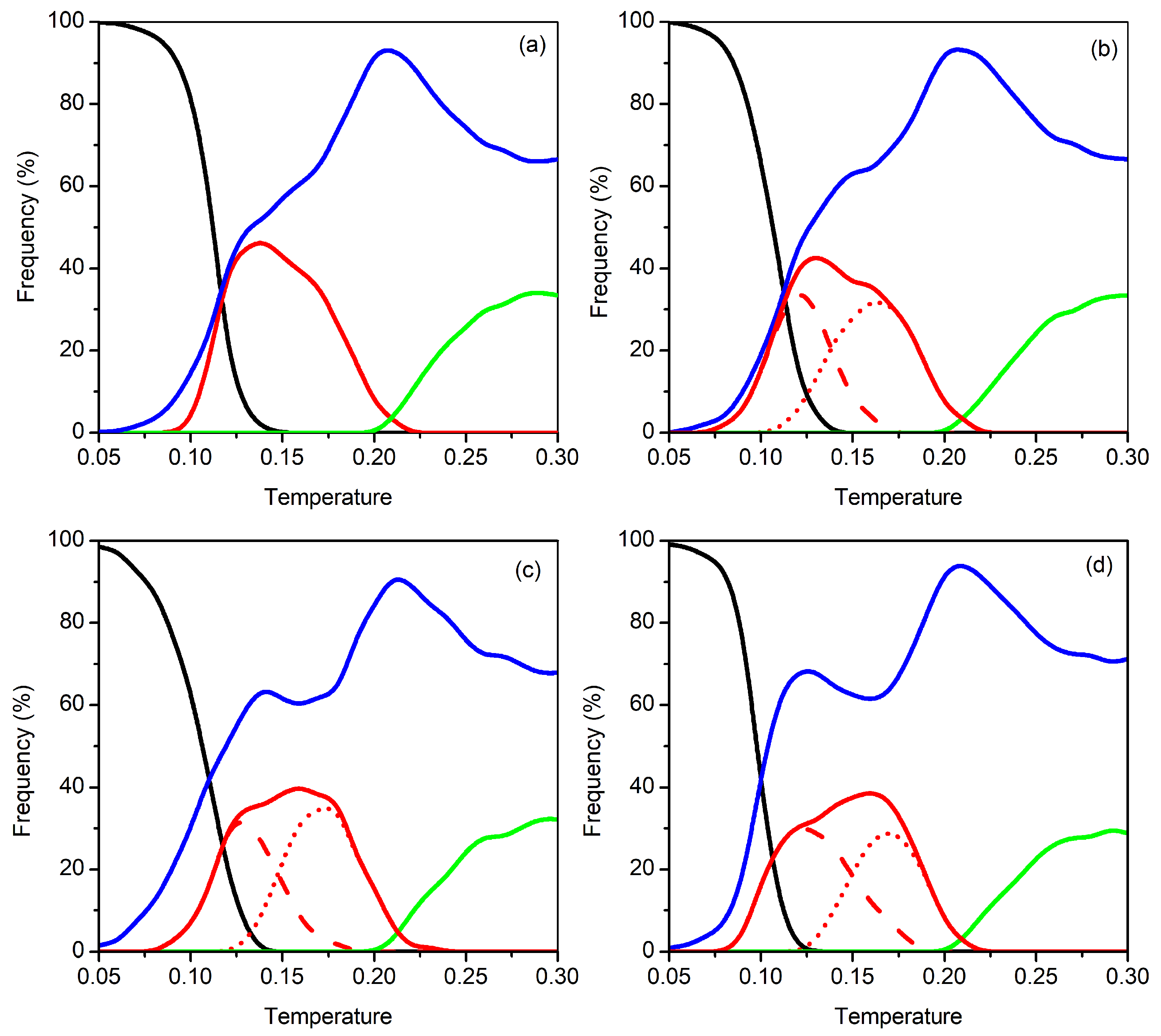
3.2. Clusters with N = 17–20 Particles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tardieu, A.; Finet, S.; Bonneté, F. Structure of the macromolecular solutions that generate crystals. J. Cryst. Growth 2001, 232, 1–9. [Google Scholar] [CrossRef]
- Piazza, R. Protein interactions and association: An open challenge for colloid science. Curr. Opin. Colloid Interface Sci. 2004, 8, 515–522. [Google Scholar] [CrossRef]
- Doye, J.; Poon, W. Protein crystallization in vivo. Curr. Opin. Colloid Interface Sci. 2006, 11, 40–46. [Google Scholar] [CrossRef]
- Gliko, O.; Pan, W.; Katsonis, P.; Neumaier, N.; Galkin, O.; Weinkauf, S.; Vekilov, P. Metastable liquid clusters in super- and undersaturated protein solutions. J. Phys. Chem. B 2007, 111, 3106–3114. [Google Scholar] [CrossRef]
- Abramo, M.C.; Caccamo, C.; Costa, D.; Pellicane, G.; Ruberto, R.; Wanderlingh, U. Effective interactions in lysozyme aqueous solutions: A small-angle neutron scattering and computer simulation study. J. Chem. Phys. 2012, 136, 035103. [Google Scholar] [CrossRef]
- Sorauf, D.; Roose-Runge, F.; Grimaldo, M.; Zanini, F.; Schweins, R.; Seydel, T.; Zhang, F.; Roth, R.; Oettel, M.; Schreiber, F. Protein cluster formation in aqueous solution in the presence of multivalent ions—A light scattering study. Soft Matter 2014, 10, 894–902. [Google Scholar] [CrossRef]
- Mani, E.; Lechner, W.; Kegel, W.K.; Bolhuis, P.G. Equilibrium and non-equilibrium cluster phases in colloids with competing interactions. Soft Matter 2014, 10, 4479–4486. [Google Scholar] [CrossRef]
- Fusco, D.; Charbonneau, P. Soft matter perspective on protein crystal assembly. Colloid Surf. B 2016, 137, 22–31. [Google Scholar] [CrossRef]
- Riest, J.; Nägele, G.; Liu, Y.; Wagner, N.J.; Godfrin, P.D. Short-time dynamics of lysozyme solutions with competing short-range attraction and long-range repulsion: Experiment and theory. J. Chem. Phys. 2018, 148, 065101. [Google Scholar] [CrossRef]
- Beck, C.; Grimaldo, M.; Braun, M.K.; Bühl, L.; Matsarskaia, O.; Jalarvo, N.H.; Zhang, F.; Roosen-Runge, F.; Schreiber, F.; Seydel, T. Temperature and salt controlled tuning of protein clusters. Soft Matter 2021, 17, 8506–8516. [Google Scholar] [CrossRef]
- Meilhac, N.; Destainville, N. Clusters of proteins in biomembranes: Insights into the roles of interaction potential shapes and of protein diversity. J. Phys. Chem. B 2011, 115, 7190–7199. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Ambrosi, D.; Girraudo, E.; Preziosi, L.; Bussolini, F. Modeling the early stages of vascular network assembly. EMBO J. 2003, 22, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Merks, R.; Brodsky, S.; Goligorsky, M.; Newman, S.; Glazier, J. Cell elongation is key to in silico replication of in vitro vasculogenesis and subsequent remodeling. Dev. Biol. 2006, 289, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Gamba, A.; Ambrosini, D.; Coniglio, A.; de Candia, A.; Talia, S.; Giraudo, E.; Serini, G.; Preziosi, L.; Bussolino, F. Percolation, morphogenesis, and burguers dynamics in blood vessels formation. Phys. Rev. Lett. 2003, 90, 118101. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Perryn, E.; Czirok, A. Network formation of tissue cells via preferential attraction to elongated structures. Phys. Rev. Lett. 2007, 98, 038102. [Google Scholar] [CrossRef] [PubMed]
- Palm, M.; Merks, R. Vascular networks due to dynamically arrested crystaline ordering of elongated cells. Phys. Rev. E 2013, 87, 012725. [Google Scholar] [CrossRef]
- Green, J.; Waters, S.; Whiteley, J.; Edelstein-Keshet, L.; Shakesheff, K.; Byrne, H. Non-local models for the formation of hepatocyte-stellate cell aggregates. J. Theor. Biol. 2010, 267, 106–120. [Google Scholar] [CrossRef]
- Phillips, C.M.; Lima, E.A.B.F.; Woodall, R.T.; Brock, A.; Yankeelov, T.E. A hybrid model of tumor growth and angiogenesis: In silico experiments. PLoS ONE 2020, 15, e0231137. [Google Scholar] [CrossRef]
- Sciortino, F.; Mossa, S.; Zaccarelli, E.; Tartaglia, P. Equilibrium Cluster Phases and Low-Density Arrested Disordered States: The Role of Short-Range Attraction and Long-Range Repulsion. Phys. Rev. Lett. 2004, 93, 055701. [Google Scholar] [CrossRef]
- Klix, C.L.; Murata, K.; Tanaka, H.; Williams, S.; Malins, A.; Royall, C.P. The role of weak charging in metstable colloidal clusters. arXiv 2009, arXiv:0905.3393. [Google Scholar]
- Perry, R.W.; Meng, G.; Dimiduk, T.G.; Fung, J.; Manoharan, V.N. Real-space studies of the structure and dynamics of self-assembled colloidal clusters. Faraday Discuss. 2012, 159, 211–234. [Google Scholar] [CrossRef][Green Version]
- Zhang, I.; Royall, C.P.; Faers, M.A.; Bartlett, P. Phase separation dynamics in colloid-polymer mixtures: The effect of interaction range. Soft Matter 2013, 9, 2076–2084. [Google Scholar] [CrossRef]
- Santos, A.P.; Pekalski, J.; Panagiotopoulos, A.Z. Thermodynamic signatures and cluster properties of self-assembly in systems with competing interactions. Soft Matter 2017, 13, 8055–8063. [Google Scholar] [CrossRef] [PubMed]
- Malescio, G.; Sciortino, F. Aggregate formation in fluids with bounded repulsive core and competing interactions. J. Mol. Liq. 2020, 303, 112601. [Google Scholar] [CrossRef]
- Valadez-Pérez, N.E.; Liu, Y.; Castañeda-Priego, R. Cluster Morphology of Colloidal Systems With Competing Interactions. Front. Phys. 2021, 9, 88. [Google Scholar] [CrossRef]
- de Hek, H.; Vrij, A. Preparation of sterically stabilized silica dispersions in nonaqueous media. J. Colloid Interface Sci. 1981, 79, 289–294. [Google Scholar] [CrossRef]
- Pathmamanoharan, C.; de Hek, H.; Vrij, A. Phase separation in mixtures of organophilic spherical silica particles and polymer molecules in good solvents. Colloid Polym. Sci. 1981, 259, 769–771. [Google Scholar] [CrossRef]
- Sperry, P.R. Morphology and mechanism in latex flocculated by volume restriction. J. Colloid Interface Sci. 1984, 99, 97–108. [Google Scholar] [CrossRef]
- Gast, A.P.; Russel, W.B.; Hall, C.K. An experimental and theoretical study of phase transitions in the polystyrene latex and hydroxyethylcellulose system. J. Colloid Interface Sci. 1986, 109, 161–171. [Google Scholar] [CrossRef]
- Smits, C.; van der Most, D.; Dhont, J.K.G.; Lekkerkerker, H.N.W. Influence of non-adsorbing polymer on the formation of colloidal crystals. Adv. Colloid Interface Sci. 1992, 42, 33–40. [Google Scholar] [CrossRef][Green Version]
- Mao, Y.; Cates, M.E.; Lekkerkerker, H.N.W. Depletion force in colloidal systems. Phys. A 1995, 222, 10–24. [Google Scholar] [CrossRef]
- Asakura, S.; Oosawa, F. On interaction between 2 bodies immersed in a solution of macromolecules. J. Chem. Phys. 1954, 22, 1255–1256. [Google Scholar] [CrossRef]
- Asakura, S.; Oosawa, F. Interaction between particles suspended in solutions of macromolecules. J. Polym. Sci. 1958, 33, 183–192. [Google Scholar] [CrossRef]
- Bianchi, E.; Blaak, R.; Likos, C. Patchy colloids: State of the art and perspectives. Phys. Chem. Chem. Phys. 2011, 13, 6397–6410. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Palis, H.; Mérindol, R.; Majimel, J.; Ravaine, S.; Duguet, E. Colloidal molecules and patchy particles: Complementary concepts, synthesis and self-assembly. Chem. Soc. Rev. 2020, 49, 1955–1976. [Google Scholar] [CrossRef]
- Mossa, S.; Sciortino, F.; Tartaglia, P.; Zaccarelli, Z. Ground-State Clusters for Short-Range Attractive and Long-Range Repulsive Potentials. Langmuir 2004, 20, 10756–10763. [Google Scholar] [CrossRef]
- Campbell, A.I.; Anderson, V.J.; van Duijneveldt, J.S.; Bartlett, P. Dynamical Arrest in Attractive Colloids: The Effect of Long-Range Repulsion. Phys. Rev. Lett. 2005, 94, 208301. [Google Scholar] [CrossRef]
- Sciortino, F.; Tartaglia, P.; Zaccarelli, E. One-Dimensional Cluster Growth and Branching Gels in Colloidal Systems with Short-Range Depletion Attraction and Screened Electrostatic Repulsion. J. Phys. Chem. B 2005, 109, 21942–21953. [Google Scholar] [CrossRef]
- Malins, A.; Williams, S.R.; Eggers, J.; Tanaka, H.; Royall, C.P. The effect of inter-cluster interactions on the structure of colloidal clusters. J. Non-Cryst. Solids 2011, 357, 760–766. [Google Scholar] [CrossRef][Green Version]
- Reese, C.E.; Guerrero, C.D.; Weissman, J.M.; Lee, K.; Asher, S.A. Synthesis of Highly Charged, Monodisperse Polystyrene Colloidal Particles for the Fabrication of Photonic Crystals. J. Colloid Interface Sci. 2000, 232, 76–80. [Google Scholar] [CrossRef]
- Shell, M.S. The relative entropy is fundamental to multiscale and inverse thermodynamic problems. J. Chem. Phys. 2008, 129, 144108. [Google Scholar] [CrossRef] [PubMed]
- Torquato, S. Inverse optimization techniques for targeted self-assembly. Soft Matter 2009, 5, 1157–1173. [Google Scholar] [CrossRef]
- Chaimovich, A.; Shell, M.S. Coarse-graining errors and numerical optimization using a relative entropy framework. J. Chem. Phys. 2011, 134, 094112. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bollinger, J.A.; Truskett, T.M. Inverse methods for material design. AIChE J. 2014, 60, 2732–2740. [Google Scholar] [CrossRef]
- Jadrich, R.B.; Bollinger, J.A.; Lindquist, B.A.; Truskett, T.M. Equilibrium cluster fluids: Pair interactions via inverse design. Soft Matter 2015, 11, 9342–9354. [Google Scholar] [CrossRef]
- Jadrich, R.B.; Lindquist, B.A.; Truskett, T.M. Probabilistic inverse design for self-assembling materials. J. Chem. Phys. 2017, 146, 184103. [Google Scholar] [CrossRef]
- Banerjee, D.; Lindquist, B.A.; Jadrich, R.B.; Truskett, T.M. Assembly of particle strings via isotropic potentials. J. Chem. Phys. 2019, 150, 124903. [Google Scholar] [CrossRef]
- Zhang, T.H.; Klok, J.; Tromp, R.H.; Groenewold, J.; Kegel, W.K. Non-equilibrium cluster states in colloids with competing interactions. Soft Matter 2012, 8, 667–672. [Google Scholar] [CrossRef]
- Klix, C.L.; Murata, K.; Tanaka, H.; Williams, S.; Malins, A.; Royall, C.P. Novel kinetic trapping in charged colloidal clusters due to self-induced surface charge organization. Sci. Rep. 2013, 3, 2072. [Google Scholar] [CrossRef]
- Jadrich, R.B.; Bollinger, J.A.; Johnston, K.P.; Truskett, T.M. Origin and detection of microstructural clustering in fluids with spatial-range competitive interactions. Phys. Rev. E 2015, 91, 042312. [Google Scholar] [CrossRef]
- Cruz, S.M.A.; Marques, J.M.C. A Detailed Study on the Low-Energy Structures of Charged Colloidal Clusters. J. Phys. Chem. B 2016, 120, 3455–3466. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.M.A.; Marques, J.M.C.; Pereira, F.B. Improved evolutionary algorithm for the global optimization of clusters with competing attractive and repulsive interactions. J. Chem. Phys. 2016, 145, 154109. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.M.A.; Marques, J.M.C. Low-energy structures of clusters modeled with competing repulsive and either long- or moderate short-range attractive interactions. Comput. Theor. Chem. 2017, 1107, 82–93. [Google Scholar] [CrossRef]
- Liu, Y.; Xi, Y. Colloidal systems with a short-range attraction and long-range repulsion: Phase diagrams, structures, and dynamics. Curr. Opin. Colloid Interface Sci. 2019, 39, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, S.; Lu, H.; Bäcklund, M.; Woodward, C.E.; Forsman, J. Polymer-Like Self-Assembled Structures from Particles with Isotropic Interactions: Dependence upon the Range of the Attraction. Langmuir 2021, 37, 6052–6061. [Google Scholar] [CrossRef]
- Ruiz-Franco, J.; Zaccarelli, E. On the Role of Competing Interactions in Charged Colloids with Short-Range Attraction. Annu. Rev. Condens. Matter Phys. 2021, 12, 51–70. [Google Scholar] [CrossRef]
- Zhang, T.H.; Groenewold, J.; Kegel, W.K. Observation of a microcrystalline gel in colloids with competing interactions. Phys. Chem. Chem. Phys. 2009, 11, 10827–10830. [Google Scholar] [CrossRef]
- Toledano, J.C.F.; Sciortino, F.; Zaccarelli, E. Colloidal systems with competing interactions: From an arrested repulsive cluster phase to a gel. Soft Matter 2009, 5, 2390–2398. [Google Scholar] [CrossRef]
- Pereira, F.B.; Marques, J.M.C.; Leitão, T.; Tavares, J. Analysis of Locality in Hybrid Evolutionary Cluster Optimization. In Proceedings of the 2006 IEEE Congress on Evolutionary Computation, Vancouver, BC, Canada, 16–21 July 2006; pp. 2270–2277. [Google Scholar]
- Pereira, F.B.; Marques, J.M.C.; Leitão, T.; Tavares, J. Designing Efficient Evolutionary Algorithms for Cluster Optimization: A Study on Locality. In Advances in Metaheuristics for Hard Optimization; Siarry, P., Michalewicz, Z., Eds.; Springer Natural Computing Series; Springer: Berlin, Germany, 2008; pp. 223–250. [Google Scholar]
- Pereira, F.B.; Marques, J.M.C. A Study on Diversity for Cluster Geometry Optimization. Evol. Intel. 2009, 2, 121–140. [Google Scholar] [CrossRef]
- Marques, J.M.C.; Pereira, F.B. An Evolutionary Algorithm for Global Minimum Search of Binary Atomic Clusters. Chem. Phys. Lett. 2010, 485, 211–216. [Google Scholar] [CrossRef]
- Morse, P.M. Diatomic molecules according to the wave mechanics. II. Vibrational levels. Phys. Rev. 1929, 34, 57–64. [Google Scholar] [CrossRef]
- Guimarães, M.N.; de Almeida, M.; Marques, J.M.C.; Prudente, F.V. A thermodynamic view on the microsolvation of ions by rare gas: Application to Li+ with argon. Phys. Chem. Chem. Phys. 2020, 22, 10882–10892. [Google Scholar] [CrossRef] [PubMed]
- Marinari, E.; Parisi, G. Simulated Tempering: A New Monte Carlo Scheme. Europhys. Lett. (EPL) 1992, 19, 451–458. [Google Scholar] [CrossRef]
- Koji, H.; Koji, N. Exchange Monte Carlo Method and Application to Spin Glass Simulations. J. Phys. Soc. Jpn. 1996, 65, 1604–1608. [Google Scholar] [CrossRef]
- Neirotti, J.P.; Calvo, F.; Freeman, D.L.; Doll, J.D. Phase changes in 38-atom Lennard-Jones clusters. I. A parallel tempering study in the canonical ensemble. J. Chem. Phys. 2000, 112, 10340–10349. [Google Scholar] [CrossRef]
- Calvo, F.; Neirotti, J.P.; Freeman, D.L.; Doll, J.D. Phase changes in 38-atom Lennard-Jones clusters. II. A parallel tempering study of equilibrium and dynamic properties in the molecular dynamics and microcanonical ensembles. J. Chem. Phys. 2000, 112, 10350–10357. [Google Scholar] [CrossRef]
- Royall, C.P.; Louis, A.A.; Tanaka, H. Measuring colloidal interactions with confocal microscopy. J. Chem. Phys. 2007, 127, 044507. [Google Scholar] [CrossRef]
- Taffs, J.; Malins, A.; Williams, S.R.; Royall, C.P. A structural comparison of models of colloid-polymer mixtures. J. Phys. Condens. Matter 2010, 22, 104119. [Google Scholar] [CrossRef]
- Okabe, T.; Kawata, M.; Okamoto, Y.; Mikami, M. Replica-exchange Monte Carlo method for the isobaric-isothermal ensemble. Chem. Phys. Lett. 2001, 335, 435–439. [Google Scholar] [CrossRef]
- Lingenheil, M.; Denschlag, R.; Mathias, G.; Tavan, P. Efficiency of exchange schemes in replica exchange. Chem. Phys. Lett. 2009, 478, 80–84. [Google Scholar] [CrossRef]
- Swendsen, R.H. How the maximum step size in Monte Carlo simulations should be adjusted. Phys. Procedia 2011, 15, 81–86. [Google Scholar] [CrossRef]
- Calvo, F. Coating Polycyclic Aromatic Hydrocarbon Cations with Helium Clusters: Snowballs and Slush. J. Phys. Chem. A 2015, 119, 5959–5970. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prudente, F.V.; Marques, J.M.C. Thermodynamic Signatures of Structural Transitions and Dissociation of Charged Colloidal Clusters: A Parallel Tempering Monte Carlo Study. Molecules 2022, 27, 2581. https://doi.org/10.3390/molecules27082581
Prudente FV, Marques JMC. Thermodynamic Signatures of Structural Transitions and Dissociation of Charged Colloidal Clusters: A Parallel Tempering Monte Carlo Study. Molecules. 2022; 27(8):2581. https://doi.org/10.3390/molecules27082581
Chicago/Turabian StylePrudente, Frederico V., and Jorge M. C. Marques. 2022. "Thermodynamic Signatures of Structural Transitions and Dissociation of Charged Colloidal Clusters: A Parallel Tempering Monte Carlo Study" Molecules 27, no. 8: 2581. https://doi.org/10.3390/molecules27082581
APA StylePrudente, F. V., & Marques, J. M. C. (2022). Thermodynamic Signatures of Structural Transitions and Dissociation of Charged Colloidal Clusters: A Parallel Tempering Monte Carlo Study. Molecules, 27(8), 2581. https://doi.org/10.3390/molecules27082581